

A chemical approach enables the rapid mapping of CoA metabolites in complex biological systems.
Abstract
Metabolic engineering uses enzymes to produce small molecules with industrial, pharmaceutical, and energy applications. However, efforts to optimize enzymatic pathways for commercial production are limited by the throughput of assays for quantifying metabolic intermediates and end products. We developed a multiplexed method for profiling CoA-dependent pathways that uses a cysteine-terminated peptide to covalently capture CoA-bound metabolites. Captured metabolites are then rapidly separated from the complex mixture by immobilization onto arrays of self-assembled monolayers and directly quantified by SAMDI mass spectrometry. We demonstrate the throughput of the assay by characterizing the cell-free synthesis of HMG-CoA, a key intermediate in the biosynthesis of isoprenoids, collecting over 10,000 individual spectra to map more than 800 unique reaction conditions. We anticipate that our rapid and robust analytical method will accelerate efforts to engineer metabolic pathways.
INTRODUCTION
Biosynthetic pathways offer new opportunities for producing molecules with medical, energy, and industrial applications at commercial scales (1). Coenzyme A (CoA) is a central molecule in metabolism and is required in more than 100 distinct reactions, serving as an obligate cofactor for 4% of known enzymes (2). Acetyl-CoA (Ac-CoA) serves as the universal precursor for numerous biosynthetic pathways including isoprenoids, fatty acids/alkanes, polyketides, bioplastics, and a number of biofuels (3). In yeast alone, Ac-CoA is involved in 34 different known metabolic reactions (1). A diverse range of bioengineering efforts have used CoA-dependent pathways; these include the semisynthetic production of the antimalarial drug artemisinin (4), the biosynthesis of methyl-butenol as a potential biofuel (5), and the bioengineering of brewer’s yeast to produce the hop flavor compounds linalool and geraniol (6). The importance of CoA metabolism has motivated the engineering of organisms such as Saccharomyces cerevisiae as “chassis” strains for overproducing Ac-CoA (7) or other short-chain acyl-CoA biosynthetic precursors (8).
While several pathways have been successfully developed at the commercial scale (9), engineering cells is a time-consuming and nonintuitive process that can require hundreds of person years to tune enzymatic pathways (10). Many approaches use an iterative design-build-test cycle to test numerous “weak hypotheses” and explore a large parameter space in which the underlying biology is not completely understood or where the complexity of the system does not yet permit rational design. Recent advances in DNA synthesis and assembly highlight new capabilities to design and build biological systems (11, 12) while emphasizing the need for additional methods to rapidly test biosynthetic reaction systems. For CoA-bound metabolites, the current state-of-the-art detection methods rely on column-based separation, followed by mass spectrometry (13–17). These methods are highly sensitive (picomol of analyte per sample) (18) and can be adapted to measure multiple acyl-CoA compounds in a single analysis (19) but have low throughput, generally requiring greater than 15 min per analysis. Colorimetric screens and intracellular metabolite-sensing circuits offer increased throughput but are typically specific for a single CoA-bound molecule and require laborious redesign to quantitate new target molecules (20).
In this work, we developed an assay based on SAMDI-MS [self-assembled monolayers for matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry] that is capable of rapidly detecting CoA-bound metabolites in high throughput. In SAMDI, biochemical assays are performed on self-assembled monolayers (SAMs) of alkanethiolates on gold, and the resulting immobilized reaction products are directly quantified using MALDI-TOF MS (21). Previously, we have shown that SAMDI can be used as a general assay platform to profile a wide range of enzyme activities in situ and in complex lysates (22–26). Here, we demonstrate how SAMDI can characterize biosynthetic pathways by immobilizing a specific class of metabolites to the surface and obtaining mass spectra to quantitate each metabolite. Specifically, we use selective bioconjugation to capture all acyl intermediates on CoA, followed by quantitative analysis by SAMDI-MS. The use of chemically defined monolayers enables the rapid isolation of all acyl-CoA species from complex lysates while simultaneously serving as the platform for detection.
An important benefit of SAMDI is the compatibility with microtiter formats and laboratory automation to allow evaluation of tens of thousands of reactions per day (22, 24, 27). Here, we use this robust and high-throughput method to characterize a cell-free reaction system engineered to produce hydroxymethylglutaryl-CoA (HMG-CoA), the biosynthetic precursor to mevalonate and isoprenoid metabolites. Cell-free engineering is a powerful approach for accelerating the design and construction of biosynthetic pathways and has the further benefit that it is compatible with assay automation (28). In cell-free reactions, biosynthetic pathways are reconstituted in crude lysates, rather than living cells. These cell-free systems are liberated from the regulatory processes that, in vivo, support cell viability and growth, allowing for the design of synthetic pathways that may be difficult in living cells (28–30). We demonstrate the combination of cell-free reactions and high-throughput characterization with SAMDI to perform thousands of biosynthetic reactions in a single experiment. This strategy, applied here for the optimization of HMG-CoA biosynthesis, greatly increases the speed at which a large reaction space can be mapped for complex networks of enzymes.
RESULTS
Developing a selective assay for CoA metabolites
We identified the thioester functional group, through which the CoA cofactor binds metabolites, as a unique chemical handle for biospecific capture. Thioesters have been used extensively to ligate peptides via the native chemical ligation (NCL) chemistry pioneered by Kent and co-workers (31). We recognized that this ligation strategy could also be used to capture acyl moieties from CoA with the specificity required to operate in complex cell lysates. A peptide with an N-terminal cysteine was used for covalent capture, immobilization, and detection of CoA metabolites. When the capture peptide is added to reactions containing CoA-bound species, it readily scavenges the acyl groups from CoA by a trans-thioesterification reaction followed by a rapid internal rearrangement to irreversibly transfer the captured analyte to the N-terminal amine of the peptide. We then applied the reaction mixture to a monolayer presenting a maleimide group, where the free thiol of the cysteine undergoes immobilization to the monolayer and the lysate can be removed by rinsing (Fig. 1A). The resulting monolayers are covalently tethered to all of the intermediates and products from the CoA biosynthesis and can be directly analyzed by SAMDI-MS.
Fig. 1. A generalized approach for capturing CoA-bound metabolites.
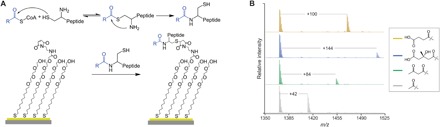
(A) A peptide with an N-terminal cysteine readily reacts with the thioester of CoA metabolites, creating a stable amide bond with the acyl group. After capture, the thiol of the peptide can then be used to immobilize the analyte and peptide onto a maleimide-presenting monolayer. (B) Five-hundred micromolar CoA conjugates of acetyl, acetoacetyl, HMG, and succinyl was reacted with the peptide CAK(Me3)SA. The resulting SAMDI spectra show that all analytes can be efficiently detected. m/z, mass/charge ratio.
NCL has previously seen limited use for in vivo bioconjugation, with this chemical strategy used to label proteins containing N-terminal cysteines with fluorophores that have thioester handles (32). However, the possibility of cross-reactivity of NCL chemistry with biological thioesters, such as acyl-CoA species and intermediates of fatty acid synthesis, has been cited as a limitation of the chemistry for use as a bioconjugation technique (33). In this work, we take advantage of this potential shortcoming, using it to develop an assay targeting acyl-CoA metabolites.
We first validated this chemical strategy for capture and detection using a variety of commercially available, biologically relevant, CoA compounds. These CoA species were each reacted with the capture peptide, of sequence CAK(Me)3SA, in phosphate-buffered saline at pH 7 and 40°C, at concentrations of 500 μM CoA species and 1 mM capture peptide for 2 hours. The reactions were then applied to SAMs presenting maleimide at a density of 20% against a background of tri(ethylene glycol) groups and analyzed by SAMDI-MS to reveal peaks that correspond to capture of the acyl groups (Fig. 1B). For the purposes of this assay, the capture peptide requires an N-terminal cysteine, and the remaining sequence does not strongly affect the reaction. The peptide used in this work was chosen because of its high ionization efficiency, a characteristic that is important for maximizing the limit of detection in mass spectrometry. We characterized the kinetics for the reaction of the peptide with both Ac-CoA and HMG-CoA (fig. S1). Both species were found to have similar second-order rate constants, which fall between 0.10 and 0.04 M−1 s−1 over the pH range of 7.5 to 6.5, where the rate increases with increased pH.
Characterization of pathway kinetics and flux
We applied this strategy to analyze the cell-free biosynthesis of HMG-CoA (Fig. 2A)—a biosynthetic precursor to isoprenoids, which represent a diverse and useful class of molecules with applications in pharmaceuticals, flavorings, fragrances, commodity chemicals, and biofuels (4, 34–37). This pathway proceeds from intermediates Ac-CoA and acetoacetyl-CoA (AA-CoA) to HMG-CoA. We assembled the biosynthetic reactions by mixing crude Escherichia coli lysates that separately overexpressed the necessary Ac-CoA acetyltransferase and HMG-CoA synthase enzymes. The cell-free reactions also allowed precise control of the concentrations of glucose substrate, buffer, salts, and cofactors (38). All cell-free reactions were done in standard 384-well microtiter plates and were quenched by addition of formic acid. For capture of CoA metabolites, the pH was adjusted to 6.5 and the capture peptide was introduced. After capture, the reaction mixtures were diluted several folds and applied to arrays of monolayers presenting maleimide functional groups using liquid-handling robotics, where captured analytes were selectively isolated from the reaction mixture (Fig. 2B). We performed the analysis using SAMDI arrays of 1536 spots, where each spot is 1 mm in diameter. The reactions from the 384-well plates were each spotted in quadruplicate so that four replicate spectra could be averaged for each reaction. For each spot, we used 0.5 μl of the reaction mixture for immobilization.
Fig. 2. A cell-free metabolic pathway from glucose to isoprenoid intermediate HMG-CoA.
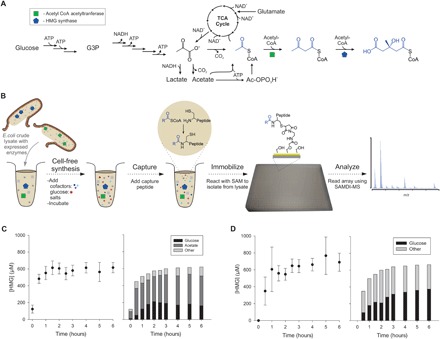
(A) Cellular overexpression of Ac-CoA acetyltransferase and HMG-CoA synthase and subsequent lysis produces enzyme-enriched lysates, which can convert glucose to HMG-CoA, as well as acetate and glutamate. The pathway includes Ac-CoA and AA-CoA intermediates. SAMDI can capture metabolites from crude lysates. NADH, reduced form of NAD+. (B) Cell-free reactions containing lysates, cofactors, salts, and substrate were performed in standard 384-well microtiter plates. Reactions were then quenched, and any CoA-bound products were captured by incubation with the sensor peptide. Complex reaction mixtures were printed onto monolayer arrays using liquid-handling robotics for isolation and detection. HMG-CoA accumulates in the reaction over time for both (C) acetate salts and (D) glutamate salts. The dominant carbon source used for HMG production was determined by feeding cell-free reactions 13C-labeled glucose and 13C-labeled acetate and monitoring isotopic incorporation into the HMG product. The concentration of cofactors ATP, NAD+, and CoA was set to 1 mM each.
We first characterized the kinetics of HMG-CoA production in these cell-free reaction systems with cofactors adenosine 5′-triphosphate (ATP), nicotinamide adenine dinucleotide (NAD+), and CoA each supplemented at 1 mM (Fig. 2, C and D). We measured the rate of HMG-CoA production in two systems containing either acetate or glutamate salts, commonly used in cell-free reactions as necessary counterions for K+, NH4+, and Mg2+ ions (39). Both systems reached similar yields of just over 600 μM HMG-CoA, although HMG-CoA accumulated more rapidly in the acetate salts, where it was detectible at the earliest reaction times. The same reaction in the glutamate salts required approximately 30 min of reaction time to observe HMG-CoA.
While glucose is the intended primary carbon source for metabolism, acetate and glutamate act as secondary sources of carbon and can also be metabolized by enzyme pathways native to E.coli and converted to HMG-CoA (Fig. 2A) (40). We explored the utilization of these different carbon sources by repeating the experiments with 13C6-glucose and quantifying the isotopic incorporation into the final product. Similarly, we used 13C2-acetate to monitor metabolic flux from acetate. HMG-CoA is composed of three, two-carbon acetyl subunits such that four possible label states (0, +2, +4, or +6 mass units) are observed. We quantified the relative abundance of each from the mass spectra and then used these values to determine the fraction of labeled carbon, f C13, and the corresponding fraction of unlabeled carbon, f C12, in the HMG-CoA produced (fig. S2).
We further used the label incorporation to calculate the concentration of HMG-CoA produced. A known amount of commercially available, unlabeled HMG-CoA was spiked into each capture reaction and the fraction of label incorporation determined before and after. The change in measured label content can be used to calculate the original concentration of HMG-CoA, as described in Eq. 1, where represents the fraction of unlabeled carbon before the spike and represents the fraction of unlabeled carbon after the spike. From these values, the initial concentration of HMG-CoA, [HMG]i, can be determined.
| (1) |
This quantification method is robust to any variations in signal intensity across spectra, as it relies on the peak splitting pattern for the HMG product, and enabled the tracking of both HMG-CoA concentration and carbon usage simultaneously. The cell-free reactions performed here contained 200 mM glucose and 150 mM either acetate or glutamate. We observed that acetate was the primary source of carbon in the acetate system, and for early time points, it was the source for the bulk of the HMG-CoA produced (Fig. 2C). With longer reaction times, glucose utilization increased and leveled off at 30% of the incorporated carbon. In the glutamate system, glutamate was also a major source of carbon, but the carbon fraction from glucose reached the higher level of 55% after 6 hours (Fig. 2, C and D). These results show that acetate metabolism and glutamate metabolism are active and that both are significant carbon sources and not just passive components of the cell-free system, consistent with previous reports (40).
Identification of an additional metabolite
It is difficult to independently and quantitatively control the concentrations of cofactors in cell-based metabolic engineering. The cell-free approach used here enables straightforward, independent optimization of buffer composition, cofactor concentrations, and carbon sources to achieve the greatest yield of biosynthetic targets. In particular, supplementation of ATP, NAD+, and CoA can markedly influence cell-free metabolism (38). As described below, we mapped the dependency of HMG-CoA synthesis over a wide range of these cofactors. However, during initial experiments, we observed an unidentified product bound to the peptide sensor that did not correspond to any of the expected CoA metabolites. This unidentified product gave a mass shift of +72 Da, and this peak increased by 3 mass units when the lysates were supplemented with 13C6-glucose. This mass and carbon content was consistent with a lactate-functionalized peptide, but no enzyme in E.coli is known to produce lactyl-CoA.
We suspected that this unidentified product was derived from glycolysis and identified the source as glyceraldehyde-3-phosphate (G3P) by biochemically reconstituting the initial steps of glycolysis using five purified enzymes (Fig. 3). When the pathway was reconstructed stepwise and fed glucose, this species appeared only when all of the enzymes necessary to produce G3P were present. We hypothesize that the peak derives from a pyruvaldehyde intermediate, which is formed from G3P and dihydroxyacetone phosphate (DHAP) through elimination of the phosphate and which is a known toxic byproduct of glycolysis (41). The enzyme that interconverts G3P and DHAP, triosephosphate isomerase, has also been shown to catalyze the formation of pyruvaldehyde from these species (42–44). Pyruvaldehyde reacts with thiols, through a thiohemiacetal rearrangement, to produce a lactyl-thioester (Fig. 4A) (45, 46). In this case, the resulting lactyl-thioester can further react to yield an N-lactyl-peptide.
Fig. 3. G3P can be detected.
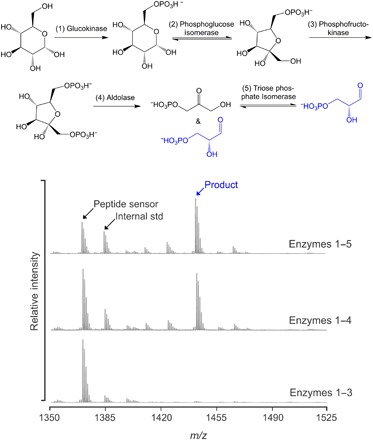
The initial steps of glycolysis were reconstituted in situ using purified enzymes and fed glucose. When sufficient enzymes are present, G3P is captured and detected.
Fig. 4. Derivatives of non–CoA bound G3P were captured by the sensor peptide.
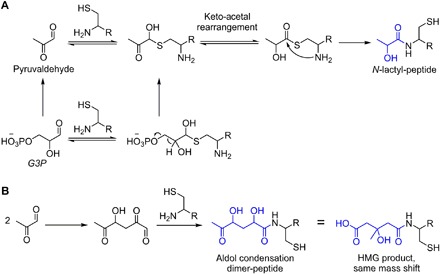
(A) A possible mechanism for detection of G3P as N-lactyl-peptide is via pyruvaldehyde, which is known to be generated from G3P and can undergo rearrangement with thiols to form lactyl-thioesters. (B) Pyruvaldehyde can also undergo pH-dependent aldol condensation to yield a six-carbon species that overlaps in mass with the desired HMG product.
We also observed a peak corresponding to the aldol condensation product of two molecules of pyruvaldehyde (Fig. 4B). This dimeric adduct results in a product with the same mass shift as HMG-CoA. However, it can be differentiated from HMG because it derives from two, three-carbon subunits and has a characteristic +3, +6 labeling pattern when isolated from lysates that are supplemented with 13C6 glucose. We found that this aldol condensation product was only observed at a pH above 7. Therefore, to exclude capture of this adduct that can interfere with HMG-CoA detection, we performed all capture reactions at pH 6.5. The thioester rearrangement still proceeds at this pH, but the aldol condensation does not occur.
Mapping metabolite levels and pathway flux
To optimize HMG-CoA production and better understand the effect of cofactor concentrations on the reaction network, we performed a high-throughput experiment that tested 768 unique cofactor conditions—where the concentrations of NAD, CoA, and ATP were systematically varied. For each condition, three replicate cell-free reactions were performed, and for each reaction, four spectra were collected and averaged, totaling more than 9000 individual spectra (Fig. 5). All cell-free reactions were allowed to proceed for 2 hours, quenched, reacted with the capture peptide, and immobilized as described above. Once immobilized, each 1536-spot array can be read by MALDI-TOF MS in just 50 min. We did not observe AA-CoA under any conditions tested. In a separate control, we supplemented reactions with commercial AA-CoA and found that it was readily converted back to Ac-CoA, suggesting that the Ac-CoA acetyltransferase favors the reverse reaction and so no significant amount of AA-CoA accumulates (fig. S3). For each spectrum, we determined the percent conversion for the three primary observed species (HMG-CoA, Ac-CoA, and G3P) using the integrated areas under the peaks (AUPs) for the unreacted capture peptide and the products. Below, Eq. 2 shows how this calculation was done for HMG, and analogous calculation was also performed for acetyl and G3P products.
| (2) |
Fig. 5. Parallel measurement of metabolites.
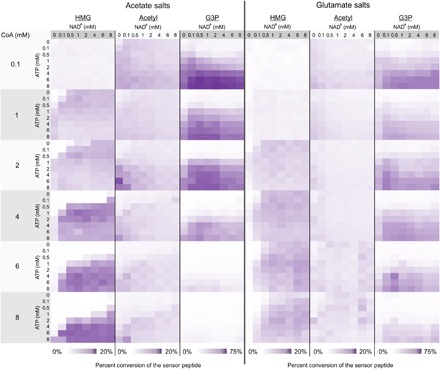
Ac-CoA, HMG-CoA, and G3P production was profiled in 768 unique cofactor conditions. Each product was quantified by calculating percent conversion relative to the unreacted peptide sensor using the area under the curve for each species. All reactions proceeded for 2 hours at 37°C.
These high-throughput experiments revealed several notable trends (Fig. 5). First, when low concentrations of CoA and high concentrations of ATP were used, G3P dominated as the major product. This is perhaps not unexpected as low CoA limits the total amount of HMG-CoA the system is capable of synthesizing, and the high ATP accelerates the early, pay-in phase of glycolysis. Second, as the concentration of CoA was increased, the amount of G3P decreased; this trade-off was much more significant when reactions were performed in the system using acetate. Thus, G3P was prominent under conditions favoring early glycolysis but where metabolic flux was unable to proceed to Ac-CoA.
We analyzed the same reaction dataset to monitor yield of and metabolic flux to HMG-CoA (Fig. 6A). As described earlier, we used 13C-labeled glucose to monitor metabolic flux from glucose across the array. For both acetate and glutamate systems, we identified the three reactions that produced the highest concentration of HMG-CoA among the set of 768 reactions. The averages of these highest yielding reactions were 6.4 ± 0.5 and 2.6 ± 0.1 mM for the acetate and glutamate systems, respectively. Across the array, the AUP for the HMG-CoA product was normalized to an internal standard present at a constant concentration across all reactions to control for variations in ionization across spectra. For this standard, we used the peptide Ac-SK(Me)3GGC, which has similar ionization efficiency to the capture peptide but lacks an N-terminal cysteine and does not overlap in mass with any species of interest. We explored the limit of detection by performing reactions with known quantities of HMG-CoA under reaction conditions identical to those used for the array (fig. S4). Under these conditions, HMG-CoA was detectible at concentrations as low as 5 μM, suggesting that the observed concentration range for the array spans roughly three orders of magnitude.
Fig. 6. HMG-CoA concentration and carbon source shifts in response to cofactor conditions.
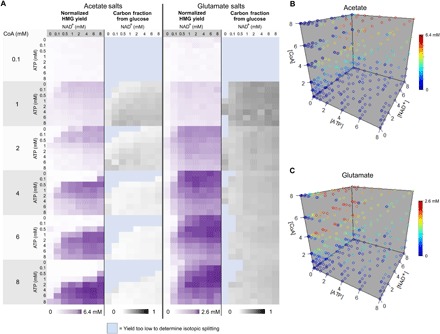
(A) To analyze HMG-CoA yield across all conditions, the dataset was normalized to an internal standard, a peptide of similar sequence to the sensor without an N-terminal Cys, present at a constant concentration across all reactions. 13C-labeled glucose was used to concurrently monitor the fraction of HMG-CoA product coming from glucose. HMG production was also visualized as four-dimensional plots for both (B) acetate and (C) glutamate systems. For the three highest yielding conditions in each system, average [HMG] was determined. In these plots, each point represents a specific concentration condition of cofactors with [ATP], [NAD+], [CoA] on the x, y, and z axes, respectively. Color of each point represents yield with the highest yield represented by red. From acetate to glutamate, the red region shifts from high CoA and high ATP to moderate CoA and low ATP.
Metabolic flux and HMG-CoA production complexly interplayed with cofactor concentrations, and we observed significantly different reaction profiles for the systems using acetate and glutamate salts. For the acetate system, higher CoA concentrations improved HMG-CoA titers but also required an associated increase in ATP concentration to achieve this maximal concentration. At low ATP concentrations, increasing available CoA inhibited HMG-CoA production (Fig. 6B). Conversely, over the 2-hour reaction time, the glutamate system reached peak HMG-CoA production at 4 mM CoA. Increasing CoA concentration further to 6 and 8 mM did not increase the production of HMG-CoA. However, this shifted the region where highest product concentrations were observed, requiring a shift to higher NAD+ concentration and somewhat higher ATP concentration to maintain the same titers (Fig. 6C).
When product concentration is highest, the fraction of carbon derived from glucose accounted for 5% of the product in the acetate salts and 22% in the glutamate salts (Fig. 6A). As shown in Fig. 3, we see significant incorporation of nonglucose carbon (i.e., acetate and glutamate) in the final product for all cofactor conditions tested. At high CoA concentrations, the glucose incorporation decreased. Counterintuitively, in the acetate system, the best cofactor conditions limit glycolytic flux in favor of optimizing the four-step reaction from acetate to HMG-CoA exclusively. These results suggest that multiple substrates are an important consideration when optimizing cofactors for crude-lysate cell-free metabolic engineering.
DISCUSSION
Nonintuitive relationships underlie the optimization of complex biological reaction networks. In many cases, understanding the complex interdependency between system variables can be ascertained only by methodical testing of a large number of reaction conditions. In this work, we describe the design of an assay that enables the high-throughput analysis of CoA using metabolic pathways. This assay combines a general strategy for biospecific capture of CoA-bound metabolites with simple purification by covalent immobilization onto SAMs that serve as the platform for detection by SAMDI-MS. We expect this strategy to be applicable to a wide range of CoA anchored metabolites with the only limitation being metabolites present at large concentration disparities, 100-fold or more, may become difficult to detect. In this work, we applied this assay to the characterization of a cell-free reaction system for the biosynthesis of HMG-CoA, demonstrating the ability to perform and analyze thousands of individual biosynthetic reactions and to map the reaction space for this complex system with respect to multiple variables.
This assay represents a new, generalizable method for understanding and optimizing biosynthetic pathways. Technologies for rapid characterization can synergize with existing high capacity methods for design and construction of biological systems; these projects can also provide large experimental datasets to inform development of improved design tools. The method described can track multiple metabolites simultaneously and unambiguously using mass spectrometry and is compatible with tracking of metabolic flux using isotopic labeling. When compared to conventional column-based analysis of CoA metabolites, this strategy offers an orders-of-magnitude increase in scale and speed of analysis, which should enable high-throughput metabolic engineering and decrease the time needed to develop high-yielding biosynthetic systems.
MATERIALS AND METHODS
Preparation of enzyme-enriched lysates
Two strains of E. coli BL21(DE3) containing plasmids pETBCS-ACAT(Eco) and pETBCS-HMGS(Sce) were grown in a 1-liter fermenter at 37°C (250 rpm) at constant pH 6.95 in rich medium [glucose (18 g liter−1), yeast extract (16 g liter−1), tryptone (10 g liter−1), NaCl (5 g liter−1), potassium phosphate dibasic (K2HPO4; 7 g liter−1), and potassium phosphate monobasic (KH2PO4; 3 g liter−1)] containing carbenicillin (100 μg ml−1; IBI Scientific, Peosta, IA). After induction with 0.1 mM isopropyl-β-d-thiogalactopyranoside at OD600 (optical density at 600 nm) of 0.6, growth continued for 4 hours at 30°C until harvest by centrifugation. See (38) for plasmid sequences and expression characterization. E. coli cells were lysed, and crude lysates were generated using methods previously described (38).
Cell-free reactions
Cell-free reactions were performed at a volume of 15 μl in 384-well plates and incubated at 37°C. The standard reaction contained the following components: 200 mM glucose, 100 mM bis-tris buffer, acetate or glutamate salts (8 mM magnesium, 10 mM ammonium, and 134 mM potassium), and 10 mM potassium phosphate [K2HPO4 (pH 7.2)]. Unless specified, reactions also included 1 mM NAD+, 1 mM ATP, and 1 mM CoA (38). All reagents and chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Two lysates, enriched in ACAT(Eco) and HMGS(Sce), were mixed together at a total protein concentration of 5 mg ml−1 each. Reactions were quenched by precipitating proteins using 2.25 μl of 10% formic acid and immediately stored at −80°C until peptide incubation. Acetic acid-2-13C was neutralized to acetate (pH 7.00) by titration with 45% (w/w) potassium hydroxide and diluted to 5 M stock concentration.
Preparation of monolayer arrays
Array plates were prepared by patterning 1536 gold spots, in a standard microtiter format, on steel plates using electron beam metal evaporation to deposit 5 nM titanium, followed by 30 nM gold. These plates were soaked in a solution of disulfide molecules in ethanol for 24 to 48 hours to form a self-assembled monolayer on the gold surfaces. The solution consisted of a mixture of tri(ethylene glycol)-alkanethiol (EG3-alkanethiol) disulfide and a mixed disulfide of EG3-alkanethiol and maleimide-terminated EG3-alkanethiol (47). The two disulfide molecules were present in a stoichiometric ratio to yield a 20% maleimide surface density, with an overall concentration of 1 mM. After the formation of this primary monolayer on the gold spots, the plates were rinsed with ethanol and dried. The array plates were then soaked in a 10 mM hexadecylphosphonic acid solution in ethanol for 15 min. The phosphonic acid terminated hydrocarbon molecules react selectively with the steel background giving it hydrophobic properties that aid with delivery of submicroliter reaction volumes to the spots.
Synthesis of peptide reagents
The peptide of the sequence CAK(Me)3SA was used in this work for capture of acyl-CoA species. This sequence was chosen after testing a small set of potential 5-mer sequences, all containing N-terminal cysteine residues. While the sequence identity did not strongly influence the reactivity toward CoA metabolites, it did affect the analysis by SAMDI-MS. We found the trimethyl-lysine residue to provide excellent ionization efficiency. Ionization efficiency is critical with respect to the assay’s limit of detection, determining how abundant a species must be for a peak to be observed. In addition, this sequence and all of its observed reaction products ionized as a single molecular ion peak, rather than a set of salt adducts commonly observed with MALDI-TOF MS. This greatly simplified the resulting spectra and their interpretation. These advantageous properties likely stem from the enforced positive charge provided by the trimethyl-lysine residue. The peptide Ac-SK(Me)3GGC was used as an internal standard for normalization of signal across reactions, as it has similar sequence and ionization efficiency to the capture peptide but is mass-resolved from all reaction species and lacks an N-terminal cysteine. Standard Fmoc (9-fluorenylmethoxycarbonyl) solid phase peptide synthesis on Rink amide resin was used to synthesize both peptides. To introduce the non-natural trimethyl-lysine residue, Fmoc-Lys(Me)3Cl was purchased from Novabiochem and used along with standard amino acid coupling conditions.
Capture of CoA-bound moieties in lysates
After completion, cell-free reactions were quenched with formic acid to denature the proteins and stop the reactions. The reactions, in 384-well plates, were centrifuged at 3500g for 15 min to pellet any precipitated protein. For the acyl-CoA capture reactions, 3 μl of this cell-free reaction was transferred to a new 384-well plate, and the following species were added, bringing the final reaction volume to 8 μl with the final concentrations as follows: 100 mM phosphate buffer at pH 6.5, 40 mM EDTA, 0.9 mM capture peptide, and 0.1 mM normalization peptide. The well plates were sealed, and the reaction mixtures were incubated at 42°C for 3 hours. It is important to choose and appropriate concentration of capture peptide. If the concentration is too high, the signal from unreacted sensor peptide may overwhelm the signal from any captured species. In this work, the total added peptide concentration was chosen to be 1 mM across all reactions, a reasonable concentration relative to the expected yield from the best cell-free reactions while also giving good dynamic range for detection of acyl-species under low-yield conditions.
Immobilization of captured products
The 8 μl of capture reactions in 384-well plates were diluted threefold to 24 μl with 100 mM phosphate buffer at pH 7.2. This serves to dilute the concentration of a peptide, which will immobilize efficiently at concentrations as low as 10 μM, and adjust the pH to an optimal range for the reaction with maleimide. A TECAN liquid-handling robot equipped with a 384-tipped head was used to print the reaction mixtures onto 1536-spot array surfaces, generating four replicates per reaction, at a volume of 0.5 μl per spot. The surfaces were placed in a humidified chamber and incubated at 37°C for 60 min to allow the cysteine-containing peptides to react with the maleimide-functionalized SAM. After reaction, the surfaces were rinsed with 1% SDS detergent, then rinsed with distilled water, and dried under a stream of nitrogen gas.
Analysis of reactions
Matrix of 2′,4′,6′-trihydroxyacetophenone (THAP) in acetonitrile (20 mg/ml) was applied directly to the 1536-spot surface. The matrix was allowed to dry, and each spot was analyzed by MALDI-TOF MS using an AB Sciex 5800 series instrument. Captured metabolites of the cell-free reactions were identified by their mass shifts relative to the unreacted peptide. Analysis of more than 10,000 spectra collected was performed using in-house, Python-based software package to automate the integration of peaks of interest from the mass spectra dataset. The software calculates the area under the curve at any mass/charge ratio value given to it over a user-defined half-width mass window. For this work, a half-width mass window of 0.2 mass units was used across all spectra analyzed. For every spectrum in this dataset, the following peaks were integrated: 1371 Da (capture peptide), 1385 Da (normalization peptide), 1413 Da (Ac-CoA reaction product), 1515 Da (HMG-CoA reaction product), and 1443 Da (pyruvaldehyde adduct). In addition, for each peak analyzed, regions immediately flanking the peak were integrated to determine a local spectral baseline, which was subtracted from the peak area. For isotopic labeling experiments, the area under the curve for various isotope peaks was also collected.
Supplementary Material
Acknowledgments
Funding: We would like to thank the Department of Energy (BER grant DE-SC0018249 to M.C.J.), the Air Force Research Laboratory Center of Excellence Grant (FA8650-15-2-5518 to M.M. and M.C.J.), the David and Lucile Packard Foundation (2011-37152 to M.C.J.), the Department of Defense, Defense Threat Reduction Agency (grant HDTRA1-15-1-0052 to M.M. and M.C.J.), and the Dreyfus Teacher-Scholar Program (to M.C.J.) for funding and support. Author contributions: All authors contributed to experimental design. P.T.O., Q.M.D., and A.K.M. performed experiments. P.T.O. and Q.M.D. analyzed the experimental data. P.T.O., M.M., Q.M.D., and M.C.J wrote the manuscript. M.M. and M.C.J. conceived and supervised the project. Competing interests: M.M. is the founder and chairman of SAMDI Tech Inc., which uses SAMDI-MS to assist clients in the pharmaceutical industry in early phase drug discovery. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw9180/DC1
Fig. S1. Second-order kinetics for the reaction of CoA metabolites with the peptide.
Fig. S2. Description of spectral analysis.
Fig. S3. The pathway intermediate, AA-CoA, is not observed.
Fig. S4. Limit of detection for HMG-CoA.
REFERENCES AND NOTES
- 1.Nielsen J., Keasling J. D., Engineering cellular metabolism. Cell 164, 1185–1197 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Leonardi R., Zhang Y.-M., Rock C. O., Jackowski S., Coenzyme A: Back in action. Prog. Lipid Res. 44, 125–153 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Krivoruchko A., Zhang Y., Siewers V., Chen Y., Nielsen J., Microbial acetyl-CoA metabolism and metabolic engineering. Metab. Eng. 28, 28–42 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Paddon C., Westfall P. J., Pitera D. J., Benjamin K., Fisher K., McPhee D., Leavell M. D., Tai A., Main A., Eng D., Polichuk D. R., Teoh K. H., Reed D. W., Treynor T., Lenihan J., Jiang H., Fleck M., Bajad S., Dang G., Dengrove D., Diola D., Dorin G., Ellens K. W., Fickes S., Galazzo J., Gaucher S. P., Geistlinger T., Henry R., Hepp M., Horning T., Iqbal T., Kizer L., Lieu B., Melis D., Moss N., Regentin R., Secrest S., Tsuruta H., Vazquez R., Westblade L. F., Xu L., Yu M., Zhang Y., Zhao L., Lievense J., Covello P. S., Keasling J. D., Reiling K. K., Renninger N. S., Newman J. D., High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 496, 528–532 (2013). [DOI] [PubMed] [Google Scholar]
- 5.George K. W., Thompson M. G., Kang A., Baidoo E., Wang G., Chan L. J. G., Adams P. D., Petzold C. J., Keasling J. D., Lee T. S., Metabolic engineering for the high-yield production of isoprenoid-based C5 alcohols in E. coli. Sci. Rep. 5, 11128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denby C. M., Li R. A., Vu V. T., Costello Z., Lin W., Chan L. J. G., Williams J., Donaldson B., Bamforth C. W., Petzold C. J., Scheller H. V., Martin H. G., Keasling J. D., Industrial brewing yeast engineered for the production of primary flavor determinants in hopped beer. Nat. Commun. 9, 965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lian J., Si T., Nair N. U., Zhao H., Design and construction of acetyl-CoA overproducing Saccharomyces cerevisiae strains. Metab. Eng. 24, 139–149 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Krink-Koutsoubelis N., Loechner A. C., Lechner A., Link H., Denby C. M., Vögeli B., Erb T. J., Yuzawa S., Jakociunas T., Katz L., Jensen M. K., Sourjik V., Keasling J. D., Engineered Production of short-chain acyl-coenzyme A esters in Saccharomyces cerevisiae. ACS Synth. Biol. 7, 1105–1115 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Nielsen J., Fussenegger M., Keasling J., Lee S. Y., Liao J. C., Prather K., Palsson B., Engineering synergy in biotechnology. Nat. Chem. Biol. 10, 319–322 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Keasling J. D., Synthetic biology and the development of tools for metabolic engineering. Metab. Eng. 14, 189–195 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Nielsen A. A. K., der B. S., Shin J., Vaidyanathan P., Paralanov V., Strychalski E. A., Ross D., Densmore D., Voigt C. A., Genetic circuit design automation. Science 352, aac7341 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Smanski M. J., Bhatia S., Zhao D., Park Y., Woodruff L. B. A., Giannoukos G., Ciulla D., Busby M., Calderon J., Nicol R., Gordon D. B., Densmore D., Voigt C. A., Functional optimization of gene clusters by combinatorial design and assembly. Nat. Biotechnol. 32, 1241–1249 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Magnes C., Suppan M., Pieber T. R., Moustafa T., Trauner M., Haemmerle G., Sinner F. M., Validated comprehensive analytical method for quantification of coenzyme A activated compounds in biological tissues by online solid-phase extraction LC/MS/MS. Anal. Chem. 80, 5736–5742 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Lu W., Clasquin M. F., Melamud E., Amador-Noguez D., Caudy A. A., Rabinowitz J. D., Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal. Chem. 82, 3212–3221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basu S. S., Mesaros C., Gelhaus S. L., Blair I. A., Stable isotope labeling by essential nutrients in cell culture for preparation of labeled coenzyme A and its thioesters. Anal. Chem. 83, 1363–1369 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmermann M., Thormann V., Sauer U., Zamboni N., Nontargeted profiling of coenzyme A thioesters in biological samples by tandem mass spectrometry. Anal. Chem. 85, 8284–8290 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Li Q., Zhang S., Berthiaume J. M., Simons B., Zhang G.-F., Novel approach in LC-MS/MS using MRM to generate a full profile of acyl-CoAs: Discovery of acyl-dephospho-CoAs. J. Lipid Res. 55, 592–602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang S., Sadilek M., Synovec R. E., Lidstrom M. E., Liquid chromatography–tandem quadrupole mass spectrometry and comprehensive two-dimensional gas chromatography–time-of-flight mass spectrometry measurement of targeted metabolites of Methylobacterium extorquens AM1 grown on two different carbon sources. J. Chromatogr. A 1216, 3280–3289 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X., Sadhukhan S., Sun S., Wagner G. R., Hirschey M. D., Qi L., Lin H., Locasale J. W., High-resolution metabolomics with Acyl-CoA profiling reveals widespread remodeling in response to diet. Mol. Cell. Proteomics 14, 1489–1500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang F., Carothers J. M., Keasling J. D., Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat. Biotechnol. 30, 354–359 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Gurard-Levin Z. A., Mrksich M., Combining self-assembled monolayers and mass spectrometry for applications in biochips. Annu. Rev. Anal. Chem. 1, 767–800 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Gurard-Levin Z. A., Scholle M. D., Eisenberg A. H., Mrksich M., High-throughput screening of small molecule libraries using SAMDI mass spectrometry. ACS Comb. Sci. 13, 347–350 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuo H.-Y., DeLuca T. A., Miller W. M., Mrksich M., Profiling deacetylase activities in cell lysates with peptide arrays and SAMDI mass spectrometry. Anal. Chem. 85, 10635–10642 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ban L., Pettit N., Li L., Stuparu A. D., Cai L., Chen W., Guan W., Han W., Wang P. G., Mrksich M., Discovery of glycosyltransferases using carbohydrate arrays and mass spectrometry. Nat. Chem. Biol. 8, 769–773 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson L. L., Berns E. J., Bugga P., George A. L. Jr., Mrksich M., Measuring Drug Metabolism Kinetics and Drug–Drug interactions using self-assembled monolayers for matrix-assisted laser desorption-ionization mass spectrometry. Anal. Chem. 88, 8604–8609 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Kightlinger W., Lin L., Rosztoczy M., Li W., DeLisa M. P., Mrksich M., Jewett M. C., Design of glycosylation sites by rapid synthesis and analysis of glycosyltransferases. Nat. Chem. Biol. 14, 627–635 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Patel K., Sherrill J., Mrksich M., Scholle M. D., Discovery of SIRT3 inhibitors using SAMDI mass spectrometry. J. Biomol. Screen. 20, 842–848 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Dudley Q. M., Karim A. S., Jewett M. C., Cell-free metabolic engineering: Biomanufacturing beyond the cell. Biotechnol. J. 10, 69–82 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hodgman C. E., Jewett M. C., Cell-free synthetic biology: Thinking outside the cell. Metab. Eng. 14, 261–269 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karim A. S., Jewett M. C., A cell-free framework for rapid biosynthetic pathway prototyping and enzyme discovery. Metab. Eng. 36, 116–126 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Dawson P. E., Muir T. W., Clark-Lewis I., Kent S. B., Synthesis of proteins by native chemical ligation. Science 266, 776–779 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Yeo D. S. Y., Srinivasan R., Uttamchandani M., Chen G. Y. J., Zhu Q., Yao S. Q., Cell-permeable small molecule probes for site-specific labeling of proteins. Chem. Commun. 0, 2870–2871 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Sletten E. M., Bertozzi C. R., Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 48, 6974–6998 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bohlmann J., Keeling C. I., Terpenoid biomaterials. Plant J. 54, 656–669 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Leavell M. D., McPhee D. J., Paddon C. J., Developing fermentative terpenoid production for commercial usage. Curr. Opin. Biotechnol. 37, 114–119 (2016). [DOI] [PubMed] [Google Scholar]
- 36.George K. W., Alonso-Gutierrez J., Keasling J. D., Lee T. S., Isoprenoid drugs, biofuels, and chemicals—Artemisinin, farnesene, and Beyond. Adv. Biochem. Eng. Biotechnol. 148, 355–389 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Li Y., Pfeifer B. A., Heterologous production of plant-derived isoprenoid products in microbes and the application of metabolic engineering and synthetic biology. Curr. Opin. Plant Biol. 19, 8–13 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Dudley Q. M., Anderson K. C., Jewett M. C., Cell-free mixing of Escherichia coli crude extracts to prototype and rationally engineer high-titer mevalonate synthesis. ACS Synth. Biol. 5, 1578–1588 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jewett M. C., Swartz J. R., Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol. Bioeng. 86, 19–26 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Jewett M. C., Calhoun K. A., Voloshin A., Wuu J. J., Swartz J. R., An integrated cell-free metabolic platform for protein production and synthetic biology. Mol. Syst. Biol. 4, 220 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allaman I., Bélanger M., Magistretti P. J., Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 9, 23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richard J. P., Kinetic parameters for the elimination reaction catalyzed by triosephosphate isomerase and an estimation of the reaction’s physiological significance. Biochemistry 30, 4581–4585 (1991). [DOI] [PubMed] [Google Scholar]
- 43.Iyengar R., Rose I. A., Concentration of activated intermediates of the fructose-1,6-bisphosphate aldolase and triosephosphate isomerase reactions. Biochemistry 20, 1223–1229 (1981). [DOI] [PubMed] [Google Scholar]
- 44.Richard J. P., Mechanism for the formation of methylglyoxal from triosephosphates. Biochem. Soc. Trans. 21, 549–553 (1993). [DOI] [PubMed] [Google Scholar]
- 45.Weber A. L., Formation of the thioester, N-acetyl, S-lactoylcysteine, by reaction of N-acetylcysteine with pyruvaldehyde in aqueous solution. J. Mol. Evol. 18, 354–359 (1982). [DOI] [PubMed] [Google Scholar]
- 46.Okuyama T., Komoguchi S., Fueno T., Reaction of thiols with phenylglyoxal to give thiol esters of mandelic acid. II. Intramolecular general-base catalysis and change in rate-determining step. J. Am. Chem. Soc. 104, 2582–2587 (1982). [Google Scholar]
- 47.Houseman B. T., Mrksich M., Towards quantitative assays with peptide chips: A surface engineering approach. Trends Biotechnol. 20, 279–281 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw9180/DC1
Fig. S1. Second-order kinetics for the reaction of CoA metabolites with the peptide.
Fig. S2. Description of spectral analysis.
Fig. S3. The pathway intermediate, AA-CoA, is not observed.
Fig. S4. Limit of detection for HMG-CoA.