

Abstract
Caffeine is commonly used in Dictyostelium to inhibit the synthesis of the chemoattractant cAMP and, therefore, its secretion and the autocrine stimulation of cells, in order to prevent its interference with the study of chemoattractant-induced responses. However, the mechanism through which caffeine inhibits cAMP synthesis in Dictyostelium has not been characterized. Here, we report the effects of caffeine on the cAMP chemoattractant signaling network. We found that caffeine inhibits phosphatidylinositol 3-kinase (PI3K) and mechanistic target of rapamycin complex 2 (mTORC2). Both PI3K and mTORC2 are essential for the chemoattractant-stimulated cAMP production, thereby providing a mechanism for the caffeine-mediated inhibition of cAMP synthesis. Our results also reveal that caffeine treatment of cells leads to an increase in cAMP-induced RasG and Rap1 activation, and inhibition of the PKA, cGMP, MyoII, and ERK1 responses. Finally, we observed that caffeine has opposite effects on F-actin and ERK2 depending on the assay and Dictyostelium strain used, respectively. Altogether, our findings reveal that caffeine considerably affects the cAMP-induced chemotactic signaling pathways in Dictyostelium, most likely acting through multiple targets that include PI3K and mTORC2.
Keywords: Dictyostelium, Chemotaxis, cAMP, Caffeine, PI3K, mTORC2
Introduction
Caffeine is a purine alkaloid that is lipid soluble, allowing it to easily cross biological membranes. In humans, caffeine acts as a non-competitive antagonist of adenosine receptors but also inhibits phosphodiesterases and several protein kinases of the phosphatidylinositol 3-kinase (PI3K)-related kinase (PIKK) family, including PI3K and mechanistic target of rapamycin (mTOR) [1, 2]. In yeasts, caffeine inhibits the mTOR complex 1 (mTORC1) [3].
In Dictyostelium, early studies of the role of cAMP in development lead to the identification of caffeine as a compound that inhibits cAMP synthesis, with a maximal effect at ~3 mM [4]. In Dictyostelium, cAMP acts both as an extracellular chemoattractant and an intracellular second messenger [5]. Caffeine was found to only marginally affect cAMP binding to its cell surface receptors but to efficiently inhibit adenylyl cyclase activity and cAMP production in an indirect and reversible fashion [4, 6]. Efforts at defining the mechanism through which caffeine inhibits cAMP synthesis in Dictyostelium lead to the suggestion that there are at least two different targets of caffeine, with at least one of them downstream from the heterotrimeric G protein Gα2, which couples to the main cAMP receptor, cAR1 [7–10]. Although the targets of caffeine in Dictyostelium remain unknown, caffeine continues to be widely used by Dictyostelium researchers to inhibit cAMP synthesis and, thereby, prevent the autocrine stimulation of cells in studies of cAMP chemoattractant signaling.
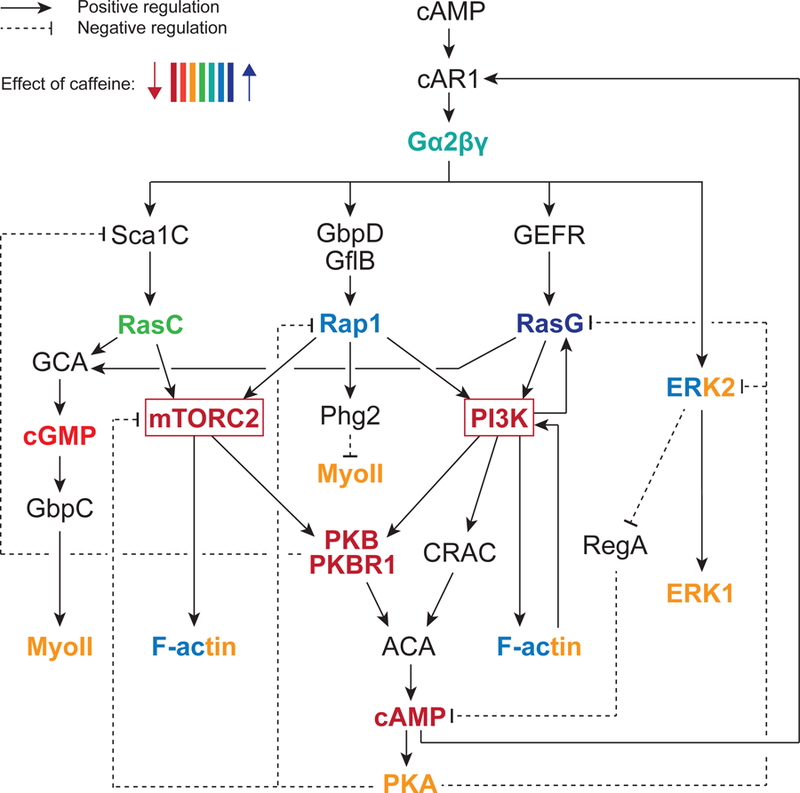
Much is now known about the cAMP chemoattractant signal transduction network in Dictyostelium, which includes central PI3K and mTOR complex 2 (mTORC2) pathways (Fig. 1) [11]. Interestingly, input from both of these pathways is necessary for adenylyl cyclase A (ACA) activation and cAMP production in response to extracellular cAMP stimulation [12–15]. In addition, we have found that many of the upstream components of the cAMP chemotactic signaling cascades are regulated by downstream kinases through negative feedback loops. We have shown that AKT/Protein Kinase B (PKB) and PKB-related kinase 1 (PKBR1) inhibit the upstream RasC-mTORC2 pathway; and that Protein Kinase A (PKA), which is activated by intracellular cAMP, negatively regulates the activity of upstream RasG, Rap1, and mTORC2 pathways (Fig. 1) [16, 17].
Fig. 1. cAMP chemoattractant signaling pathways in Dictyostelium.

cAMP binding to its chemoattractant receptor cAMP receptor 1 (cAR1), coupled to the heterotrimeric G protein Gα2βγ, induces the activation of multiple signaling cascades, including the RasC, Rap1, and RasG-mediated pathways. The chemotactic effectors phosphatidylinositol 3-kinase (PI3K) and mechanistic Target of Rapamycin Complex 2 (mTORC2) play central roles in the cAMP chemoattractant signaling network. The different signaling cascades interact through crosstalks and many of the components are also linked through feedback loops. ACA, adenylyl cyclase A. CRAC, cytosolic regulator of adenylyl cyclase. ERK1, extracellular regulated kinase 1. ERK2, extracellular-regulated kinase 2. GbpC, cGMP binding protein C. GbpD, cGMP binding protein D. GCA, guanylyl cyclase A. GEFR, guanine nucleotide exchange factor R. GflB, guanine nucleotide exchange factor-like protein B. MyoII, myosin II. F-actin, filamentous actin. Phg2, phagocytosis protein 2. PKA, protein kinase A. PKB, protein kinase B. PKBR1, PKB-related kinase 1. Sca1C, scaffold 1 complex.
Dictyostelium is a widely used experimental model for studying cell migration, chemotaxis, and chemoattractant signaling pathways, and much of what we know today about the signaling pathways and mechanisms implicated in the directed migration of eukaryotic cells was originally discovered in Dictyostelium [18]. However, since caffeine is widely used in Dictyostelium chemotaxis studies without knowledge of its mechanism of action, we wondered if caffeine could be changing the chemotactic responses and, thus, affect the interpretation of the data obtained in its presence. Therefore, the present study was undertaken to characterize the effects of caffeine on cAMP chemoattractant signal transduction in Dictyostelium. Our findings suggest that caffeine inhibits PI3K and mTORC2, which explains many of the effects of caffeine on chemoattractant signaling, including its inhibition of cAMP synthesis. In addition, our work shows that caffeine differentially affects the F-actin polymerization response and ERK2 activity depending on the assay or the Dictyostelium strain used, respectively.
Materials and methods
Reagents
cAMP sodium salt monohydrate, 2’-deoxyadenosine-5’-monophosphate (2’-deoxy-cAMP) disodium salt, caffeine powder, protein kinase A (PKA), and anti-Flag M2 were from Sigma-Aldrich (St. Louis, MO, USA). H2B was from Roche-Genentech (San Francisco, CA, USA) and Geneticin was purchased from Life Technologies (Grand Island, NY, USA). Torin2 was purchased from ApexBio (Houston, TX, USA). Phospho-p70 S6 kinase (Thr389; 1A5), phospho-Akt substrate (110B7), phospho-(Ser/Thr) PKA substrate, pan-phospho-PKC (zeta Thr410; 190D10), and phospho-p44/42 MAPK (Erk1/2; Thr202/Tyr204) antibodies were from Cell Signaling Technology (Danvers, MA, USA). Pan-Ras antibody (Ab-3; RAS10) was from Calbiochem/EMD Millipore (Billerica, MA, USA). ERK1 antibody (C16; sc-93) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). EMD Millipore/Novagen T7.Tag™ monoclonal antibody and antibody agarose, as well as EMD Millipore/Calbiochem PANSORBIN™ cells, were purchased from Fisher Scientific (Waltham, MA, USA). The Rap1 (directed against amino acids 169–182 of Dictyostelium Rap1) was custom-made by ProSci Incorporated (Poway, CA, USA). HRP-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA). PKB and PKBR1 antibodies, Lifeact-GFP, GFP-MyoII, and PHcrac-GFP constructs were gifts from Rick Firtel and were previously described [19–22]. Flag-tagged RasC construct was reported elsewhere [17]. T7-tagged Pianissimo (T7-Pia) was cloned by ligating AvrII digested restriction sites into the compatible SpeI site of the extrachromosomal vector pDM304, which was obtained from the Dicty Stock Center [23] (depositor: Douwe Veltman; GenBank Accession Number EU912539), using the following primers: T7-Pia Forward, AAGTGCCTAGGAAAAAATGGCATCAATGACAGGTGGTCAACAAATGGGTAG AATGACAAGTTCTGATAGTAGTGTAAATACTACATCG; Pia reverse, AAGTGCCTAGGTTAATTTAAATCATGATATGGATCAGATGAAAATATTGCAA CATC.
Cell culture and strains used
Dictyostelium cells were grown attached to substrate in axenic HL5 medium (ForMedium, Hunstanton, Norfolk, UK) at 22°C and transformants were generated by electroporation. Transformed cells were selected in 20 μg/ml Geneticin and confirmed by immunoblot. The wild-type strain used was AX3, and we also used AX2 where indicated. piaA null cells were generously provided by Peter Devreotes (Johns Hopkins University, Baltimore, MD) and described elsewhere [14]. For all assays, cells were developed by pulsing with 30 nM cAMP every 6 min for 5.5 h in 12 mM Na/K phosphate buffer (pH 6.1) at a confluency of 5X106 cells/ml in a shaking suspension culture. Prior assays, developed cells were washed twice with 12 mM Na/K phosphate buffer followed by 30 min incubation with or without 5 mM caffeine and stimulation with 1 μM cAMP.
Biochemical assays and immunoblots
F-actin measurements using Phalloidin staining, PKB and PKBR1 kinase assays, Ras-GTP and Rap1-GTP pull-down assays were performed as previously described [20–22, 24–26]. Phosphorylation of PKB and PKBR1, ERK1 and ERK2, PKB substrates, and PKA substrates from total cell lysates was detected by immunoblot. Activation loop (AL) phosphorylation of PKB (T278) and PKBR1 (T309) was detected using pan-phospho-PKC antibody diluted 1:1000. Hydrophobic motif (HM) phosphorylation of PKB (T435) and PKBR1 (T470) was detected using phospho-p70 S6 kinase antibody diluted 1:1000. Phospho-ERK1 and -ERK2 were detected using phospho-p44/42 MAPK antibody diluted 1:5000. PKB and PKA substrates phosphorylation was detected using phospho-Akt substrate and phospho-(Ser/Thr) PKA antibodies, respectively, diluted 1:2000 as described previously [17].
cAMP was measured using a protocol adapted from [27]. Briefly, developed cells were stimulated with 10 μM 2’-deoxy-cAMP for the time indicated, deproteinized using perchloric acid (1.75%) precipitation followed by neutralization with 50% bicarbonate. Samples were then incubated with 0.01 μCi [3H]cAMP and 5 ug of PKA in assay buffer (100 mM K2HPO4/KH2PO4, 10 mM EDTA, 2 mg/ml BSA, 3 mM NaN3, pH 7.0) for 2 h in ice water. Reactions were stopped by adding equal volume of 5% charcoal and incubating 1 min before centrifuging to sediment the charcoal. The supernatants were then transferred to scintillation vials for counting. Results were calculated as previously described [27].
cGMP was measured using a cGMP Enzyme Imunoassay Kit (Sigma-Aldrich). After cAMP stimulation, samples were deproteinized using trichloroacetic acid (10%) precipitation followed by neutralization with 50% bicarbonate. Samples were then diluted 1:10 with the assay buffer before cGMP was measured according to the manufacturer’s protocol.
To measure mTORC2 kinase activity in vitro, 1×108 developed T7-Pia/piaA null and piaA null cells cells were stimulated or not with 1 μM cAMP for 10 s and lysed in lysis buffer (final concentrations: 20 mM HEPES pH 7.5, 60 mM NaCl, 0.5 mM EDTA, 0.15% CHAPS, 5 mM Na-pyrophosphate, 5 mM β-glycerophosphate, 25 mM NaF, 0.25 mM Na-orthovanadate, 2.5 μg/ml aprotinin, and 2.5 μg/ml leupeptin) for 30 min at 4°C with shaking. Lysates were cleared by centrifugation at 20,000 x g 10 min. Supernatents were collected, proteins quantified and adjusted to have 2 μg/μl x 1 ml lysates. Lysates were then precleared with PANSORBIN™ cells 30 min at 4°C before incubation with T7.Tag™ antibody agarose for 4 h at 4°C with rocking. Immunoprecipiates were washed four times in lysis buffer followed by one wash in kinase buffer (25 mM HEPES, 0.1 M potassium acetate, 1 mM MgCl2). The purification of mTORC2 by T7-Pia pull-down was confirmed by mass spectrometry. Immunopurified mTORC2 was then incubated or not with 5 mM caffeine or 1 μM Torin2 for 30 min at 22°C before assaying the kinase activity of mTORC2 using piaA null cell lysates as substrate. This “substrate” piaA null cell lysates was produced by mechanical disruption of the cells using Polytron homogenization. For the kinase reaction, immunopurified mTORC2 was incubated in 15 μl kinase buffer containing 1.5 mM ATP and 30 μg piaA null cell lysates, for 15 min at 22°C with frequent mixing. Kinase reactions were stopped by adding 45 μl of SDS-PAGE sample buffer and the in vitro mTORC2-mediated phosphorylation of PKB and PKBR1 in the piaA null cell lysates was detected by immunoblot using the phospho-p70 S6 kinase antibody.
Imaging
Assessment of global responses of fluorescent reporters to cAMP stimulation, and image acquisition were performed as previously described [16, 25, 26, 28]. Images were acquired using a Marianas Spinning Disk Confocal Workstation (Intelligent Imaging Innovations, Inc., Denver, CO, USA) equipped with an Evolve™ 512 EMCCD camera (Photometrics, Tucson, AZ, USA), and image analysis was performed using the Slidebook software (Intelligent Imaging Innovations, In., Denver, CO, USA). Analysis of fluorescent reporter translocations between the cytosol and the cell cortex was performed by measuring changes in fluorescent intensities in the cytosol normalized to each cell’s basal levels, as described previously [29]. To assess the reversibility of caffeine inhibition of PI3K, developed cells plated on 35 mm dishes, with coverslips for imaging, were treated with caffeine for 30 min, washed twice with NaK phosphate buffer, and then stimulated with cAMP while imaging the PHcrac-GFP translocation response ~10 min after caffeine removal.
Results
Caffeine inhibits PI3K and mTORC2 activity
Since caffeine inhibits PI3K and mTOR in mammalian cells, and PI3K and mTORC2 play key roles in cAMP chemoattractant signaling in Dictyostelium, we started by assessing the effect of caffeine on these two kinases. For PI3K, we evaluated the effect of caffeine on the cAMP-induced, PI3K-mediated production of phosphatidylinositol (3,4,5)-triphosphate [PI(3,4,5)P3] using the PI(3,4,5)P3 fluorescent reporter PHcrac-GFP [30, 31]. PHcrac-GFP is mostly cytosolic in resting cells and translocates to the plasma membrane of control cells upon cAMP stimulation, reflecting the production of PI(3,4,5)P3 and, thereby, PI3K activation (Fig. 2a). By contrast, only a fraction of the PI(3,4,5)P3 reporter was recruited to the plasma membrane in caffeine-treated cells. However, the cAMP-induced translocation of PHcrac-GFP in caffeine-treated cells was recovered shortly after the removal of caffeine (Fig. 2b). Therefore, these observations suggest that caffeine inhibits the cAMP-induced activation of PI3K in a reversible manner.
Fig. 2. Caffeine inhibits PI3K and mTORC2.

a and b, cAMP-induced PI(3,4,5)P3 response in control and caffeine-treated cells before and after caffeine removal. The cytosolic fluorescence intensity of the the PI(3,4,5)P3 reporter, consisting of the plextrin homology (PH) domain of the cytosolic regulator of adenylyl cyclase (CRAC) fused to GFP (PHcrac-GFP), was quantified, normalized to basal levels and plotted as a function of time after cAMP stimulation. Data in a represent the mean fluorescence intensity ± SEM of 294 control cells and 344 caffeine-treated cells from six different experiments. Data in b represent the mean fluorescence intensity ± SEM of 64 control, 83 caffeine-treated, and 58 caffeine-treated then washed cells from a representative experiment. c and d, cAMP-induced phosphorylation of PKB and PKBR1 at their hydrophobic motif (HM; mTORC2 site) and activation loop (AL; PDK1 site) in control cells and in caffeine-treated cells before and after caffeine removal. In d, caffeine-treated cells that were washed to remove caffeine were stimulated for 10 s with cAMP at 2, 5, or 10 min after the washes. e, cAMP-induced mTORC2 kinase activity. The kinase activity of immunopurified mTORC2 was assessed using PKB- and PKBR1-containing cell lysates as substrates. PKB and PKBR1 HM phosphorylation were revealed by immunoblot. f, cAMP-induced PKB and PKBR1 kinase activity in control and caffeine-treated cells. The kinase activity of immunopurified PKB and PKBR1 was assessed using H2B as substrate. H2B phosphorylation was detected by autoradiography, and PKB and PKBR1 were revealed by immunoblot. g, cAMP-induced phosphorylation of PKB and PKBR1 cellular substrates in control and caffeine-treated cells was detected by immunoblot using an antibody directed against a PKB-phosphorylated substrate motif (P-PKBS). Immunoblots and autoradiography data are representative of at least three independent experiments. CS, Coomassie blue staining.
To investigate if caffeine inhibits mTORC2 in Dictyostelium, we assessed the effect of caffeine on the cAMP-induced and mTORC2-mediated PKB and PKBR1 phosphorylation [32, 33]. In Dictyostelium, mTORC2 phosphorylates PKB and PKBR1 at their C-terminal hydrophobic motif (HM; PKBT435 and PKBR1S470) and this phosphorylation is required for the PDK1-mediated phosphorylation of the PKB kinases in their activation loop (AL; PKBT278 and PKBR1T309) [33]. PI3K is also implicated in promoting PKB activation through the production of PI(3,4,5)P3 that mediates the recruitment of PKB to the plasma membrane [34]. PKBR1 is anchored at the membrane through myristylation and its phosphorylation and activation is independent of PI3K [33]. As expected, control cells display strong and transient cAMP-induced PKB- and PKBR1-AL and -HM phosphorylation (Fig. 2c). On the other hand, caffeine-treated cells exhibit considerably reduced cAMP-stimulated PKB and PKBR1 phosphorylation at both the AL and HM sites. In addition, similar to the PI3K response, we observed the rescue of PKB and PKBR1 phosphorylation shortly after the removal of caffeine, although the phosphorylation of PKB at its AL and HM sites is not fully recovered (Fig. 2d). Together, these observations suggest that caffeine inhibits the cAMP-induced activation of mTORC2 in a mostly reversible manner.
Since other intracellular factors control the phosphorylation state of the PKB kinases in addition to mTORC2, we also more directly tested the effect of caffeine on the kinase activity of mTORC2 in vitro. Using the mTORC2 specific component Pianissimo (Pia) tagged to T7 and expressed in piaA null cells (Online Resource 1 a and b), we purified mTORC2 from developed cells stimulated with cAMP. Purified mTORC2 was then incubated with 5 mM caffeine, or 1 μM of the mTOR inhibitor Torin2 as control (Online Resource 1 c), before incubation with piaA null cell lysates serving as a source of PKB and PKBR1 substrates [35]. The kinase activity of purified mTORC2 was then evaluated by revealing its phosphorylation of PKB and PKBR1 by immunoblot. We observed that caffeine treatment of immunopurified mTORC2 inhibits its kinase activity towards PKB and PKBR1 in vitro to a similar extent as Torin2 (Fig. 2e). Therefore, this result suggests that caffeine directly inhibits mTORC2.
We then assessed the kinase activity of PKB and PKBR1 as well as their downstream phosphorylation of substrates in cells. As could be expected, caffeine-treated cells display considerably reduced cAMP-induced PKB and PKBR1 kinase activity (Fig. 2f), as well as reduced PKB and PKBR1 cellular substrate phosphorylation, as detected by immunoblot using an anti-phospho-PKB substrate (P-PKBS) antibody (Fig. 2g). The phosphorylated protein detected by the P-PKBS antibody at ~25 kDa that is unaffected by caffeine treatment has previously been shown to be phosphorylated independently of the PKB kinases [34]. Altogether, these observations suggest that, similar to findings in mammalian cells, caffeine inhibits PI3K and mTORC2 in Dictyostelium, and that this leads to a decrease in PKB and PKBR1 signaling.
Effects of caffeine on other chemotactic pathways
We then investigated the effect of caffeine on other pathways, whether they are already known to be linked or not to PI3K and mTORC2 signaling in Dictyostelium, to better characterize the outcome of caffeine treatment on cAMP signaling. We compared the activity of Protein Kinase A (PKA; activated by cellular cAMP) and Extracellular-Regulated Kinases 1 and 2 (ERK1 and ERK2), as well as the production of the second messenger cGMP, in cells treated or not with 5 mM caffeine. With the lack of a readily available commercial PKA kinase activity assay that can be used with Dictyostelium, we assessed PKA activation by evaluating the phosphorylation of PKA substrates by immunoblot using a phospho-PKA substrate antibody. We stimulated the cells with cAMP and, to circumvent the potential intake of extracellular cAMP that could directly bind and activate intracellular PKA, we also assessed PKA activity in response to stimulation with the cAMP analog 2’-dcAMP, which is an agonist for cAR1 but cannot bind and activate PKA directly [36]. We observed that caffeine-treated cells display reduced cAMP- and 2’-dcAMP-induced phosphorylation of cellular PKA substrates compared to non-treated cells (Fig. 3a, 3b). This result indicates that caffeine treatment of cells inhibits PKA activation in response to cAMP chemoattractant stimulation, consistent with the caffeine-mediated inhibition of cAMP production (Fig. 3c) [4].
Fig. 3. Caffeine differentially affects the PKA, ERK, and cGMP responses. a and b, cAMP.

(a)- and 2’-deoxy-cAMP (b)-induced phosphorylation of PKA cellular substrates in control and caffeine-treated cells was detected by immunoblot using an antibody directed against a PKA-phosphorylated substrate motif (P-PKAS). c, 2’-deoxy-cAMP-induced cAMP production in control and caffeine-treated cells. Data from four independent experiments are shown, along with the mean ± SD. d, cAMP-induced ERK1 and ERK2 phosphorylation in control and caffeine-treated AX2 and AX3 cells was detected using anti-phospho-p42/p44 antibody. e, cAMP-induced cGMP production in control and caffeine-treated cells. Data from three independent experiments are shown, along with the mean ± SD. Immunoblot data are representative of at least three independent experiments. CS, Coomassie blue staining.
To determine the effect of caffeine on the cAMP-induced activation of ERK1 and ERK2, we evaluated the kinases’ phosphorylation as a measure of their activity, as previously reported [37]. Interestingly, when we first assessed ERK1 and ERK2 phosphorylation, we reproducibly observed differences in their profiles whether we used AX2 or AX3 wild-type strains (Fig. 3d). For ERK1, we observed much higher phosphorylation levels in AX3 than in AX2 cells, particularly in resting, non-stimulated cells. However, ERK1 is similarly inhibited by caffeine in both AX2 and AX3 cells: basal ERK1 phosphorylation levels are considerably reduced and the cAMP-induced phosphorylation of ERK1 is delayed in caffeine-treated cells compared to control cells (Fig. 3d). For ERK2, whereas its phosphorylation profile is similar in both AX2 and AX3 untreated cells, the effect of caffeine is completely different: caffeine-treated AX2 cells display reduced cAMP-induced ERK2 phosphorylation while caffeine-treated AX3 cells consistently display increased and prolonged cAMP-induced ERK2 phosphorylation compared to control cells (Fig. 3d). Together, these observations suggest that caffeine inhibits the cAMP-induced ERK1 activation and that the effect of caffeine on ERK2 considerably differs with the strain used.
We then defined the effect of caffeine on the cAMP-induced production of cGMP, which is also associated with the chemotactic response to cAMP in Dictyostelium [38]. Whereas little is known about the cGMP pathway in Dictyostelium, evidence suggest the potential involvement of small GTPases RasC and RasG in promoting cGMP production in response to cAMP chemoattractant stimulation (Fig. 1) [39]. Comparing caffeine-treated and non-treated cells, we measured the cGMP levels in resting conditions (basal) as well as 5 and 12 sec after cAMP stimulation (Fig. 3e). As previously reported, control cells display a cAMP-induced increased in cGMP reaching ~25 times over basal levels at 12 sec after stimulation [39]. Caffeine-treated cells also produced cGMP in response to cAMP stimulation, but only up to ~10 times over basal levels (Fig. 3e). Thus, this observation indicates that caffeine treatment of Dictyostelium cells reduces the cAMP-induced cGMP response.
Effects of caffeine on cytoskeleton regulation
Since PI3K and mTORC2 pathways regulate F-actin, and cGMP regulates Myosin II (MyoII) in chemotaxing Dictyostelium cells, we next assessed the effect of caffeine on the cAMP-induced F-actin polymerization and MyoII assembly responses. To measure F-actin, we used two different assays: 1) the imaging of adherent cells expressing the F-actin reporter Lifeact-GFP [40]; and 2) the TRITC-Phalloidin labeling of F-actin in cells in suspension. As previously reported, non-treated cells display a typical biphasic cAMP-stimulated F-actin polymerization response with a first sharp peak at ~5 sec, which associated with initial actin reorganization (cringe response), and a second broader peak at ~ 40–60 sec, which is associated with pseudopod protrusions (Fig. 4a, 4b) [41]. Interestingly, we observed opposite effects of caffeine on the F-actin responses in the two different assays. In Lifeact-GFP imaging, caffeine-treated cells display reduced translocation of the F-actin reporter to the cell cortex upon cAMP stimulation, suggesting reduced F-actin polymerization (Fig. 4a). On the other hand, measurement of F-actin using TRITC-Phalloidin labeling shows increased F-actin polymerization in caffeine-treated compared to non-treated cells (Fig. 4b). We do not know why caffeine has different effects on F-actin polymerization depending on the assay used, but different possibilities are considered in the Discussion section below.
Fig. 4. Caffeine differentially affects the F-actin and MyoII responses.

a, Relative cytosolic fluorescence intensity of the F-actin reporter Lifeact-GFP in control and caffeine-treated cells stimulated with cAMP for the indicated time. Data represent the mean fluorescence intensity ± SEM of 251 control cells and 340 caffeine-treated cells from five independent experiments. b, cAMP-induced F-actin polymerization in control and caffeine-treated cells, measured by Phalloidin labeling at the indicated time after stimulation. Data represent the mean ± SD of 4 independent experiments. c, Relative cytosolic fluorescence intensity of GFP-MyoII in control and caffeine-treated cells stimulated with cAMP for the indicated time. Data represent the mean fluorescence intensity ± SEM of 258 control cells and 273 caffeine-treated cells from four independent experiments.
To assess the effect of caffeine on MyoII dynamics in response to cAMP stimulation, we used cells expressing GFP-fused myosin heavy chain A (GFP-MyoII) [28]. As reported previously, GFP-MyoII localizes to the cell cortex in resting cells and translocates to the cytosol upon cAMP stimulation, which is then followed by an increase in cortical GFP-MyoII, with a peak ~40–50 sec after cAMP stimulation, before returning back to basal levels (Fig. 4c). By comparison to non-treated cells, caffeine-treated cells displayed a greater initial displacement of cortical GFP-MyoII to the cytosol, and then directly and slowly returned to basal levels at the cell cortex ~50 sec post-stimulus. Thus, this result suggests that caffeine decreases the cAMP-induced assembly of MyoII at the cell cortex.
Effects of caffeine on the upstream Ras and Rap1 responses
The Ras and Rap1 GTPases are activated early in the cAMP chemoattractant-induced signaling pathways, upstream of PI3K and mTORC2. However, RasC, RasG, and Rap1 are also linked to PI3K and mTORC2 through feedback loops (Fig. 1) [16, 22, 42, 43]. We therefore assessed the effect of caffeine on the cAMP-induced activation of RasC, RasG, and Rap1. For RasC, cAMP stimulation induces its transient activation with a peak at 5–10 s and a return to basal levels after ~40 s (Fig. 5a). We did not observe any meaningful effect of caffeine on basal nor cAMP-stimulated RasC activity. For RasG, control cells display a 5 s peak of cAMP-induced activation with a rapid return to basal levels by 20 s after stimulation (Fig. 5b). By contrast, while we observed similar basal RasG activity levels in caffeine-treated cells compared to control cells, cAMP stimulation produced a ~2 fold stronger RasG activation response in caffeine-treated cells. After this initial response, RasG activity is then reduced by ~50% at 20 s after cAMP stimulation and remains at this level at least until 60 s after stimulation. For Rap1, cAMP stimulation of control cells induces its activation with a peak at ~ 5–10 s and a ~50% reduction in activity at ~20 s that persists afterwards at least until 60 s after stimulation (Fig. 5c). In caffeine-treated cells, we observed increased basal levels as well as cAMP-stimulated Rap1 activity at 5, 10, and 20 s after stimulation, with a return to basal levels after 40 s. Altogether, these observations show that treating Dictyostelium cells with caffeine, while not affecting RasC activity, leads to stronger RasG and Rap1 responses and changes their profiles.
Fig. 5. Caffeine potentiates the cAMP-induced activation of RasG and Rap1, but not RasC.

cAMP-induced activation of RasC (a), RasG (b), and Rap1 (c) in control and caffeine-treated cells. Active RasC, RasG, and Rap1, were pulled down with GST-Byr2(RBD), GST-Raf1(RBD), and GST-RalGDS(RBD), respectively, and revealed by immunoblotting. For RasC, cells expressing FLAG-RasC were used and RasC was revealed by immunoblotting using anti-FLAG antibody. Pan-Ras antibody was used to reveal RasG, and our custom Rap1 antibody (described previously [17]) was used to reveal Rap1. Immunoblots shown are representative of at least three independent experiments. Graphs show quantified data from three RasC, five RasG, and three Rap1 experiments, expressed as percentage of the maximal response in control cells, along with the means ± SD.
Discussion
We have characterized the effect of caffeine on chemoattractant signaling in Dictyostelium and our findings suggest that it includes the inhibition of PI3K and mTORC2. Both enzymes are necessary for ACA activation in response to chemoattractant stimulation, thereby explaining the inhibitory effect of caffeine on cAMP synthesis (Fig. 6). In addition, considering the known cAMP chemoattractant signaling pathways, caffeine inhibition of PI3K and mTORC2 also explains several of the other observed effects of caffeine in Dictyostelium. These effects include caffeine’s inhibition of PKB, PKBR1, and PKA activity, as well as its positive effect on RasG and Rap1, possibly due to the PKA-mediated negative feedback loops (Fig. 6). In addition, the observation that caffeine mostly affects the amplitude of the responses measured and not their timing, could indicate that some PI3K and mTORC2 are still active in cells treated with 5 mM caffeine and that substrates are in excess, and thus, that PI3K and mTORC2 signaling may not, or minimally, be govern by a threshold. On the other hand, the lack of an effect of caffeine on RasC activity, as well as its effects on cGMP, ERK1, ERK2, F-actin and MyoII are more intriguing and suggest either that PI3K and/or mTORC2 play other, unknown roles in the chemotactic signaling network or that caffeine also targets other proteins in the network.
Fig. 6. Caffeine differentially affects many cAMP chemoattractant signaling effectors in Dictyostelium.
Our findings suggest that caffeine inhibits mTORC2 and PI3K in Dictyostelium, leading to reduced activation of PKB, PKBR1, and cAMP production. We then propose that the caffeine inhibition of these two pathways lead to the observed reduced PKA activity and, consequently, the increased activity of Rap1 and RasG, which are negatively regulated by PKA [17]. The cAMP-stimulated production of cGMP is also considerably inhibited by caffeine, but through an unknown mechanism. In turn, the decrease in cGMP together with the increased Rap1 activity likely contributes to the observed decrease in MyoII assembly in caffeine-treated cells. Intriguingly, we obtained conflicting results concerning the effect of caffeine on F-actin polymerization depending on the assay used, and on ERK2 activity depending on the Dictyostelium strain used (depicted by the dual color). Finally, ERK1 activity is reduced and the cAMP-induced activation of the heterotrimeric G protein Gα2βγ is potentiated in caffeine treated cells, both through unknown mechanisms, whereas RasC activity is unaffected. A color scheme is used to depict the effects of caffeine on the chemotactic effectors, with dark red signifying the most inhibitory and dark blue the potentiation effects.
We were surprised to observe that caffeine treatment of cells inhibits the cAMP-induced production of cGMP, since a previous study reports no effects of caffeine on the cGMP response [4]. This discrepancy may lie in the fact that different Dictyostelium strains were used. This is highly possible since we observed, for example, that the effect of caffeine on the ERK2 response varies considerably between the AX2 and AX3 strains. Indeed, whereas caffeine inhibits the basal and early cAMP-induced phosphorylation of ERK1 in both AX2 and AX3 cells, the cAMP-induced ERK2 phosphorylation response is reduced in AX2 but increased in AX3 cells treated with caffeine. The mechanisms through which caffeine affects ERK1 and ERK2 activity are unknown, but the potentiating effect of caffeine on ERK2 in AX3 cells could partly be due to reduced PKA activity, which was previously suggested to negatively regulate ERK2 [44]. Although evidence suggest that intracellular cAMP-PKA signaling may not mediate adaptation of ERK2 as previously expected [45], a role for PKA in regulating the extent of ERK2 activation is not excluded.
Another unexpected observation is the effect of caffeine on the cAMP-induced F-actin polymerization. First, the underlying cause for the opposite effects of caffeine on the F-actin response measured by Lifeact-GFP imaging versus Phalloidin labeling is unknown, but we speculate that this is due to the different cellular conditions. The imaging is performed on attached cells whereas Phalloidin labeling is performed after stimulation of cells in suspension. This is an important observation, as it suggests that responses to chemoattractant stimulation of Dictyostelium cells may considerably vary whether the cells are in suspension or attached to a substrate. In addition, the increased cAMP-induced F-actin polymerization observed in cells in suspension is very counterintuitive since the expected targets of caffeine, PI3K and mTORC2, are involved in promoting the F-actin response [13, 46] (Fig. 6). Therefore, this observation suggests that, under these conditions, there is another caffeine target responsible for this effect. On the other hand, the inhibitory effect of caffeine on MyoII assembly likely results from the decrease in cGMP production in caffeine-treated cells.
In addition to the observations presented herein, in a previous study, we showed that caffeine also affects the cAR1-mediated heterotrimeric G protein activation, specifically increasing the potency of the cAMP-induced G protein subunit dissociation [47]. We don’t think that the effect of caffeine on G protein subunit dissociation plays a role in the other effects of caffeine reported therein, since all the experiments were performed using saturating concentrations of the cAMP stimulus (1 μM) and that, at this concentration, caffeine displays no significant effect on G protein subunit dissociation. The mechanism through which caffeine affects G protein activation remains unknown, including whether it involves PI3K and/or mTORC2, but this observation suggests that caffeine affects a regulator of G protein activation.
Early studies of the effect of caffeine in Dictyostelium have shown that caffeine inhibits endocytosis, particularly pinocytosis at 1.5–6 mM caffeine, in addition to cAMP production [48]. Caffeine-mediated inhibition of PI3K could partly explain the effect of caffeine on pinocytosis since PI3K is involved in this process [49]. Previous studies also suggest that caffeine inhibits the Dictyostelium phosphodiesterase 4 (PDE4), the extracellular PDE responsible for degradation of secreted cAMP during development [7]. Caffeine also inhibits PDEs in human cells and could inhibit intracellular PDEs in Dictyostelium as well, but these are unlikely major targets of caffeine since cAMP and cGMP levels are reduced, and not increased as would be expected from PDE inhibition, in caffeine-treated cells [1]. However, there are likely other proteins and cAMP chemoattractant responses that are affected by caffeine that we haven’t tested for. For example, we expect that the activation of Rho GTPases may be inhibited, since some of them are known to lie downstream from PI3K [11].
In conclusion, our study reveals that caffeine inhibits PI3K and mTORC2 activity and downstream signaling in Dictyostelium, and that caffeine also differentially affects the cAMP-induced production of cGMP, the activation of RasG, Rap1, ERK1, and ERK2, as well as the F-actin and MyoII responses. Although there are likely several caffeine targets responsible for the observed effects, given our observations and evidence from mammalian studies that caffeine directly inhibits PIKK family kinases, we propose that the effect of caffeine in Dictyostelium involves its direct targeting of PI3K and mTORC2. Therefore, these effects need to be considered when caffeine is used in studies of the Dictyostelium chemoattractant signaling network.
Supplementary Material
Acknowledgements
We are grateful to the Dicty Stock Center and its material depositors for providing cells and DNA constructs.
Funding: This study was funded by a Research Scholar Grant 127940-RSG-15-024-01-CSM from the American Cancer Society to P.G.C. M.S. was supported by NIH T32 grant GM008804 and P.L. was supported by a U.S. Public Health Service grant GM037830.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Bode AM, Dong Z (2007) The enigmatic effects of caffeine in cell cycle and cancer. Cancer Lett 247:26–39. 10.1016/j.canlet.2006.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pohanka M (2015) The perspective of caffeine and caffeine derived compounds in therapy. Bratisl Med J 116:520–530 [DOI] [PubMed] [Google Scholar]
- 3.Reinke A, Chen JCY, Aronova S, Powers T (2006) Caffeine targets TOR complex I and provides evidence for a regulatory link between the FRB and kinase domains of Tor1p. J Biol Chem 281:31616–31626. 10.1074/jbc.M603107200 [DOI] [PubMed] [Google Scholar]
- 4.Brenner M, Thoms SD (1984) Caffeine blocks activation of cyclic AMP synthesis in Dictyostelium discoideum. Dev Biol 101:136–146. doi: 0012-1606(84)90124-6 [pii] [DOI] [PubMed] [Google Scholar]
- 5.Reymond CD, Schaap P, Véron M, Williams JG (1995) Dual role of cAMP during Dictyostelium development. Experientia 51:1166–1174. 10.1007/BF01944734 [DOI] [PubMed] [Google Scholar]
- 6.Theibert A, Devreotes PN (1983) Cyclic 3’, 5’-AMP relay in Dictyostelium discoideum: adaptation is independent of activation of adenylate cyclase. J Cell Biol 97:173–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alvarez-Curto E, Weening KE, Schaap P (2007) Pharmacological profiling of the Dictyostelium adenylate cyclases ACA, ACB and ACG. Biochem J 401:309–316. 10.1042/BJ20060880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pupillo M, Klein P, Vaughan R, et al. (1988) cAMP receptor and G-protein interactions control development in Dictyostelium. In: Cold Spring Harbor Symposia on Quantitative Biology pp 657–665 [DOI] [PubMed]
- 9.Kumagai A, Pupillo M, Gundersen R, et al. (1989) Regulation and function of G alpha protein subunits in Dictyostelium. Cell 57:265–275. doi: 0092-8674(89)90964-1 [pii] [DOI] [PubMed] [Google Scholar]
- 10.Kumagai a, Hadwiger J a, Pupillo M, Firtel R a (1991) Molecular genetic analysis of two G alpha protein subunits in Dictyostelium. J Biol Chem 266:1220–8 [PubMed] [Google Scholar]
- 11.Devreotes PN, Bhattacharya S, Edwards M, et al. (2017) Excitable Signal Transduction Networks in Directed Cell Migration. Annu Rev Cell Dev Biol 33:103–125. 10.1146/annurev-cellbio-100616-060739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim CJ, Spiegelman GB, Weeks G (2001) RasC is required for optimal activation of adenylyl cyclase and Akt/PKB during aggregation. EMBO J 20:4490–4499. 10.1093/emboj/20.16.4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S, Comer FI, Sasaki A, et al. (2005) TOR complex 2 integrates cell movement during chemotaxis and signal relay in Dictyostelium. Mol Biol Cell 16:4572–4583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen MY, Long Y, Devreotes PN (1997) A novel cytosolic regulator, Pianissimo, is required for chemoattractant receptor and G protein-mediated activation of the 12 transmembrane domain adenylyl cyclase in Dictyostelium. Genes Dev 11:3218–3231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comer FI, Parent CA (2006) Phosphoinositide 3-kinase activity controls the chemoattractant-mediated activation and adaptation of adenylyl cyclase. Mol Biol Cell 17:357–366. 10.1091/mbc.E05-08-0781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charest PGG, Shen Z, Lakoduk A, et al. (2010) A Ras signaling complex controls the RasC-TORC2 pathway and directed cell migration. Dev Cell 18:737–749. 10.1016/j.devcel.2010.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scavello M, Petlick ARAR, Ramesh R, et al. (2017) Protein kinase A regulates the Ras, Rap1 and TORC2 pathways in response to the chemoattractant cAMP in Dictyostelium. J Cell Sci 130:1545–1558. 10.1242/jcs.177170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Müller-Taubenberger A, Kortholt A, Eichinger L (2013) Simple system - substantial share: The use of Dictyostelium in cell biology and molecular medicine. Eur. J. Cell Biol 92:45–53 [DOI] [PubMed] [Google Scholar]
- 19.Bastounis E, Meili R, Alonso-Latorre B, et al. (2011) The SCAR/WAVE complex is necessary for proper regulation of traction stresses during amoeboid motility. Mol. Biol. Cell 22:3995–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeon TJ, Lee D-J, Merlot S, et al. (2007) Rap1 controls cell adhesion and cell motility through the regulation of myosin II. J Cell Biol 176:1021–1033. 10.1083/jcb.200607072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meili R, Ellsworth C, Lee S, et al. (1999) Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium. EMBO J 18:2092–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sasaki AT, Chun C, Takeda K, Firtel RA (2004) Localized Ras signaling at the leading edge regulates PI3K, cell polarity, and directional cell movement. J Cell Biol 167:505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fey P, Dodson RJ, Basu S, Chisholm RL (2013) One stop shop for everything Dictyostelium: DictyBase and the Dicty Stock Center in 2012. Methods Mol Biol 983:59–92. 10.1007/978-1-62703-302-2-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Insall RH, Borleis J, Devreotes PN (1996) The aimless RasGEF is required for processing of chemotactic signals through G-protein-coupled receptors in Dictyostelium. Curr Biol 6:719–729 [DOI] [PubMed] [Google Scholar]
- 25.Sasaki AT, Janetopoulos C, Lee S, et al. (2007) G protein-independent Ras/PI3K/F-actin circuit regulates basic cell motility. J Cell Biol 178:185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang S, Charest PGG, Firtel RAA (2008) Spatiotemporal regulation of Ras activity provides directional sensing. Curr Biol 18:1587–1593. 10.1016/j.cub.2008.08.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Haastert PJ (2006) Analysis of signal transduction: formation of cAMP, cGMP, and Ins(1,4,5)P3 in vivo and in vitro. Methods Mol Biol 346:369–392. 10.1385/1-59745-144-4:369 [DOI] [PubMed] [Google Scholar]
- 28.Chung CY, Firtel RA (1999) PAKa, a putative PAK family member, is required for cytokinesis and the regulation of the cytoskeleton in Dictyostelium discoideum cells during chemotaxis. J Cell Biol 147:559–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeda K, Shao D, Adler M, et al. (2012) Incoherent feedforward control governs adaptation of activated ras in a eukaryotic chemotaxis pathway. Sci Signal 5: 10.1126/scisignal.2002413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dormann D, Weijer G, Parent CA, et al. (2002) Visualizing PI3 kinase-mediated cell-cell signaling during Dictyostelium development. Curr Biol 12:1178–1188. doi: S0960982202009508 [pii] [DOI] [PubMed] [Google Scholar]
- 31.Parent CA, Blacklock BJ, Froehlich WM, et al. (1998) G protein signaling events are activated at the leading edge of chemotactic cells. Cell 95:81–91. doi: S0092-8674(00)81784-5 [pii] [DOI] [PubMed] [Google Scholar]
- 32.Kamimura Y, Devreotes PN (2010) Phosphoinositide-dependent protein kinase (PDK) activity regulates phosphatidylinositol 3,4,5-trisphosphate-dependent and - independent protein kinase B activation and chemotaxis. J Biol Chem 285:7938–7946. 10.1074/jbc.M109.089235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao X-HH, Buggey J, Kimmel AR, et al. (2010) Chemotactic activation of Dictyostelium AGC-family kinases AKT and PKBR1 requires separate but coordinated functions of PDK1 and TORC2. J Cell Sci 123:983–992. 10.1242/jcs.064022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamimura Y, Xiong Y, Iglesias PA, et al. (2008) PIP3-independent activation of TorC2 and PKB at the cell’s leading edge mediates chemotaxis. Curr Biol 18:1034–1043. 10.1016/j.cub.2008.06.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Q, Wang J, Kang S a, et al. (2011) Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl) benzo[h][1,6]naphthyridin-2(1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. J Med Chem 54:1473–80. 10.1021/jm101520v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomer RHRH, Armstrong DD, Leichtling BHBH, Firtel RARA (1986) cAMP induction of prespore and prestalk gene expression in Dictyostelium is mediated by the cell-surface cAMP receptor. Proc Natl Acad Sci USA 83:8624–8628. 10.1073/pnas.83.22.8624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwebs DJ, Hadwiger JA (2015) The Dictyostelium MAPK ERK1 is phosphorylated in a secondary response to early developmental signaling. Cell Signal 27:147–155. 10.1016/j.cellsig.2014.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Haastert PJ, Kuwayama H (1997) cGMP as second messenger during Dictyostelium chemotaxis. FEBS Lett 410:25–28 [DOI] [PubMed] [Google Scholar]
- 39.Veltman D, Van Haastert PJM (2003) Regulation of Dictyostelium guanylyl cyclases. Protist 154:33–42. 10.1078/143446103764928477 [DOI] [PubMed] [Google Scholar]
- 40.Riedl J, Crevenna AH, Kessenbrock K, et al. (2008) Lifeact: a versatile marker to visualize F-actin. Nat Methods 5:605–607. 10.1038/nmeth.1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hall AL, Warren V, Dharmawardhane S, Condeelis J (1989) Identification of actin nucleation activity and polymerization inhibitor in ameboid cells: Their regulation by chemotactic stimulation. J Cell Biol 109:2207–2213. 10.1083/jcb.109.5.2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sasaki AT, Janetopoulos C, Lee S, et al. (2007) G protein-independent Ras/PI3K/F-actin circuit regulates basic cell motility. J Cell Biol 178: . 10.1083/jcb.200611138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Charest PGG, Firtel RAA (2006) Feedback signaling controls leading-edge formation during chemotaxis. Curr Opin Genet Dev 16:339–347. 10.1016/j.gde.2006.06.016 [DOI] [PubMed] [Google Scholar]
- 44.Knetsch MLW, Epskamp SJP, Schenk PW, et al. (1996) Dual role of cAMP and involvement of both G-proteins and ras in regulation of ERK2 in Dictyostelium discoideum. EMBO J 15:3361–3368 [PMC free article] [PubMed] [Google Scholar]
- 45.Brzostowski JA, Kimmel AR (2006) Nonadaptive Regulation of ERK2 in Dictyostelium: Implications for Mechanisms of cAMP Relay10.1091/mbc.E06–05-0376. Mol Biol Cell 17:4220–4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeda K, Sasaki AT, Ha H, et al. (2007) Role of PI3 kinases in chemotaxis in Dictyostelium. J Biol Chem 282:11874–11884 [DOI] [PubMed] [Google Scholar]
- 47.Tariqul Islam AFM, Yue H, Scavello M, et al. (2018) The cAMP-induced G protein subunits dissociation monitored in live Dictyostelium cells by BRET reveals two activation rates, a negative effect of caffeine and potential role of microtubules. Cell Signal 48:25–37. 10.1016/j.cellsig.2018.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonzalez C, Klein G, Satre M (1990) Caffeine, an inhibitor of endocytosis in dictyostelium discoideum amoebae. J Cell Physiol 144:408–415. 10.1002/jcp.1041440307 [DOI] [PubMed] [Google Scholar]
- 49.Zhou K, Pandol S, Bokoch G, Traynor-Kaplan AE (1998) Disruption of Dictyostelium PI3K genes reduces [32P]phosphatidylinositol 3,4 bisphosphate and [32P]phosphatidylinositol trisphosphate levels, alters F-actin distribution and impairs pinocytosis. J Cell Sci 111 ( Pt 2):283–94 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.