

Abstract
The expression of DNA-dependent protein kinase catalytic subunit (DNA-PKc) is highly variable in smokers and reduced enzyme activity has been associated with risk for lung cancer. An in vitro model of lung pre-malignancy was used to evaluate the role of double-strand break DNA repair capacity in transformation of hTERT/CDK4 immortalized human bronchial epithelial cells (HBECs) and reprograming of the epigenome. Here we show that knockdown of DNA-PKc to levels simulating haploinsufficiency dramatically reduced DNA repair capacity following challenge with bleomycin and significantly increased transformation efficiency of HBEC lines exposed weekly for 12 weeks to this radiomimetic. Transformed HBEC lines with wild type or knockdown of DNA-PKc showed altered expression of more than 1,000 genes linked to major cell regulatory pathways involved in lung cancer. While lung cancer driver mutations were not detected in transformed clones, more than 300 genes that showed reduced expression associated with promoter methylation in transformed clones or predictive for methylation in malignant tumors were identified. These studies support reduced DNA repair capacity as a key factor in the initiation and clonal expansion of pre-neoplastic cells and double-strand break DNA damage as causal for epigenetic mediated silencing of many lung cancer-associated genes. The fact that DNA damage, repair, and epigenetic silencing of genes are causal for many other cancers that include colon and prostate extends the generalizability and impact of these findings.
Keywords: DNA-PKc, epigenome, DNA repair capacity, pre-malignancy, lung cancer
Introduction
Lung cancer is responsible for more than 1.5 million deaths globally [1]. Adenocarcinoma (AdC) and squamous cell carcinoma (SCC) are the two main histologic types that account for more than 70% of cases . Suboptimal DNA repair capacity (DRC) and polymorphisms that reduce the activity of genes involved in double strand break repair (DSBR) have been associated with increased risk for lung cancer [2, 3]. Tobacco carcinogens induce single and double strand breaks (DSBs) in DNA and can promote step-wise accumulation of genetic and epigenetic alterations propagated through deficient DRC that in turn increases the risk of lung cancer associated with smoking [4].
The two main pathways that repair double strand DNA breaks are non-homologous end joining (NHEJ) and homologous recombination (HR) [5, 6]. NHEJ involves a direct ligation of broken ends while HR uses genetic information from the homologous DNA sequence. NHEJ repair occurs throughout the cell cycle but dominates in G0/G1 phase and early G2. Components of the NHEJ pathway include DNA-dependent protein kinase catalytic subunit (DNA-PKc), ATP-dependent DNA ligase IV (LIG4), X-ray repair cross-complementing protein 4 (XRCC4), and the Lupus Ku autoantigen protein 70/80 complex (Ku70/Ku80), while components of the HR pathway include RAD50 homolog (RAD50), meiotic recombination 11 homolog A (MRE11A), DNA repair protein XRS2 and X-ray repair cross-complementing protein 3 (XRCC3) [7].
DNA damage signaling in the NHEJ pathway involves the association of DNA-PKc with Ku70/80 to form an active hetero dimer called DNA-PK, which is a critical component of the NHEJ DNA repair pathway [8, 9]. A case-control study from our group showed that reduced DNA-PKc activity in peripheral mononuclear cells was associated with an increased risk for lung cancer suggesting an important role for this enzyme in cancer susceptibility [10]. Importantly, we showed that DNA-PKc expression and activity in peripheral mononuclear cells and bronchial epithelial cells, a precursor of SCC, was highly correlated. Furthermore, the role of DNA-PKc as a potential modifier for lung cancer risk was supported by the fact that cell growth in bronchial epithelial cells exposed to bleomycin, a radiomimetic, was directly associated with enzyme activity.
Driver mutations in oncogenes such as K-ras and EGFr and mutations in tumor suppressor genes such as p53 are clearly important factors in the progression of lung cancer to malignancy [11, 12]. However, epigenetic deregulation involving the methylation of cytosine to form 5-methylcytosine in conjunction with histone modifications and nucleosome remodeling in gene promoters to silence transcription is a key step in initiation and premalignancy affecting hundreds of genes involved in all aspects of cell regulation [13–15]. This supposition is supported by many studies and exemplified by the silencing of the tumor suppressor p16 by promoter hypermethylation at the earliest histologic stage of SCC (basal cell hyperplasia) with increased frequency seen during histologic progression [16]. We provided the first potential link between double-strand break DNA damage and methylation through studying AdCs from plutonium-exposed workers and controls [17]. The frequency for p16 methylation in AdCs from the exposed workers increased as a function of plutonium lung dose. Subsequent studies also demonstrated a highly significant association between DSBR capacity measured in lymphocytes and the propensity for gene methylation detected in sputum from cancer-free smokers from the Lovelace Smokers Cohort [2]. A second study in the same cohort identified dietary factors including folate, leafy green vegetables, and multivitamin use as protective against the acquisition of gene methylation, possibly through their antioxidant effects that could modulate DNA repair and/or the reduce DNA damage induced by tobacco-derived carcinogens [18]. A strong mechanistic link between DSBs and induction of de novo methylation has also been established through in vitro studies [19–21]. For example, Mortusewicz et al. [20] found that cytosine DNA-methyltransferase 1 (DNMT1) is rapidly recruited to sites of DSBs in mammalian cells following laser microirradiation. Cuozzo et al. [21] generated a recombinant plasmid containing a 1-SCE1 restriction site within one copy of two inactivated tandem repeated green fluorescent protein (GFP) genes that was introduced into HeLa or mouse embryonic stem cells. The restriction endonuclease 1-Sce1 was added to the cells to induce a DSB in the 5′ copy of the GFP gene. Rapid gene silencing associated with homologous recombination and DNA hypermethylation of the recombinant gene was detected that could be blocked by treatment with the demethylating agent, 5-aza-deoxycytidine. Chromatin immunoprecipitation revealed that DNMT1 was bound specifically to the homologous-recombined GFP DNA. Together, these studies substantiate chronic DNA damage and reduced DRC as important determinants for inducing gene methylation.
Our group has identified key molecular changes driving transformation and clonal outgrowth of pre-neoplastic cells during exposure to carcinogens in tobacco smoke using an in vitro model of hTERT/CDK4 immortalized human bronchial epithelial cells (HBECs) [22]. Transformation is epigenetically driven through increased DNMT1 protein, can be attenuated by overexpression of the de novo cytosine methyltransferase DNMT3b, and involves epithelial to mesenchymal transition [22–24]. The goal of this study was to stably knock down DNA-PKc in HBECs to levels simulating haploinsufficiency and to evaluate the effect of reduced DRC on transformation following 12 weekly exposures to bleomycin. Transformed clones were evaluated for genome-wide effects on mutation, methylation, and transcription. Genomic changes associated with DNA-PKc deficiency were validated in tumor-derived cell lines, primary tumors, and The Cancer Genome Atlas (TCGA) dataset along with functional studies of some identified novel genes.
Materials and Methods
Cell culture, carcinogen exposure, and tumor specimens
HBEC2 and HBEC3 immortalized with hTERT and CDK4 were obtained from Drs. Shay and Minna, Southwestern Medical Center, Dallas, TX and cell culture conditions have been described [22]. This model is superior to SV40 that inactivates the p53 and RB genes because in our model the cells do not go through a crisis that induces genomic instability prior to immortalization, and the HBECs retain a G1S checkpoint for responding to DNA damage. Primary bronchial epithelial cells undergo senescence after approximately 6 passages, negating the ability to study early changes in lung cancer development and to address important questions such as those in this study regarding the effect of reduced double-strand break repair capacity on transformation and reprogramming of the genome. HBEC2 and HBEC3 were exposed to 0.5 units/L bleomycin for 4hrs, once a week for 12 weeks, and propagated in keratinocyte specific medium (KSM) containing 15% FBS. Tumor-derived cell lines were obtained from the American Type Culture Collection (Manassas, VA). Cell line authentication has been performed by Genetica DNA laboratories within the last year. The Institutes’ Ethical Committees acting in accordance with US Common Rule guidelines approved this study, and all human samples were obtained with written informed consent from participating patients. Primary lung tumors and distant normal tissue were obtained as previously described [25].
pSilencer shRNA, expression plasmids, and transfections
ShRNA targeting DNA-PKc was cloned into pSilencer (pSil) vector (Life Technologies, Carlsbad, CA) while expression plasmids for IRF6 and PKP1 were obtained from Origene (Rockville, MD), and GeneCopoeia (Rockville, MD) respectively. HBECs or tumor lines were transfected using the Neon Transfection System (Life Technologies, Carlsbad, CA). Following selection with zeocin or hygromycin, stable clones were identified. Plasmid-driven gene expression was validated at the RNA and/or protein level, and clones were selected expressing either empty vector (pSil) or shRNA targeting DNA-PKc. Cell lines were transfected with plasmids expressing IRF6 and PKP1, and stable clones were isolated.
DNA repair capacity
HBECs in the log phase of growth were treated with bleomycin for 1 hour and cytochalasin for 27 hours. DRC was measured by the cytokinesis-block micronucleus assay as previously described [22]. The number of micronuclei per 1000 bi-nucleated cells was scored and is inversely related to DRC.
Soft agar colony formation
HBECs, A549 or H23 cells harvested from cultures were plated in triplicate in soft agar at a density of 2 × 103 cells per well in six-well plates. The colonies were fixed with methanol, stained with trypan blue and counted after 14 to 21 days.
DNA extraction and genetic analysis
DNA was extracted and quantified as described [25]. Mutation analysis was performed at the Winship Cancer Institute of Emory University using a combination of mutation assays as described [26]. These assays include the SNaPshot (Life Technologies, Grand Island, NY) multiplex PCR with single base primer extension method developed [26] the TruSight (Illumina Inc, San Diego, CA) tumor target amplicon-based library preparation kit, the HaloPlex Cancer NGS (Agilent Technologies, Santa Clara, CA), and the Fluidigm 48.48 Access Array custom designed for lung cancer related mutations . DNA samples from HBEC parent and transformed lines were used for the different sample preparations for the libraries and next generation sequencing was done on the Illumina MiSeq . Sequence alignment, index and primer trimming, and variant calls were performed using onboard MiSeq software. FASTQ files were analyzed and variants were detected using the various manufacturers’ recommended software.
Gene expression analysis
Total RNA was isolated with TRIZOL reagent (Life Technologies, Grand Island, NY) and reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Life Technologies, Grand Island, NY). Quantitative real-time PCR (RT-qPCR) was carried out using TaqMan assays (Life Technologies, Grand Island, NY) and gene expression results were normalized to PCNA or to GAPDH using the 2−ΔΔCt method.
Identification of methylated genes from the TCGA dataset
A goal for our studies was to identify globally the effect of double-strand DNA break associated transformation on the induction of global gene silencing mediated through cytosine methylation in the promoter region. In order to provide a comprehensive gene list, we analyzed the TCGA dataset generated with the Illumina HM450K Beadchip that interrogated 811 adenocarcinomas and squamous cell carcinomas for genome-wide methylation changes [27, 28]. The establishment of a comprehensive list of candidate methylated genes is complicated by several factors that include tissue heterogeneity (normal cells in the tumor), heterogeneity of levels of CG methylation associated with silencing expression across genes, and the reduction in mean change in beta value between tumor and normal for an individual CG probe based on the percentage of tumors that are methylated for that probe in the complete dataset. Based on these issues four parameters were defined prior to analysis. These were the interrogation of methylation in the promoter region defined by 1500 base pairs beyond the transcriptional start site (TSS) extending through exon 1, average beta value for each CG in normal tissue ≤0.2 (n=74 [beta value range from 0 to 1 being fully methylated]), average beta value in lung cancer greater than in normal tissue with FDR ≤0.05, and a significant (FDR ≤0.05) inverse correlation between CG beta value and gene expression. There were 13,321 genes interrogated by both HM450K in the promoter regions defined above and RNA-seq with ≥5 reads per million sequences for evaluating expression. A total of 3579 genes with 11,768 probes were identified as methylated with reduced expression ≥50%. Of the 3579 genes, 2198, 1586, and 1195 have 2 or more, 3 or more, and 4 or more CGs with acquired methylation and reduced expression. A 50% reduction in gene expression was associated with a median beta value of 0.31 (range: 0.14, 0.66). Our laboratory has confirmed methylation of 115 genes in primary lung tumors and cell lines through expression changes and methylation assessed via sequencing, combined bisulfite restriction analysis, and/or methylation specific PCR [25, 29, 30]. Our strategy for identifying methylated genes in TCGA identified 90 of these genes (78%), thus validating this approach.
Methylation and expression arrays
Bisulfite modified DNA (1μg) isolated from transformed clones and controls were hybridized to the Infinium HumanMethylation450K Beadchip (Illumina, San Diego, CA) for methylation arrays. Idat files were exported from Genome Studio and preprocessed using the noob (normal-exponential out-of-band) method for background correction with dye-bias normalization from the minfi package in R (3.3.2) to generate β-values for the 485,577 probes on the arrays . Total RNA was isolated, converted to cRNA and hybridized using the Illumina Whole-Genome Human HT-12 v4.0 Gene Expression BeadChip. Raw expression data were processed using the lumi package in R (v 3.3.2) for variance stabilizing transformation and robust spline normalization and log2 fold changes were calculated to identify differentially expressed genes. Genome wide datasets were deposited (GEO 121434). Qiagen Ingenuity Pathway Analysis software was used to identify pathways and networks statistically over-represented in the lists of differentially expressed genes.
Cytotoxicity assay
The MTT (3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide) assay was used to determine cell viability as an indicator for the relative sensitivity of the cells to DNA-PKc deficiency. Cells growing in the logarithmic phase were seeded in 96-well plates (1 × 103 per well), allowed to attach overnight and then 72 hr later, twenty-five microliters of MTT (5 mg/ml; Sigma) was added to each well 2h post-treatment, color formation was quantified by a spectrophotometric plate reader (VersaMax; Molecular Devices) at 570 nm wavelength after solubilizing in 200 μl of dimethyl sulfoxide.
Methylation-specific PCR
Methylation-specific PCR (MSP) was used to assess promoter methylation of IRF6 and PKP1 in lung AdC and SCC derived cell lines and primary tumors. Genes were scored as methylated if a PCR product was detected. Methylation-specific primer sequences and PCR conditions are available upon request.
Statistical analysis
All data are presented as mean ±SD. Statistical analyses were performed using a paired two-tailed test. The p-values were calculated using Fisher’s exact tests. Our analytic strategy for the HM450K arrays focused on the methylation status of 186,493 CpG oligonucleotide probes within 1500 base pairs 5’ of the TSS and extending through exon 1. Methylation analysis was restricted to probes in control cell lines with β-value <0.2. Probes were considered to be methylated when the difference between transformed clones and control cell lines (Δβ) was > 0.1. Statistical analyses were conducted in SAS 9.4 and R 3.3.2.
Results
Variation in DNA-PKc expression in bronchial epithelial cell lines from cancer-free current and former smokers
Our prior studies demonstrated variation in DNA-PKc activity in bronchial epithelial cells from smokers, but did not evaluate gene expression levels [10]. To better inform our strategy for knockdown of this gene in immortalized human bronchial epithelial cell (HBECs) lines, the expression of DNA-PKc was quantified in bronchial epithelial cells from 20 cancer-free current or former smokers and normalized to GADPDH. The highest level of expression was determined using the 2−ΔΔCt method and set to 1 and values for the remaining subjects determined relative to that value. DNA-PKc expression varied considerably with mean levels of approximately 0.45 seen across subjects that did not differ significantly by smoking status or sex (Fig. 1).
Figure 1.

DNA-PKc-expression varies in normal bronchial epithelial cells (BECs) from cancer-free current and former smokers. Relative DNA-PKc expression was determined from 20 BECs isolated from cancer-free individuals, and normalized to GAPDH. DNA-PKc-expression was stratified by current smoking status (A) and sex (B). Smoking status was not available for one subject.
Generation and characterization of DNA-PKc deficient cells
HBECs with reduced DNA-PKc expression were generated using the pSilencer (p-Sil) construct with siRNA targeting the gene. Based on the distribution of expression seen in smokers, the goal was to identify clones that simulated haploinsufficiency, rather than complete loss of expression. Two clones, HBEC2 DNA-PKc and HBEC3 DNA-PKc selected from transfected HBEC2 and HBEC3 showed 75% and 50% reduction in transcription, respectively, and were further characterized (Fig. 2A). Western blots confirmed reduced DNA-PKc expression in the selected clones (Fig. 2B). The two lower bands seen on the Western blot are the result of splice variants, neither of which are active enzymatically [31]. To determine whether DNA-PKc deficiency affects DRC, DNA-PKc deficient HBECs were treated with bleomycin (0.25 units/L for 4h) and DRC was assessed by the cytokinesis-block micronucleus assay. DNA-PKc deficiency significantly reduced DRC in both HBECs as evident by a 300-to-400% increase number of micronuclei (Fig. 2C). Although DRC was compromised in DNA-PKc-deficient HBECs, this was not sufficient to induce spontaneous transformation, as no colonies were formed on soft agar.
Figure 2.
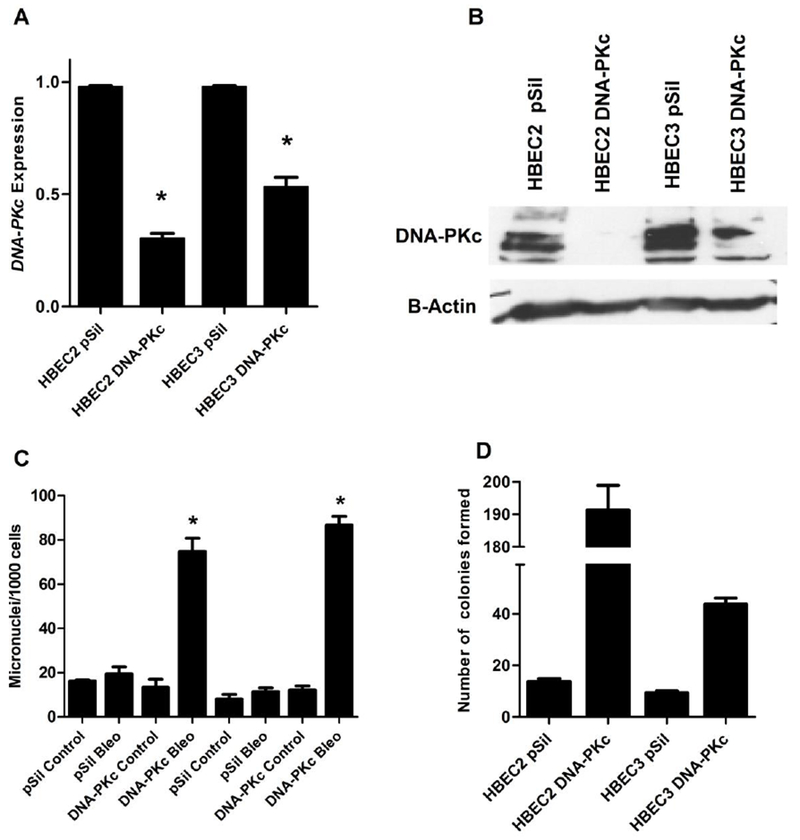
Characterization of HBECs deficient in DNA-PKc expression. (A) Levels of DNA-PKc expression in selected clones relative to p-Silencer and normalized to GAPDH. (B) Western blot showing reduced DNA-PKc expression in HBEC clones with gene knockdown compared to p-Sil. (C) DNA Repair capacity is decreased in HBECs with knockdown of DNA-PKc. HBECs were treated with 0.25U/L bleomycin for 1 hour, and subsequently with cytochalasin for 48 hrs. Cells were harvested and analyzed for DRC measured by the cytokinesis-block micronucleus assay. (D) DNA-PKc deficiency enhances transformation of HBECs following treatment with bleomycin once a week for 12 weeks. Error bars are the mean ±SD and p<0.05 using a paired, two-tailed t test.
DNA-PKc deficiency increases carcinogen-induced transformation efficiency
The two HBEC clones deficient in DNA-PKc (HBEC2 DNA-PKc and HBEC3 DNA-PKc) and control HBECs transfected with empty plasmid (HBEC pSil) were treated with vehicle or 0.25 units/L bleomycin once a week for 12 weeks to define the effects of decreased DNA-PKc on sensitivity to transformation. No transformation was observed in vehicle treated HBEC-pSil or HBEC DNA-PKc knockdown cell lines. Treatment of HBEC2- and HBEC3-pSil cell lines with bleomycin for 12 weeks induced modest transformation with formation of 10–14 colonies (Fig. 2D). In contrast, when DNA-PKc expression was reduced, bleomycin treatment resulted in 45 and 190 colonies in HBEC2 DNA-PKc and HBEC3 DNA-PKc clones, a 4.3- and 14-fold increase (p<0.05) in transformation efficiency, respectively, compared to the empty plasmid control after 12 weeks of bleomycin treatment (Fig. 2D). No transformation was observed in cells treated with bleomycin for 4 weeks. Furthermore, transformed HBEC clones recovered from soft agar did not form tumors when inoculated into nude mice.
Targeted next generation sequencing (NGS)
The transformed clones, p-Sil, and DNA-PKc knockdown cell lines were evaluated for cancer driver mutations by NGS. The 20 genes selected for inclusion in this panel were based on two recent publications that identified frequent mutation of these genes in lung cancer and nominated these genes as cancer drivers because of the properties of the mutations [27, 32]. The genes evaluated included ALK, BRAF, CDKN2A, EFGR, ERBB2, KEAP1, KRAS, MARK1, MET, MYC, NF1, NRAS, PIK3CA, PTEN, Ret, RIT1, SETD2, SMARCA4, STK11, and TP53. No nonsynonymous mutations were detected in any cell line or transformed clone.
Global reprogramming of the transcriptome in transformed clones
The effect of transformation on global gene expression (log2 fold change ≥50%) was assessed in the HBEC2 and HBEC3 lines using Illumina Whole-Genome Expression BeadChip. We identified 1633 differentially expressed genes in HBEC2/HBEC3 bleomycin transformed (Bleo-T) cell lines (Fig. 3A). A similar number of differentially expressed genes (n=1669) were identified in DNA-PKc Bleo-T cell lines. A total of 892 genes were commonly altered across Bleo-T and DNA-PKc Bleo-T cell lines. Pathway analysis for the 892 common genes showed highly significant alterations in molecular and cellular functions involved in cellular movement, cell death, organismal injury, cancer, and cellular development (Fig. 3B). With respect to the 741 genes unique to Bleo-T, the top five pathways were cell death, cell cycle, morphology, assembly, and DNA replication and repair (Fig. 3C). In contrast, the top pathways altered by differential expression of 777 genes unique to DNA-PKc Bleo-T were cell movement, death, assembly, growth, and development (Fig. 3D).
Figure 3.
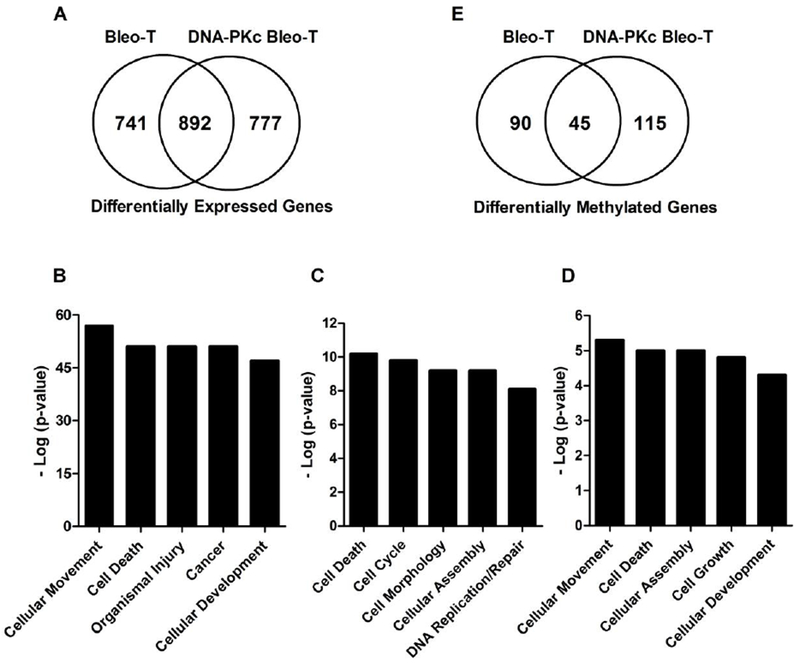
Bleomycin-induced transformation affects genome-wide expression and methylation. (A) Venn diagram summarizing common and unique genes with differential expression fold change ≥50% in HBEC pSil Bleo-T and HBEC DNA-PKc Bleo-T cells compared to vehicle controls. (B) Common major pathways significantly altered in response to gene expression changes across transformed cell lines are ranked by −1 × log10 P values. (C) Pathways altered in p-Sil Bleo-T cells. (D) Pathways altered in DNA-PKc Bleo-T cells. (E) Venn diagram summarizing common and unique methylated genes in HBEC pSil Bleo-T and HBEC DNA-PKc Bleo-T cells compared to vehicle controls.
Gene methylation and expression profiles in DNA-PKc normal and knockdown bleomycin transformed clones
The pre-malignancy model developed through exposure of HBECs to DNA damaging carcinogens is epigenetically driven based on the requirement for DNMT1 and acceleration of transformation by overexpression of the de novo methyltransferase DNMT3b [22, 24]. Epigenetic-mediated silencing occurs through chromatin remodeling via histone modifications and cytosine DNA methylation. Chromatin remodeling involves in part, trimethylation at histone H3 Lysine27 (H3K27me3) and dimethylation of histone H3 Lysine9 (H3K9me2) to reduce transcription and may precede cytosine methylation in CpG islands within promoter regions [23]. In addition, methylation of scattered CpG sites within a CpG island denoted as “methylation seeds” is proposed to serve as a precursor to dense methylation that leads to transcriptional silencing of the gene [33, 34]. Therefore, to determine the role of the epigenome in transformation induced by bleomycin we first assessed whether genes with reduced expression in DNA-PKc normal and knockdown Bleo-T clones were methylated in malignant tumors by assessing their status in our list of methylated genes with reduced expression identified through interrogation of the TCGA dataset described under Methods. Second, for the genes found to be methylated in TCGA, methylation status was assessed by comparing the change in their beta value for the CGs interrogated by the HM450K across the promoter region in the transformed clones to the vehicle treated control cell lines.
From the transcriptome analysis described above, 987 and 1,046 genes whose expression was reduced by more than 50% in the Bleo-T and DNA-PKc Bleo-T clones respectively were evaluated for methylation status in TCGA. Two hundred and thirty-seven of these genes altered in Bleo-T and 239 genes altered in DNA-PKc Bleo-T clones were methylated in TCGA tumors (369 unique genes, Supplementary Table 1). Evaluating their status in the transformed clones revealed that 135 of 237 and 159 of 239 genes were methylated in Bleo-T and DNA-PKc Bleo-T (Supplementary Table 2). There were 45 genes in common among the transformed cell lines (Fig. 3E). Several of the genes commonly or uniquely methylated and transcriptionally repressed across clones are potent tumor suppressors. Common genes included CCND2 [cyclinD2], CDKN1A [p21], FOXA1, SFRP1, SOX15 and unique genes were MLH1 (Bleo-T), CDH13 [H-cadherin], KRT19, and WNT3a (DNA-PKc Bleo-T). A representation of the extent of individual gene methylation assessed by number of affected CGs and heterogeneity with respect to change in beta value compared to control for two of the common genes SFRP1 and SOX15 and two novel genes selected for further characterization below that were common (PKP1) across transformed clones versus unique (IRF6) to DNA-PKc bleomycin transformed clones is shown in Table 1.
Table 1.
Heterogeneity for methylated probes within a sample of gene promoters in bleomycin-induced transformed clones
| β-value1 |
|||
|---|---|---|---|
| HM450K Probe ID | Gene | Bleo-Transformed | DNA-PKc Bleo-Transformed |
| cg16030177 | IRF6 | NM | 0.23 |
| cg21951975 | IRF6 | NM | 0.19 |
| cg22029157 | IRF6 | NM | 0.19 |
| cg09009380 | PKP1 | 0.30 | NM |
| cg17463149 | PKP1 | NM | 0.19 |
| cg20869257 | PKP1 | 0.23 | 0.28 |
| cg01074584 | SFRP1 | NM | 0.30 |
| cg10406295 | SFRP1 | NM | 0.55 |
| cg13398291 | SFRP1 | 0.76 | NM |
| cg15839448 | SFRP1 | NM | 0.43 |
| cg21517947 | SFRP1 | NM | 0.42 |
| cg22418909 | SFRP1 | NM | 0.59 |
| cg24319902 | SFRP1 | NM | 0.48 |
| cg01029592 | SOX15 | NM | 0.34 |
| cg02256874 | SOX15 | 0.23 | 0.48 |
| cg0682989 | SOX15 | 0.55 | 0.43 |
| cg06812840 | SOX15 | NM | 0.29 |
| cg07488259 | SOX15 | NM | 0.52 |
| cg13159929 | SOX15 | 0.95 | NM |
| cg13483263 | SOX15 | NM | 0.29 |
| cg14187123 | SOX15 | NM | 0.78 |
| cg15910588 | SOX15 | 0.30 | 0.74 |
NM = not methylated
A CG was considered methylated when the change in β-value was ≥0.1 compared to vehicle control β-values that were 0.01–0.2.
Functional studies of selected genes silenced in DNA-PKc-deficient transformed cells
Two genes, plakophilin1 (PKP1) and interferon regulatory factor 6 (IRF6), were selected for further functional analyses because they were both methylated with associated reduced expression in the HBEC3 DNA-PKc Bleo-T clones and their role in lung cancer has not been previously described. RTq-PCR verified the loss of expression with a 47% and >95% reduction of both genes in Bleo-T and DNA-PKc Bleo-T clones, respectively (Fig. 4). PKP1 expression levels were also reduced 35 to 47% after 4 wks of bleomycin treatment. Methylation specific PCR was used to determine the prevalence for methylation of these genes in tumor-derived cell lines and primary tumors. PKP1 was methylated in 2½3 (91%) cell lines and in 26/31 (84%) of tumors while IRF6 was methylated in 10/23 (43%) cell lines and in 13/31 (42%) of tumors (Supplementary Table 3).
Figure 4.
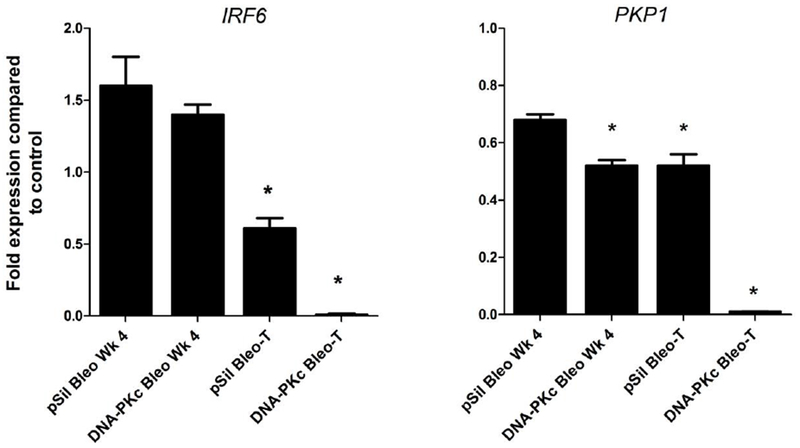
Transcriptional silencing of IRF6 and PKP1. Expression of IRF6 and PKP1 in HBEC3 p-Sil Bleo-T and DNA-PKc-deficient cells was analyzed after 4 weeks of bleomycin treatment (p<0.05), and in transformed cells (p<0.005) compared to vehicle controls and normalized to GAPDH. Error bars are the mean ±SD and p <0.05 using a paired, two-tailed t test.
Given the high prevalence of methylation of PKP1 and IRF6, studies were extended to define the role of these genes in cellular function and transformation. PKP1 and IRF6 were re-expressed through stable integration of expression plasmids in HBEC3 DNA-PKc Bleo-T, A549 and H23 cell lines. Selection of clones for further studies was based on showing physiological increase in expression that ranged from 0.7 to 4-fold compared to parental cell lines. The re-expressed clones containing PKP1 and IRF6 demonstrated tumor suppressor properties that included a significant (P<0.05) reduction of the number of colonies on soft agar and decreased cell viability. One clone each for PKP1 and IRF6 from A549 and H23 that showed the most significant effects on cell growth along with clones from HBEC3 DNA-PKc were selected for studies on cell cycle effects. Moderate, yet statistically significant decrease in the percentage of cells in the S-phase of the cell cycle was seen (Fig. 5A–C).
Figure 5.

Stable re-expression of IRF6 and PKP1 significantly suppress soft agar colony formation, growth, and cell cycle progression. IRF6 and PKP1 were stably re-expressed in A549, H23, and HBEC3 DNA-PKc Bleo-T cells and 1–2 clones/cell line were characterized. (A) Re-expression of IRF6 and PKP1 markedly reduced colony formation compared to transformed control cells in A549, H23 and HBEC3 DNA-PKc Bleo-T. (B) Re-expression of IRF6 and PKP1 reduced cell growth measured by MTT assay in all cell lines with exception of IRF6-re-expressing clone 6-7 in A549 cells and PKP1 re-expressing clone in HBEC3 DNA-PKc Bleo-T. (C) Reexpression of IRF6 and PKP1 moderately increases accumulation of cells in the S phase of the cell cycle. Error bars are the mean ±SD and p <0.05.
Genome-wide transcription patterns of A549, H23, and HBEC3 DNA-PKc Bleo-T reexpressing IRF6 and PKP1 were compared to parent cells to identify pathways regulated in part by either gene. Re-expression of PKP1 resulted in increased expression (50% or more) of 642, 1168, and 763 genes while 135, 275 and 271 genes were decreased in H23, A549 and HBEC3 DNA-PKc Bleo-T, respectively (not shown). Re-expression of IRF6 resulted in increased expression (50% or more) of 433, 1871, and 271 genes while 58, 698 and 117 genes were decreased in H23, A549 and HBEC3 DNA-PKc Bleo-T, respectively (not shown). Fourteen and 25% of genes with altered expression in response to re-expression of IRF6 and PKP1, respectively were also altered in parental HBEC DNA-PKc Bleo-T clones. Ingenuity Pathway Analyses of all genes affected by re-expression of IRF6 or PKP1 revealed several important cell functions across cell lines. The affected cellular pathways included cell cycle, growth, death, survival, morphology, movement and ATM signaling (Supplementary Tables 4 and 5).
Discussion
This study demonstrates that deficiency in DRC due to reduced expression of DNA-PKc greatly enhances transformation of lung epithelial cells in concert with reprogramming of the epigenome. It is not possible to accurately extrapolate these in vitro findings to predict the extent of the contribution of reduced DNA-PKc-dependent repair for the induction of human lung cancer. However, reduced DNA-PKc enzyme activity is associated with increased risk for lung cancer and its expression is highly variable in bronchial epithelial cells from smokers, and its reduced expression likely plays a major role in initiation and clonal expansion of pre-neoplastic cells through acquiring persistent levels of DNA damage in response to chronic exposure to carcinogens in tobacco smoke. This pre-malignancy model has enabled a comprehensive identification of hundreds of genes whose expression is significantly altered in response to transformation induced by double-strand DNA breaks, many of which show reduced expression that is associated with or predictive for promoter hypermethylation in transformed clones and in malignant primary tumors, respectively. While genes well established as tumor suppressors were altered in response to double-strand break DNA damage, novel genes not previously associated with lung cancer were identified. Two of these genes (IRF6 and PKP1) characterized in detail are silenced early during transformation of HBECs, are commonly methylated in lung tumors, and exert growth inhibitory and tumor suppressor effects in vivo through modulating multiple cancer-related pathways that regulate cell cycle, growth, death, and survival. Their involvement in carcinogenesis is not limited to lung as IRF6 functions as a DNA binding protein to suppress cell proliferation, cell growth, and reverse cancer stem cell properties in nasopharyngeal carcinoma and as a tumor suppressor in head and neck squamous cell carcinoma [35, 36]. PKP1 was shown to be important in stabilizing desmosome assembly involved in cytoskeletal integrity and has been implicated in skin cancer [37–39].
DNA-PKc is recognized as a key component of the NHEJ repair pathway for DNA damage response and repair [40, 41]. Our studies support the premise that its deficiency is rate limiting for repair of double-strand break damage as evident by the large increase in micronuclei following exposure of HBECs with reduced DNA-PKc protein levels to bleomycin. Furthermore, compromised DRC secondary to reduced DNA-PKc activity has been associated with an increased risk for lung cancer by our group and others and plays a role in pre-malignant field cancerization, defined by the presence of multiple genetic and epigenetic alterations throughout normal and cytologically altered cells in the lungs of smokers; and further characterized through association with increased prevalence of a gene methylation biomarker panel in sputum from smokers predictive for cancer [30]. The targeting of gene silencing mediated through chromatin remodeling and promoter cytosine hypermethylation is mechanistically plausible given the role of DNMT1 as the major maintenance cytosine-methyltransferase that maintains the epigenetic code during replication and following repair of DNA damage [20, 42]. The repressive histone modifications H3K27me3 and H3K9me2 are increased adjacent to double-strand breaks to affect chromatin conformation to impede transcription within the damaged region to allow for repair [43]. When a double-strand break is repaired, the epigenetic code is lost as there is no longer a hemi-methylated daughter strand for DNMT1 to copy. This can lead to the observed seeds of methylation and concomitant retention of the repressive chromatin histone changes to initiate epigenetic silencing [2, 19, 33, 34]. While we did not conduct ChIP-sequencing for the repressive chromatin marks in the transformed clones, transcriptionally repressed and methylated genes in these clones showed considerable heterogeneity for both number of CG sites methylated and increase in level of methylation (beta value) compared to vehicle controls. The increase in density of methylation within some gene promoters seen in malignant tumors compared to the transformed pre-malignant clones further supports this progressive acquisition of hypermethylation.
Bleomycin forms single and double-strand breaks with both likely contributing to transformation that was not genetically mediated in so far that point mutations in driver genes associated with lung cancer were not detected [44]. This finding is consistent with our initial studies in this model where chemical-induced transformation induced by methylnitrosourea, an alkylating agent that forms the promutagenic adduct O6methylguanine and/or benzo(a)pyrene diol expoxide 1 (BPDE1) to form a N2-deoxyguanonsine adduct did not induce mutation in K-ras or p53 [22]. The O6-methylguanine adduct repaired by O6-methylguanine-DNA methyltransferase and the N2-deoxyguanonsine adduct removed through nucleotide excision repair can lead to mutation in K-ras and p53, respectively [45, 46]. Furthermore, and consistent with prior studies, the transformed clones did not form tumors in nude mice. A recent study corroborates this conclusion through showing that cigarette smoke-induced transformation of HBEC3 was epigenetically driven and the subsequent introduction of the K-ras gene led to tumor growth in nude mice [47]. Thus, our model with support now from others shows that epigenetically mediated gene repression dominates the early molecular events in the spectrum of alterations underlying lung pre-malignancy and the likelihood that genetic mutations in oncogenes and tumor suppressors are mostly involved in progression to malignancy [47–49].
The reduction in DNA-PKc resulted in elevated genomic instability as seen by the marked elevation in micronuclei following exposure to bleomycin that in turn manifested in enhanced transformation compared to repair proficient cells. While there were differences in gene expression profiles between Bleo-T and DNA-PKc Bleo-T clones, the pathways and magnitude of effect based on p-value were similar, a finding consistent with the fact that both the pharmacological agent and the genetic alteration impact repair of double-strand breaks. There were differences with respect to genes that were methylated that could impact transformation. SFRP1, Wnt3a, and KRT19 all function in the Wnt signaling pathway important for lung cancer development and their combined loss in DNA-PKc knock down cells could augment transformation [50]. In addition, there were many examples of greater magnitude for loss of gene-specific expression in DNA-PKc Bleo-T compared to Bleo-T clones such as seen with IRF6 and PKP1. Genomic instability arises from many different processes beyond epigenetic modifications that include mitochondrial gene reprogramming, energy balance, and centrosome amplification leading to aneuploidy that could contribute to the heightened increase in transformation efficiency [51].
This pre-malignancy model has enabled a comprehensive identification of more than 300 genes targeted for epigenetically mediated silencing in association with double-strand DNA breaks and reduced DRC. The annotation of methylated genes with reduced expression in TCGA facilitated uncovering genes methylated and silenced during transformation, while validating these and other genes with reduced expression in transformed clones as methylated in primary lung tumors. The loss of function of these genes affects many cellular regulatory pathways that include cell cycle, signaling, growth, and communication; apoptosis and repair, thereby substantiating a key role for tobacco carcinogen-induced double-strand DNA breaks in the initiation of lung cancer.
Supplementary Material
Acknowledgement
Data generated by The Cancer Genome Atlas (TCGA) project established by the NCI and NHGRI were used to validate part of our findings. The dbGaP accession number for TCGA data is phs000178.v8.p7. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at http://cancergenome.nih.gov/. In addition, the gene methylation and expression datasets were deposited at NCBI Geo with accession number 121434. We acknowledge the following technical support from Kieu Do and Randy Willink.
Funding
This work was supported largely by National Institute of Health grant (R01 ES015262 to SAB) and in part by [U10 CA180950 to SR] and [P30CA11800 to C. Willman] from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary Tables 1 – 5 related to this article can be found online.
Competing Financial Interest: None
REFERENCES
- [1].Siegel RL, Miller KD, Jemal A, Cancer Statistics, CA Cancer J Clin, 67 (2017) 7–30. [DOI] [PubMed] [Google Scholar]
- [2].Leng S, Stidley CA, Willink R, Bernauer A, Do K, Picchi MA, Sheng X, Frasco MA, Van Den Berg D, Gilliland FD, Zima C, Crowell RE, Belinsky SA, Double-strand break damage and associated DNA repair genes predispose smokers to gene methylation, Cancer Res, 68 (2008) 3049–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shen H, Spitz MR, Qiao Y, Guo Z, Wang LE, Bosken CH, Amos CI, Wei Q, Smoking, DNA repair capacity and risk of nonsmall cell lung cancer, Int J Cancer, 107 (2003) 84–88. [DOI] [PubMed] [Google Scholar]
- [4].Zhao H, Albino AP, Jorgensen E, Traganos F, Darzynkiewicz Z, DNA damage response induced by tobacco smoke in normal human bronchial epithelial and A549 pulmonary adenocarcinoma cells assessed by laser scanning cytometry, Cytometry A, 75 (2009) 840–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Weterings E, Chen DJ, The endless tale of non-homologous end-joining, Cell Res, 18 (2008) 114–124. [DOI] [PubMed] [Google Scholar]
- [6].Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA, DNA repair pathways as targets for cancer therapy, Nat Rev Cancer, 8 (2008) 193–204. [DOI] [PubMed] [Google Scholar]
- [7].Bressan DA, Baxter BK, Petrini JH, The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae, Mol Cell Biol, 19 (1999) 7681–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shrivastav M, Miller CA, De Haro LP, Durant ST, Chen BP, Chen DJ, Nickoloff JA, DNA-PKcs and ATM co-regulate DNA double-strand break repair, DNA Repair (Amst), 8 (2009) 920–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dong J, Ren Y, Zhang T, Wang Z, Ling CC, Li GC, He F, Wang C, Wen B, Inactivation of DNA-PK by knockdown DNA-PKcs or NU7441 impairs non-homologous end-joining of radiation-induced double strand break repair, Oncol Rep, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Auckley DH, Crowell RE, Heaphy ER, Stidley CA, Lechner JF, Gilliland FD, Belinsky SA, Reduced DNA-dependent protein kinase activity is associated with lung cancer, Carcinogenesis, 22 (2001) 723–727. [DOI] [PubMed] [Google Scholar]
- [11].Bongiorno PF, Whyte RI, Lesser EJ, Moore JH, Orringer MB, Beer DG, Alterations of K-ras, p53, and erbB-2/neu in human lung adenocarcinomas, J Thorac Cardiovasc Surg, 107 (1994) 590–595. [PubMed] [Google Scholar]
- [12].Cai G, Wong R, Chhieng D, Levy GH, Gettinger SN, Herbst RS, Puchalski JT, Homer RJ, Hui P, Identification of EGFR mutation, KRAS mutation, and ALK gene rearrangement in cytological specimens of primary and metastatic lung adenocarcinoma, Cancer Cytopathol, 121 (2013) 500–507. [DOI] [PubMed] [Google Scholar]
- [13].Shanmugam MK, Arfuso F, Arumugam S, Chinnathambi A, Jinsong B, Warrier S, Wang LZ, Kumar AP, Ahn KS, Sethi G, Lakshmanan M, Role of novel histone modifications in cancer, Oncotarget, 9 (2018) 11414–11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Reik W, Dean W, Walter J, Epigenetic reprogramming in mammalian development, Science, 293 (2001) 1089–1093. [DOI] [PubMed] [Google Scholar]
- [15].Jones PA, Baylin SB, The epigenomics of cancer, Cell, 128 (2007) 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E, Baylin SB, Herman JG, Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis, Proceedings of the National Academy of Sciences of the United States of America, 95 (1998) 11891–11896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Belinsky SA, Klinge DM, Liechty KC, March TH, Kang T, Gilliland FD, Sotnic N, Adamova G, Rusinova G, Telnov V, Plutonium targets the p16 gene for inactivation by promoter hypermethylation in human lung adenocarcinoma, Carcinogenesis, 25 (2004) 1063–1067. [DOI] [PubMed] [Google Scholar]
- [18].Stidley CA, Picchi MA, Leng S, Willink R, Crowell RE, Flores KG, Kang H, Byers T, Gilliland FD, Belinsky SA, Multivitamins, folate, and green vegetables protect against gene promoter methylation in the aerodigestive tract of smokers, Cancer Res, 70 (2010) 568–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].O’Hagan HM, Mohammad HP, Baylin SB, Double strand breaks can initiate gene silencing and SIRTl-dependent onset of DNA methylation in an exogenous promoter CpG island, PLoS Genet, 4 (2008)e1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H, Recruitment of DNA methyltransferase I to DNA repair sites, Proceedings of the National Academy of Sciences of the United States of America, 102 (2005) 8905–8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, Messina S, Iuliano R, Fusco A, Santillo MR, Muller MT, Chiariotti L, Gottesman ME, Avvedimento EV, DNA damage, homology-directed repair, and DNA methylation, PLoS Genet, 3 (2007) e110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [22].Damiani LA, Yingling CM, Leng S, Romo PE, Nakamura J, Belinsky SA, Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells, Cancer Res, 68 (2008) 9005–9014. [DOI] [PubMed] [Google Scholar]
- [23].Tellez CS, Juri DE, Do K, Bernauer AM, Thomas CL, Damiani LA, Tessema M, Leng S, Belinsky SA, EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells, Cancer Res, 71 (2011) 3087–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Teneng I, Tellez CS, Picchi MA, Klinge DM, Yingling CM, Snider AM, Liu Y, Belinsky SA, Global identification of genes targeted by DNMT3b for epigenetic silencing in lung cancer, Oncogene, 34 (2015) 621–630. [DOI] [PubMed] [Google Scholar]
- [25].Tessema M, Yu YY, Stidley CA, Machida EO, Schuebel KE, Baylin SB, Belinsky SA, Concomitant promoter methylation of multiple genes in lung adenocarcinomas from current, former and never smokers, Carcinogenesis, 30 (2009) 1132–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Su Z, Dias-Santagata D, Duke M, Hutchinson K, Lin YL, Borger DR, Chung CH, Massion PP, Vnencak-Jones CL, Iafrate AJ, Pao W, A platform for rapid detection of multiple oncogenic mutations with relevance to targeted therapy in non-small-cell lung cancer, J Mol Diagn, 13 (2011) 74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].T.C.G.A.R.N. TCGA, Comprehensive genomic characterization of squamous cell lung cancers, Nature, 489 (2012) 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].N. Cancer Genome Atlas Research, Comprehensive molecular profiling of lung adenocarcinoma, Nature, 511 (2014) 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Belinsky SA, Unmasking the lung cancer epigenome, Annu Rev Physiol, 77 (2015) 453–474. [DOI] [PubMed] [Google Scholar]
- [30].Leng S, Do K, Yingling CM, Picchi MA, Wolf HJ, Kennedy TC, Feser WJ, Baron AE, Franklin WA, Brock MV, Herman JG, Baylin SB, Byers T, Stidley CA, Belinsky SA, Defining a gene promoter methylation signature in sputum for lung cancer risk assessment, Clin Cancer Res, 18 (2012) 3387–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Convery E, Shin EK, Ding Q, Wang W, Douglas P, Davis LS, Nickoloff JA, Lees-Miller SP, Meek K, Inhibition of homologous recombination by variants of the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs), Proceedings of the National Academy of Sciences of the United States of America, 102 (2005) 1345–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, Stojanov P, Cibulskis K, Choi K, de Waal L, Sharifnia T, Brooks A, Greulich H, Baneiji S, Zander T, Seidel D, Leenders F, Ansen S, Ludwig C, Engel-Riedel W, Stoelben E, Wolf J, Goparju C, Thompson K, Winckler W, Kwiatkowski D, Johnson BE, Janne PA, Miller VA, Pao W, Travis WD, Pass HI, Gabriel SB, Lander ES, Thomas RK, Garraway LA, Getz G, Meyerson M, Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing, Cell, 150 (2012) 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Moriguchi K, Yamashita S, Tsujino Y, Tatematsu M, Ushijima T, Larger numbers of silenced genes in cancer cell lines with increased de novo methylation of scattered CpG sites, Cancer Lett, 249 (2007) 178–187. [DOI] [PubMed] [Google Scholar]
- [34].Song JZ, Stirzaker C, Harrison J, Melki JR, Clark SJ, Hypermethylation trigger of the glutathione-S-transferase gene (GSTP1) in prostate cancer cells, Oncogene, 21 (2002) 1048–1061. [DOI] [PubMed] [Google Scholar]
- [35].Xu L, Huang TJ, Hu H, Wang MY, Shi SM, Yang Q, Lin F, Qiang YY, Mei Y, Lang YH, Li CZ, Peng LX, Zheng LS, Huang JL, Li XJ, Zhang SJ, Qian CN, Huang BJ, The developmental transcription factor IRF6 attenuates ABCG2 gene expression and distinctively reverses sternness phenotype in nasopharyngeal carcinoma, Cancer Lett, 431 (2017) 230–243. [DOI] [PubMed] [Google Scholar]
- [36].Botti E, Spallone G, Moretti F, Marinari B, Pinetti V, Galanti S, De Meo PD, De Nicola F, Ganci F, Castrignano T, Pesole G, Chimenti S, Guerrini L, Fanciulli M, Blandino G, Karin M, Costanzo A, Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas, Proceedings of the National Academy of Sciences of the United States of America, 108 (2011) 13710–13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Boyce AE, McGrath JA, Techanukul T, Murrell DF, Chow CW, McGregor L, Warren LJ, Ectodermal dysplasia-skin fragility syndrome due to a new homozygous internal deletion mutation in the PKP1 gene, Australas J Dermatol, 53 (2012) 61–65. [DOI] [PubMed] [Google Scholar]
- [38].Zheng R, Bu DF, Zhu XJ, Compound heterozygosity for new splice site mutations in the plakophilin 1 gene (PKP1) in a Chinese case of ectodermal dysplasia-skin fragility syndrome, Acta Derm Venereol, 85 (2005) 394–399. [DOI] [PubMed] [Google Scholar]
- [39].Hernandez-Martin A, Torrelo A, Ciria S, Colmenero I, Aguilar A, Grimalt R, Gonzalez-Sarmiento R, Ectodermal dysplasia-skin fragility syndrome: a novel mutation in the PKP1 gene, Clin Exp Dermatol, 38 (2013) 787–790. [DOI] [PubMed] [Google Scholar]
- [40].Chen BP, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, Yajima H, Lobrich M, Shiloh Y, Chen DJ, Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break, J Biol Chem, 282 (2007) 6582–6587. [DOI] [PubMed] [Google Scholar]
- [41].Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K, Botvinick E, Qin J, Chen DJ, Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks, J Biol Chem, 280 (2005) 14709–14715. [DOI] [PubMed] [Google Scholar]
- [42].Leonhardt H, Page AW, Weier HU, Bestor TH, A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei, Cell, 71 (1992) 865–873. [DOI] [PubMed] [Google Scholar]
- [43].Chen Y, Zhu WG, Biological function and regulation of histone and non-histone lysine methylation in response to DNA damage, Acta Biochim Biophys Sin (Shanghai), 48 (2016) 603–616. [DOI] [PubMed] [Google Scholar]
- [44].Sidik K, Smerdon MJ, Bleomycin-induced DNA damage and repair in human cells permeabilized with lysophosphatidylcholine, Cancer Res, 50 (1990) 1613–1619. [PubMed] [Google Scholar]
- [45].Belinsky SA, Devereux TR, Maronpot RR, Stoner GD, Anderson MW, Relationship between the formation of promutagenic adducts and the activation of the K-ras protooncogene in lung tumors from A/J mice treated with nitrosamines, Cancer Res, 49 (1989) 5305–5311. [PubMed] [Google Scholar]
- [46].Kucab JE, van Steeg H, Luijten M, Schmeiser HH, White PA, Phillips DH, Arlt VM, TP53 mutations induced by BPDE in Xpa-WT and Xpa-Null human TP53 knock-in (Hupki) mouse embryo fibroblasts, Mutat Res, 773 (2015) 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vaz M, Hwang SY, Kagiampakis I, Phallen J, Patil A, O’Hagan HM, Murphy L, Zahnow CA, Gabrielson E, Velculescu VE, Easwaran HP, Baylin SB, Chronic Cigarette Smoke-Induced Epigenomic Changes Precede Sensitization of Bronchial Epithelial Cells to Single-Step Transformation by KRAS Mutations, Cancer Cell, 32 (2017) 360–376 e366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pulling LC, Divine KK, Klinge DM, Gilliland FD, Kang T, Schwartz AG, Bocklage TJ, Belinsky SA, Promoter hypermethylation of the O6-methylguanine-DNA methyltransferase gene: more common in lung adenocarcinomas from never-smokers than smokers and associated with tumor progression, Cancer Res, 63 (2003) 4842–4848. [PubMed] [Google Scholar]
- [49].Hirano T, Franzen B, Kato H, Ebihara Y, Auer G, Genesis of squamous cell lung carcinoma. Sequential changes of proliferation, DNA ploidy, and p53 expression, Am J Pathol, 144 (1994) 296–302. [PMC free article] [PubMed] [Google Scholar]
- [50].Stewart DJ, Wnt signaling pathway in non-small cell lung cancer, J Natl Cancer Inst, 106 (2014) djt356. [DOI] [PubMed] [Google Scholar]
- [51].Ferguson LR, Chen H, Collins AR, Connell M, Damia G, Dasgupta S, Malhotra M, Meeker AK, Amedei A, Amin A, Ashraf SS, Aquilano K, Azmi AS, Bhakta D, Bilsland A, Boosani CS, Chen S, Ciriolo MR, Fujii H, Guha G, Halicka D, Helferich WG, Keith WN, Mohammed SI, Niccolai E, Yang X, Honoki K, Parslow VR, Prakash S, Rezazadeh S, Shackelford RE, Sidransky D, Tran PT, Yang ES, Maxwell CA, Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition, Semin Cancer Biol, 35 Suppl (2015) S5–S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.