

Abstract
In non-alcoholic fatty liver disease (NAFLD), triglycerides accumulate within the liver because the rates of fatty acid accrual by uptake from plasma and de novo synthesis exceed elimination by mitochondrial oxidation and secretion as VLDL-triglycerides. Thioesterase superfamily member 2 (Them2) is an acyl-CoA thioesterase that catalyzes the hydrolysis of fatty acyl-CoAs into free fatty acids plus CoASH. Them2 is highly expressed in the liver, as well as other oxidative tissues. Mice globally lacking Them2 are resistant to diet-induced obesity and hepatic steatosis, and exhibit improved glucose homeostasis. These phenotypes are attributable, at least in part, to roles of Them2 in the suppression of thermogenesis in brown adipose tissue and insulin signaling in skeletal muscle. To elucidate the hepatic function of Them2, we created mice with liver-specific deletion of Them2 (L-Them2−/−). Whereas L-Them2−/− mice were not protected against excess weight gain, hepatic steatosis or glucose intolerance, they exhibited marked decreases in plasma triglyceride and apolipoprotein B100 concentrations. These were attributable to reduced rates of VLDL secretion owing to decreased incorporation of plasma-derived fatty acids into triglycerides. The absence of hepatic steatosis in L-Them2−/− mice fed chow was explained by compensatory increases in rates of fatty acid oxidation and by decreased de novo lipogenesis in high fat fed mice. Consistent with a role for Them2 in hepatic VLDL secretion, THEM2 levels were increased in livers of obese patients with NAFLD characterized by simple steatosis. Conclusion: Them2 functions in the liver to direct fatty acids towards triglyceride synthesis for incorporation into VLDL particles. When taken together with its functions in brown adipose and muscle, these findings suggest Them2 as target for the management of NAFLD and dyslipidemia.
Keywords: Fatty acid metabolism, fatty acyl-CoA, fatty acid oxidation, triglycerides, very low-density lipoprotein, dyslipidemia, nonalcoholic fatty liver disease, obesity
Introduction
Alterations in hepatic fatty acid (FA) metabolism contribute to the development of metabolic disorders, including non-alcoholic fatty liver disease (NAFLD), type 2 diabetes and atherosclerotic cardiovascular disease. Long-chain FAs accrue in the liver by uptake from the plasma, de novo synthesis, or the turnover of complex lipids (1). Within the hepatocyte, free FA molecules experience one of several metabolic fates, including complex lipid biosynthesis and oxidation, but also may regulate intracellular processes, such as protein localization, intracellular signaling, and transcription factor activation (2). Although the mechanisms that control the dispositions of long-chain FAs are incompletely understood, their entry into nearly all metabolic pathways requires activation to fatty acyl-CoAs. This reaction is catalyzed by long-chain acyl-CoA synthetases (ACSLs) and is reversed by acyl-CoA thioesterases (ACOTs) (2, 3). Multiple ACSL and ACOT isoforms are expressed in cells, and emerging evidence suggest key roles for these enzymes in the regulation of intracellular FA channeling (2).
Thioesterase superfamily member 2 (Them2; synonymous, ACOT13) is a mitochondria-associated ACOT with substrate specificity for medium- and long-chain acyl-CoAs (4). It is robustly expressed in oxidative tissues, including liver, kidney, heart, and brown adipose tissue (BAT) (4). Them2 was identified as an interacting partner for phosphatidylcholine transfer protein (PC-TP) in the liver (5). Mechanistic studies in cultured mouse hepatocytes revealed both PC-TP-dependent and -independent functions for Them2 (6–8). Under conditions simulating fasting, Them2/PC-TP complex promotes FA oxidation in vitro. However, when cells are cultured under high glucose concentrations, Them2 in the absence of PC-TP expression is sufficient to induce glucose oxidation and lipogenesis (7). The complex of Them2 and PC-TP also suppresses insulin signaling (6), which serves to promote hepatic glucose production during fasting (7). In BAT, Them2 appears to limit rates of lipolysis, and its global ablation leads to decreased lipid droplet sizes and enhanced thermogenesis (9).
In mouse liver, high fat feeding increases Them2 mRNA expression, but decreases steady-state levels of the protein modestly (10). Them2−/− mice are protected against diet-induced obesity and exhibit decreased adiposity, reduced hepatic glucose production, enhanced hepatic insulin sensitivity, and resistance to high fat diet-induced hepatic steatosis (10). In the setting of overnutrition, hepatic Them2 promotes the channeling of saturated FA into ER membrane phospholipids, which reduce membrane fluidity and promote calcium efflux into the cytosol (8). This activates the unfolded protein response, leading to increases in hepatic insulin resistance and glucose production (8). Although these observations underscore the key contributions of Them2 to the control of hepatic FA metabolism, it remains unclear how Them2 in the liver contributes to whole-body nutrient homeostasis.
To elucidate the metabolic roles of hepatic Them2 in vivo, we generated mice with liver-specific deletion of Them2. Our results demonstrate that, without influencing body weight, adiposity or glucose homeostasis, Them2 in liver promotes the hepatic secretion of triglyceride (TG)-enriched very low-density lipoprotein (VLDL) by channeling exogenous FA towards TG synthesis.
Material and Methods
Animals and diets
Liver specific Them2 knockout mice (L-Them2−/−) were created by a Cre/loxP strategy (11). To generate Them2 floxed mice, homozygous FLPeR mice B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/RainJ (Stock #009086; The Jackson Laboratory, Bar Harbor, ME, USA) were mated with homozygous B6Dnk;B6N-Acot13tm1a(EUCOMM)Wtsi/Ieg mice (EM:04103; European Mouse Mutant Archive, Munich, Germany), which contain a cassette composed of two loxP sites flanking Them2 exon 2. Duplex PCR was performed to distinguish wild type and Flox alleles using the following primers: 5’- TTATGAGTACATTGTAGCTCAGACAC-3’ (forward) and 5’- GTGCGCTACAACCATGATCTCT-3’ (reverse). Homozygous Them2 floxed mice were backcrossed two times to C57BL/6J mice (Stock #000664; The Jackson Laboratory). The established homozygous Them2 floxed mice were then crossed with heterozygous B6.Cg-Tg(Alb-cre)21Mgn/J (Stock #003574; The Jackson Laboratory) to yield L-Them2−/− mice (Them2Flox/Flox-Alb-creTg/Tg and Them2Flox/Flox-Alb-creTg/0), as well as littermate controls (Them2Flox/Flox-Alb-cre0/0). Mice were maintained on the mixed 6J/6N genetic background. The presence of Alb-cre allele was determined by PCR analysis using the primers specified by the Jackson Laboratory. Mice were housed in a barrier facility on a 12 h light/dark cycle. Male mice were weaned at 3–4 w of age and fed normal chow diet (PicoLab Rodent Diet 20; LabDiet, St. Louis, MO, USA). Alternatively, 5 w old male mice were fed a high fat diet (60 % calories from fat; Research Diets Inc., New Brunswick, NJ, USA) for 12 w. Following a 6 h fast, 17 w old mice were sacrificed and plasma was collected from cardiac puncture. Tissues were harvested for immediate use or snap frozen in liquid nitrogen and stored at −80oC. Animal use and euthanasia protocols were approved by Weill Cornell Medical College.
Primary hepatocyte isolation
Hepatocytes were isolated from 12 w old chow fed mice and cultured as previously described (7). Isolated hepatocytes were plated at the density of 4 × 105 cells on 35 mm Primaria plates (Corning, Tewksbury, MA, USA) with 2 mL Williams’ Medium E (Sigma-Aldrich) containing 10 % fetal bovine serum (FBS) and 1 % penicillin-streptomycin. Cells were allowed to attach overnight and used for experimental procedures within 24 h after plating.
Hepatic triglyceride secretion rates
Following a 5 h fasting, the lipoprotein lipase inhibitor Tyloxapol (500 mg/kg bw) (Sigma-Aldrich) was administered via retro-orbital injection (12). Tail tip blood samples (25 μL) were collected into microtubes containing EDTA prior to Tyloxapol injection and at regular intervals for up to 4 h. Plasma TG concentrations were determined using the enzymatic assay described above. Rates of hepatic TG secretion were calculated from the time-dependent linear increases in plasma TG concentration, assuming a plasma volume of 3.5 % of body weight (12).
Human liver samples
Liver biopsy specimens from subjects undergoing weight loss surgery were obtained from the NAFLD Biorepository of the Massachusetts General Hospital (Boston, MA, USA). Subjects underwent fasting blood samples within 8 months of their liver biopsy and the presence of comorbid diseases was assessed by their treating physician. Liver biopsies were reviewed by single blinded hepatopathologist and assigned a score for grade of steatosis, hepatocyte ballooning, and lobular inflammation (13). Steatosis was defined as the presence of >5 % steatosis without evidence of hepatocyte ballooning, lobular inflammation, or fibrosis. Nonalcoholic steatohepatitis (NASH) was defined as lobular inflammation ≥ 1, hepatocyte ballooning ≥ 1, and steatosis grade ≥1 (13). NAFLD activity score (NAS) was assessed for each subject. Patients gave informed consent at the time of recruitment and studies were approved by the Partners Health Care Human Research Committee.
Statistical analyses
Statistical significance was determined using two-tailed unpaired Student’s t-test or one-way ANOVA followed by Tukey’s post-test for comparisons between 2 or among 3 groups, respectively. Differences were considered significant at P < 0.05. All statistical analyses were performed using GraphPad Prism 7 (GraphPad Software). Data are presented as mean values with error bars representing S.E.M.
Results
Generation of mice lacking Them2 selectively in liver
To evaluate the hepatic function(s) of Them2, we generated L-Them2−/− mice, with littermates lacking Cre allele used as controls. Them2 protein was absent in livers of L-Them2−/− mice and unaffected in other tissues (Suppl. Fig. 1A). Them2 mRNA was undetectable in livers of L-Them2−/− mice and the expression of other Acot genes was largely unchanged (Suppl. Fig. 1B), suggesting that the loss of Them2 was not compensated by upregulation of other ACOTs. Moreover, long-chain acyl-CoA thioesterase activity in livers of L-Them2−/− mice exhibited a 39 % increase in apparent Km values for palmitoyl-CoA compared to control (Suppl. Fig. 1C). Whereas no genotype-related changes were observed in mRNA level of the Them2-interacting protein PC-TP (Suppl. Fig. 2A) (6), there was a 2-fold increase in steady-state protein level (Suppl. Fig. 2B).
In contrast to mice lacking Them2 globally (10), L-Them2−/− mice fed a regular chow diet exhibited similar sizes and body compositions as control mice (Fig. 1A and B). Nor were there differences in energy expenditure, ambulatory activity, food consumption or fecal caloric content between genotypes (Suppl. Fig. 3). Liver-specific deletion of Them2 did not affect glucose homeostasis compared with controls (Suppl. Fig 4A-D) notwithstanding modest differences in the mRNA and protein expression of rate-determining gluconeogenic enzymes (Suppl. Fig. 4E and F).
Figure 1. Liver- of Them2 reduces hepatic free fatty acid concentrations in chow fed mice.

(A) Body weights (Control, n = 5; L-Them2−/−, n = 18). (B) Fat and lean masses of 15 w old mice (Control, n = 5; L-Them2−/−, n = 18). (C) Liver masses of 17 w old mice (Control, n = 11; L-Them2−/−, n = 13). (D) Representative light microscopic images of H&E-stained liver sections. CV, central vein; PV, portal vein; BD, bile duct. (Control, n = 3; L-Them2−/−, n = 3). (E) Hepatic concentrations of acyl-CoA molecular species (Control, n = 5; L-Them2−/−, n = 6). (F) Hepatic concentrations of free fatty acids, triglycerides, phospholipids, and cholesterol (Control, n = 10; L-Them2−/−, n = 13 – 16). (G) Rates of [14C]palmitate uptake in primary cultured hepatocytes (Control, n = 4; L-Them2−/−, n = 4). (H) Hepatic activities of long-chain acyl-CoA synthetase (ACSL) (Control, n = 3; L-Them2−/−, n = 3). Data are presented as means ± SEM. *P < 0.05 versus control. ***P < 0.001 versus control.
Reduced free fatty acid concentrations in livers of L-Them2−/− mice
Livers from L-Them2−/− mice were normal in size and exhibited no histological changes compared to control animals (Fig. 1C and D). Liver-specific deletion of Them2 tended to increase (80 – 280 % increase, P = 0.05 – 0.22) a broad range of long-chain acyl-CoA species (Fig. 1E). In keeping with the accumulation of fatty acyl-CoA, hepatic free FA concentrations were reduced by 30 % in L-Them2−/− mice (Fig. 1F). However, steady-state concentrations of TG, phospholipids, free cholesterol, and cholesteryl-esters in the liver were unaffected (Fig. 1F).
No effect of Them2 expression on the hepatic uptake, activation and binding of fatty acids
To determine whether decreases in hepatic free FA concentrations could be a result of reduced rates of FA uptake, we measured FA transporter mRNA expression in livers, as well as rates of uptake of exogenous palmitate into primary cultured hepatocytes. Despite a 68 % increase in mRNA expression of Cd36 (Suppl. Fig. 5), no changes were observed in the rates of [14C]palmitate uptake by hepatocytes isolated from L-Them2−/− mice in comparison to control cells (Fig. 1G). Because reduced steady-state FA concentrations in livers of L-Them2−/− mice might also have resulted from increased conversion to fatty acyl-CoAs, we determined the influence of Them2 expression on hepatic ACSLs. Whereas the mRNA levels of Acsl4 and Acsl5 were upregulated in livers of L-Them2−/− mice by 21 and 37 %, respectively (Suppl. Fig. 5), hepatic ACSL activity remained unaffected (Fig. 1H). Because intracellular binding might have influenced interconversion rates of FA and fatty acyl-CoAs, we also measured the mRNA levels of key cytosolic lipid-binding proteins. There were no appreciable changes in mRNA expression of acyl-CoA-binding protein (Acbp) or fatty acid-binding protein 1 (Fabp1) (Suppl. Fig. 5). Collectively, these findings indicate that decreased concentrations of free FA and increased levels of long-chain acyl-CoA in the liver were attributable to the loss of hepatic Them2 activity.
Increased rates of fatty acid oxidation in livers of L-Them2−/− mice
Because FA and fatty acyl-CoAs derivatives are endogenous ligands for transcription factors that control nutrient metabolism (14, 15), we examined whether the observed changes in their concentrations altered hepatic genes related to lipid metabolism. Whereas mRNA levels of Pgc1α, Pparγ, and Srebp1c exhibited 40 – 61 % increases in the absence of hepatic Them2 expression, we did not observe systematic changes in the transcriptional profile of their downstream lipid-related gene targets (Suppl. Fig. 5).
We next determined whether changes in hepatic acyl-CoA and free FA concentrations directly influenced the control of major lipid metabolic pathways. Because cultured primary hepatocytes isolated from Them2−/− mice exhibit decreased rates of FA oxidation (7), we hypothesized that similar effects would be observed in livers of L-Them2−/− animals. In contrast to expectations, FA oxidation rates in L-Them2−/− livers were increased by 32 % compared with littermate controls (Fig. 2A). Hepatic TG hydrolase activity was 14 % lower in L-Them2−/− mice (Fig. 2B), indicating that increased oxidation of FA is not explained by increased mobilization of hepatic TG stores.
Figure 2. Increased rates of fatty acid oxidation in livers of chow fed L-Them2−/− mice.

(A) Rates of fatty acid oxidation in livers is represented as the sum of [14C]acid soluble metabolites and [14C]CO2 produced from [14C]palmitic acid. [14C]acid soluble metabolites production accounted for > 98 % of total [14C]palmitic acid degradation products. (Control, n = 6; L-Them2−/−, n = 8). (B) Hepatic triglyceride hydrolase activities (Control, n = 6; L-Them2−/−, n = 6). (C) Representative immunoblots of hepatic protein expression (left panel) and densitometric quantifications (middle and right panels). HSP90 was used to control for unequal loading (Control, n = 6; L-Them2−/−, n = 6). Data are presented as means ± SEM. *P < 0.05 versus control. **P < 0.01 versus control.
Mitochondrial β-oxidation is regulated by AMPK, which phosphorylates and inactivates acetyl-CoA carboxylase (ACC) leading to reduced concentrations of malonyl-CoA, an allosteric inhibitor of carnitine palmitoyltransferase 1 (CPT1) (16). As shown in Fig. 2C, no changes were observed in phosphorylated/total ratios of AMPK or CPT1 protein levels. Although hormone-sensitive lipase mRNA (Hsl) levels were decreased by 30 % (Suppl. Fig. 5), the ratios of phosphorylated/total HSL were not altered in livers of L-Them2−/− mice (Fig. 2C). These findings are supportive of a role of Them2 in the liver in limiting FA entry in oxidative pathways.
Reduced lipogenesis from exogenous fatty acids in hepatocytes lacking Them2
The observation that hepatic steady-state concentrations of complex lipids remained unchanged despite increased rates of FA oxidation in L-Them2−/− mice prompted us to evaluate whether lipid oxidation would be counteracted by decreased rates of lipogenesis. The first and rate-limiting step in the synthesis of glycerolipids is the esterification of long-chain acyl-CoA to glycerol-3-phosphate by the isoforms of glycerol-3-phosphate acyltransferase (GPAT) (17). Notwithstanding changes in mRNA levels of Gpat2 (77 % increase), Gpat3 (35 % decrease), as well as the downstream glycerolipid biosynthetic enzymes Lipin1 (2.6-fold increase), and diacylglycerol acyltransferase 2 (Dgat2) (36 % increase) in livers of L-Them2−/− mice (Suppl. Fig. 5), hepatic GPAT activity remained unchanged (Fig. 3A).
Figure 3. Decreased lipogenesis from exogenous fatty acids in Them2−/− hepatocytes.
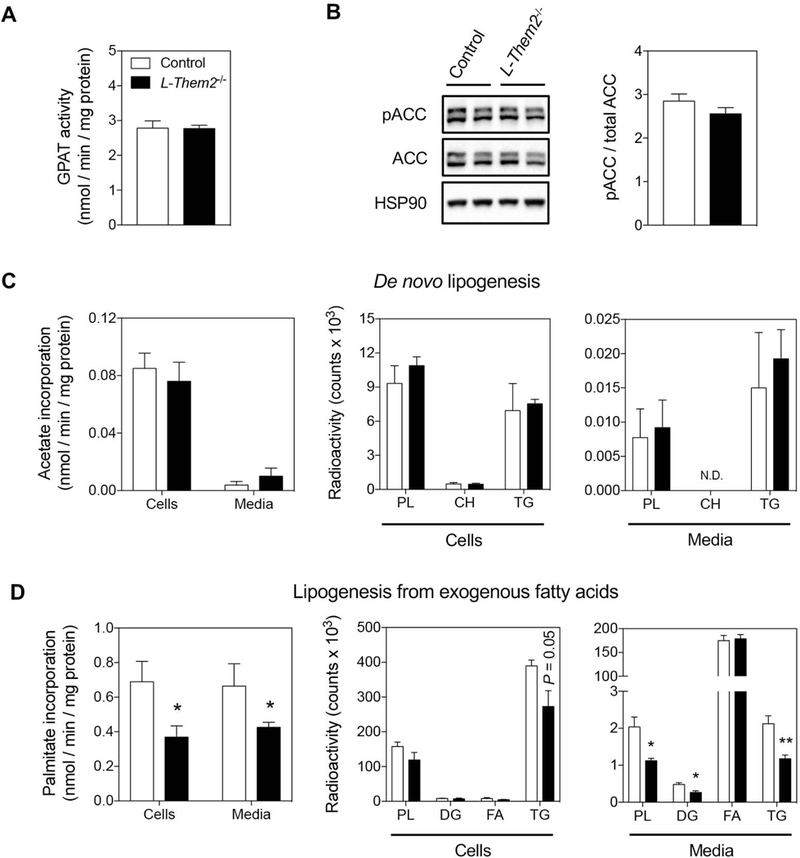
(A) Glycerol-3-phosphate acyltransferase (GPAT) activities in livers (Control, n = 4; L-Them2−/−, n = 4). (B) Representative immunoblots from mouse livers. HSP90 was used to control for unequal loading (Control, n = 6; L-Them2−/−, n = 6). (C and D) Rates of lipogenesis were determined in primary hepatocytes as the incorporation of (C) [14C]acetate (de novo lipogenesis) or (D) [14C]palmitate (lipogenesis from exogenous fatty acids) into lipids extracted from cells or culture media (left panels). Lipid classes were analyzed by TLC (middle and right panels). PL, phospholipid; CH, cholesterol; TG, triglyceride; DG, diglyceride; FA, fatty acid. (Control, n = 4; L-Them2−/−, n = 5). Data are presented as means ± SEM. *P < 0.05 versus control. **P < 0.01 versus control.
To distinguish between the esterification of endogenous and exogenous FA pools into complex lipids, an in vitro approach was used. Cultured primary hepatocytes from control and L-Them2−/− mice were incubated in the presence of [14C]acetate (a precursor for de novo FA synthesis) or [14C]palmitate, and the specific lipogenic activity was calculated from the radiolabeled substrates incorporated into cellular and secreted lipids (Fig. 3C and D). In keeping with the absence of changes in AMPK phosphorylation in livers of L-Them2−/− mice (Fig. 2C), there were no changes in phosphorylation of ACC (Fig. 3B), nor in mRNA levels of acetyl-CoA carboxylase (Acaca) and fatty acid synthase (Fasn) (Suppl. Fig. 5). And despite a 2.2-fold increase in stearoyl-CoA desaturase-1 (Scd1) (Suppl. Fig. 5), rates of de novo lipogenesis were unchanged in primary hepatocytes lacking Them2 (Fig. 3C). However, these cells exhibited a marked reduction (46 %) in the rates of lipogenesis derived from exogenous FA (Fig. 3D). This reduction was reflected by a 30 % decrease in esterification of [14C]palmitate to intracellular TGs, and by 45–48 % decreases in secretion of phospholipid, diacylglycerol and TG into the media (Fig. 3D). Collectively, these data suggest that a compensatory increase in hepatic FA oxidation rates occurs in L-Them2−/− mice in response to the accumulation of exogenously-derived fatty acyl-CoAs that are not esterified to form complex lipids.
Decreased rates of hepatic triglyceride secretion in L-Them2−/− mice
Current concepts suggest that endogenously synthesized FAs are primarily incorporated into TG that are stored in lipid droplets, whereas exogenously supplied FAs are esterified to TG molecules that are incorporated into VLDL (18–20). We therefore examined whether reduced lipogenesis derived from exogenous FA resulted in abnormal VLDL metabolism in L-Them2−/− mice. Liver-specific loss of Them2 resulted in a marked decrease (41 %) in the concentrations of TG in the plasma, whereas phospholipids, free cholesterol, cholesteryl-esters and free FA levels remained unchanged (Fig. 4A). There was a selective reduction (51 %) in VLDL-TG in L-Them2−/− mice, with no differences observed in the concentrations of LDL- and HDL-TG (Fig. 4B). Concurrently, liver-specific deletion of Them2 resulted in a 21 % decrease in steady-state plasma levels of apolipoprotein B100 (apoB100), the main protein component of VLDL, while apoB48, which is present in both VLDL and chylomicrons of rodents (21), remained unchanged (Fig. 4C). Indicative of reduced VLDL formation, L-Them2−/− mice exhibited a 22 % decrease in hepatic TG secretion rates (Fig. 4D).
Figure 4. Reduced VLDL-triglyceride secretion in chow fed L-Them2−/− mice.

(A) Plasma lipid concentrations (Control, n = 3–11; L-Them2−/−, n = 8 – 15). (B) Plasma lipoproteins were fractionated by FPLC and triglyceride concentrations were determined (Control, n = 6; L-Them2−/−, n = 5). (C) Representative immunoblots of plasma proteins (left panel) and densitometric quantifications (right panel). Coomassie staining was used to control for unequal loading (Control, n = 10 – 12; L-Them2−/−, n = 12). (D) Rates of triglyceride secretion were determined following retro-orbital administration of the lipoprotein lipase inhibitor Tyloxapol (500 mg/kg bw) (Control, n = 5; L-Them2−/−, n = 9). Data are presented as means ± SEM. *P < 0.05 versus control. **P < 0.01 versus control.
To ascertain the mechanisms underlying the reduction in VLDL secretion, we investigated the regulation of genes involved in VLDL biogenesis (1). mRNA levels of ApoB, microsomal triglyceride transfer protein (Mtp), Sec24c, and Sec24d were decreased in livers from L-Them2−/− mice by 14 – 24 %, whereas mRNA levels of Cidea and Sar1b were increased by 678 and 28 %, respectively (Fig. 5A). However, there were no differences in protein expression levels of Cidea, MTP, and Sar1b in liver homogenates (Fig. 5B). Under the current experimental conditions, we were unable to detect hepatic levels of apoB proteins in the liver, possibly owing to their rapid degradation due to misfolding or insufficient lipidation (22). Collectively, these findings indicate that decreased VLDL-TG secretion in L-Them2−/− mice is primarily attributable to the reduced availability of FA substrates for TG incorporation into VLDL particles.
Figure 5. No effect of hepatic Them2 on the expression of genes that control VLDL assembly and secretion.

(A) Relative mRNA expression of hepatic genes (Control, n = 4 – 6; L-Them2−/−, n = 4 – 7). (B) Representative immunoblots of hepatic proteins (left panel) and densitometric quantifications (right panel). HSP90 was used to control for unequal loading (Control, n = 6; L-Them2−/−, n = 6). Data are presented as means ± SEM. *P < 0.05 versus control. **P < 0.01 versus control. ***P < 0.001 versus control.
Reduced plasma VLDL-triglyceride concentrations in L-Them2−/− mice fed a high fat diet
We next examined the influence of hepatic Them2 expression on lipid metabolism in response to a high fat diet. Liver-specific deletion of Them2 did not influence growth, body composition or liver size (Fig. 6A-C). Similarly, there were no genotype-related changes in energy balance (Suppl. Fig. 3) or glucose homeostasis (Suppl. Fig. 4). Livers of high fat fed L-Them2−/− mice generally exhibited downregulation (17 – 48 %) of Acot genes compared with their high fat fed counterparts (Suppl. Fig. 6). Whereas hepatic concentrations of long-chain acyl-CoA species tended to increase (61 – 509 %, P = 0.08 – 0.19) (Suppl. Fig. 7A), there were no differences in steady-state concentrations of TG, free FA, phospholipids, free cholesterol or cholesteryl-esters in the liver (Fig. 6D and E, and Suppl. Fig. 7B). Additionally, there were no genotype-related changes in hepatic ACSL activity, FA oxidation, lipolysis or glycerolipid synthesis (Suppl. Fig. 7C-F).
Figure 6. Liver-specific deletion of Them2 reduces plasma VLDL-triglyceride concentrations in high fat fed mice.

(A) Body weights (Control, n = 6; L-Them2−/−, n = 15). (B) Fat and lean masses measured after 10 w of high fat feeding (Control, n = 6; L-Them2−/−, n = 15). (C) Liver masses measured after 12 w of high fat feeding (Control, n = 9; L-Them2−/−, n = 11). (D) Representative light microscopic images of H&E-stained liver section. CV, central vein; PV, portal vein; BD, bile duct. (Control, n = 3; L-Them2−/−, n = 3). (E) Hepatic triglyceride concentrations (Control, n = 10; L-Them2−/−, n = 16). (F) Plasma lipid concentrations (Control, n = 6–12; L-Them2−/−, n = 11–17). (G) Plasma lipoproteins were fractionated by FPLC and triglyceride concentrations were determined (Control, n = 3; L-Them2−/−, n = 3). (H) Rates of triglyceride secretion were determined following retro-orbital administration of the lipoprotein lipase inhibitor Tyloxapol (Control, n = 8; L-Them2−/−, n = 6). Data are presented as means ± SEM. *P < 0.05 versus control. **P < 0.01 versus control.
The differences observed in VLDL-TG metabolism in L-Them2−/− mice fed a regular chow diet prompted us to evaluate whether similar changes would persist in the setting of overnutrition. High fat fed L-Them2−/− mice exhibited a 44 % decrease in steady-state plasma TG concentrations, which was accompanied by 23 % and 25 % increases in the plasma concentrations of phospholipids and cholesteryl-esters, respectively (Fig. 6F). The differences in plasma TG concentrations were attributable to reduced VLDL-TG concentrations (Fig. 6G). This occurred in the absence of appreciable alterations steady-state plasma concentrations of apoB100 and apoB48 (Fig. 6H) or the hepatic expression of proteins involved in VLDL assembly and secretion (Suppl. Fig. 8). In keeping with a role of Them2 in channeling FA towards glycerolipid biosynthesis and secretion, we observed a 24 % decrease in the rates of VLDL-TG secretion in L-Them2−/− mice (Fig. 6I). The absence of an increase in steady-state hepatic TG concentrations in the setting of reduced rates VLDL-TG secretion was not attributable to a compensatory increase in rates of FA oxidation (Suppl. Fig. 7D). Rather, we observed 17 – 40 % decreases in mRNA levels for key transcription factors that promote lipogenesis, including both Chrebp isoforms, Lxra, and Srebp1c (Suppl. Fig. 6). There were also trends towards decreased Acaca expression (20 % decrease, P = 0.1) (Suppl. Fig. 6) and increased phosphorylation of ACC (20 % increase, P = 0.08) (Suppl. Fig. 7G). These findings suggest that, under conditions of high fat feeding, the secretory defect of VLDL-TG that is observed in the absence of Them2 is compensated by reduced de novo lipogenesis.
Increased levels of THEM2 protein in human livers with steatosis
To gain insights into the potential role of THEM2 in human hepatic lipid metabolism, we analyzed liver samples from obese subjects with normal livers, simple steatosis or NASH (Suppl. Table 4). Whereas no significant changes were observed in THEM2 mRNA levels (Fig. 7A), THEM2 protein was increased in livers with steatosis compared with normal livers or NASH (60 % and 46 % increase, respectively) (Fig. 7B). The hypertriglyceridemia that is commonly associated with NAFLD has a complex pathophysiology that is generally attributable to a combination of hepatic overproduction of VLDL particles and decreased rates of lipoprotein lipase-mediated intravascular metabolism (23). The finding of increased hepatic THEM2 expression in the setting of hepatic steatosis is consistent with its observed role in VLDL production in mice. However, in the small sample size after exclusion of patients taking lipid-lowering therapies, we were unable to discern a correlation between THEM2 expression in liver and plasma TG concentrations (Fig. 7C).
Figure 7. Increased THEM2 expression levels in livers of obese subjects with simple steatosis but not NASH.

(A) Relative human THEM2 mRNA expression levels in livers from obese patients (Normal, n = 6; Steatosis, n = 6; NASH, n = 5). (B) Representative immunoblots (left panel) and densitometric quantifications (right panel) of human liver proteins. β-ACTIN was used to control for unequal loading (Normal, n = 5; Steatosis, n = 6; NASH, n = 6). (C) Correlation between hepatic THEM2 expression and plasma triglyceride concentrations for patients not receiving lipid lowering medications (Normal, n = 4; Steatosis, n = 4; NASH, n = 4). Data are presented as means ± SEM. *P < 0.05 versus other groups.
Discussion
This study has identified a key function of Them2 in the liver in the control of FA metabolism, which in turn regulates plasma VLDL-TG concentrations. Our current findings support a model in which Them2 in the liver directs plasma-derived FA towards the synthesis of TG for incorporation into VLDL particles, leading to increased VLDL-TG secretion (Fig. 8).
Figure 8. Proposed model for the role of Them2 in hepatic VLDL secretion.

In hepatocytes, free fatty acids (FAs) obtained from the plasma or de novo synthesis are activated to form fatty acyl-CoAs by long-chain acyl-CoA synthetases (ACSLs). At the mitochondria, Them2 selectively hydrolyzes plasma-derived long-chain acyl-CoA. Under fasting conditions, newly-formed FAs may be directed to mitochondrial ACSLs, which in turn traffic fatty acyl-CoAs towards the glycerol-3-phosphate pathway for diacylglycerol acyltransferase 1 (DGAT1)-mediated triglyceride (TG) synthesis. Newly-formed TGs are then packaged into very-low density lipoprotein (VLDL) particles and secreted into plasma. By this mechanism, Them2 sustains the export of triglycerides from the liver into plasma during fasting. In the setting of overnutrition (not shown) when the flux of FA from adipose tissue to the liver and rates of hepatic de novo lipogenesis are high, Them2-mediated increases in VLDL production rates may serve to mitigate, at least in part, the intrahepatic accumulation of excess TG. Relative magnitude of flux is represented by line thickness.
We previously demonstrated that Them2−/− mice exhibit decreased body weights along with enhanced energy expenditures, despite increases in caloric intake (9, 10). This was attributed, at least in part, to roles for Them2 in restricting FA trafficking to mitochondria for oxidation and thermogenesis in BAT (9). By these mechanisms, Them2 functions within BAT to limit adaptive thermogenesis (9). In addition, studies in vitro have demonstrated that Them2 expression in hepatocytes suppresses insulin signaling, while promoting FA oxidation and gluconeogenesis (6–8, 10). Based on these observations, we ascribed the enhanced insulin sensitivity and improved glucose homeostasis observed in Them2−/− mice primarily to functions of Them2 within the liver (6–8, 10). However, our current findings indicate that the liver-specific loss of Them2 is insufficient to recapitulate the changes in energy or glucose homeostasis caused by systemic Them2 ablation, either in chow or a high fat fed mice (10), instead implicating Them2 expression in extrahepatic tissue(s) as the main driver of these phenotypes. Although increased BAT thermogenesis presumably contributes to improving hepatic metabolism, we have observed that the selective ablation of Them2 in skeletal or cardiac muscle reduces basal blood glucose levels and prevents diet-induced glucose intolerance (24). It is also possible that distinct genetic backgrounds of L-Them2−/− compared with Them2−/− mice (6–8, 10) could have contributed to the observed differences (25, 26).
Mice with liver-specific disruption of Them2 revealed unanticipated functions of this enzyme in VLDL metabolism. There were marked reductions in VLDL-TG secretion both in chow and high fat fed L-Them2−/− mice, which resulted in decreased steady-state plasma TG concentrations. In support of a role of hepatic Them2 in the hydrolysis of fatty acyl-CoAs in vivo, chow fed L-Them2−/− mice exhibited increases in the hepatic concentrations of long-chain acyl-CoA species, along with decreased intracellular free FA concentrations. The blunting of this effect in high fat fed animals most likely reflects contributions from other sources, such as increased hepatic uptake of plasma adipose tissue-derived free FA in the context of diet-induced insulin resistance (1), and the downregulation of other Acot isoforms in the livers of high fat fed L-Them2−/− mice, which presumably reflects an adaptive response that prevented the accumulation of more toxic free FA molecules in the liver.
The synthesis of TG and other glycerolipids occurs via the glycerol-3-phosphate pathway in most mammalian cell types (17). Although Them2 expression had no impact on hepatic GPAT activity, the initial and rate-limiting step in glycerolipid synthesis (17), reduced rates of glycerolipid synthesis were observed in primary hepatocytes lacking Them2 when exogenous FA, but not endogenous FA, were available as the substrate. In the absence of changes in expression levels of proteins that control VLDL assembly or secretion, the most likely mechanism whereby Them2 regulates VLDL secretion is by controlling the availability of TG for the lipidation of apoB and assembly into nascent VLDL particles. This is consistent with observations in mice and humans that plasma FA are the principal source of VLDL-TG in both fasting and fed states (27). In further support of this hypothesis, studies of DGAT enzymes, which catalyze the final step in TG synthesis, have revealed that different pools of FAs experience different metabolic fates with respect to TG synthesis (19, 20). Whereas DGAT2 incorporates fatty acyl-CoA molecules derived from de novo synthesis into TG for storage in cytoplasmic lipid droplets, DGAT1 exhibits substrate preference for exogenously-derived FA, which are utilized for the synthesis of TG that are incorporated into VLDL particles (19). Based on our current findings, we propose that Them2 acts upstream of GPAT isoforms and DGAT1 to provide a selected pool of exogenous FA for incorporation into VLDL and secretion from the liver into the plasma (Fig. 8). Because DGAT1 is also able to utilize diacylglycerol and fatty acyl-CoA generated from the hydrolysis of lipid droplet-associated TG, albeit to a lesser extent (19), it is also possible that the reduced rates of lipid droplet lipolysis in chow fed L-Them2−/− mice have contributed to the observed reductions in VLDL-TG secretion rates.
Another potential mechanism whereby Them2 controls VLDL secretion is through the activity of mTORC1. Postprandial activation of hepatic mTORC1 promotes VLDL-TG secretion by stimulating phosphocholine cytidylyltransferase α (CCTα) activity, the rate-limiting step in the synthesis of phosphatidylcholines (PC), which are utilized as the amphipathic molecules that stabilize the TG core of VLDL (28). In this connection, in cultured mouse hepatocytes under nutrient restriction, changes in PC composition of cellular membranes lead to the formation of Them2/PC-TP complex that directly suppresses mTORC1 signaling by interacting with tuberous sclerosis complex 2 (TSC2) to stabilize the TSC1-TSC2 complex (6). However, when FA are abundant, the Them2/PC-TP complex functions to promote the synthesis of PC for ER membranes (8). Taken together, these findings provide a plausible mechanism by which Them2/PC-TP interactions are modulated in response to nutritional status and may function to: (i) sustain PC synthesis and facilitate the lipidation of nascent VLDL particles within the ER when lipid substrates are available, and (ii) suppress mTORC1 signaling, leading to reduced CCTα activity and decreased VLDL secretion upon more severe nutrient deprivation, potentially contributing to the hepatic steatosis associated with prolonged fasting (26).
A reduced capacity to secrete VLDL owing to genetic mutations/polymorphisms (29–31) or pharmacologic interventions (32) can contribute to hepatic steatosis. Notwithstanding, the marked reduction in VLDL-TG secretion observed in L-Them2−/− mice did not cause excess lipid accumulation within the liver because FA were oxidized rather than stored as TG in lipid droplets in chow fed animals, whereas de novo FA synthesis appeared to be suppressed in the setting of overnutrition. In humans with hepatic steatosis, increases in free FA flux from adipose tissue to the liver and in the rates of hepatic de novo lipogenesis stimulate the production and secretion of VLDL particles (33, 34). In the absence of compensatory increases in rates of FA oxidation, the synthesis of hepatic TG most likely exceeds the capacity of increased VLDL production rates to prevent intrahepatic lipid accumulation (33, 34). Suggestive of a role in the compensatory increases in VLDL production, THEM2 levels were highest in livers of obese subjects with steatosis. In support of this possibility, hepatic Them2 levels were shown to be markedly reduced in rats following a gastric bypass surgery, which ultimately leads to decreased body weight and decreased hepatosteatosis (35). The observation that THEM2 expression returned to basal levels in livers of NASH patients may reflect an inflammation-induced failure of this compensatory mechanism.
The current findings define a role for Them2 in the liver in the trafficking of plasma-derived FA towards VLDL production. When taken in context with the improvements in hepatic steatosis and glucose homeostasis associated with its systemic disruption, Them2 may be an attractive target for the management of NAFLD and diet-induced dyslipidemia.
Supplementary Material
Acknowledgements
The authors thank Corey Holman and Curtis J. Bare (Weill Cornell Medical College) for assistance with the metabolic monitoring experiments, and Stephanie A. Osganian (Massachusetts General Hospital) for assistance with the human liver samples.
Financial Support: This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases grants R37 DK048873 and R01 DK056626 to D.E.C., and K23 DK099422 to K.E.C., a Georgia Clinical & Translational Science Alliance of the National Institutes of Health grant UL1TR002378 to E.A.O., and an American Heart Association postdoctoral fellowship 18POST33990445 to M.A-B.. M.A-B. was also the recipient of an American Liver Foundation Non-Alcoholic Steatohepatitis Fatty Liver Disease Postdoctoral Research Fellowship Award. This work was supported in part by the Weill Cornell Metabolic Phenotyping Core and by the Emory Integrated Lipidomics Core, which is subsidized by the Emory University School of Medicine.
Abbreviations:
- ACC
acetyl-CoA carboxylase
- ACOT
acyl-CoA thioesterase
- ACSL
long-chain acyl-CoA synthetase
- ApoB
apolipoprotein B
- BAT
brown adipose tissue
- CCTα
phosphocholine cytidylyltransferase α
- CPT1
carnitine palmitoyltransferase 1
- DGAT
diacylglycerol acyltransferase
- FA
fatty acid
- GPAT
glycerol-3-phosphate acyltransferase
- HSL
hormone-sensitive lipase
- MTP
microsomal triglyceride transfer protein
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PC
phosphatidylcholine
- PC-TP
phosphatidylcholine transfer protein
- TG
triglyceride
- Them2
thioesterase superfamily member 2
- VLDL
very low-density lipoprotein
Footnotes
Additional methods are described in Supplementary Information.
Conflict of Interest: The authors declare no competing interests.
References
- 1.Alves-Bezerra M, Cohen DE. Triglyceride metabolism in the liver. Compr Physiol 2017;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grevengoed TJ, Klett EL, Coleman RA. Acyl-CoA metabolism and partitioning. Annual Review of Nutrition 2014;34:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tillander VE, Alexson SE, Cohen DE. Deactivating fatty acids: acyl-CoA thioesterase-mediated control of lipid metabolism. Trends in Endocrinology and Metabolism 2017;28:473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei J, Kang HW, Cohen DE. Thioesterase superfamily member 2 (Them2)/acyl-CoA thioesterase 13 (Acot13): a homotetrameric hotdog fold thioesterase with selectivity for long-chain fatty acyl-CoAs. Biochemical Journal 2009;421:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanno K, Wu MK, Agate DA, Fanelli BK, Wagle N, Scapa EF, Ukomadu C, et al. Interacting proteins dictate function of the minimal START domain phosphatidylcholine transfer protein/StarD2. Journal of Biological Chemistry 2007;282:30728–30736. [DOI] [PubMed] [Google Scholar]
- 6.Ersoy BA, Tarun A, D’Aquino K, Hancer NJ, Ukomadu C, White MF, Michel T, et al. Phosphatidylcholine transfer protein interacts with thioesterase superfamily member 2 to attenuate insulin signaling. Sci Signal 2013;6:ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawano Y, Ersoy BA, Li Y, Nishiumi S, Yoshida M, Cohen DE, Jul;34(13):2396–408 MCB. Thioesterase superfamily member 2 (Them2) and phosphatidylcholine transfer protein (PC-TP) interact to promote fatty acid oxidation and control glucose utilization. Molecular and Cellular Biology 2014;34:2396–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ersoy BA, Maner-Smith KM, Li Y, Alpertunga I, Cohen DE. Thioesterase-mediated control of cellular calcium homeostasis enables hepatic ER stress. J Clin Invest 2018;128:141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang HW, Ozdemir C, Kawano Y, LeClair KB, Vernochet C, Kahn CR, Hagen SJ, et al. Thioesterase superfamily member 2/Acyl-CoA thioesterase 13 (Them2/Acot13) regulates adaptive thermogenesis in mice. Journal of Biological Chemistry 2013;288:33376–33386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang HW, Niepel MW, Han S, Kawano Y, Cohen DE. Thioesterase superfamily member 2/acyl-CoA thioesterase 13 (Them2/Acot13) regulates hepatic lipid and glucose metabolism. FASEB Journal 2012;26:2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kos CH. Cre/loxP system for generating tissue-specific knockout mouse models. Nutr Rev 2004;62:243–246. [DOI] [PubMed] [Google Scholar]
- 12.Tietge UJ, Bakillah A, Maugeais C, Tsukamoto K, Hussain M, Rader DJ. Hepatic overexpression of microsomal triglyceride transfer protein (MTP) results in increased in vivo secretion of VLDL triglycerides and apolipoprotein B. J Lipid Res 1999;40:2134–2139. [PubMed] [Google Scholar]
- 13.Corey KE, Misdraji J, Gelrud L, Zheng H, Chung RT, Krauss RM. Nonalcoholic steatohepatitis is associated with an atherogenic lipoprotein subfraction profile. Lipids Health Dis 2014;13:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jump DB, Tripathy S, Depner CM. Fatty acid-regulated transcription factors in the liver. Annual Review of Nutrition 2013;33:249–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura MT, Yudell BE, Loor JJ. Regulation of energy metabolism by long-chain fatty acids. Progress in Lipid Research 2014;53124–144. [DOI] [PubMed] [Google Scholar]
- 16.Foster DW. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest 2012;122:1958–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coleman RA, Mashek DG. Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chemical Reviews 2011;111:6359–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villanueva CJ, Monetti M, Shih M, Zhou P, Watkins SM, Bhanot S, Farese RV Jr. Specific role for acyl CoA:Diacylglycerol acyltransferase 1 (Dgat1) in hepatic steatosis due to exogenous fatty acids. Hepatology 2009;50:434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wurie HR, Buckett L, Zammit VA. Diacylglycerol acyltransferase 2 acts upstream of diacylglycerol acyltransferase 1 and utilizes nascent diglycerides and de novo synthesized fatty acids in HepG2 cells. FEBS Journal 2012;279:3033–3047. [DOI] [PubMed] [Google Scholar]
- 20.Qi J, Lang W, Geisler JG, Wang P, Petrounia I, Mai S, Smith C, et al. The use of stable isotope-labeled glycerol and oleic acid to differentiate the hepatic functions of DGAT1 and −2. J Lipid Res 2012;53:1106–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blanc V, Xie Y, Luo J, Kennedy S, Davidson NO. Intestine-specific expression of Apobec-1 rescues apolipoprotein B RNA editing and alters chylomicron production in Apobec1−/− mice. J Lipid Res 2012;53:2643–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher E, Lake E, McLeod RS. Apolipoprotein B100 quality control and the regulation of hepatic very low density lipoprotein secretion. J Biomed Res 2014;28:178–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen DE, Fisher EA. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin Liver Dis 2013;33:380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imai N, Cohen DE. Muscle-specific metabolic regulation by thioesterase superfamily member 2 in the pathogenesis of nonalcoholic fatty liver disease. Hepatology 2018;68:738. [Google Scholar]
- 25.Andrikopoulos S, Massa CM, Aston-Mourney K, Funkat A, Fam BC, Hull RL, Kahn SE, et al. Differential effect of inbred mouse strain (C57BL/6, DBA/2, 129T2) on insulin secretory function in response to a high fat diet. J Endocrinol 2005;187:45–53. [DOI] [PubMed] [Google Scholar]
- 26.Geisler CE, Hepler C, Higgins MR, Renquist BJ. Hepatic adaptations to maintain metabolic homeostasis in response to fasting and refeeding in mice. Nutr Metab (Lond) 2016;13:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. Journal of Clinical Investigation 2005;115:1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinn WJ 3rd, Wan M, Shewale SV, Gelfer R, Rader DJ, Birnbaum MJ, Titchenell PM mTORC1 stimulates phosphatidylcholine synthesis to promote triglyceride secretion. J Clin Invest 2017;127:4207–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raabe M, Veniant MM, Sullivan MA, Zlot CH, Bjorkegren J, Nielsen LB, Wong JS, et al. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J Clin Invest 1999;103:1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol 2014;25:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. Journal of Biological Chemistry 2016;291:10659–10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cuchel M, Rader DJ. Microsomal transfer protein inhibition in humans. Curr Opin Lipidol 2013;24:246–250. [DOI] [PubMed] [Google Scholar]
- 33.Adiels M, Taskinen MR, Packard C, Caslake MJ, Soro-Paavonen A, Westerbacka J, Vehkavaara S, et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 2006;49:755–765. [DOI] [PubMed] [Google Scholar]
- 34.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 2010;51:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sridharan GV, D’Alessandro M, Bale SS, Bhagat V, Gagnon H, Asara JM, Uygun K, et al. Multi-omic network-based interrogation of rat liver metabolism following gastric bypass surgery featuring SWATH proteomics. Technology (Singap World Sci) 2017;5:139–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.