

Abstract
Glycosaminoglycans (GAGs) are very complex, natural anionic polysaccharides. They are polymers of repeating disaccharide units of uronic acid and hexosamine residues. Owing to their template-free, spatiotemporally-controlled, and enzyme-mediated biosyntheses, GAGs possess enormous polydispersity, heterogeneity, and structural diversity which often translate into multiple biological roles. It is well documented that GAGs contribute to physiological and pathological processes by binding to proteins including serine proteases, serpins, chemokines, growth factors, and microbial proteins. Despite advances in the GAG field, the GAG–protein interface remains largely unexploited by drug discovery programs. Thus, non-saccharide glycosaminoglycan mimetics (NSGMs) have been rationally developed as a novel class of sulfated molecules that modulate GAG–protein interface to promote various biological outcomes of substantial benefit to human health. In this review, we describe the chemical, biochemical, and pharmacological aspects of recently reported NSGMs and highlight their therapeutic potentials as structurally and mechanistically novel anti-coagulants, anti-cancer agents, anti-emphysema agents, and anti-viral agents. We also describe the challenges that complicate their advancement and describe ongoing efforts to overcome these challenges with the aim of advancing the novel platform of NSGMs to clinical use.
Keywords: Glycosaminoglycans, Sulfated molecules, Non-saccharide glycosaminoglycan mimetics, Anticoagulants, Anticancers, Antivirals
1. INTRODUCTION
Glycosaminoglycans (GAGs) are complex, linear polysaccharides that are generally made up of repeating units of uronic acid (D-glucuronic/L-iduronic acid) and hexosamine (D-glucosamine/D-galactosamine. Although they are present on essentially all animal cell surfaces, GAGs can be found intracellularly as well as extracellularly. With one exception, they are usually covalently associated with proteins to form proteoglycans. Owing to their carboxylate and sulfate groups, GAGs are highly negatively charged. GAGs are also a highly diverse class of molecules which differ from one another in the type of monomeric units, linkages between these monomeric units, position of sulfate groups, and degree of sulfation. These differences arise from GAG biosynthesis which is very complex to the extent that even a particular class of GAGs ends up as a complex heterogeneous mixture of species with different molecular weights [1–4].
Considering components of the disaccharide repeating units and the type of the glycosidic bonds involved, there are four types of GAGs which are heparin/heparan sulfate, chondroitin/dermatan sulfates (A, B, C, D, and E), hyaluronic acid/hyaluronan, and keratan sulfates (I and II) (Figure 1) [1,4]. Heparin and heparan sulfate are similar GAGs having the same disaccharide repeating units (D-glucuronic acid and L-iduronic acid as the uronic acid units and glucosamine as the hexosamine unit) and the glycosidic bond of β-(1–4). However, in heparin, L-iduronic acid is the predominant uronic acid residue whereas D-glucuronic acid is the most abundant uronic acid residue in heparan sulfate, and this ultimately results in heparin being significantly more sulfated than heparan sulfate. In fact, heparin is the most highly charged and most acidic biomacromolecule known in human physiology [5–7]. Chondroitin sulfates A, C, D and E are predominantly composed of the disaccharide repeating unit of D-glucuronic acid and N-aceylgalactosamine residues. Chondroitin sulfate B (also known as dermatan sulfate) is predominantly composed of the disaccharide repeating unit of L-iduronic acid and N-aceylgalactosamine residues. Chondroitin sulfates A, C, D, and E have alternating β-(1–4) and β-(1–3)-glycosidic bonds whereas chondroitin sulfate B possesses the alternating glycosidic bonds of β-(1–4) and α-(1–3). Chondroitin sulfates predominantly have the sulfate groups at position-4 (as in A and B) or position-6 (as in C) of the galactosamine residues, however, chondroitin sulfates D and E possess additional sulfate groups and are known as oversulfated chondroitin sulfates. Chondroitin sulfate D possesses sulfate groups at positon-2 of the glucuronic acid residue and position-6 of the galactosamine residue. Chondroitin sulfate E has its sulfate groups at positions-4 and −6 of the galactosamine residue [8]. Hyaluronic acid, the only GAG that is not covalently linked to proteins, has the disaccharide repeating unit of D-glucuronic acid and N-acetylglucosamine linked by the alternating β-(1–4) and β-(1–3)-glycosidic bonds. It is also the only GAG that does not have sulfate moieties, and thus, it owes its negative charge to the carboxylate groups, making it the simplest and least charged among all GAGs [4,9]. For keratan sulfate, there are at least two types known: type I which is found in the cornea and type II which is found in skeletal tissues. The first type has D-galactose and D-glucosamine-6-O-sulfate as the repeating disaccharide unit whereas the second type involves D-galactose and D-galactosamine-6-O-sulfate as the repeating disaccharide unit [4,10].
Figure 1.

The chemical structures of the predominant repeating disaccharide units and the glycosidic bonds based on which GAGs have been classified into heparin/heparan sulfate, chondroitin sulfates, hyaluronic acid/hyaluronan, and keratan sulfates. Heparin is the most acidic biomacromolecule in human physiology whereas hyaluronic acid is the only GAG that lacks sulfate groups. The chemical structures of keratan sulfates are not shown.
At the molecular level, GAGs are known to interact with several proteins and receptors, and thus, modulate a host of biological processes. By interacting with coagulation enzymes and their physiological inhibitors, GAGs have an important role in the coagulation process [11]. It is worth mentioning that the use of GAGs for therapeutic purposes started with the use of heparin as an anticoagulant in 1935 following its discovery in 1916 [12]. GAGs are also known to interact with growth factors and their receptors, and thus, affect processes such as cell growth, proliferation, and migration among others [13]. These interactions also make certain GAGs important players in cancer biology where they are involved in the initiation and growth of tumor cells [14, 15]. Interestingly, other GAG–protein interactions are known to drive tumor suppression [15]. Inflammation is another process known to be modulated by GAGs [16,17]. GAGs including heparin/heparan sulfate, chondroitin sulfates, and hyaluronic acid have been shown to reduce inflammation in a number of different cell types by interacting with chemokines, selectins, cytokines, and proteases [18–20]. GAGs have also been shown to be involved in the pathogenesis of infectious diseases via their direct interaction with viruses, bacteria, fungi and parasites [21, 22]. For example, by interacting with these pathogens, GAGs facilitate the viral invasion of host cells as well as the spread of infection. Enveloped viruses in particular have been shown to attach to their target cells with the aid of heparan sulfate [23, 24]. Conversely, other GAGs including heparin have been found to inhibit viruses including herpes simplex virus (HSV) and human immunodeficiency virus (HIV) due to their structural similarity to heparan sulfate [25, 26]. Accordingly, the numerous biological functions of GAGs qualify them to be at the center of many drug discovery programs for the development of novel treatments for various human diseases [27].
2. GAG–PROTEIN INTERFACE AS A PLATFORM FOR DRUG DISCOVERY
To date, hundreds of GAG-binding proteins have been identified. These include serine proteases, serine protease inhibitors (serpins), growth factors, lipolytic enzymes, extracellular matrix proteins, viral coat proteins, and transcription factors [28, 29]. GAG-binding proteins usually have clusters of basic amino acids on their surface, a feature that has commonly been employed in the prediction of GAG-binding proteins and GAG-binding sites [30]. The clusters of basic amino acids are interspaced by hydrophobic patches which introduce differences in the topology of GAG-binding sites from one GAG-binding protein to another (Figure 2), and ultimately determines the type and the chain length of binding GAGs [30]. For instance, a protein surface with widely spaced cationic residues may have better interaction with a GAG sequence with a lower sulfation level, such as heparan sulfate, than with a highly sulfated GAG like heparin, and vice versa [31]. Due to their highly charged nature, GAGs generally interact with a number of their binding partners using non-specific cooperating electrostatic binding, which is dependent on charge density of the GAGs [1–4]. These interactions are between the highly acidic sulfate groups and the basic side chains of Arg and Lys, and in certain cases His residues that are exposed on the protein surface [31]. Of these basic amino acids, the electrostatic interaction with Arg residues is the strongest (~2.5-fold greater than that with Lys) [32, 33]. Figure 3 depicts the interactions between the sulfate groups of a heparin pentasaccharide sequence known as DEFGH 1 and the Lys and Arg residues in the heparin-binding site of antithrombin (ATIII) [34].
Figure 2.

Cartoon representations and electrostatic potential surface maps of GAG-binding sites of four serine proteases [plasmin (3UIR) (A), thrombin (1XMN) (B), factor Xa (2GD4) (C), and factor XIa (1ZHM) (D)]. Basic residues in each site are shown as spheres (carbon atom is depicted in green color) and the active site serine is shown as spheres (orange color). The electrostatic potential surface was calculated using APBS tool. Electropositive surface is coded blue, while electronegative surface is in red. These sites are targeted by different sulfated NSGMs. The maps exhibit strong positive charge density for all four proteases but with significant structural differences, which justify the potential of developing selective NSGM-based inhibitors.
Figure 3.
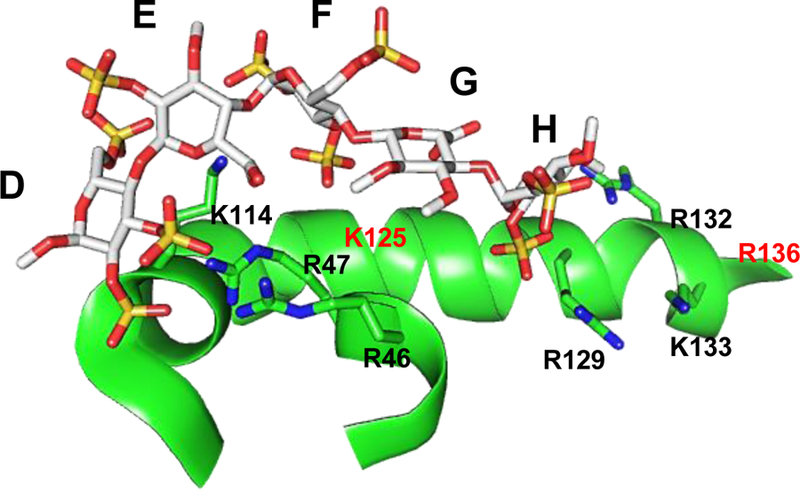
The crystal structure of heparin pentasaccharide DEFGH binding to the heparin-binding site on ATIII (PDB ID: 1E03) showing the interacting partners i.e. the negatively charged groups of sulfate/carboxylate of DEFGH with the positively charged amino acids of ATIII i.e. Arg and Lys residues.
As far as the nature of the GAG-binding sites and the types of interaction involved between GAGs and their protein partners, Cardin and Weintraub postulated the existence of consensus sequences for GAG recognition and identified two patterns of amino acid sequences for GAG-binding sites, XBBBXXBX and XBBXBX, where B and X are basic and non-basic amino acids, respectively [35]. Subsequently, Sobel et al. identified another consensus sequence, XBBBXXBBBXXBBX [36]. Further, Margalit et al. suggested that a 20 Å separation between certain basic amino acid residues was important for the interaction of GAG-binding proteins with GAGs [37]. The contribution of charged interactions to the overall binding energy of GAGs to GAG-binding proteins varies greatly from one GAG–protein interaction to the other. In some cases, the contribution of ionic interactions to the total binding energy has been found to be as high as 85% [38], whereas in other cases, non-ionic interactions such as van der Waals forces, hydrogen bonding, and hydrophobic interactions, greatly outweigh contributions from ionic interactions [39]. A typical example is the binding of brain natriuretic peptide to heparin, where the ionic component of the interaction was found to be only 6% of the total binding energy [32]. This, however, does not suggest that these GAG–protein interactions would be possible in the absence of the negatively charged groups on the GAGs, as charge is important for steering the interactions. Importantly, the variability in the contribution of various types of interactions to GAG–protein binding has contributed to our understanding of the specificity component of GAG–protein interactions. The following section provides details about specific GAG–protein interactions which have been used to discover, design, and develop GAG mimetics with potential to treat various pathological conditions.
2.A). SERINE PROTEASE INHIBITORS (SERPINS)
Serpins are the natural inhibitors of serine proteases and include α1-antitrypsin, antithrombin III (ATIII), and heparin cofactor II (HCII) among others [40]. Serpins play a vital role in maintaining homeostasis in several very important physiological processes including inflammation, coagulation, and digestion [41]. The unique mechanism of inhibition utilized by serpins involves a significant conformational change which results in the reactive site loop, a sequence of amino acids of the serpin, interacting with the active site serine of the protease. Cleavage of the loop and its insertion into the active site of the protease results in its irreversible inhibition [42, 43]. The most studied GAG–protein interaction is that of the heparins with ATIII. ATIII is a natural inhibitor of several serine proteases in the coagulation cascade including thrombin, factor IXa (FIXa), factor Xa (FXa), factor XIa (FXIa), and factor XIIa (FXIIa) [29]. Heparins exert their anticoagulant activities by activating ATIII and to some extent HCII, and thus accelerating their inhibition of the coagulation proteases. Specifically, ATIII inhibits thrombin and FXa slowly in the absence of heparin, however, the inhibition rates are increased by more than 300-fold in the presence of heparin [44]. This is the rationale underlying the clinical use of heparins [42, 44–46].
In the case of FXa, a specific pentasaccharide sequence in heparin, known as DEFGH 1 (Figures 3 and 4) was found to be sufficient to bring about clinically relevant ATIII-mediated inhibition [47]. Mechanistically, the binding of this sequence to an anion-binding site on ATIII involves a two-step process. The first step is the recognition step to form a low-affinity initial recognition complex and this is followed by a conformational change in ATIII which dramatically increases the exposure of a 15-residue protease recognition sequence containing the cleavable bond. This conformational change leads to about 300-fold enhancement in the inhibition rate of FXa [45, 46]. In the case of thrombin, however, a longer sequence of about 18 saccharide units is required for the acceleration of its inhibition by ATIII [48]. In this case, it has been shown that a bridging mechanism, in which both ATIII and thrombin bind simultaneously to the same heparin chain is required. This also results in over 1000-fold increase in the rate of inhibition. [44, 49, 50].
Figure 4.

The chemical structures of saccharide and nonsaccharide ATIII activators. A) Structure-activity relationship studies revealed that the trisaccharide unit (DEF; 2) from the nonreducing end of the pentasaccharide (DEFGH; 1) is critical for both the initial recognition and the conformational activation processes. The trisaccharide DEF binds to ATIII with a KD value of 2 µM (pH 6.0) and accelerates ATIII-mediated inhibition of FXa nearly 300-fold which is equivalent to the acceleration obtained by the pentasaccharide DEFGH. B) The first generation of flavonoid-based sulfated NSGMs (3–8) was computationally designed to promote ATIII-mediated FXa inhibition. First generation molecules bind to ATIII with KD values of 3.5 – 26 µM (pH 6.0) and accelerate FXa inhibition by 8–22-fold.
Full length heparin and the heparin pentasaccharide bind with nanomolar affinity to ATIII (Table 1) [51–55]. Biochemical and crystallography studies indicated the presence of a highly positively charged region known as the pentasaccharide-binding site on ATIII. Following several studies spanning a period of over a decade, the heparin-binding site of ATIII was identified by Ersdal-Badju et al. using alanine scanning mutagenesis of basic residues previously shown to interact with heparin [56, 57]. Subsequently, the determination of the crystal structure of ATIII in complex with heparin pentasaccharide DEFGH clearly confirmed the heparin-binding site of ATIII as including Lys 11, Arg13, Arg46, Arg47, Lys114, Lys125, and Arg129 (Figure 3) to which the carboxylate and sulfate groups of the pentasaccharide sequence bind with no significant contribution from the saccharide backbone. [49, 58]. Specifically, Lys114 is deemed the most important residue for the interaction. Structure-activity relationship studies revealed that the trisaccharide unit DEF 2 (Figure 4) is very important for the initial recognition step as well as for the conformational activation process. Interestingly, the trisaccharide DEF was found to accelerate ATIII-facilitated inhibition of FXa nearly 300-fold which is similar to the acceleration obtained by the pentasaccharide DEFGH. This suggested that residues G and H are important for increasing the binding affinity under physiological conditions but not as important for the conformational activation process [42, 45, 46]. Full length heparin has been shown to also engage the Lys136 residue [59]. Similar to ATIII, HCII is important for coagulation. HCII is a thrombin-specific serpin whose activity is similarly accelerated in the presence of dermatan sulfate and heparin by >1000-fold [60]. Heparin and dermatan sulfate have been shown to have micromolar affinity for HCII (Table 1) [52, 55].
Table 1.
Binding affinity of various GAG ligands to different proteins.
| Protein | GAG-ligand | Binding affinity | Methoda | Reference |
|---|---|---|---|---|
| Antithrombin | Heparin (High affinity) | 10 – 20 nM | FS | [51] |
| Heparin | 24.7 ± 6.6 nM | FS | [52] | |
| 10 ± 3 nM | FS | [53] | ||
| α-Antithrombin | Heparin | 24 ± 1nM | FS | [54] |
| Pentasaccharide | 36 ± 11 nM | FS | [53] | |
| α-Antithrombin | Pentasaccharide | 63 ± 10 nM | FS | [54] |
| HCII | Heparin | 25 – 35 µM | FS | [55] |
| Heparin | 13.1 ± 2.1 µM | FS | [52] | |
| LMWH | 62.6 µM | FS | [52] | |
| Dermatan sulfate | 71 µM | FS | [52] | |
| Dermatan sulfate | 236 – 291 µM | FS | [55] | |
| Thrombin | Heparin | 20 – 200 nM | FS | [55] |
| Dermatan sulfate | 1 – 5.8 µM | FS | [55] | |
| Heparin | 420 ± 140 nM | SPR | [79] | |
| FIXa | Heparin | 4.0 µM | FS | [52] |
| LMWH | 6.4 µM | FS | [52] | |
| Heparin | 17.8 nM | SPR | [79] | |
| FXIa | Heparin | 8.6 ± 1.1 nM | SPR | [79] |
| FXIa (Cat. Domain) | Heparin | 11.2 ± 1.1 nM | SPR | [79] |
| FXa | Heparin | 331 ± 43 nM | SPR | [79] |
| FXIIa | Heparin | 12 ± 1.4 nM | SPR | [79] |
| Activated protein C | Heparin | 66 ± 8 nM | SPR | [79] |
| Human neutrophil elastase | Heparin | 3.3 ± 1.0 nM | SHA | [224] |
| LMWH | 89 ± 12 nM | SHA | [225] | |
| Plasmin | Heparin | 10 nM | FS | [226] |
| Heparin | 6.7 ± 0.8 µM | FS | [103] | |
| IL8 | Heparin | 37 ± 4.7 µM | FS | [227] |
| Heparin | 5.5 µM | ACE | [112] | |
| Heparan sulfate | 5.5 µM | ACE | [112] | |
| Heparin oligosaccharides | 0.39 – 2.63 µM | ITC | [111] | |
| Platelet factor 4 | Heparin | 27 nM | ACE | [112] |
| Heparin | 20 nM | FS | [228] | |
| Heparin | 70 nM | RBA | [228] | |
| Heparan sulfate | 112 nM | ACE | [112] | |
| Heparin | 30 nM | RBA | [229] | |
| Dextran sulfate | 60 nM | RBA | [229] | |
| Groα | Heparin | 250 nM | ACE | [112] |
| FGF1 | Heparin 16-mer | 1.2 ± 0.1 µM | ITC | [117] |
| FGF2 | LMWH (3 kD) | 470 ± 20 nM | ITC | [116] |
| LMWH (5 kD) | 1.3 ± 0.28 µM | ITC | [139] | |
| Glycoprotein D | Heparan sulfate | 1.7 ± 0.9 µM | ACE | [230] |
| Heparin octasaccharide | 18 µM | CCE | [231] | |
| Glycoprotein 120 | Heparin | 220 nM | SPR | [232] |
ACE: Affinity Co-Electrophoresis; CCE: Capillary Co-Electrophoresis; FS: Fluorescence Spectroscopy; ITC: Isothermal Titration Calorimetry; RBA: Radioligand Binding Assay; SHA: Substrate Hydrolysis Assay; SPR: Surface Plasmon Resonance.
Among the multiple GAG–serpin interaction platforms, heparin–ATIII interface has served as a novel platform to design and develop new anticoagulants that address the different issues related to the clinical use of heparins. Particularly, sulfated flavonoids [61–65] and tetrahydroisoquinolines (THIQs) [66–68] were designed as ATIII activators and indirect FXa inhibitors. Details are provided in subsequent sections.
2.B). SERINE PROTEASES
Serine proteases are a functionally diverse class of enzymes with wide ranging presence in living organisms to serve diverse sets of biological functions [69, 70]. For instance, thrombin, FXa, FIXa, and FXIa are coagulation serine proteases whereas plasmin and neutrophil elastase are serine proteases important for fibrinolysis and inflammation, respectively. Activity of thrombin, FXa, FIXa, and FXIa physiologically contributes to hemostasis and clot formation, yet pathologically to thromboembolic diseases [71]. Activity of plasmin is also physiologically important for hemostasis and clot lysis, yet pathologically to bleeding disorders and potentially to angiogenesis and metastasis [72]. Activity of neutrophil elastase contributes physiologically to a powerful host defensive mechanism at inflammatory sites, yet pathologically it is a destructive enzyme that leads to serious diseases as in the case of emphysema [73–75]. GAGs have been shown to interact with a number of serine proteases as depicted in Table 1. A key feature of these serine proteases is the presence of positively charged exosites to which GAGs bind, and subsequently, modulate function through a conformational change at the active site i.e. allosteric modulation [76, 77].
For instance, GAGs are reported to bind to thrombin with micro-and nanomolar affinities. As revealed by multiple high-resolution crystal structures, the heparin-binding site of thrombin is the exosite II in which Arg233, Lys236, and Lys240 are the important residues for heparin interaction (Figure 2B) [78–82]. FXa also binds with nano-molar affinity to heparin, and the heparin-binding site has been found to include the residues Arg93, Lys96, Arg125, Arg165, Lys169, Lys236, and Arg240, with Lys236 and Lys240 contributing the most to the interaction (Figure 2C) [79, 83]. Human FXIa is the only coagulation enzyme that exists as a homodimer and has been shown to have two different heparin-binding sites, one located on the catalytic domain and the other on the Apple 3 domain (Figure 2D) [79, 84, 85]. Plasma serpins such as ATIII may physiologically inhibit FXIa. Yet, the proteolytic function of FXIa can also be inhibited by sulfated GAGs including heparin. Heparin can inhibit FXIa either directly by allosteric modulation or charge neutralization through binding to cationic amino acids in the catalytic domain (Lys539, Lys535, Arg532, Arg530, and Lys529), or indirectly by enhancing the serpin-facilitated template inhibition mechanism in which the serpin and the A3 domain of FXIa (Lys255, Lys253, and Lys252) bind to the same heparin sequence forming an inhibitory FXIa-serpin-heparin complex [71].
Another serine protease that interacts with GAGs is plasmin which is the major enzyme in the degradation of fibrin clots. Particularly, plasmin binds to heparin, and though the exact heparin-binding site on plasmin remains unknown, it is thought to be located on the catalytic domain (Figure 2A) [72]. The serpin ATIII inhibits plasmin activity, and in this case too, heparin has been shown to accelerate the process [86–87]. Human neutrophil elastase, a serine protease that contributes to the pathophysiology of emphysema, interacts with GAGs and a significant difference between this interaction and other GAG-serine protease interactions is that neutrophil elastase is inhibited directly and potently by GAGs [88–90]. Although the binding of heparin to human neutrophil elastase has been shown to be in the nano-molar range, the exact binding site has not been established definitively [91]. Binding affinities of various GAGs to serine proteases and serine protease inhibitors as measured by different biochemical and biophysical tools are provided in Table 1.
Using the above GAG–serine protease interfaces, multiple sulfated/sulfonated nonsaccharide glycosaminoglycan mimetics (NSGMs) have been designed and studied. For instance, sulfated benzofuran and coumarin-based monomers, dimers, and trimers [92–97] were designed as allosteric inhibitors of thrombin, and thus, as anticoagulants. Sulfated galloids [98–100] and quinazolinone dimers [101] were developed as allosteric inhibitors of FXIa, and thus, as anticoagulants. Sulfated flavonoid dimers were also developed as allosteric inhibitors of plasmin [102, 103], and thus, as antifibrinolytics and potentially anticancer agents. Details are provided in subsequent sections.
2.C). CHEMOKINES AND GROWTH FACTORS
Chemokines are a large family of proteins with molecular weights of 8–12 kDa. They have important roles in inflammation, angiogenesis, and metastasis among others [104–107]. Chemokines are known to interact with GAGs and these interactions are known to modulate chemokine interactions and responses [108, 109]. Since the most of chemokines are basic proteins, their binding with GAGs involves nonspecific electrostatic interactions, however, specific chemokine–GAG interactions are known to exist. This is demonstrated by the fact that different chemokines do not bind a particular GAG with the same affinity, and also that a specific chemokine binds different GAG fractions with varying affinities [110] which emphasizes the significance of the length and the sulfation patterns of GAG sequences. Platelet factor 4 (PF4), interleukin 8 (IL-8), macrophage inflammatory protein 1-alpha (MIP-1α), growth-regulated oncogene-alpha (GROα), and neutrophil activating peptide-2 (NAP-2) are examples of chemokines that bind GAGs [111, 112]. Binding affinities of GAGs to chemokines as measured by biochemical and biophysical tools are provided in Table 1.
The growth factors represent another family of heparan sulfate-binding proteins [113]. Growth factors that have been shown to interact with heparan-sulfate GAGs include fibroblast growth factors (FGFs), vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF) [114]. Of the growth factors, the interactions of FGFs with heparan sulfate GAGs are the best studied to date. The FGFs with their receptors regulate a number of important biological processes including cell proliferation, migration and differentiation among others. Not surprisingly, aberrant behavior in FGFs and their receptors is known to be a contributor to many cancers [115]. It is known that heparan sulfate proteoglycans mediate the interaction of FGFs and FGFRs resulting in the activation of the receptors [113]. Additionally, binding of FGFs to these GAGs makes them thermo-and protease-resistant and also promotes their oligomerization [113]. As with other GAG-binding proteins, a cluster of basic amino acids form part of the GAG-binding sites of the FGFs. FGF1 and FGF2 are fibroblast growth factors that bind GAGs with high nano-molar to low miro-molar affinities (Table 1) [116–118].
2.D). VIRAL GLYCOPROTEINS
A key step in viral infections is the initial contact of the viral envelope glycoproteins with the cell-surface heparan sulfate [119]. A number of GAG-binding glycoproteins have been identified as important contributors to this step. In herpes simplex virus (HSV), glycoproteins gB, gC and gD have been shown to be important for viral binding entry [120]. Though structural information is sparse, heparan sulfate bearing a 3-O-sulfate group has been shown to be required for binding to glycoprotein gD, resulting in the inception of viral entry [121].
The entry of HIV-1 into cells is also mediated by glycoprotein gp120 which is known to bind both heparin and heparan sulfate [122]. Four GAG binding domains of the protein, all rich in basic amino acids have been identified [123]. These binding domains, found in the V2 and V3 loops, the C-terminal domain and the co-receptor binding domain, are all important for biological function [123]. A study of the interaction of different GAG species and oligosaccharides with gp120 reveals that longer and more sulfated GAG species have a higher binding affinity [122]. A hexasaccharide was shown to be the minimum GAG length required for gp120 binding. Loss of 2-O-, 3-O-and or N-sulfates led to the loss of binding affinity, indicating their importance. Heparin and heparan sulfate were found to be better binders than dermatan sulfate and chondroitin sulfate [122]. Binding of gD to heparin and heparan sulfate was found to be in the micro-molar range whereas binding to gp120 was much stronger (Table 1). Several sulfated NSGMs were found to demonstrate antiviral activities including sulfated galloids [124], sulfated flavonoglycosides [125], and xanthone glycosides [125]. Details are provided in subsequent sections.
3. GLYCOSAMINOGLYCAN MIMETICS AND THE RATIONALE FOR NON-SACCHARIDE GLYCOSAMINOGLYCAN MIMETICS (NSGMs)
As earlier discussed, natural GAGs are complex heterogeneous molecules which interact with a multitude of proteins. However, the structural features that facilitate these interactions remain not fully understood, except for the heparin−ATIII interaction [46–59]. A fundamental reason for the lack of understanding is the exceptional structural diversity of GAG sequences which not only differ in their length but are also randomly modified through sulfation, epimerization, and acetylation. Added to these variations are the multiple helical structures assumed by the polymeric GAG sequences [126]. Furthermore, the source of natural GAGs frequently adds to the complexity of their structures [127] as well as to the risk of having contaminants such as super-sulfated GAGs and/or viruses which further complicate their therapeutic use and may lead to serious adverse effects [128–132].
To address the issues of natural GAGs, multiple strategies have been exploited to enhance our understanding of GAG – protein platforms, and subsequently, fully realize their therapeutic potentials. Among these strategies are bioengineering production of GAGs [133–136], advanced computational techniques [137–141], and chemical and chemo-enzymatic synthesis of saccharide-based GAG mimetics [142,143]. Despite promises, the cost, the challenges with large scale production, and the need for optimization and standardization of the production of all involved biosynthetic enzymes need serious consideration before bioengineered heparin can clinically be used [144]. Furthermore, computational chemistry-based approaches to explain specificity aspects of GAG–protein interactions have met limited success [138–142, 145]. A potential reason for this marginal success is that GAG sequences are flexible because of the gyrational motions of sulfate groups [146] which translates into an enormous conformational space that is difficult to be comprehensively searched. Attempts have also been made to synthesize homogeneous sequences of GAGs. Nevertheless, most of the syntheses have been found to require several steps [143, 147], making it extremely difficult to construct a library of GAG sequences with good diversity. For instance, heparin octasaccharides that block HSV entry into cells were synthesized in more than 45 steps [148]. Other approaches have exploited the microarray technology [149, 150], yet identification of GAG sequences that exhibit activity has been challenging. Therefore, GAG mimetics that are not based on a saccharide scaffold, herein referred to as non-saccharide GAG mimetics (NSGMs), offer significant advantages [151, 152].
NSGMs are molecules that have a non-sugar backbone and sulfates, sulfonates, carboxylates and/or phosphates as the negatively charged groups. NSGMs are a promising platform for the development of molecules that regulate the activity of GAG–binding proteins for a number of reasons [151, 152]. First, these molecules will be easier to synthesize and obtain as homogenous species in high yields and with high purity than saccharide-based GAG mimetics. Also, the synthetic ease will be helpful in developing structural analogs and hence chemical libraries that will aid in understanding their structure-activity relationships and their binding partners. Moreover, computational studies of NSGMs will be easier to handle compared to GAGs as they have significantly fewer chiral centers and rotatable bonds. Another added advantage to these NSGMs has to do with their sites of interaction. Since GAGs bind to proteins at allosteric sites rather than the active sites, NSGMs are likely to demonstrate allosteric mechanism of action. Allostery presents several advantages over orthostery. First, allosteric sites are less conserved than orthosteric sites, and thus, targeting allosteric sites is likely to result in more selective molecules [153–155]. Second, in orthosteric inhibition, there is a total loss of activity, whereas in allosteric inhibition there can be submaximal inhibitor efficacy. Consequently, allosteric inhibitors can regulate rather than totally inhibit activity. This is certainly beneficial when targeting multifunctional proteins for which only partial inhibition is sufficient to promote the desired pharmacology while avoiding side effects. For example, submaximal allosteric inhibition of thrombin is anticipated to provide sufficient prophylaxis against thrombosis without causing bleeding [95, 97].
3.A). SULFATED NSGM-BASED ALLOSTERIC MODULATORS OF COAGULATION AND FIBRINOLYSIS PROTEINS
Allosteric Activators of Antithrombin (ATIII), an Anticoagulant Serpin
Despite the clinical efficacy of heparin-based therapy, its animal origin is problematic. Furthermore, unfractionated heparin and low molecular weight heparin therapy is associated with many complications including the elevated risk of bleeding, osteoporosis, thrombocytopenia, patient response variations, and the risk of contamination [156]. Therefore, several efforts have been focusing on the design of synthetic nonsaccharide ATIII activators to eventually inhibit FXa. Using the hydropathic interaction (HINT) technique, (−)-epicatechin sulfate (ECS) 3 (Figure 4) was designed to mimic the trisaccharide DEF sequence. Kinetic studies indicated that ECS binds ATIII with KD values of 10.5 and 66 µM at pH 6.0, I 0.025, and pH 7.4, I 0.035, respectively, (Table 2) which compared well with 2 and 80 µM, respectively, observed for DEF. However, ECS accelerated the ATIII inhibition of FXa only 8-fold as compared to the ~300-fold observed with DEF [61].
Table 2.
Binding affinity of various sulfated NSGMs to different proteins.a
| Protein | NSGM | Binding affinity | Scaffold (N=Sulfate/Sulfonate) | Reference |
|---|---|---|---|---|
| Antithrombin | 3 (ECS) | 10.5 – 66 µM | Flavonoid monomer (N=5) | [61] |
| 4 ((+)-CS) | 3.5 – 17.9 µM | Flavonoid monomer (N=5) | [62] | |
| 5 ((±)-CS)) | 26.1 µM | Flavonoid monomer (N=5) | [62] | |
| 6 (SQ) | 17 µM | Flavonoid monomer (N=5) | [63] | |
| 7 (SMo) | 1.8 µM | Flavonoid monomer (N=5) | [63] | |
| 9 | 320 µM | THIQ (N=5) | [66] | |
| 10 | 1322 µM | THIQ (N=4) | [67] | |
| 11 | 81 µM | THIQ (N=4) | [68] | |
| 12 | 394 µM | THIQ (N=4) | [68] | |
| 13 | 1 – 2.3 µM (pH=6) | Polyacrylic acid (N=0) | [157] | |
| 34 – 180 µM (pH=7.4) | ||||
| Thrombin | 31 | 0.143 µM | Coumarin dimer (N=1) | [97] |
| FXIa | 32 (SPGG2) | 0.4 µM | Pentagalloyl pyranoside (N~10) | [98] |
| 33 | 37 µM | Quinazolinone dimer (N=2) | [101] | |
| 34 | 38 µM | Quinazolinone dimer (N=2) | [101] | |
| Plasmin | 36 | 0.7 µM | Flavonoid dimer (N=8) | [103] |
| FXIIIa | 37 | 25.3 µM | Flavonoid trimer (N=11) | [179] |
| CCL5 | 48 | 20 µM | Benzoic acid (N=1) | [200] |
All values were measured by Fluorescence Spectroscopy.
Subsequently, two more analogs of ECS were synthesized and tested for ATIII activation. (+)-Catechin sulfate ((+)-CS) 4 was found to bind to ATIII with KD values of 3.5 µM (pH 6.0) and 17.9 µM (pH 7.4) (Table 2) and accelerated ATIII-mediated inhibition of FXa by 20.8-fold at pH 6. The racemic mixture (±)-catechin sulfate ((±)-CS) 5 bound ATIII with a KD value of 26.1 µM (pH 6.0) (Table 2) and accelerated FXa inhibition by ATIII by 8.5-fold. All the three activators exhibited a free energy of binding of 6.0–7.5 kcal/mol to ATIII. Furthermore, salt effect studies indicated that ECS exhibits one ion-pair interaction whereas (+)-CS exhibits two ionic interactions which are significantly fewer than the 4–5 interactions observed with the corresponding saccharide-based activators under similar conditions. This suggested that nonionic forces contributed 55–73% of the binding energy in the case of nonsaccharide activators compared to 40–60% in the case of DEFGH. Moreover, competition studies with a tetrasaccharide EFGH” and low affinity heparin indicated that the small nonsaccharide activators may bind in the extended heparin-binding site and not in the pentasaccharide-binding site [62]. The pentasulfated quercetin (SQ) 6 and its regioisomer, the pentasulfated morin (SMo) 7 (Figure 4) were also found to bind ATIII with KD values of 17 and 1.8 µM (Table 2), respectively, at pH 6.0. They also accelerated the ATIII-mediated inhibition of FXa by 17.5-and 21.8-fold, respectively [63]. In a similar study, the pentasulfated flavonoids indirectly inhibited FXa by 82–89% whereas the tetrasulfated and trisulfated flavonoids inhibited FXa by 40–60% and 0–12%, respectively [64]. Hydropathic interaction analyses of the nonsaccharidic activators binding to ATIII predicted more favorable interaction of most of the above activators, except (−)-catechin sulfate ((−)-CS) 8 (Figure 4), with the activated form of ATIII than with the native ATIII form supporting the fact that these molecules work as ATIII activators [65].
Motivated by the above results, a second generation of sulfated NSGM ATIII activators was introduced based on the tetrahydroquinoline (THIQ) scaffold [66–68]. A pharmacophore-based approach was used to mimic the trisaccharide DEF. Initially, four sulfated nonsaccharide molecules were synthesized and tested for their indirect inhibition of FXa. The four molecules possessed a 6,7-disulfated THIQ moiety that was acylated at N2-position by four different sulfated benzoyl moieties. The four molecules were found to bind to ATIII with KD values of 320 – >1000 µM and accelerated ATIII-mediated inhibition of FXa by 2.3–30-fold at pH 7.4. Specifically, the tetrasulfated THIQ 9 (Figure 5) bound ATIII with a KD value of 320 µM (Table 2) and accelerated ATIII-mediated inhibition of FXa by 30-fold [66, 67]. To further enhance the biochemical profile of these molecules, eight additional sulfated N-acyl THIQ analogs were synthesized and tested for their ATIII-mediated inhibition of FXa [68]. The molecules varied in three aspects: 1) the position of the two sulfate groups on the THIQ moiety (6,7-, 5,8-, and 5,6-disulfated); 2) the presence or absence of a carboxylate group at position-3; and 3) the linker length between the THIQ bicyclic moiety and the 2,5-disulfated benzene monocyclic moiety (2-atom, 3-atom, and 4-atom linkers). The eight molecules were found to bind to ATIII with KD values of 81–3883 µM and accelerate AT-mediated inhibition of FXa by 8.3– 79.4-fold at pH 6.0. Specifically, the tetrasulfated THIQ 10 (Figure 5) bound ATIII with KD value of 1322 µM and accelerated ATIII-mediated inhibition of FXa by 79.4-fold [68]. Furthermore, THIQs 11 and 12 (Figure 5) bound ATIII with KD values of 81 and 394 µM (Table 2), respectively, and accelerated ATIII-mediated inhibition of FXa by 53.2 and 67.1-fold, respectively [68].
Figure 5.
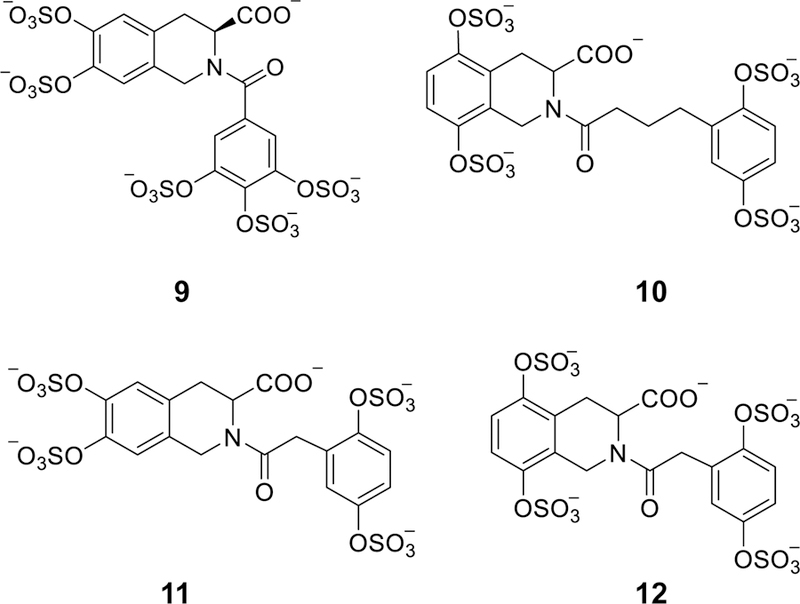
The chemical structures of the second generation of THIQ-based ATIII activators (9–12). Pharmacophore approach was exploited to design this generation. Structural variations were as follows: positions of the two sulfate groups on THIQ scaffold (5,6-, 6,7-, and 5,8-), the presence or absence of carboxylate group at position-3, the linker length between the bicyclic and monocyclic units (1-, 2-, 3-, and 4-atom linkers), and the number and position of sulfate groups on the monocyclic unit (2 or 3 sulfates) and (2,5-disulfate, 3,4-disulfate, and 3,4,5-trisulfate groups). Considering the acceleration potential, THIQ-N-acyl derivative 10 accelerated ATIII inhibition of FXa by ~80-fold, only 3.75-fold less than that achieved by DEF unit under similar condition.
Along those lines, a series of polyacrylic acid polymers 13 (Figure 6A) having different average molecular weight, polydispersity, number of monomers, and polymer length were tested with respect to their potential to accelerate ATIII-mediated inhibition of thrombin and FXa [157]. Polyacrylic acid polymers carry a charge density of about −0.46 charges/Å, which is similar to the average charge density of heparin. The polymers were found to bind ATIII with KD values of 1.0–2.3 µM at pH 6.0 and 65–180 µM at pH 7.4 (Table 2). In one hand, the polymers accelerated ATIII-mediated FXa inhibition much better at pH 6.0 (15–284-fold) than at pH 7.4 (6.2–17-fold). In the other hand, they accelerated ATIII-mediated thrombin inhibition in comparable fashion at the two pHs; pH 6.0 (24–1392-fold) and at pH 7.4 (114–1109-fold). The results demonstrated that polyacrylic acid polymers accelerate thrombin inhibition by ATIII in a chain length-dependent fashion at the two pHs. However, FXa inhibition acceleration was chain length-dependent only at pH 6.0 [157]. To better understand the mechanism of polyacrylic acid polymers’ acceleration of ATIII-mediated thrombin inhibition, a series of biochemical experiments were performed [158]. Competitive binding studies with a heparin tetrasaccharide and low-affinity heparin as well as molecular modeling studies revealed that polyacrylic acid polymers bound ATIII in both the extended heparin-and the pentasaccharide-binding sites. The salt-dependence of the KD of the polyacrylic acid polymer-ATIII interaction indicated the formation of five ionic interactions. Furthermore, the bell-shaped profile of the measured rate constant for ATIII-mediated thrombin inhibition as a function of polyacrylic acid polymer concentration confirmed the bridging mechanism of inhibition [158]. Overall, this work demonstrated that NSGMs lacking sulfate groups can effectively activate ATIII.
Figure 6.

The chemical structures of polymeric NSGMs. A) Polyacrylic acid polymers 13 were found to bind ATIII with KD values of 1.0–2.3 µM at pH 6.0 and 34–180 µM at pH 7.4. The polymers accelerated ATIII-mediated FXa inhibition much better at pH 6.0 (15– 284-fold) than at pH 7.4 (6.2–17-fold). The polymers also accelerated ATIII-mediated thrombin inhibition in comparable fashion at the two pHs; pH 6.0 (24–1392-fold) and pH 7.4 (114–1109-fold). Polyacrylic acid polymers have opened up an opportunity to developing orally bioavailable, carboxylate-based ATIII activators. B) Chemo-enzymatically prepared oligomers of 4-hydroxycinnamic acids, known as dehydrogenation polymers (DHPs) displayed interesting anti-coagulant (thrombin inhibitors and ATIII activators), anti-fibrinolytic (plasmin inhibitors), and anti-emphysema (inhibitors of pulmonary elastolysis, oxidation, and inflammation) properties. The oligomers were prepared by peroxidase-catalyzed oxidative coupling of caffeic 14 (CDSO3), ferulic 15 (FDSO3), and sinapic 16 (SDSO3) acids, followed by sulfation using SO3–NEt3 complex. Various analytical studies suggested that the DHPs are heterogeneous and polydisperse preparations that are composed of inter-monomer linkages similar to those found in natural lignins including β−5, β-O-4, β-β and β−5. DHPs were later named as low molecular weight lignins (LMWLs).
In similar fashion, chemo-enzymatically prepared oligomers of 4-hydroxycinnamic acids, known as dehydrogenation polymers (DHPs), displayed interesting anticoagulant properties. The oligomers were prepared by peroxidase-catalyzed oxidative coupling of caffeic 14, ferulic 15, and sinapic 16 acids (Figure 6B), followed by sulfation using SO3–NEt3 complex [159]. Various analytical studies suggested that the DHPs are heterogeneous, polydisperse preparations with great similarity to natural lignins. A 2-fold increase in prothrombin time (PT) required 98–212 µg/mL plasma concentration of unsulfated DHPs. This concentration decreased to 42–105 µg/mL for sulfated DHPs. Likewise, doubling of activated partial thromboplastin time (APTT) required about 25–40 µg/mL and about 13–23 µg/mL of unsulfated and sulfated DHPs, respectively. Detailed mechanistic studies revealed that unsulfated DHPs directly inhibit FXa and thrombin with IC50 values of 0.4–2.8 µg/mL and 0.19–1.16 µg/mL, respectively. However, sulfated DHPs were found to inhibit FXa and thrombin directly and indirectly. Directly, sulfated DHPs inhibited FXa and thrombin with IC50 values of 0.11–0.84 µg/mL (32–239 nM) and 0.07–0.33 µg/mL (20–94 nM), respectively. Indirectly, ATIII inhibited FXa and thrombin in the presence of sulfated DHPs with IC50 values of 0.19–0.56 µg/mL (55–132 nM) and 0.16–0.31 µg/mL (46–73 nM), respectively. The sulfated DHPs did not directly inhibit factor VIIa at the highest concentration tested (23.6 µM), while directly inhibited FIXa with IC50 of 1.7–>28.5 µM [158]. DHPs also inhibited FIXa in the presence of ATIII with IC50 values of 1.1–1.7 µM. Likewise, DHPs also inhibited FVIIa in the presence of ATIII with IC50 values of 0.356– >23.6 µM. Fluorescence binding studies indicated that sulfated DHPs bind to ATIII with micromolar affinity under physiological conditions. Salt-dependent fluorescence binding studies also indicated that the non-ionic component of the ATIII-sulfated DHP interaction was 80–87%. Competitive binding studies with full length heparin and heparin pentasaccharide indicated that sulfated DHPs bind to both the pentasaccharide-binding site as well as the extended heparin-binding site of ATIII [160].
A small library of flavonoid-O-glycosides were also designed and tested for anticoagulant activity [161]. Four flavonoid-O-glycosides (17–20) (Figure 7) doubled the clotting time in APTT assay at micromolar concentrations. The flavonoside 19 was the most potent and doubled the clotting time at a concentration of 66 μM. The corresponding sulfated aglycone increased the APTT by 1.65-fold at 500 µM. The flavonoside 19 also doubled the PT and TT at concentrations of 649 µM and 877 µM, respectively. The anticoagulant activity of the reported flavonosides was further confirmed in human whole blood using thrombo-elastography technique and molecule 19 was effective at a concentration of 125 µM. Flavonosides 19 and 20 also demonstrated antiplatelet activity in collagen/epinephrine-induced aggregation at concentrations of 125 and 625 µM, respectively. In chromogenic substrate hydrolysis assays, none of the tested flavonosides demonstrated significant effect on thrombin activity at the highest concentrations tested, whereas flavonosides 17 and 18 were ATIII activators and flavonosides 19 and 20 were direct competitive inhibitors of FXa. In vivo clotting times, chemical stability, and hepatotoxicity were all time-dependently evaluated in mice following the intraperitoneal administration of 150 μmol/kg from flavonosides 17 or 19. Results indicated that these molecules effectively prolonged the APTT, PT, and TT of mice plasma over the period of 120 min, exhibited significant chemical stability over 3 hrs, and demonstrated no acute hepatotoxicity. In another study, flavonoside 19 dose-dependently decreased the levels of activated protein C and prothrombin in citrated human plasma. Particularly, the molecule decreased the level of activated protein C by ~3-fold and the level of prothrombin by ~10-fold at a concentration of 1 mM [161]. Furthermore, all four flavonosides 17-20 exhibited antiplatelet activity via inhibition of arachidonic acid and adenosine diphosphate-induced platelet aggregation with different potencies [161]. These results led to the conclusion that flavonosides can be developed as dual anticoagulant-antiplatelet agents to treat venous as well as arterial thromboses. Likewise, mangiferin heptasulfate 21 [162], 3,6-(O-β-glucopyranosyl)xanthone octasulfate 22 [162], and trans-resveratrol 3-β-D-glucopyranoside hexasulfate 23 [163] (Figure 7) also exhibited dual anticoagulant and antiplatelet activities comparable to the above flavonosides with significant chemical stability and no signs of acute hepatotoxicity under the reported test conditions. Molecule 21 was found to be a direct inhibitor of FXa, while molecule 22 inhibited FXa directly as well as indirectly by activating ATIII. All three molecules 21-23 also exhibited similar antiplatelet effects to flavonosides 17-20.
Figure 7.

The chemical structures of flavonoid-based (17-20), xanthone-based (21 and 22), and resveratrol-based (23) GAG mimetics. The seven mimetics demonstrated significant antithrombotic activity by inhibiting the coagulation proteins and/or inhibiting the arachidonic acid-and adenosine diphosphate-induced platelet aggregation. Molecules 19-21 are direct FXa inhibitors whereas molecules 17, 18, and 22 exhibit indirect inhibition of FXa by activating ATIII.
Allosteric Inhibitors of Thrombin, a Coagulation Serine Protease
Thrombin (factor IIa) is a pivotal serine protease in the common coagulation pathway. It catalyzes the conversion of fibrinogen to soluble fibrin monomers which are crosslinked into insoluble mesh by the action of a thrombin-activated factor XIII (FXIII). Furthermore, thrombin also activates several other factors including factors V, VIII, and XI in multiple feedback activations meant to facilitate rapid clot formation [164, 165]. Thrombin also binds to platelet surface glycoproteins including glycoprotein Ibα (GPIbα) that subsequently facilitates platelet activation by cleaving protease activated receptors on the platelet surface to eventually induce aggregation [166]. In contrast to its prothrombotic role, thrombin also possesses an anticoagulant role. Thrombin–thrombomodulin complex activates protein C which subsequently cleaves factors Va and VIIIa [167].
Structurally, thrombin is a serine protease belonging to the trypsin superfamily with multiple ligand-binding domains. These domains are the active site, the sodium-binding domain, and the anion-binding exosites I and II. Most of the drug design and development efforts have been targeting thrombin’s active site by small monovalent, competitive peptidomimetics including argatroban and dabigatran etexilate. However, another class of molecules, the peptidic hirudins, are bivalent inhibitors of thrombin that bind to both the active site and the exosite I. Heparins are known to occupy exosite II in the ATIII-mediated inhibition of thrombin through the bridging mechanism [156]. It was reasoned that allosteric regulators of thrombin may be designed to target exosite II so as to lead to partial inhibition of thrombin. This tactic was proposed to selectively disrupt thrombin’s procoagulant role but not other physiological roles. Thus, it was hypothesized that exosite II-mediated allosteric regulation of thrombin by sulfated NSGMs may potentially lead to effective anticoagulants with limited likelihood of bleeding consequences.
Therefore, the three previously synthesized variants of DHPs, namely CDSO3 14, FDSO3 15, and SDSO3 16 (Figure 6B), were further studied for their direct inhibition of thrombin among other coagulation proteins including factors Xa, IXa, and VIIa [168]. All three sulfated DHPs displayed a 2–3-fold selectivity for direct thrombin inhibition (18–94 nM) over that of FXa (34–244 nM). The DHPs were 17– 300-fold selective for thrombin over factors IXa and VIIa. Furthermore, Michaelis–Menten kinetics indicated that CDSO3 inhibited thrombin directly by an allosteric mechanism. Competitive binding studies with bovine heparin, fluorescein-labeled hirudin peptide, heparin octasaccharide, and enoxaparin suggested that CDSO3 preferentially binds in or near the anion-binding exosite II of thrombin. Subsequently, Arg93 and Arg175 in exosite 2 were identified as the most important residues for DHPs (renamed as low molecular weight lignins (LMWLs)) binding followed by Arg165, Lys169, Arg173, and Arg233 [169].
DHPs represent a highly diverse library of structures arising from multiple inter-monomeric linkages including β−5, β-O-4, β-β and β−5, variable chain length, variable carboxylation and sulfation levels of chains, and multiple chiral centers. Therefore, to enhance the biochemical profiles of DHPs, a small library of seventeen β−5–like monomeric benzofuran derivatives was synthesized and screened against thrombin and FXa [92]. From this library, only seven benzofuran monomers were found to inhibit thrombin and/or FXa. The minimal concentration necessary to promote inhibition was found to be about 400 μM. Structure-activity relationship studies indicated that all ester containing molecules are inactive. Whereas no inhibitor displayed more than 40% inhibition of thrombin at a concentration of 2.9 mM, some inhibited FXa better (>40%) at 2.6 mM with the 3-carboxylate and 6-sulfate units being the most important functional groups. Michaelis–Menten kinetics revealed an allosteric inhibition mechanism by sulfated benzofuran monomers [92].
To further enhance the allosteric thrombin inhibition profile of sulfated benzofuran-based molecules, another library of 15 sulfated monomers and 13 sulfated dimers with different charged, polar, and hydrophobic substituents was synthesized and tested [93]. Of the 28 potential inhibitors, only eleven molecules exhibited reasonable inhibition of human α-thrombin under physiological condition with IC50 values of 7.3–375 µM and variable efficacies of 14–95%. Structure–activity relationship studies indicated that sulfation at position-5 of the benzofuran scaffold was essential for targeting thrombin. A tert-butyl 5-sulfated benzofuran derivative 24 (Figure 8) was found to be the most potent benzofuran dimer-based thrombin inhibitor with an IC50 of 7.3 µM and efficacy of 84% under physiologically relevant conditions. Michaelis–Menten studies showed an allosteric inhibition phenomenon. Sulfated benzofuran dimer 24 doubled APTT at a concentration of 355 µM but exhibited no effect on PT at the highest concentration tested of 2.1 mM [93].
Figure 8.

The chemical structures of various allosteric inhibitors of thrombin. A) Monosulfated benzofuran dimers (24-26) and trimers (27) were designed considering the structural and mechanistic aspects of LMWLs. Extensive biochemical work revealed that sulfated NSGMs bind to exosite II of thrombin in similar fashion to heparin, yet in contrast to heparin, they allosterically disrupt the active site’s catalytic triad. Trimer 27 is the most potent inhibitor in this class of sulfated NSGMs with IC50 value of 0.67 µM and efficacy of 79%. A 2-fold increase in APTT and PT required 139 and 568 μM of inhibitor 27, respectively. B) A sulfated β-O4 lignin (SbO4L) 28 was intuitively designed as a mimic of the sulfated tyrosine sequence of GPIbα. Extensive biochemical studies established that this polymeric sulfated NSGMs is an allosteric, exosite II-binding inhibitor of human thrombin and a competitive inhibitor of GPIbα-mediated platelet aggregation. SbO4L has presented unique anticoagulant and antiplatelet properties which were confirmed by studies in human plasma, human whole blood, FeCl3-induced carotid arterial thrombosis model in mice, and Rose Bengal–laser injury model of arterial thrombosis in mice. C) The chemical structures of a new generation of sulfated coumarin dimers (29–31) that partially and allosterically modulate the catalytic activity of human thrombin. This was established by extensive biochemical studies. Dimer 31 is the most potent inhibitor with an IC50 value of 0.2 µM (KD=143 nM) and efficacy of 47%.
In subsequent studies, sulfated benzofuran dimer 25 (Figure 8) inhibited thrombin with an IC50 value of 6.2 µM [94]. Biochemical studies revealed that the presence of unfractionated heparin decreased its inhibition potency by 4-fold, whereas heparin octasaccharide and hirudin peptide did not affect the potency of thrombin inhibition. The presence of γ′ fibrinogen peptide, which is known to bind to Arg93, Arg97, Arg173, Arg175, and other residues, led to a significant loss of affinity which correlates with the ideal Dixon−Webb competitive profile. Mutation of several Arg and Lys residues of exosite II with alanine did not change thrombin inhibition potency, except for Arg173, which exhibited about 22-fold decrease in inhibition potency. Molecular modeling studies suggested a hydrophobic patch around Arg173 as a plausible binding site for sulfated benzofuran dimer 25. MTT-based cellular toxicity studies indicated that sulfated benzofuran dimers are essentially nontoxic to human alveolar basal epithelial A549 cell line at concentrations as high as 250 mg/kg [94].
Another set of sulfated benzofuran derivatives was recently reported by Afosah et al. [95]. In the new series, 16 sulfated benzofuran dimers and 12 sulfated benzofuran monomers were studied. Particularly, sulfated benzofuran dimer 26 (Figure 8) displayed the best inhibition potency using the chromogenic substrate hydrolysis assay with IC50 value of 1.8 µM and efficacy of 58%. The inhibitor was found to bind to thrombin with a KD value of 3.7 µM using spectrofluorimetry. Michaelis-Menten kinetics, competition with unfractionated heparin and hirudin peptide, and mutagenesis studies indicated that this molecule inhibits thrombin by a non-competitive, partial allosteric mechanism with Arg233 and Lys235 residues of exosite II being the most important for binding and inhibition. The dimer 26 also inhibited thrombin-mediated fibrinogen cleavage with a maximum efficacy of ~70 ± 8%. In fact, results with inhibitor 26 indicated that partial allosterism against the macromolecule fibrinogen (∆Y = 55–70%) can be achieved by a sulfated small molecule (MW < 800). This partial allosterism of thrombin has been proposed as a means to maintain basal level of a pro-clotting response, and thus, to minimize the risk of major bleeding, which complicates the use of currently used direct oral anticoagulants.
A generation of sulfated benzofuran trimers and tetramers have also been introduced [96]. Monosulfated benzofuran tri-and tetramers exhibited a wide range of IC50 values of 0.67– >500 µM. Among these, trimer 27 (Figure 8) was found to be ~10-fold more potent than dimer 25 with IC50 of 0.67 µM and efficacy of 79%. Michaelis−Menten kinetics indicated an allosteric mechanism of inhibition. Competitive studies using heparin octasaccharide, unfractionated heparin, γ′-fibrinogen peptide, and a hirudin peptide indicated recognition of exosite II in a manner different from that of the parent sulfated benzofuran dimers. Alanine scanning mutagenesis of several positively charged residues of thrombin’s exosite II revealed Arg233 as the most important residue for binding of this generation of sulfated benzofurans. In human plasma, the 2-fold increase in APTT and PT was brought about by 139 and 568 μM of inhibitor 27, respectively.
A sulfated β-O4 lignin (SbO4L) 28 (Figure 8) was also introduced as an allosteric, exosite II-binding inhibitor of human thrombin and a structural mimic of the sulfated tyrosine sequence of GPIbα [170]. In contrast to DHPs (or sulfated LMWLs), SbO4L involves only one type of inter-monomer linkage (β−O4). Using the chromogenic substrate hydrolysis assay, SbO4L inhibited human α-thrombin with an IC50 of 0.17 μg/mL (∼14 nM), while FXa and FXIa were inhibited at 90−500 μg/mL SbO4L (∼10−55 μM), indicating a substantially selective inhibition. FVIIa−recombinant tissue factor complex was inhibited at 9.4 μg/mL SbO4L. Furthermore, the activity of human leukocyte elastase, cathepsin G, kallikrein, activated protein C, porcine pancreatic elastase, trypsin, and chymotrypsin remained unaffected in the presence of 81 μg/mL of SbO4L, suggesting at least 475-fold selectivity. However, SbO4L inhibited human plasmin with an IC50 of 0.38 μg/mL. Similar to other sulfated LMWLs introduced previously, Michaelis–Menten kinetics indicated that SbO4L is a noncompetitive, allosteric inhibitor of human thrombin. Competitive studies revealed an ideal competition with full-length heparin and GPIbα. Studies with site-directed thrombin mutants revealed that SbO4L binds to Arg233, Lys235, and Lys236 of exosite II. A series of biochemical studies also revealed that exosite II binding by SbO4L significantly reduces access to thrombin’s active site and that SbO4L–thrombin interaction is 60% electrostatic [171].
In human plasma, SbO4L doubled the clotting time at a concentration of 68 μg/mL (7.5 µM) in PT assay. This concentration is substantially lower than that was required by a generic LMWH (142 μg/mL or 31.6 μM) and a clinically used LMWH (enoxaparin, 339 μg/mL or 75 μM). Likewise, SbO4L doubled the clotting time in APTT assay at a concentration of 20 μg/mL (2.2 µM) which is similar to the concentration of 5.9 μg/mL (1.3 μM) for generic LMWH and 5.4 μg/mL (1.2 μM) for enoxaparin. These results demonstrate that SbO4L is an effective anticoagulant in human plasma and approximately as potent as LMWHs. Furthermore, SbO4L was also found to prevent thrombin-induced platelet activation and aggregation [170].
In another set of studies, it was found that SbO4L inhibited fibrinogen cleavage by thrombin with an IC50 of 0.19 µg/mL [172]. SbO4L inhibition of thrombin was found to be reversible by the action of protamine sulfate. Moreover, SbO4L showed good anticoagulant activity in human whole blood using thrombo-elastography technique. In fact, SbO4L and enoxaparin caused very similar changes in thrombo-elastography parameters, yet at different dosage levels. SbO4L was effective in the range of 5–150 µg/mL, whereas enoxaparin was effective in the range of 1–5 µg/mL. SbO4L also showed good antiplatelet potential in human whole blood hemostatic analysis. In FeCl3-induced carotid arterial thrombosis model in mice, 250–500 µg/animal of SbO4L prevented occlusion in about 35–50% of cases studied. IV administration of 1 mg of SbO4L prevented occlusion in 100% of cases. In the Rose Bengal–laser injury model of arterial thrombosis in mice, the time to occlusion increased by ~ 70% with a 0.5-mg dose of SbO4L. Lastly, tail bleeding studies demonstrated that SbO4L does not increase bleeding at a dose of 1 mg/animal. In comparison, a 0.3-mg/animal dose of enoxaparin increased the bleeding time and blood volume loss [171]. Owing to the above studies, it can be said that SbO4L is a dual anticoagulant and antiplatelet agent with apparent limited effect on hemostasis.
A new set of sulfated coumarin molecules were recently introduced as partial, allosteric inhibitors of thrombin [97]. Few sulfated monomeric coumarins inhibited thrombin, however most of sulfated dimeric coumarins exhibited good inhibitory potencies (IC50 < 20 μM). Particularly, inhibitors 29–31 (Figure 8) inhibited thrombin with IC50 values of 0.5, 1.0, and 0.2 µM, respectively. Inhibitor 31 was 815-fold and 155-fold selective for thrombin over factors Xa and IXa, respectively. Inhibitor 31 was also found to bind to thrombin with a KD value of 143 nM (Table 2). Michaelis-Menten kinetics, competitive inhibition studies, and site-directed mutagenesis studies confirmed thrombin’s exosite II as the binding site for inhibitor 31. Stern-Volmer quenching of active site-labeled fluorophore revealed that sulfated coumarin dimer 31 induces moderate structural changes in the active site with an efficacy level of 47%. ATIII inactivation of thrombin was compromised in the presence of inhibitor 31 suggesting a catalytic dysfunction of the active site being induced by this submaximal, allosteric inhibitor of thrombin. The advantage of this NSGM seems to be similar to that of inhibitor 26, in term of the limited bleeding expected to accompany the anticoagulant activity.
Few other synthetic, nonsaccharide, sulfonated styrene-containing polymers and co-polymers have also been reported to demonstrate anticoagulant activity by targeting thrombin. Structural and mechanistic aspects of these polymeric molecules were recently reviewed by Nahain et al. [173].
Allosteric Inhibitors of Factor XIa, a Coagulation Serine Protease
Factor XIa is a homodimer serine protease in the intrinsic coagulation cascade. FXIa’s main physiological function is reported to be the amplification of the coagulation cascade via activation of FIX [174–177]. Several experimental, epidemiological, and clinical observations suggested that inhibiting FXIa may lead to effective anticoagulants with no serious impact on the hemostatic process. Thus, such inhibitors would be devoid of bleeding complications that are typically associated with all currently used anticoagulants [174–177]. Accordingly, to design and develop effective anticoagulants with no substantial risk of internal bleeding, several sulfated NSGMs that allosterically inhibit FXIa by targeting the GAG-binding site on the catalytic domain have been reported.
The discovery of sulfated pentagalloyl glucopyranoside (SPGG) as a potent, allosteric, and selective inhibitor of human FXIa was reported by Al-Horani et al. [98, 99]. A specific chemical species of SPGG called SPGG2 32 (Figure 9) inhibited FXIa with an IC50 value of 550 nM (KD=0.4 µM) (Table 2). SPGG2 was found to be at least 200-fold selective against a series of related serine proteases including factors IIa, VIIa, IXa, Xa, XIIa, trypsin, and chymotrypsin. At the saturating concentration of 3 µM, SPGG2 also inhibited FIX activation by FXIa. Michaelis–Menten kinetics as well as competition studies with full-length heparin, polymeric polyphosphates, and GPIbα using the wild type FXIa or a FXIa only containing the catalytic domain revealed that SPGG2 binds to or near the anion-binding site on the catalytic domain suggesting an allosteric inhibition mechanism. It was also found that SPGG2 is at least 200–300-fold selective over plasma kallikrein, activated protein C, and FXIIIa [100]. SPGG2 inhibition of FXIa was dose-dependently reversed by protamine, polybrene, bovine albumin, and the zymogen FXI suggesting several reversal strategies. In normal human plasma APTT and PT assays, SPGG2 doubled the clotting time in the APTT assay at a concentration of 45 µM, whereas, the concentration required to double the clotting time in the PT assay was found to be more than 370 µM. The APTT EC2X were 114 µM and 40 µM in human plasmas deficient of FXI and ATIII/HCII, respectively. This indicated that the anticoagulant activity of SPGG2 principally stems from directly inhibiting the intrinsic pathway of coagulation and not from activating serpins to indirectly inhibit coagulation proteases [100]. Furthermore, SPGG2 displayed good anticoagulant activity in human blood when tested by the thrombo-elastography technique [100].
Figure 9.
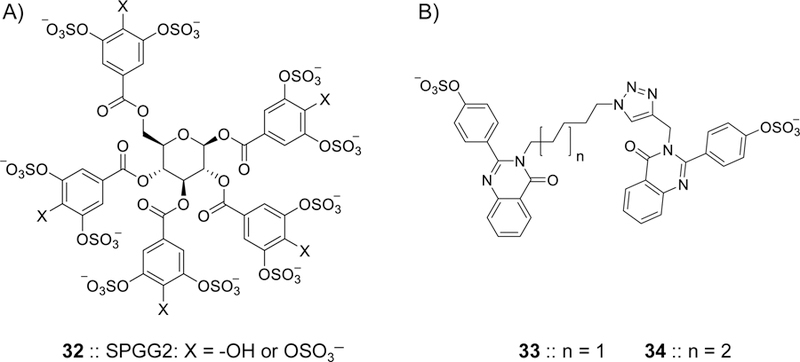
Chemical structures of allosteric inhibitors of FXIa. A) SPGG2 32 is a sulfated galloid-based NSGM. SPGG2 variant is the first small molecule allosteric inhibitor of FXIa. SPGG2 inhibited human FXIa with an IC50 value of 0.5 µM and a selectivity index of at least 200-fold against a range of coagulation, fibrinolysis, and digestive proteins. Extensive biochemical studies established the structural and mechanistic properties of this molecule. SPGG2 binds to the anion-binding site in the catalytic domain of FXIa. Its anticoagulant activity was confirmed in human normal and deficient plasmas as well as in human whole blood using thrombo-elastography technology. B) Sulfated quinazolinone dimers 33 and 34 were reported as allosteric inhibitors of FXIa. They were structurally designed using the dual-element recognition approach. Sulfated quinazolinone homodimers were identified to have micromolar potencies toward FXIa and significant selectivity against thrombin, FXa, chymotrypsin, and trypsin. In different studies, NSGM 32 inhibited the HSV-1 entry into multiple cell lines demonstrating antiviral activity that was comparable to acyclovir.
Another strategy to develop FXIa allosteric inhibitors was the dual-element recognition strategy. Using this strategy, sulfated quinazolinone homodimers were developed with micromolar inhibitory potencies toward human FXIa. The most potent inhibitors identified were 33 and 34 (Figure 9) with IC50 values of 59 µM (KD= 37 µM) and 52 µM (KD= 38 µM) (Table 2), respectively, and substantial selectivity over FXa, thrombin, chymotrypsin, and trypsin [101]. Other allosteric inhibitors of FXIa that have been reported in the context of thrombin and/or FXa inhibition are the benzofuran trimer 27 (Figure 8) [96] and the sulfated coumarin dimer 31 (Figure 8) [97]. The monosulfated benzofuran trimer 27 was found to be a potent FXIa inhibitor with an IC50 value of 0.82 µM. Michaelis-Menten kinetics revealed a noncompetitive mechanism of action for benzofuran 27. Competition studies with full-length heparin, fluorescence quenching studies, and computational studies suggested a site near Lys255 on the A3 domain as a putative binding site for this inhibitor. The binding site in the A3 domain was further supported by using a form of FXIa lacking the A3 domain. The sulfated coumarin 31 also inhibited FXIa with an IC50 value of 31 µM. However, these two molecules 27 and 31 were more potent inhibitors of thrombin.
Allosteric Inhibitors of Plasmin, a Fibrinolysis Serine Protease
Plasmin is a serine protease that demonstrates trypsin-like broad specificity. Intravascular plasmin has a central role in fibrinolysis, and therefore, plasmin inhibitors are used as antifibrinolytic agents. Cell surface plasmin is important for the degradation of extracellular matrix, and thus, for tissue remodeling, metastasis, chemotaxis, and tissue repair and wound healing. Accordingly, plasmin inhibitors may also show anti-inflammatory and anticancer activities [72]. Structurally, plasmin contains a strong electropositively charged region in the catalytic domain to which heparin potentially binds. Previous studies have indicated that heparin directly and allosterically modulates the catalytic activity of plasmin [178–181] The previously synthesized LMWLs were found to directly bind to and allosterically inhibit human plasmin. Chromogenic substrate hydrolysis assays indicated that CDSO3 14 (Figure 6) potently inhibits human plasmin with an IC50 value of 0.24 μM and efficacy of ~100%. FDSO3 15 and SDSO3 16 were 3-fold and 5-fold less potent, respectively [182].
To discover specific sulfated, homogeneous NSGMs that allosterically inhibit plasmin, an in-house diverse library of 55 sulfated NSGMs with varying positions and number of sulfate groups was screened against full-length human plasmin by a chromogenic substrate-based assay. A pentasulfated flavonoid-quinazolinone dimer 35 (Figure 10) was identified to be the most potent sulfated inhibitor of plasmin with an IC50 value of 45 μM and efficacy of 100% [102]. Michaelis-Menten kinetics indicated an allosteric inhibition of plasmin by sulfated NSGMs. Studies also revealed that the most potent NSGM-based inhibitors are selective for plasmin over FXa and thrombin. Subsequent development of this class of molecules led to sulfated diflavonoid 36 (KD= 0.7 µM) (Figure 10) (Table 2). This molecule inhibited human full-length plasmin with an IC50 value of 7.2 µM and displayed selectivity indices of >277-, 70-, 26-, >138-, >61-, and 27-fold over factors IXa, Xa, XIa, XIIa, trypsin, and chymotrypsin, respectively [103]. This inhibitor was also found to be a reversible, uncompetitive inhibitor of human plasmin. Competition with full-length heparin, salt-dependent studies, and molecular modeling studies suggested that this inhibitor may recognize an anion-binding site in the catalytic domain of human plasmin. More importantly, molecule 36 also inhibited the physiologic function of plasmin i.e. clot lysis under physiological conditions in a dose-dependent manner with an IC50 value of 8.8 µM and efficacy of 69%.
Figure 10.

The chemical structures of flavonoid-based sulfated NSGMs that allosterically inhibit human plasmin and human FXIIIa. Pentasulfated flavonoid-quinazolinone heterodimer 35 was initially discovered as allosteric and marginally selective inhibitor of human plasmin. Subsequent efforts led to the identification of octasulfated flavonoid homodimer 36 inhibited human full-length plasmin with an IC50 value of 7.2 µM (KD=0.7 µM) and displayed significant selectivity against factors Xa, XIa, IXa, XIIa, trypsin, and chymotrypsin. Sulfated diflavonoid 36 also inhibited the physiologic function of plasmin i.e. clot lysis under physiological conditions in a dose-dependent manner with an IC50 value of 8.8 µM and efficacy of 69%. Interestingly, homologation of the sulfated diflavonoid 36 led to the undecasulfated flavonoid trimer 37 which was the first allosteric inhibitor of FXIIIa, a coagulation transglutaminase. NSGM 37 inhibited FXIIIa with an IC50 value of 36 µM (KD = 25.3 µM) and efficacy of 98%. The molecule exhibited good selectivity against thrombin, FXa, and papain. It also inhibited the physiological function of FXIIIa as measured by the cross-linking of fibrin monomers.
Allosteric Inhibitors of Factor XIIIa, a Coagulation Transglutaminase
The concept of sulfated NSGMs-mediated allosteric inhibition of enzymes was also applied to transglutaminases, particularly FXIIIa [183]. FXIIIa catalyzes the last step in the coagulation cascade by cross-linking the α-and γ-chains of fibrin leading to the formation of the insoluble mesh of blood clot. In addition, FXIIIa also cross-links α2-plasmin inhibitor to fibrin which subsequently protects the newly formed blood clot from lysis by plasmin. In a model of ligation-based thrombosis of the mouse inferior vena cava, FXIII-deficiency led to a substantial decrease in thrombus weight [184, 185]. Moreover, heterologous FXIII gene knockout in the mouse was reported to lack extreme bleeding [186, 187]. These results suggest that FXIIIa can be considered as a therapeutic target to develop new anticoagulants for the prevention and/or treatment venous thromboembolism and other thrombotic diseases. Thus, a library of 22 variably sulfated NSGMs and GAGs were screened against FXIIIa using a bisubstrate-based trans-glutamination assay under physiological conditions. An undecasulfated quercetin trimer 37 (Figure 10) was identified as a reversible, moderate inhibitor of FXIIIa that showed an allosteric inhibition mechanism with an IC50 value of 36 µM (KD = 25.3 µM) (Table 2) and efficacy of 98%. The molecule exhibited selectivity indices of 10–30-fold over FXa, thrombin, and papain. The flavonoid trimer 37 also inhibited the physiological function of FXIIIa as measured by the effect on cross-linking of fibrin monomers. The IC50 of fibrin cross-linking inhibition was measured to be 76.3 µM with an efficacy of ~100% under near physiological conditions of 25°C and pH 7.4 [183]. As a result, the trimer 37 serves as a lead inhibitor of human FXIIIa for further drug development efforts.
3.B). SULFATED NSGM-BASED INHIBITORS OF ANGIOGENESIS AND CANCER STEM CELLS
Heparan sulfate proteoglycans are important facilitators in tumor-associated angiogenesis. Heparan sulfate proteoglycans act as co-receptors for a variety of pro-and anti-angiogenic factors including FGFs, VEGF, and endostatin [188]. In fact, several randomized clinical trials suggest that heparin may have direct anti-tumor effects. Examples of studies investigating the therapeutic effect of LMWHs in cancer patients have included 1) enoxaparin in RASTEN (NCT00717938) and SYRINGES (NCT00771563) for small cell lung cancer and for advanced non-small cell lung cancer, respectively; 2) tinzaparin in TILT(NCT00475098) and PERIOP-01 (NCT01455831) for non-small cell lung cancer and for colorectal cancer, respectively; 3) 2,3-desulfated heparin in PGPC1 (NCT01461915) for pancreas cancer; and 4) nadroparin in LMWHCR (NCT00951574) for colorectal cancer [189]. Considering these facts, a library of 18 sulfated NSGMs was screened in an in vitro assay of tumor-associated angiogenesis [188]. This exercise led to the identification of molecules 38 – 42 (Figure 11) as potent inhibitors of angiogenesis at concentrations of 100 µM. The pharmacophoric requirements appear to include two sulfate groups separated by an average distance of 7.5 Å.
Figure 11.
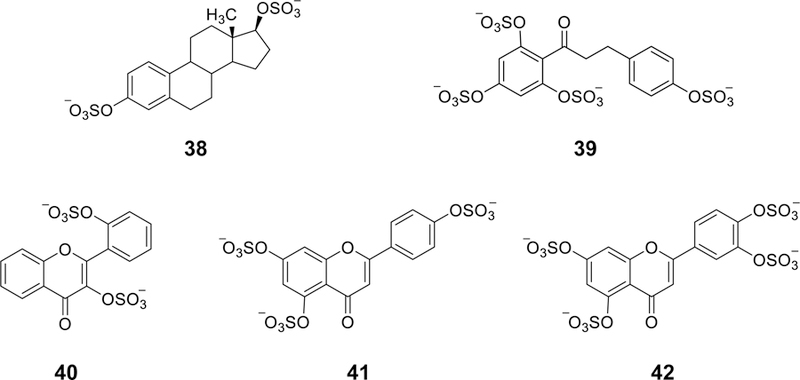
The chemical structures of sulfated NSGMs (38–42) that inhibited angiogenesis in an in vitro tube formation assay. Minimal ‘pharmacophore’ appears to be two sulfate groups at an optimal distance of 5–10 Å.
An emerging approach for cancer treatment is the targeting of cancer stem cells which contribute to cancer relapse. Knowing that sulfated GAGs play critical roles in cancer cell growth, invasion, and metastasis, sulfated NSGMs were projected to inhibit/antagonize cancer stem cells [190]. Thus, a library of 53 NSGMs from 12 chemical classes were screened against colon cancer cell lines (HT29 & HCT116) in monolayer and spheroid conditions. A dual screening protocol i.e. monolayer versus spheroid growth inhibition and primary versus secondary spheroid growth was exploited to identify three NSGMs 32, 43, and 44 (Figures 9 and 12), which inhibited spheroid growth by >50% in both screens, as selective inhibitors of colon cancer stem cells. Two NSGMs 32 and 44 downregulated several cancer stem cell markers and self-renewal factors. Particularly, NSGM 44 inhibited spheroid growth but not monolayer growth with an IC50 value of 58 μM suggesting selective targeting of cancer stem cells. Early studies suggested that NSGM 44 inhibited β-catenin transcriptional activity and activated p38MAPK only in spheroids. Pharmacological inhibition of p38 activation resulted in near complete reversal of all the morphological and molecular changes observed with this molecule. In contrast, NSGM 43 enhanced cellular differentiation. Very recently, the in vivo selective inhibitory activity of molecule 44 toward cancer stem cells via p38 MAP kinase activation was reported [191].
Figure 12.
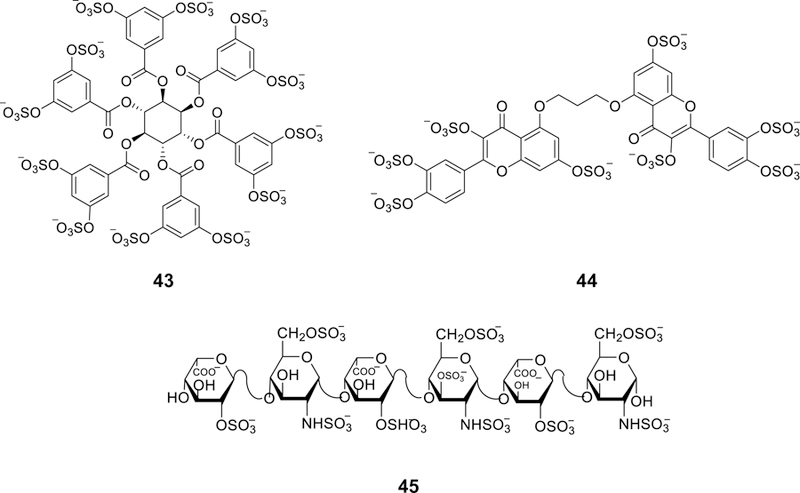
The chemical structures of sulfated NSGMs 43 and 44, two molecules identified in a dual-filter screening protocol as selective inhibitors/antagonists of colon cancer stem cells in HT29 and HCT116 cell lines. Molecule 43 is a dodecasulfated hexasubstituted inositol derivative whereas molecule 44 is an octasulfated flavonoid homodimer derivative. These molecules, in addition to 32, downregulated several cancer stem cell markers and self-renewal factors. A three-step molecular dynamics-based algorithm indicated that the dimeric flavonoid-based NSGM 44 mimics hexameric GAG sequences including HS06 45 in the protein-bound state which selectively inhibited cancer stem cells self-renewal and induced apoptosis in colorectal, pancreatic, and breast cancer stem cells through a specific early and sustained activation of p38α/β MAPK.
To further understand the mechanism underlying the anti-cancer stem cell activity of NSGM 44, a 3-step molecular dynamics algorithm was developed to compare sulfated GAGs with sulfated NSGMs. Using interactions with FGF-related proteins, the algorithm predicted the GAG structure-mimicking property of NSGMs. The study concluded that the dimeric flavonoid-based NSGM 44 mimics hexameric GAG sequences in the protein-bound state and this contributed to its anti-cancer stem cell properties [192]. In fact, a non-anticoagulant heparan sulfate hexasaccharide (HS06) 45 (Figure 12) was subsequently identified to selectively inhibit cancer stem cell self-renewal and to induce apoptosis in colorectal, pancreatic, and breast cancer stem cells. HS06 inhibition of the cancerous stem cells was promoted by a specific early and sustained activation of p38α/β mitogen activated protein kinase [193]. Overall, these findings put forward NSGMs as a viable concept to design and develop effective stem cell-targeting anti-cancer agents.
3.C). SULFATED NSGM-BASED ANTI-EMPHYSEMA AGENTS
Emphysema is a lung disease that is characterized by progressive obstruction of airflow. Unfortunately, no current drug prevents or cures the disease or reduces its progression [194]. The absence of effective therapy for emphysema is mainly attributed to the complexity of its pathophysiology which involves the pathogenic triad of inflammation, oxidative stress, and elastolysis in lung cells [73–75]. To open-up a potential therapeutic avenue for emphysema, the anti-inflammatory, anti-oxidative, and anti-human neutrophil elastase activities of unsulfated and sulfated LMWLs of caffeic acid (CD and CDSO3; 14), ferulic acid (FDSO3; 15), and sinapic acid (SDSO3; 16) (Figure 6) were studied [194].
In vitro anti-elastase assay indicated that the monomer caffeic acid failed to inhibit neutrophil elastase at the highest concentration tested of 100 µM, while its unsulfated oligomer CD and sulfated oligomer CDSO3 concentration-dependently inhibited neutrophil elastase with IC50 values of 2.82 and 0.43 µM, respectively. Likewise, FDSO3 and SDSO3 inhibited neutrophil elastase with IC50 values of 0.55 and 0.72 µM, respectively. CDSO3 also inhibited human sputum elastase with IC50 of 0.11 µM. Mechanistically, CDSO3 was found to inhibit human neutrophil elastase in an apparent mixed mechanism. CDSO3 also inhibited human sputum elastase-induced degradation of insoluble macromolecular elastin-congo red at a concentration range of 0.1–0.5 µM in a concentration-dependent fashion [194]. In vitro anti-oxidative activity assessment was performed using the chemical antioxidant assay kit and H2O2-stimulated A549 cells. In the former assay, CDSO3, FDSO3, and SDSO3 demonstrated IC50 values of 3.5, 5.1, 5.5 µM, respectively. Furthermore, 10 µM CDSO3 protected the human alveolar A549 cells by about 70% from H2O2-induced decrease in the rGSH level. The in vitro cellular anti-inflammatory activities of LMWLs were determined with the TNFα-induced NFkB– Luciferase activity and IL-8 release in the transfected and non-transfected human lung Calu-3 epithelial cell systems, respectively. Unsulfated caffeic acid-based LMWL and FDSO3 both inhibited the TNFα-induced NFkB–Luciferase activity with IC50 value of 50 µM, whereas CDSO3 more potently demonstrated anti-inflammatory activity in this assay with an IC50 value of 10 µM. CDSO3 also inhibited the IL-8 release triggered by different stimuli including TNFα, H2O2, and okadaic acid by 65–83% at a concentration of 10 µM [194]. To establish the efficacy of CDSO3 as potential treatment for emphysema, it was tested in human sputum elastase/cigarette smoke extract-induced emphysema model in rats. In this model, administration of 30 or 100 µg/kg dose of CDSO3 into the lung was found to significantly attenuate by 43–93% the human sputum elastase/cigarette smoke extract-induced barrier damage, elastolytic activity, oxidative stress, inflammatory response, luminal neutrophil infiltration and tissue myeloperoxidase activity, functional impairment of exercise endurance, and airspace enlargement, in both interventional and preventive dosing protocols. Overall, such triple-action of CDSO3 appears to be promising in developing a novel treatment for emphysema, particularly that this molecule is not peptidic polymer which may allow longer lung retention and thus therapeutic longevities can be achieved upon local pulmonary delivery [195]. Very recently, it was reported that 10 µM of CDSO3 inhibited lung endothelial HMVEC-L and epithelial A549 cell death induced by VEGF receptor antagonist (SU5416), trichostatin A-mediated histone deacetylase inhibition, and cigarette smoke extract by 65–99%, which were all substantially opposed by the addition of inhibitors of hypoxia-inducible factor-1α or excess Fe2+ [196]. CDSO3 also inhibited H2O2-and elastase-induced cell death by 95 and 92%, respectively. In the rat model of SU5416-induced apoptotic emphysema, 60 μg/kg of CDSO3 produced 61–77% interventions against exercise endurance impairment, oxidative lung damage, and airspace enlargement. This dose also normalized the cleaved caspase-3 apoptotic marker, significantly stimulated the VEGF signaling, and significantly elevated the hypoxia-inducible factor-1α and VEGF expression by more than 1.5-fold. All these results highlighted the translational aspects of the polymeric NSGM CDSO3 in inhibiting lung cell death and the subsequent development of apoptotic emphysema [196].
3.D). SULFATED NSGM-BASED ANTIMICROBIAL AGENTS
It is well reported that the entry of HSV-1 into host cells involves its glycoprotein D (gD) binding to heparan sulfate. Recently, it was hypothesized that HSV-1 entry to host cells could be competitively inhibited using NSGMs [124]. Indeed, screening a library of 13 sulfated saccharides and NSGMs led to the identification of a group of molecules that bound gD with high affinities. The saccharides included fondaparinux, sucrose octasulfate, unfractionated heparin, and chondroitin sulfate A. The NSGM group included flavonoid-based agents, benzofuran-based agents, and glycoside-based agents. Particularly, the NSGMs displayed variable binding affinities spanning the range of 8 nM to 2.22 μM. Octasulfated flavonoid-based NSGMs showed affinities of 22 and 105 nM, which were much higher than those of disulfated benzofuran-based NSGMs (0.78–2.22 µM). The glycoside NSGMs had 6–15 sulfate groups and were the most potent with KD values of 8–112 nM. Particularly, NSGM 32 (Figure 9) was found to bind gD with an affinity value of 10 nM. Stoichiometry of 32−gD complex, measured using spectrofluorimetry, was found to be 1.7:1.0 suggesting the possible engagement of two anion-binding sites simultaneously. Colorimetric studies on HSV-1 entry into HFF-1, HeLa, and VK2/E6E7 cells led to IC50 values of 0.49 µM (HFF-1), 0.43 µM (HeLa), and 1.0 µM (VK2/E6E7). These activities were found to compare well with the activities of unfractionated heparin. More importantly, the level of inhibition of HSV-1 cellular entry was 12-fold better than that detected previously with heparin octasaccharides. Not only that, but NSGM 32 also inhibited the HSV-1 KOS-promoted plaque formation in Vero cells at 10 and 100 µM concentrations suggesting a powerful antiviral activity. This activity was similar to that of acyclovir, a currently used anti-HSV-1 drug [122]. Lastly, cellular toxicity of 32 was measured using lactate dehydrogenase activity released upon damage of plasma membrane. The study indicated that 32 is completely nontoxic to human cells at concentrations as high as 100 μM.
Sulfated molecules 17, 21, and 22 (Figure 7) were also identified by in silico screening to inhibit Epstein–Barr virus (EBV) infection which is associated with multiple human malignancies, including Burkitt lymphoma [125]. Computationally, the immediate-early lytic protein Zta was proposed to be the preferred target for these sulfated small molecules and the latent Epstein–Barr virus nuclear antigen 1 (EBNA1) protein could be a secondary target for molecule 21. Using Raji cells (cells derived from B-lymphocytes from young patient diagnosed with Burkitt lymphoma), the antiviral effects of the three sulfated molecules were established by studying their effects on EBV DNA loading, latent viral protein expression, and viral reactivation induced by 12-O-tetradecanoylphorbol-13-acetate. At concentrations of 20 µM, all the tested molecules significantly decreased the EBV DNA present in Raji cells indicating that they are targeting EBV in its latent state or interfering with episome maintenance. All three molecules also significantly decreased the expression of the latent membrane protein-1, a viral oncoprotein, in the Raji cell line but had no effect on viral early-antigen diffuse protein expression or viral reactivation. The research group also hypothesized that the sulfated molecules may interfere with the virus entry as described for other sulfated compounds. Molecule 21 was indicated to be the most effective considering that it decreases the latent membrane protein-1 expression more than 50% at 20 µM. Molecular modeling studies suggested that sulfated molecule 21 occupies the DNA-binding sites on the two proteins Zta and EBNA1. Therefore, the sulfated xanthone-based molecule 21 was proposed to lead the efforts to develop more clinically relevant inhibitors for EBV [125].
Other NSGMs that have exhibited substantial antiviral activity by blocking the viral entry stage are the natural product zosteric acid 46 (Figure 13) and its analogues. Zosteric acid is isolated from the temperate marine eelgrass, Zostera marina. A library of zosteric acid derivatives was evaluated for their anti-viral properties against dengue virus (DENV) in a focus forming unit reduction assay. In this assay, zosteric acid exhibited moderate potency with an IC50 value of ~2.3 mM against DENV-2 with no evidence of toxicity to the monkey kidney cell line LLCMK-2 over the concentrations tested [197].
Figure 13.

The chemical structures of the monosulfated zosteric acid 46 and trisulfated gallic acid 47, both of which are NSGMs which demonstrate significant antifouling activities. Zosteric acid also inhibits the viral entry of dengue virus into host cells. The figure also depicts the chemical structure of 3-carboxy-4-hydroxybenzenesulfonate 48 which potentially inhibits CCL5-mediated leukocyte recruitment to the peritoneal cavity in the thiogallate-induce peritonitis mice model. Chemical structures of acamprosate 49, tramiprosate 50, eprodisate 51, and suramin 52 are also depicted.
Zosteric acid has also been reported to inhibit biofilm formation by multiple micro-organisms [198]. Although the mechanism appears to be species-specific, several studies indicate the necessity of the sulfate group for the antifouling activity of zosteric acid through interfering with the surface hydrophilicity, blocking the sulfate-binding of microbial surface sensors, or inhibiting the polymerization of the microbial extracellular matrix [198]. Inspired by these findings, a diverse library of 13 sulfated molecules were synthesized and tested for antifouling activity using the anti-settlement activity of mussel plantigrade post-larvae and bacterial growth inhibition towards four biofilm-forming bacterial strains [199]. In this study, the sulfated molecules with the most potent anti-settlement activity were sulfated molecules 47 (trisulfated gallic acid) (Figure 13), 19 (rutin persulfate), and 22 (3,6-bis(β-D-glucopyranosyl) xanthone persulfate) (Figure 7) with EC50 values of 17.65 μM, 22.59 μM, and 23.19 µM, respectively. Importantly, the corresponding nonsulfated precursors of these molecules lacked the anti-settlement activity at comparable concentrations suggesting that the sulfate group is an essential structural criterion for this type of activity. Molecule 19 also significantly inhibited the growth of the biofilm-forming Vibrio harveyi (EC20 = 7.7 µM) with no effect on Cobetia marina, Pseudoalteromonas atlantica, and Halomonas aquamarina. All the three bioactive sulfated molecules were also found to be non-ecotoxic as indicated by the lack of toxicity against Artemia salina (<10% death at 250 μM) and Vibrio fischeri (LC50 > 1000 μg/mL). Overall, this study highlights the potential of sulfated molecules as effective, non-ecotoxic antifouling agents.
Lastly, NSGMs, particularly sulfonated mimetics, are also being studied for other activities. For instance, sulfonated NSGMs may have the potential to disrupt GAG–chemokine interactions, and thus, promote anti-inflammatory activity. In this regard, Severin et al. reported that the sulfonated NSGM 48, which was identified via a virtual screening exercise, inhibits heparin binding to chemokine ligand-5 (CCL5) with an IC50 value of 320 µM (KD=20 µM) [200]. Molecule 48 also selectively inhibited the binding of the chemokine CCL5 to its receptor CCR1 (but not CCR5) with an IC50 value of 6.7 µM. In the mouse model of thioglycollate-induced peritonitis, molecule 48 inhibited the leukocyte recruitment to the peritoneum in a dose-dependent manner with an EC50 value of 0.06 µg/animal providing a proof-of-concept that NSGMs can be developed as anti-inflammatory agents. Likewise, two polysulfonated polymers, which were produced by polymerization of vinyl-sulfonic acid, and copolymerization of vinyl-sulfonic acid with 1-vinyl-2-pyrrolidone were tested for potential prevention of malaria infection [201]. Vinyl-sulfonic acid reproducibly decreased midgut oocyst development by 99% in mosquitoes fed with Plasmodium falciparum and Plasmodium berghei. Experiments suggested that vinyl-sulfonic acid binds to the circumsporozoite-and TRAP-related protein, a protein that is expressed by ookinetes and is required for midgut invasion. This interaction significantly inhibits a key step of parasite development, and consequently, compromises subsequent events essential for mosquito-to-human transmission. This work lays the foundation for a novel NSGM-based prophylactic strategy against malarial infections.
4. CHALLENGES AND FUTURE DIRECTIONS
Despite the promise of NSGMs in treating multiple pathologies, much work remains to bring sulfated NSGMs into clinical use. Advancements in their manufacturing process, their structural biology aspects, and their route of administration are specifically needed. Although NSGMs are easier to chemically synthesize than sulfated polysaccharides, chemical sulfation of small scaffolds can be challenging [202]. Nearly all sulfated molecules are highly water soluble which makes their isolation in highly pure form difficult due to the presence of inorganic salts. Susceptibility of sulfate groups to hydrolysis under acidic conditions and high temperatures may also restrain their manufacturing process as well as their characterization by high resolution mass spectrometers. Furthermore, given the highly ionized nature of sulfate groups, few functional group transformations can successfully be accomplished in the presence of sulfate group which ultimately forces the synthetic scheme to include sulfation as the last reaction. Importantly, these challenges increase exponentially in the synthesis of polysulfated scaffolds, particularly with the added need to achieve regioselectivity and/or to drive the reaction to completion to avoid the formation of heterogenous mixtures of partially sulfated species. Therefore, an important step toward advancing sulfated NSGMs into the clinical use is to invent new technologies to make them under/by more “user-friendly” conditions. This will allow significant level of chemical maneuverability and regioselectivity to be achieved, robust characterization to be reported, and pure quantitative amount of NSGMs to be produced. In this arena, using NSGMs as organic salts of protonated tertiary amines, for example triethylammonium salts, has been exploited with variable levels of success, yet the nature of amine used for each class of NSGMs may require optimization [146].
From the perspective of drug design, reported NSGMs were discovered by medium-throughput screening of in-house libraries of small sulfated molecules [97, 98,102, 124, 183] or computational docking and scoring of virtual libraries [68]. Ligand-based computational approaches were also exploited in the design of early generations of ATIII activators [66] and the advanced design of plasmin inhibitors [103]. Yet, understanding of sulfated NSGMs interactions with the corresponding binding sites on the targeted proteins at an atomic level continues to be lacking. Understanding atomic level interactions is important to help engineer structural modifications that lead to enhanced binding affinity and specificity. Protocols to co-crystallize sulfated NSGMs with the corresponding proteins are yet to be developed. Although this may appear to be difficult to achieve given the salt-dependent nature of NSGM-protein interactions, conditions under which co-crystal structures of heparins with proteins [49, 50, 81, 82, 203–208] have been obtained can be exploited as a starting point. The use of current advancements in structural biology including automation and nanotechnology can also help in achieving this goal. Furthermore, the development of NSGM-based electrophilic probes which can form radical-initiated covalent bond(s) with specific amino acid(s) in the putative binding site may further enhance the possibility of obtaining the first co-crystal structure of NSGM-protein complex.
The fact that NSGMs are negatively charged means that they are less likely to be passively absorbed following oral administration, and perhaps, it means that they are more suitable for parenteral administration. While there is an urgent need for anticoagulants to parenterally be used in hospital settings, for example during surgery, oral administration is more convenient to treat chronic illnesses in out-clinic patients. In this arena, multiple strategies can be exploited to enhance the oral bioavailability of NSGMs. Similar to heparins, 1) conjugation of NSGMs with cholesterol, bile acids such as deoxycholic acid, or fatty acids such as palmitic acid, stearic acid, myristic acid, and lauric acid [209–212]; 2) formulation with penetration enhancers including L-arginine [206] and 18β-glycyrrhetinic acid [214] among others; or 3) encapsulation of NSGMs in micro and nanoparticles [215] are all strategies that may help the oral use of NSGMs for systemic indications without disturbing their biological activity.
Few hundreds of sulfated NSGMs have been chemically synthesized and characterized and biochemically evaluated for various in vitro and ex vivo activities. Yet, all sulfated NSGMs reported thus far are aryl-O-sulfonates suggesting that the chemical space of NSGMs can further be expanded by developing multiple bioisosteric scaffolds involving aryl-N-sulfonates, aryl-N,O-sulfonates, aryl-C-sulfonates, and aryl-sulfonates as well as their corresponding aliphatic congeners. Evidence for the need for bio-isosteric modifications to achieve druggability is demonstrated by the clinical availability of acamprosate and suramin as well as the clinical potential of tramiprosate and eprodisate (Figure 13). Acamprosate 49 is aliphatic sulfonate derivative that is clinically used to treat alcohol dependence and it apparently does so by blocking glutaminergic N-methyl-D-aspartate receptors and activating gamma-aminobutyric acid type A receptors [216]. Trampiprosate 50 was investigated in advanced clinical trials as a potential treatment for Alzheimer’s disease. It is anti-amyloidogenic agent which binds to soluble β-amyloid protein, and subsequently, inhibits the formation and the deposition of GAG-induced neurotoxic aggregates [217]. Eprodisate 51 was evaluated to protect renal function in patients with AA amyloidosis. It inhibits the polymerization of amyloid fibrils and the deposition of the fibrils in tissues by antagonizing the interactions between amyloidogenic proteins and GAGs [218]. Suramin 52 is aryl-sulfonate derivative that is clinically used in the treatment of first-stage African trypanosomiasis (sleeping sickness) caused by Trypanosoma brucei without CNS involvement [219]. Suramin and its analogs have also demonstrated multiple pharmacological effects including anti-cancer activities [220], antiviral activities [221], anti-autism activity [222], and a potential to treat coronary artery disease by targeting proprotein convertase subtilisin/kexin type 9 (PCSK9) [223]. Therefore, bioisosteric modifications appear to be necessary so as to enhance the structural diversity of the available pool of sulfated NSGMs, and thus, to accelerate the discovery of much needed clinical candidates.
ACKNOWLEDGMENTS
RAAH is supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under grant numbers 5 P20 GM103424-15 and 3 P20 GM103424-15S1. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
ABBREVIATIONS
- APTT
Activated Partial Thromboplastin Time
- ATIII
Antithrombin
- CCL5
Chemokine Ligand-5
- DENV
Dengue Virus
- DHPs
Dehydrogenation Polymers
- EBNA1
Epstein–Barr Virus Nuclear Antigen 1
- EBV
Epstein–Barr Virus
- FXa
Factor Xa
- FIXa
Factor IXa
- FXIa
Factor XIa
- FXIIa
Factor XIIa
- FXIIIa
Factor XIIIa
- FGF
Fibroblast Growth Factor
- GAG
Glycosaminoglycan
- HCII
Heparin Cofactor II
- HGF
Hepatocyte growth factor
- HINT
Hydropathic Interaction
- HIV
Human Immunodeficiency Virus
- HSV
Herpes Simplex Virus
- IL-8
Interleukin 8
- LMWLs
Low Molecular Weight Lignins
- MIP-1α
Macrophage inflammatory protein 1-alpha
- NAP-2
Neutrophil Activating Peptide-2
- NSGM
Non-Saccharide Glycosaminoglycan Mimetic
- PF4
Platelet Factor 4
- PT
Prothrombin Time
- Serpin
Serine Protease Inhibitor
- SPGG
Sulfated Pentagalloyl Glucopyranoside
- THIQ
Tetrahydroisoquinoline
- VEGF
Vascular endothelial growth factor
Footnotes
CONFLICT OF INTEREST
Authors declare no competing financial conflict of interest.
REFERENCES
- [1].Gandhi NS; Mancera RL The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des, 2008, 72, 455–482. [DOI] [PubMed] [Google Scholar]
- [2].Esko JD; Kimata K; Lindahl U Proteoglycans and Sulfated Glycosaminoglycans, In: Essentials of Glycobiology; Varki Ajit, Executive Editor, Cummings Richard D, Esko Jeffrey D, Stanley Pamela, Hart Gerald W, Aebi Markus, Darvill Alan G, Kinoshita Taroh, Packer Nicolle H, Prestegard James H, Schnaar Ronald L, and Seeberger Peter H., Ed.; Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press, 2009; pp. 784. [PubMed] [Google Scholar]
- [3].Imberty A; Lortat-Jacob H; Pérez S Structural view of glycosaminoglycan-protein interactions. Carbohydr. Res, 2007, 342, 430–439. [DOI] [PubMed] [Google Scholar]
- [4].Pomin VH; Mulloy B Glycosaminoglycans and proteoglycans. Pharmaceuticals (Basel), 2018, 11(1), 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sasisekharan R; Venkataraman G Heparin and heparan sulfate: Biosynthesis, structure and function. Curr. Opin. Chem. Biol, 2000, 4, 626–631. [DOI] [PubMed] [Google Scholar]
- [6].Capila I; Linhardt RJ Heparin-protein interactions. Angew. Chem. Int. Ed. Engl, 2002, 41, 391–412. [DOI] [PubMed] [Google Scholar]
- [7].Mulloy B; Hogwood J; Gray E; Lever R; Page CP Pharmacology of heparin and related drugs. Pharmacol. Rev, 2016, 68(1), 76–141. [DOI] [PubMed] [Google Scholar]
- [8].Mizumoto S; Yamada S; Sugahara K Molecular interactions between chondroitin-dermatan sulfate and growth factors/receptors/matrix proteins. Curr. Opin. Struct. Biol, 2015, 34, 35–42. [DOI] [PubMed] [Google Scholar]
- [9].Monslow J; Govindaraju P; Puré E Hyaluronan – A functional and structural sweet spot in the tissue microenvironment. Front. Immunol, 2015, 6, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Caterson B; Melrose J Keratan sulfate, a complex glycosaminoglycan with unique functional capability. Glycobiology, 2018, 28(4), 182–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bourin MC; Lindahl U Glycosaminoglycans and the regulation of blood coagulation. Biochem. J, 1993, 289, 313–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rabenstein DL Heparin and heparan sulfate: structure and function. Nat. Prod. Rep, 2002, 19, 312–331. [DOI] [PubMed] [Google Scholar]
- [13].Ruoslahti E; Yamaguchi Y Proteoglycans as modulators of growth factor activities. Cell, 1991, 64, 867–869. [DOI] [PubMed] [Google Scholar]
- [14].Sasisekharan R; Shriver Z; Venkataraman G; Narayanasami U Roles of heparan-sulphate glycosaminoglycans in cancer. Nat. Rev. Cancer, 2002, 2, 521–528. [DOI] [PubMed] [Google Scholar]
- [15].Yip GW; Smollich M; Götte M Therapeutic value of glycosaminoglycans in cancer. Mol. Cancer Ther, 2006, 5, 2139–2148. [DOI] [PubMed] [Google Scholar]
- [16].Lever R; Smailbegovic A; Page C Role of glycosaminoglycans in inflammation. Inflammopharmacology, 2001, 9, 165–169. [Google Scholar]
- [17].Gozzo AJ; Nunes VA,; Carmona AK; Nader HB; von Dietrich CP; Silveira VL; Shimamoto K; Ura N; Sampaio MU; Sampaio CA; Araújo MS Glycosaminoglycans affect the action of human plasma kallikrein on kininogen hydrolysis and inflammation. Int. Immunopharmacol, 2002, 2, 1861–1865. [DOI] [PubMed] [Google Scholar]
- [18].Holzmann J; Zemann BNA; Schabus R; Marlovits S; Cowburn R; Huettinger M Assorted effects of TGF b and chondroitinsulfate on p38 and ERK½ activation levels in human articular chondrocytes stimulated with LPS. OsteroArthritis Cartil, 2006, 14, 519–525. [DOI] [PubMed] [Google Scholar]
- [19].Yasuda T Hyaluronan inhibits cytokine production by lipopolysaccharide-stimulated U937 macrophages through down-regulation of NF-κB via ICAM-1. Inflamm. Res, 2007, 56, 246–253. [DOI] [PubMed] [Google Scholar]
- [20].Nelson BRM; Cecconi O; Roberts WG; Aruffo A; Linhardt RJ; Bevilacqua MP Heparin oligosaccharides bind L-and P-selectin and inhibit acute inflammation. Blood, 1993, 82, 3253–3258. [PubMed] [Google Scholar]
- [21].Jinno A; Park PW Role of glycosaminoglycans in infectious disease. Methods Mol. Biol, 2015, 1229, 567–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Aquino RS; Park PW Glycosaminoglycans and infection. Front. Biosci. (Landmark Ed), 2016, 21, 1260–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tiwari V; O’Donnell C; Copeland RJ; Scarlett T; Liu J; Shukla D Soluble 3-O-sulfated heparan sulfate can trigger herpes simplex virus type 1 entry into resistant Chinese hamster ovary (CHO-K1) cells. J.Gen.Virol, 2007, 88, 1075–1079. [DOI] [PubMed] [Google Scholar]
- [24].Choudhary S; Marquez M; Alencastro F; Spors F; Zhao Y; Tiwari V Herpes simplex virus type-1 (HSV-1) entry into human mesenchymal stem cells is heavily dependent on heparan sulfate. J. Biomed. Biotechnol, 2011, 2011, 264350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Witvrouw M; De Clercq E Sulfated polysaccharides extracted from sea algae as potential antiviral drugs. Gen. Pharmacol, 1997, 29, 497–511. [DOI] [PubMed] [Google Scholar]
- [26].Kim M; Yim JH; Kim SY; Kim HS; Lee WG; Kim SJ; Kang PS; Lee CK In vitro inhibition of influenza A virus infection by marine microalga-derived sulfated polysaccharide p-KG03, Antiviral Res 2012, 93, 253–259. [DOI] [PubMed] [Google Scholar]
- [27].Volpi N Therapeutic applications of glycosaminoglycans. Curr.Med.Chem, 2006, 13, 1799–810. [DOI] [PubMed] [Google Scholar]
- [28].Zhang L Glycosaminoglycan (GAG) biosynthesis and GAG-binding proteins. Prog. Mol. Biol. Transl. Sci, 2010, 93, 1–17. [DOI] [PubMed] [Google Scholar]
- [29].Jackson RL; Busch SJ; Cardin AD Glycosaminoglycans: molecular properties, protein interactions, and role in physiological processes. Physiol. Rev, 1991, 71, 481–539. [DOI] [PubMed] [Google Scholar]
- [30].Hileman RE; Fromm JR; Weiler JM; Linhardt RJ Glycosaminoglycan-protein interactions: Definition of consensus sites in glycosaminoglycan binding proteins. BioEssays, 1998, 20, 156–167. [DOI] [PubMed] [Google Scholar]
- [31].Fromm JR; Hileman RE; Caldwell EE; Weiler JM; Linhardt RJ Pattern and spacing of basic amino acids in heparin binding sites. Arch. Biochem. Biophys 1997, 343(1), 92–100. [DOI] [PubMed] [Google Scholar]
- [32].Hileman RE; Jennings RN; Linhardt RJ Thermodynamic analysis of the heparin interaction with a basic cyclic peptide using isothermal titration calorimetry. Biochemistry, 1998, 37, 15231–15237. [DOI] [PubMed] [Google Scholar]
- [33].Fromm JR; Hileman RE; Caldwell EEO; Weiler JM; Linhardt RJ Differences in the interaction of heparin with arginine and lysine and the importance of these basic amino acids in the binding of heparin to acidic fibroblast growth factor. Arch. Biochem. Biophys 1995, 323, 279–287. [DOI] [PubMed] [Google Scholar]
- [34].Mccoy AJ; Pei XY; Skinner R; Abrahams J-P; Carrell RW Structure of beta-antithrombin and the effect of glycosylation on antithrombin’s heparin affinity and activity. J Mol Biol, 2003, 326(3), 823–33. [DOI] [PubMed] [Google Scholar]
- [35].Cardin AD; Weintraub HJ Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis, 1989, 9, 21–32. [DOI] [PubMed] [Google Scholar]
- [36].Sobel M; Soler DF; Kermode JC; Harris RB Localization and characterization of a heparin binding domain peptide of human von Willebrand factor. J. Biol. Chem, 1992, 267, 8857–62. [PubMed] [Google Scholar]
- [37].Margalit H; Fischer N; Ben-Sasson SA Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J. Biol. Chem, 1993, 268, 19228–19231. [PubMed] [Google Scholar]
- [38].Fath MA; Wu X; Hileman RE; Linhardt RJ; Kashem MA; Nelson RM; Wright CD; Abraham WM Interaction of secretory leukocyte protease inhibitor with heparin inhibits proteases involved in asthma. J. Biol. Chem, 1998, 273, 13563–13569. [DOI] [PubMed] [Google Scholar]
- [39].Malik A; Ahmad S Sequence and structural features of carbohydrate binding in proteins and assessment of predictability using a neural network. BMC Struct. Biol, 2007, 7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pratt CW General features of the heparin-binding serpins antithrombin, heparin cofactor II and protein C inhibitor. Blood Coagul. Fibrinolysis, 1997, 4, 479–490. [DOI] [PubMed] [Google Scholar]
- [41].Al-Horani RA Serpin regulation of fibrinolytic system: implications for therapeutic applications in cardiovascular diseases. Cardiovasc. Hematol. Agents Med. Chem, 2014, 12(2), 91–125. [DOI] [PubMed] [Google Scholar]
- [42].Gettins PGW Serpin structure, mechanism, and function. Chem. Rev, 2002, 102, 4751–4803. [DOI] [PubMed] [Google Scholar]
- [43].Huntington JA; Read RJ; Carrell RW Structure of a serpin-protease complex shows inhibition by deformation. Nature, 2000, 407, 923–6. [DOI] [PubMed] [Google Scholar]
- [44].Desai UR New Antithrombin-based anticoagulants. Med. Res. Rev, 2004, 24, 151–181. [DOI] [PubMed] [Google Scholar]
- [45].Desai UR; Petitou M; Björk I; Olson ST Mechanism of heparin activation of antithrombin: evidence for an induced-fit model of allosteric activation involving two interaction subsites. Biochemistry, 1998, 37(37), 13033–41. [DOI] [PubMed] [Google Scholar]
- [46].Olson ST; Richard B; Izaguirre G; Schedin-Weiss S; Gettins PG Molecular mechanisms of antithrombin-heparin regulation of blood clotting proteinases. A paradigm for understanding proteinase regulation by serpin family protein proteinase inhibitors. Biochimie, 2010, 92(11), 1587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Choay J; Petitou M; Lormeau JC; Sinaÿ P; Casu B; Gatti G Structure-activity relationship in heparin: A synthetic pentasaccharide with high affinity for antithrombin III and eliciting high anti-factor Xa activity. Biochem. Biophys. Res. Commun, 1983, 116, 492–499. [DOI] [PubMed] [Google Scholar]
- [48].Lane DA; Denton J; Flynn AM; Thunberg L; Lindahl U Anticoagulant activities of heparin oligosaccharides and their neutralization by platelet factor 4. Biochem J, 1984, 218, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Johnson DJ; Li W; Adams TE; Huntington JA Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J 2006, 25(9), 2029–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Li W; Johnson DJ; Esmon CT; Huntington JA Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat. Struct. Mol. Biol, 2004, 11, 857–862. [DOI] [PubMed] [Google Scholar]
- [51].Nordenman B; Danielsson A; Björk I The binding of low-affinity and high-affinity heparin to antithrombin. Fluorescence studies. Eur J Biochem 1978, 90(1), 1–6. [DOI] [PubMed] [Google Scholar]
- [52].Weitz JI; Young E; Johnston M; Stafford AR; Fredenburgh JC; Hirsh J Vasoflux, a new anticoagulant with a novel mechanism of action. Circulation, 1999, 99(5), 682–9. [DOI] [PubMed] [Google Scholar]
- [53].Olson ST; Björk I; Sheffer R; Craig PA; Shore JD; Choay J Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J. Biol. Chem, 1992, 267(18), 12528–38. [PubMed] [Google Scholar]
- [54].Turk B; Brieditis I; Bock SC; Olson ST; Björk I The oligosaccharide side chain on Asn-135 of alpha-antithrombin, absent in beta-antithrombin, decreases the heparin affinity of the inhibitor by affecting the heparin-induced conformational change. Biochemistry, 1997, 36(22), 6682–91. [DOI] [PubMed] [Google Scholar]
- [55].Verhamme IM; Bock PE; Jackson CM The preferred pathway of glycosaminoglycan-accelerated inactivation of thrombin by heparin cofactor II. J. Biol. Chem, 2004, 279, 9785–9795. [DOI] [PubMed] [Google Scholar]
- [56].Petitou M; Casu B; Lindahl U 1976–1983, a critical period in the history of heparin: The discovery of the antithrombin binding site. Biochimie, 2003, 85, 83–89. [DOI] [PubMed] [Google Scholar]
- [57].Ersdal-Badju E; Lu A; Zuo Y; Picard V; Bock SC Identification of the antithrombin III heparin binding site. J. Biol. Chem, 1997, 272, 19393–19400. [DOI] [PubMed] [Google Scholar]
- [58].Jin L; Abrahams JP; Skinner R; Petitou M; Pike RN; Carrell RW The anticoagulant activation of antithrombin by heparin. Med. Sci, 1997, 94, 14683–14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Arocas V; Turk B; Bock SC; Olson ST; Björk I The region of antithrombin interacting with full-length heparin chains outside the high-affinity pentasaccharide sequence extends to LYS136 but not to Lys139. Biochemistry, 2000, 39, 8512–8518. [DOI] [PubMed] [Google Scholar]
- [60].Tollefsen DM; Pestka CA; Monafo WJ Activation of heparin cofactor II by dermatan sulfate. J. Biol. Chem, 1983, 258, 6713–6716. [PubMed] [Google Scholar]
- [61].Gunnarsson GT; Desai UR Designing small, nonsugar activators of antithrombin using hydropathic interaction analyses. J. Med. Chem, 2002, 45(6), 1233–43. [DOI] [PubMed] [Google Scholar]
- [62].Gunnarsson GT; Desai UR Interaction of designed sulfated flavanoids with antithrombin: lessons on the design of organic activators. J. Med. Chem, 2002, 45(20), 4460–70. [DOI] [PubMed] [Google Scholar]
- [63].Gunnarsson GT; Desai UR Exploring new non-sugar sulfated molecules as activators of antithrombin. Bioorg. Med. Chem. Lett, 2003, 13(4), 679–83. [DOI] [PubMed] [Google Scholar]
- [64].Gunnarsson GT; Riaz M; Adams J; Desai UR Synthesis of per-sulfated flavonoids using 2,2,2-trichloro ethyl protecting group and their factor Xa inhibition potential. Bioorg. Med. Chem, 2005, 13(5), 1783–9. [DOI] [PubMed] [Google Scholar]
- [65].Gunnarsson GT; Desai UR Hydropathic interaction analyses of small organic activators binding to antithrombin. Bioorg. Med. Chem, 2004,12(3), 633–40. [DOI] [PubMed] [Google Scholar]
- [66].Raghuraman A; Liang A; Krishnasamy C; Lauck T; Gunnarsson GT; Desai UR On designing non-saccharide, allosteric activators of antithrombin. Eur. J. Med. Chem, 2009, 44(6), 2626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Liang A; Raghuraman A; Desai UR Capillary electrophoretic study of small, highly sulfated, non-sugar molecules interacting with antithrombin. Electrophoresis, 2009, 30(9), 1544–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Al-Horani RA; Liang A; Desai UR Designing nonsaccharide, allosteric activators of antithrombin for accelerated inhibition of factor Xa. J. Med. Chem, 2011, 54(17), 6125–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Di Cera E Serine proteases. IUBMB Life, 2009, 61, 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hedstrom L Serine protease mechanism and specificity. Chem. Rev, 2002, 102, 4501–4523. [DOI] [PubMed] [Google Scholar]
- [71].Al-Horani RA; Afosah DK Recent advances in the discovery and development of factor XI/XIa inhibitors. Med. Res. Rev, 2018, doi: 10.1002/med.21503. [DOI] [PMC free article] [PubMed]
- [72].Al-Horani RA; Desai UR Recent advances on plasmin inhibitors for the treatment of fibrinolysis-related disorders. Med. Res. Rev, 2014, 34(6), 1168–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Tuder RM; Voelkel NF Pathobiology of emphysema. In: Chronic obstructive lung diseases, vol 2, Voelkel NF; MacNee W, Ed.; BC Decker: Hamilton, Ontario, Canada, 2008, pp 63–75. [Google Scholar]
- [74].Sabroe I; Parker LC; Calverley PM; Dower SK; Whyte MK Pathological networking: a new approach to understanding COPD. Postgrad. Med J, 2008, 84, 259–64. [DOI] [PubMed] [Google Scholar]
- [75].Fischer BM; Pavlisko E; Voynow JA Pathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammation. Int. J. Chron. Obstruct. Pulmon. Dis, 2011, 6, 413e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bock PE; Panizzi P; Verhamme IMA Exosites in the substrate specificity of blood coagulation reactions. J. Thromb. Haemost, 2007, 5, 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].O’Brien LA; Stafford AR; Fredenburgh JC; Weitz JI Glycosaminoglycans bind factor Xa in a Ca2+-dependent fashion and modulate its catalytic activity, Biochemistry, 2003, 42, 13091–13098. [DOI] [PubMed] [Google Scholar]
- [78].Sheehan JP; Sadler JE Molecular mapping of the heparin-binding exosite of thrombin (antithrombin III/serine proteases). Biochemistry, 1994, 91, 5518–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Badellino KO; Walsh PN Localization of a heparin binding site in the catalytic domain of factor XIa. Biochemistry, 2001, 40, 7569–80. [DOI] [PubMed] [Google Scholar]
- [80].Gan ZR; Li Y; Chen Z; Lewis SD; Shafer JA Identification of basic amino acid residues in thrombin essential for heparin-catalyzed inactivation by antithrombin III. J. Biol. Chem, 1994, 269, 1301–1305. [PubMed] [Google Scholar]
- [81].Carter WJ; Cama E; Huntington JA Crystal structure of thrombin bound to heparin. J. Biol. Chem, 2005, 280, 2745–2749. [DOI] [PubMed] [Google Scholar]
- [82].Li W; Adams TE; Nangalia J; Esmon CT; Huntington JA Molecular basis of thrombin recognition by protein C inhibitor revealed by the 1.6-A structure of the heparin-bridged complex. Proc. Natl. Acad. Sci. USA, 2008, 105(12), 4661–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Rezaie AR Identification of basic residues in the heparin-binding exosite of factor Xa critical for heparin and factor Va binding. J. Biol. Chem, 2000, 275, 3320–3327. [DOI] [PubMed] [Google Scholar]
- [84].Zhao M; Abdel-Razek T; Sun MF; Gailani D Characterization of a heparin binding site on the heavy chain of factor XI. J. Biol. Chem, 1998, 273, 31153–31159. [DOI] [PubMed] [Google Scholar]
- [85].Ho DH; Badellino K; Baglia FA; Walsh PN A binding site for heparin in the apple 3 domain of factor XI. J. Biol. Chem, 1998, 273, 16382–16390. [DOI] [PubMed] [Google Scholar]
- [86].Chander A; Atkinson HM; Stevic I; Berry LR; Kim PY; Chan AKC Interactions of heparin and a covalently-linked antithrombin-heparin complex with components of the fibrinolytic system. Thromb. Haemost, 2013, 110, 1180–1188. [DOI] [PubMed] [Google Scholar]
- [87].Bauer PI; Pozsgay M; Machovich R; Elödi P; Horváth I The interaction of heparin with human plasmin. Int. J. Biochem, 1983, 15, 871–874. [DOI] [PubMed] [Google Scholar]
- [88].Kostoulas G; Hörler D; Naggi A; Casu B; Baici A Electrostatic interactions between human leukocyte elastase and sulfated glycosaminoglycans: physiological implications. Biol. Chem, 1997, 378, 1481–1489. [DOI] [PubMed] [Google Scholar]
- [89].Volpi N Inhibition of human leukocyte elastase activity by chondroitin sulfates. Chem. Biol. Interact, 1997, 105, 157–167. [DOI] [PubMed] [Google Scholar]
- [90].Walsh RL; Dillon TJ; Scicchitano R; McLennan G Heparin and heparan sulphate are inhibitors of human leucocyte elastase. Clin. Sci, 1991, 81, 341–346. [DOI] [PubMed] [Google Scholar]
- [91].Spencer JL; Stone PJ; Nugent MA New insights into the inhibition of human neutrophil elastase by heparin. Biochemistry, 2006, 45, 9104–9120. [DOI] [PubMed] [Google Scholar]
- [92].Verghese J; Liang A; Sidhu PP; Hindle M; Zhou Q; Desai UR First steps in the direction of synthetic, allosteric, direct inhibitors of thrombin and factor Xa. Bioorg. Med. Chem. Lett, 2009, 19(15), 4126–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Sidhu PS; Liang A; Mehta AY; Abdel Aziz MH; Zhou Q; Desai UR Rational design of potent, small, synthetic allosteric inhibitors of thrombin. J. Med. Chem 2011, 54(15), 5522–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Abdel Aziz MH; Sidhu PS; Liang A; Kim JY; Mosier PD; Zhou Q; Farrell DH; Desai UR Designing allosteric regulators of thrombin. Monosulfated benzofuran dimers selectively interact with Arg173 of exosite 2 to induce inhibition. J. Med. Chem 2012, 55(15), 6888–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Afosah DK; Verespy S 3rd.; Al-Horani RA; Boothello RS; Karuturi R; Desai UR A small group of sulfated benzofurans induces steady-state submaximal inhibition of thrombin. Bioorg. Med. Chem. Lett, 2018, 28(6), 1101–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Sidhu PS; Abdel Aziz MH; Sarkar A; Mehta AY; Zhou Q; Desai UR Designing allosteric regulators of thrombin. Exosite 2 features multiple subsites that can be targeted by sulfated small molecules for inducing inhibition. J. Med. Chem, 2013, 56(12), 5059–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Verespy S 3rd.; Mehta AY; Afosah D; Al-Horani RA; Desai UR Allosteric partial inhibition of monomeric proteases. Sulfated coumarins induce regulation, not just inhibition, of thrombin. Sci. Rep, 2016, 6, 24043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Al-Horani RA; Ponnusamy P; Mehta AY; Gailani D; Desai UR Sulfated penta-galloylglucoside is a potent, allosteric, and selective inhibitor of factor XIa. J. Med. Chem 2013, 56(3), 867–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Al-Horani RA; Desai UR Designing allosteric inhibitors of factor XIa. Lessons from the interactions of sulfated pentagalloylglucopyranosides. J. Med. Chem 2014, 57(11), 4805–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Al-Horani RA; Gailani D; Desai UR Allosteric inhibition of factor XIa. Sulfated non-saccharide glycosaminoglycan mimetics as promising anticoagulants. Thromb. Res, 2015, 136(2), 379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Karuturi R; Al-Horani RA; Mehta SC; Gailani D; Desai UR Discovery of allosteric modulators of factor XIa by targeting hydrophobic domains adjacent to its heparin-binding site. J. Med. Chem, 2013, 56(6), 2415–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Al-Horani RA; Karuturi R; White DT; Desai UR Plasmin regulation through allosteric, sulfated, small molecules. Molecules, 2015, 20(1), 608–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Afosah DK; Al-Horani RA; Sankaranarayanan NV; Desai UR Potent, selective, allosteric inhibition of human plasmin by sulfated non-saccharide glycosaminoglycan mimetics. J. Med. Chem, 2017, 60(2), 641–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Luster AD Chemokines - Chemotactic cytokines that mediate inflammation. N. Engl. J. Med, 1998, 338, 436–445. [DOI] [PubMed] [Google Scholar]
- [105].Wells TN; Power CA; Proudfoot AE Definition, function and pathophysiological significance of chemokine receptors. Trends Pharmacol. Sci, 1998, 19, 376–380. [DOI] [PubMed] [Google Scholar]
- [106].Rollins BJ Chemokines. Blood, 1997, 90, 909–928. [PubMed] [Google Scholar]
- [107].Rossi D; Zlotnik A The biology of chemokines and their receptors. Annu. Rev. Immunol, 2000, 18, 217–242. [DOI] [PubMed] [Google Scholar]
- [108].Kuschert GS; Coulin F; Power CA; Proudfoot AE; Hubbard RE; Hoogewerf AJ; Wells TN Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry, 1999, 38, 12959–12968. [DOI] [PubMed] [Google Scholar]
- [109].Johnson Z; Proudfoot AE; Handel TM Interaction of chemokines and glycosaminoglycans: A new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev, 2005, 16, 625–636. [DOI] [PubMed] [Google Scholar]
- [110].Asada M; Shinomiya M; Suzuki M; Honda E; Sugimoto R; Ikekita M; Imamura T Glycosaminoglycan affinity of the complete fibroblast growth factor family. Biochim. Biophys. Acta, 2009, 1790(1), 40–48. [DOI] [PubMed] [Google Scholar]
- [111].Kuschert GS; Coulin F; Power CA; Proudfoot AE; Hubbard RE; Hoogewerf AJ; Wells TNC Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry, 1999, 38, 12959–12968. [DOI] [PubMed] [Google Scholar]
- [112].Witt DP; Lander AD Differential binding of chemokines to glycosaminoglycan subpopulations. Curr. Biol, 1994, 4, 394–400. [DOI] [PubMed] [Google Scholar]
- [113].Ornitz DM; Itoh N Fibroblast growth factors. Genome Biol, 2001, 2, reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Shute J (2012) Glycosaminoglycan and Chemokine/Growth Factor Interactions. In: Lever R, Mulloy B, Page C (eds) Heparin - A Century of Progress. Handbook of Experimental Pharmacology, vol 207 Springer, Berlin, Heidelberg. [Google Scholar]
- [115].Turner N; Grose R Fibroblast growth factor signalling: from development to cancer, Nat. Rev. Cancer, 2010, 10, 116–129. [DOI] [PubMed] [Google Scholar]
- [116].Pantoliano MW; Horlick RA; Springer BA; Van Dyk DE; Tobery T; Wetmore DR; Lear JD; Nahapetian AT; Bradley JD; Sisk WP Multivalent ligand-receptor binding interactions in the fibroblast growth factor system produce a cooperative growth factor and heparin mechanism for receptor dimerization. Biochemistry, 1994, 33, 10229–10248. [DOI] [PubMed] [Google Scholar]
- [117].Brown A; Robinson CJ; Gallagher JT; Blundell TL Cooperative heparin-mediated oligomerization of fibroblast growth Factor-1 (FGF1) precedes recruitment of FGFR2 to ternary complexes. Biophys. J, 2013, 104, 1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Thompson LD; Pantoliano MW; Springer BA Energetic characterization of the basic fibroblast growth factor-heparin interaction: Identification of the heparin binding domain. Biochemistry, 1994, 33, 3831–3840. [DOI] [PubMed] [Google Scholar]
- [119].WuDunn D; Spear PG Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol, 1989, 63, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Shukla D; Spear PG Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Invest, 2001, 108, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, et al. , A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [DOI] [PubMed] [Google Scholar]
- [122].Matos PM; Andreu D; Santos NC; Gutiérrez-Gallego R Structural requirements of glycosaminoglycans for their interaction with HIV-1 envelope glycoprotein gp120. Arch. Virol, 2014, 159, 555–560. [DOI] [PubMed] [Google Scholar]
- [123].Crublet E; Andrieu JP; Vivès RR; Lortat-Jacob H; The HIV-1 envelope glycoprotein gp120 features four heparan sulfate binding domains, including the co-receptor binding site, J. Biol. Chem 2008, 283, 15193–15200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Gangji RN; Sankaranarayanan NV; Elste J; Al-Horani RA; Afosah DK; Joshi R; Tiwari V; Desai UR Inhibition of herpes simplex virus-1 entry into human cells by non-saccharide glycosaminoglycan mimetics. ACS Med. Chem. Lett, 2018, 9(8), 797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Lima RT; Seca H; Palmeira A; Fernandes MX; Castro F; Correia-da-Silva M; Nascimento MS; Sousa E; Pinto M; Vasconcelos MH Sulfated small molecules targeting eBV in Burkitt lymphoma: from in silico screening to the evidence of in vitro effect on viral episomal DNA. Chem. Biol. Drug Des, 2013, 81(5), 631–44. [DOI] [PubMed] [Google Scholar]
- [126].Raman R; Sasisekharan V; Sasisekharan R Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem. Biol, 2005, 12(3), 267–77. [DOI] [PubMed] [Google Scholar]
- [127].Pervin A; Gallo C; Jandik KA; Han XJ; Linhardt RJ Preparation and structural characterization of large heparin-derived oligosaccharides. Glycobiology, 1995, 5, 83–95. [DOI] [PubMed] [Google Scholar]
- [128].Pan J; Qian Y; Zhou X; Pazandak A; Frazier SB; Weiser P; Lu H; Zhang L Oversulfated chondroitin sulfate is not the sole contaminant in heparin. Nat. Biotechnol, 2010, 28, 203–207. [DOI] [PubMed] [Google Scholar]
- [129].Guerrini M; Beccati D; Shriver Z; Naggi A; Viswanathan K; Bisio A; Capila I; Lansing JC; Guglieri S; Fraser B; Al-Hakim A; Gunay NS; Zhang Z; Robinson L; Buhse L; Nasr M; Woodcock J; Langer R; Venkataraman G; Linhardt RJ; Casu B; Torri G; Sasisekharan R Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nat. Biotechnol, 2008, 26, 669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Blossom DB; Kallen AJ; Patel PR; Elward A; Robinson L; Gao G; Langer R; Perkins KM; Jaeger JL; Kurkjian KM; Jones M; Schillie SF; Shehab N; Ketterer D; Venkataraman G; Kishimoto TK; Shriver Z; McMahon AW; Austen KF; Kozlowski S; Srinivasan A; Turabelidze G; Gould CV; Arduino MJ; Sasisekharan R Outbreak of adverse reactions associated with contaminated heparin. N. Engl. J. Med, 2008, 359, 2674–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Kishimoto TK; Viswanathan K; Ganguly T; Elankumaran S; Smith S; Pelzer K; Lansing JC; Sriranganathan N; Zhao G; Galcheva-Gargova Z; Al-Hakim A; Bailey GS; Fraser B; Roy S; Rogers-Cotrone T; Buhse L; Whary M; Fox J; Nasr M; Dal Pan GJ; Shriver Z; Langer RS; Venkataraman G; Austen KF; Woodcock J; Sasisekharan R Contaminated heparin associated with adverse clinical events and activation of the contact system. N. Engl. J. Med, 2008, 358, 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Liu H; Zhang Z; Linhardt RJ Lessons learned from the contamination of heparin. Nat. Prod. Rep, 2009, 26, 313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].DeAngelis PL Glycosaminoglycan polysaccharide biosynthesis and production: Today and tomorrow. Appl. Microbiol. Biotechnol, 2012, 94, 295–305. [DOI] [PubMed] [Google Scholar]
- [134].Oduah EI; Linhardt RJ; Sharfstein ST Heparin: Past, present, and future, Pharmaceuticals, 2016, 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Ernst B; Magnani JL From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov, 2009, 8, 661–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Magnani JL; Ernst B Glycomimetic drugs--a new source of therapeutic opportunities. Discov.Med, 2009, 8, 247–252. [PubMed] [Google Scholar]
- [137].Sankaranarayanan NV; Sarkar A; Desai UR; Mosier PD Designing “high-affinity, high-specificity” glycosaminoglycan sequences through computerized modeling. Methods Mol Biol, 2015, 1229, 289–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Sankaranarayanan NV; Desai UR Toward a robust computational screening strategy for identifying glycosaminoglycan sequences that display high specificity for target proteins. Glycobiology, 2014, 24, 1323–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Raghuraman A; Mosier PD; Desai UR Finding a needle in a haystack: Development of a combinatorial virtual screening approach for identifying high specificity heparin/heparan sulfate sequence(s). J. Med. Chem, 2006, 49, 3553–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Sarkar A; Desai UR A simple method for discovering druggable, specific glycosaminoglycan-protein systems. Elucidation of key principles from heparin/heparan sulfate-binding proteins. PLoSOne, 2015, 10, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Agostino M; Gandhi NS; Mancera RL Development and application of site mapping methods for the design of glycosaminoglycans. Glycobiology, 2014, 24, 840–851. [DOI] [PubMed] [Google Scholar]
- [142].Mende M; Bednarek C; Wawryszyn M; Sauter P; Biskup MB; Schepers U; Bräse S Chemical synthesis of glycosaminoglycans. Chem. Rev, 2016, 116(14), 8193–255. [DOI] [PubMed] [Google Scholar]
- [143].DeAngelis PL; Liu J; Linhardt RJ Chemoenzymatic synthesis of glycosaminoglycans: recreating, re-modeling and re-designing nature’s longest or most complex carbohydrate chains. Glycobiology, 2013, 23(7), 764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Fu L; Suflita M; Linhardt RJ Bioengineered heparins and heparan sulfates. Adv. Drug Deliv. Rev, 2016, 97, 237–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Samsonov SA; Pisabarro MT Computational analysis of interactions in structurally available protein-glycosaminoglycan complexes. Glycobiology, 2016, 26, 850–861. [DOI] [PubMed] [Google Scholar]
- [146].Al-Horani RA; Karuturi R; Verespy S 3rd.; Desai UR Synthesis of glycosaminoglycan mimetics through sulfation of polyphenols. Methods Mol. Biol, 2015, 1229, 49–67. [DOI] [PubMed] [Google Scholar]
- [147].Koester DC; Holkenbrink A; Werz DB Recent advances in the synthesis of carbohydrate mimetics. Synthesis (Stuttg), 2010, 3217–3242.
- [148].Hu YP; Lin SY; Huang CY; Zulueta MM; Liu JY; Chang W; Hung SC Synthesis of 3-O-sulfonated heparan sulfate octasaccharides that inhibit the herpes simplex virus type 1 host-cell interaction. Nat. Chem, 2011, 3, 557–563. [DOI] [PubMed] [Google Scholar]
- [149].de Paz JL; Seeberger PH Deciphering the glycosaminoglycan code with the help of microarrays. Mol. Biosyst, 2008, 4, 707–711. [DOI] [PubMed] [Google Scholar]
- [150].Yin J; Seeberger PH Applications of heparin and heparan sulfate microarrays. Methods Enzymol, 2010, 478, 197–218. [DOI] [PubMed] [Google Scholar]
- [151].Desai UR The promise of sulfated synthetic small molecules as modulators of glycosaminoglycan function. Future Med. Chem, 2013, 5, 1363–6. [DOI] [PubMed] [Google Scholar]
- [152].Correia-da-Silva M; Sousa E; Pinto MM Emerging sulfated flavonoids and other polyphenols as drugs: nature as an inspiration. Med. Res. Rev, 2014, 34(2), 223–79. [DOI] [PubMed] [Google Scholar]
- [153].Shen A Allosteric regulation of protease activity by small molecules. Mol. Biosyst, 2010, 6, 1431–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Merdanovic M; Mönig T; Ehrmann M; Kaiser M Diversity of allosteric regulation in proteases. ACS Chem. Biol, 2013, 8, 19–26. [DOI] [PubMed] [Google Scholar]
- [155].Hauske P; Ottmann C; Meltzer M; Ehrmann M; Kaiser M Allosteric regulation of proteases. Chembiochem, 2008, 9, 2920–8. [DOI] [PubMed] [Google Scholar]
- [156].Henry BL; Desai UR Anticoagulants: Drug discovery and development. In: Burger’s medicinal chemistry Rotella D; Abraham DJ; Ed., 7th ed., John Wiley and Sons: New York; 2010, pp. 365–408. [Google Scholar]
- [157].Monien BH; Desai UR Antithrombin activation by nonsulfated, non-polysaccharide organic polymer. J. Med. Chem, 2005, 48(4), 1269–73. [DOI] [PubMed] [Google Scholar]
- [158].Monien BH; Cheang KI; Desai UR Mechanism of poly(acrylic acid) acceleration of antithrombin inhibition of thrombin: implications for the design of novel heparin mimics. J. Med. Chem, 2005, 48(16), 5360–8. [DOI] [PubMed] [Google Scholar]
- [159].Monien BH; Henry BL; Raghuraman A; Hindle M; Desai UR Novel chemo-enzymatic oligomers of cinnamic acids as direct and indirect inhibitors of coagulation proteinases. Bioorg. Med. Chem, 2006, 14(23), 7988–98. [DOI] [PubMed] [Google Scholar]
- [160].Henry BL; Connell J; Liang A; Krishnasamy C; Desai UR Interaction of antithrombin with sulfated, low molecular weight lignins: opportunities for potent, selective modulation of antithrombin function. J. Biol. Chem, 2009, 284(31), 20897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Correia-da-Silva M; Sousa E; Duarte B; Marques F; Carvalho F; Cunha-Ribeiro LM; Pinto MM Flavonoids with an oligopolysulfated moiety: a new class of anticoagulant agents. J. Med. Chem, 2011, 54(1), 95–106. [DOI] [PubMed] [Google Scholar]
- [162].Correia-da-Silva M; Sousa E; Duarte B; Marques F; Carvalho F; Cunha-Ribeiro LM; Pinto MM Polysulfated xanthones: multipathway development of a new generation of dual anticoagulant/antiplatelet agents. J. Med. Chem, 2011, 54(15), 5373–84. [DOI] [PubMed] [Google Scholar]
- [163].Correia-da-Silva M; Sousa E; Duarte B; Marques F; Carvalho F; Cunha-Ribeiro LM; Pinto MM Dual anticoagulant/antiplatelet persulfated small molecules. Eur. J. Med. Chem, 2011, 46(6), 2347–58. [DOI] [PubMed] [Google Scholar]
- [164].Versteeg HH; Heemskerk JW; Levi M; Reitsma PH New fundamentals in hemostasis. Physiol. Rev, 2013, 93, 327–58. [DOI] [PubMed] [Google Scholar]
- [165].von dem Borne PA; Meijers JC; Bouma BN Feedback activation of factor XI by thrombin in plasma results in additional formation of thrombin that protects fibrin clots from fibrinolysis. Blood, 1995, 86, 3035–42. [PubMed] [Google Scholar]
- [166].De Candia E; Hall SW; Rutella S; Landolfi R; Andrews RK; De Cristofaro R Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of PAR-1 on intact platelets. J. Biol. Chem, 2001, 276, 4692–8. [DOI] [PubMed] [Google Scholar]
- [167].Esmon CT The roles of protein C and thrombomodulin in the regulation of blood coagulation. J. Biol. Chem, 1989, 264, 4743–6. [PubMed] [Google Scholar]
- [168].Henry BL; Monien BH; Bock PE; Desai UR A novel allosteric pathway of thrombin inhibition: Exosite II mediated potent inhibition of thrombin by chemo-enzymatic, sulfated dehydropolymers of 4-hydroxycinnamic acids. J. Biol. Chem, 2007, 282(44), 31891–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Abdel Aziz MH; Mosier PD; Desai UR Identification of the site of binding of sulfated, low molecular weight lignins on thrombin. Biochem. Biophys. Res. Commun 2011, 413(2), 348–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Mehta AY; Thakkar JN; Mohammed BM; Martin EJ; Brophy DF; Kishimoto T; Desai UR Targeting the GPIbα binding site of thrombin to simultaneously induce dual anticoagulant and antiplatelet effects. J. Med. Chem, 2014, 57(7), 3030–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Mehta AY; Desai UR Substantial non-electrostatic forces are needed to induce allosteric disruption of thrombin’s active site through exosite 2. Biochem. Biophys. Res. Commun, 2014, 452(3), 813–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Mehta AY; Mohammed BM; Martin EJ; Brophy DF; Gailani D; Desai UR Allosterism-based simultaneous, dual anticoagulant and antiplatelet action: allosteric inhibitor targeting the glycoprotein Ibα-binding and heparin-binding site of thrombin. J. Thromb Haemost, 2016, 14(4), 828–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Nahain AA; Ignjatovic V; Monagle P; Tsanaktsidis J; Ferro V Heparin mimetics with anticoagulant activity. Med. Res. Rev, 2018, 38(5), 1582–1613. [DOI] [PubMed] [Google Scholar]
- [174].Al-Horani RA; Desai UR Factor XIa inhibitors: A review of the patent literature. Expert Opin. Ther. Pat, 2016, 26(3), 323–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].He R; Chen D; He S Factor XI: hemostasis, thrombosis, and antithrombosis. Thromb Res, 2012, 129(5), 541–50. [DOI] [PubMed] [Google Scholar]
- [176].Emsley J; McEwan PA; Gailani D Structure and function of factor XI. Blood, 2010, 115(13), 2569–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Gailani D; Smith SB Structural and functional features of factor XI. J. Thromb. Haemost, 2009, 7 (Suppl 1), 75–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Smith GF; Sundboom JL Heparin and protease inhibition. II. The role of heparin in the ATIII inactivation of thrombin, plasmin, and trypsin. Thromb. Res, 1981, 22, 115–133. [DOI] [PubMed] [Google Scholar]
- [179].Jordan RE; Oosta GM; Gardner WT; Rosenberg RD The binding of low molecular weight heparin to hemostatic enzymes. J. Biol. Chem; 1980, 255, 10073–10080. [PubMed] [Google Scholar]
- [180].Yomtova VM; Stambolieva NA; Blagoev BM Kinetic study of the effect of heparin on the amidase activity of trypsin, plasmin and urokinase. Thromb. Haemost, 1983, 49, 199–203. [PubMed] [Google Scholar]
- [181].Bauer PI; Pozsgay M; Machovich R; Elodi P; Horvath I The interaction of heparin with human plasmin. Int. J. Biochem, 1983, 15, 871–874. [DOI] [PubMed] [Google Scholar]
- [182].Henry BL; Abdel Aziz M; Zhou Q; Desai UR Sulfated, low-molecular-weight lignins are potent inhibitors of plasmin, in addition to thrombin and factor Xa: Novel opportunity for controlling complex pathologies. Thromb. Haemost, 2010, 103(3), 507–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Al-Horani RA; Karuturi R; Lee M; Afosah DK; Desai UR Allosteric inhibition of factor XIIIa. Non-saccharide glycosaminoglycan mimetics, but not glycosaminoglycans, exhibit promising inhibition profile. PLoS One, 2016, 11(7), e0160189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Aleman MM; Byrnes JR; Wang JG; Tran R; Lam WA; Di Paola J; Mackman N; Degen JL; Flick MJ; Wolberg AS Factor XIII activity mediates red blood cell retention in venous thrombi. J. Clin. Invest, 2014, 124, 3590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Byrnes JR; Duval C; Wang Y; Hansen CE; Ahn B; Mooberry MJ; Clark MA; Johnsen JM; Lord ST; Lam WA; Meijers JC; Ni H; Ariëns RA; Wolberg AS Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin α-chain crosslinking. Blood, 2015, 126, 1940–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Flick MJ; Du X; Witte DP; Jirousková M; Soloviev DA; Busuttil SJ; Plow EF; Degen JL Leukocyte engagement of fibrin (ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J. Clin. Invest 2004, 113, 1596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Lauer P; Metzner HJ; Zettlmeissl G; Li M; Smith AG; Lathe R; Dickneite G Targeted inactivation of the mouse locus encoding coagulation factor XIII-A: hemostatic abnormalities in mutant mice and characterization of the coagulation deficit. Thromb. Haemost, 2002, 88, 967–74. [PubMed] [Google Scholar]
- [188].Raman K; Karuturi R; Swarup VP; Desai UR; Kuberan B Discovery of novel sulfonated small molecules that inhibit vascular tube formation. Bioorg. Med. Chem. Lett, 2012, 22(13), 4467–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Belting M Glycosaminoglycans in cancer treatment. Thromb. Res, 2014, 133 Suppl 2, S95–101. [DOI] [PubMed] [Google Scholar]
- [190].Patel NJ; Karuturi R; Al-Horani RA; Baranwal S; Patel J; Desai UR; Patel BB Synthetic, non-saccharide, glycosaminoglycan mimetics selectively target colon cancer stem cells. ACS Chem Biol, 2014, 9(8), 1826–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Boothello RS; Patel NJ; Sharon C; Abdelfadiel EI; Morla S; Brophy DF; Lippman HR; Desai UR; Patel BB A unique non-saccharide mimetic of heparin hexasaccharide inhibits colon cancer stem cells via p38 MAP kinase activation. Mol. Cancer Ther, 2018, doi: 10.1158/1535-7163. [DOI] [PMC free article] [PubMed]
- [192].Nagarajan B; Sankaranarayanan NV; Patel BB; Desai UR A molecular dynamics-based algorithm for evaluating the glycosaminoglycan mimicking potential of synthetic, homogenous, sulfated small molecules. PLoS One, 2017, 12(2), e0171619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Patel NJ; Sharon C; Baranwal S; Boothello RS; Desai UR; Patel BB Heparan sulfate hexasaccharide selectively inhibits cancer stem cells self-renewal by activating p38 MAP kinase. Oncotarget, 2016,7(51), 84608–84622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Saluja B; Thakkar JN; Li H; Desai UR; Sakagami M Novel low molecular weight lignins as potential anti-emphysema agents: In vitro triple inhibitory activity against elastase, oxidation and inflammation. Pulm. Pharmacol. Ther, 2013, 26(2), 296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Saluja B; Li H; Desai UR; Voelkel NF; Sakagami M Sulfated caffeic acid dehydropolymer attenuates elastase and cigarette smoke extract-induced emphysema in rats: sustained activity and a need of pulmonary delivery. Lung, 2014,192(4), 481–92. [DOI] [PubMed] [Google Scholar]
- [196].Truong TM; Li H; Dhapare S; Desai UR; Voelkel NF; Sakagami M Sulfated dehydropolymer of caffeic acid: In vitro anti-lung cell death activity and in vivo intervention in emphysema induced by VEGF receptor blockade. Pulm. Pharmacol.Ther, 2017, 45, 181–190. [DOI] [PubMed] [Google Scholar]
- [197].Rees CR; Costin JM; Fink RC; McMichael M; Fontaine KA; Isern S; Michael SF In vitro inhibition of dengue virus entry by p-sulfoxy-cinnamic acid and structurally related combinatorial chemistries. Antiviral Res, 2008, 80(2), 135–42. [DOI] [PubMed] [Google Scholar]
- [198].Vilas-Boas C; Sousa E; Pinto M; Correia-da-Silva M An antifouling model from the sea: a review of 25 years of zosteric acid studies. Biofouling, 2017, 33(10), 927–942. [DOI] [PubMed] [Google Scholar]
- [199].Almeida JR; Correia-da-Silva M; Sousa E; Antunes J; Pinto M; Vasconcelos V; Cunha I Antifouling potential of Nature-inspired sulfated compounds. Sci. Rep, 2017, 7, 42424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Severin IC; Soares A; Hantson J; Teixeira M; Sachs D; Valognes D; Scheer A; Schwarz MK; Wells TN; Proudfoot AE; Shaw J Glycosaminoglycan analogs as a novel anti-inflammatory strategy. Front Immunol, 2012, 3, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Mathias DK; Pastrana-Mena R; Ranucci E; Tao D; Ferruti P; Ortega C; Staples GO; Zaia J; Takashima E; Tsuboi T; Borg NA; Verotta L; Dinglasan RR A small molecule glycosaminoglycan mimetic blocks Plasmodium invasion of the mosquito midgut. PLoS Pathog, 2013, 9(11), e1003757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Al-Horani RA; Desai UR Chemical sulfation of small molecules - Advances and challenges. Tetrahedron, 2010, 66(16), 2907–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Liang WG; Triandafillou CG; Huang TY; Zulueta MM; Banerjee S; Dinner AR; Hung SC; Tang WJ Structural basis for oligomerization and glycosaminoglycan binding of CCL5 and CCL3. Proc. Natl. Acad. Sci. USA, 2016, 113(18), 5000–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Wu L; Viola CM; Brzozowski AM; Davies GJ Corrigendum: Structural characterization of human heparanase reveals insights into substrate recognition. Nat. Struct. Mol. Biol, 2016, 23(1): 91. [DOI] [PubMed] [Google Scholar]
- [205].Dasgupta J; Bienkowska-Haba M; Ortega ME; Patel HD; Bodevin S; Spillmann D; Bishop B; Sapp M; Chen XS Structural basis of oligosaccharide receptor recognition by human papillomavirus. J. Biol. Chem, 2011, 286(4), 2617–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Johnson DJ; Langdown J; Huntington JA Molecular basis of factor IXa recognition by heparin-activated antithrombin revealed by a 1.7-A structure of the ternary complex. Proc. Natl. Acad. Sci. USA, 2010, 107(2), 645–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Pellegrini L; Burke DF; von Delft F; Mulloy B; Blundell TL Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature, 2000, 407(6807), 1029–34. [DOI] [PubMed] [Google Scholar]
- [208].Schlessinger J; Plotnikov AN; Ibrahimi OA; Eliseenkova AV; Yeh BK; Yayon A; Linhardt RJ; Mohammadi M Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 2000, 6(3), 743–50. [DOI] [PubMed] [Google Scholar]
- [209].Khatun Z; Nurunnabi M; Cho KJ; Lee Y-K Imaging of the GI tract by QDs loaded heparin–deoxycholic acid (DOCA) nanoparticles. Carbohydr. Polym, 2012, 90, 1461–1468. [DOI] [PubMed] [Google Scholar]
- [210].Kim SK; Lee DY; Kim CY; Nam JH; Moon HT; Byun Y A newly developed oral heparin derivative for deep vein thrombosis: Non-human primate study. J. Control. Release, 2007, 123, 155–163. [DOI] [PubMed] [Google Scholar]
- [211].Kim SK; Vaishali B; Lee E; Lee S; Lee Y-K; Kumar TS; Moon HT; Byun Y Oral delivery of chemical conjugates of heparin and deoxycholic acid in aqueous formulation. Thromb. Res, 2006, 117, 419–427. [DOI] [PubMed] [Google Scholar]
- [212].Lee Y-K; Kim S; Byun Y Oral delivery of new heparin derivatives in rats. Pharm. Res, 2000, 17, 1259–1264. [DOI] [PubMed] [Google Scholar]
- [213].Motlekar NA; Srivenugopal KS;Wachtel MS; Youan BB Modulation of gastrointestinal permeability of low-molecular-weight heparin by L-arginine: In vivo and in vitro evaluation. J. Pharm. Pharmacol, 2006, 58, 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Motlekar NA; Srivenugopal KS; Wachtel MS; Youan BBC Evaluation of the oral bioavailability of low molecular weight heparin formulated with glycyrrhetinic acid as permeation enhancer. Drug Dev. Res, 2006, 67, 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Neves AR; Correia-da-Silva M; Sousa E; Pinto M Strategies to overcome heparins’ low oral bioavailability. Pharmaceuticals (Basel), 2016, 9(3), pii: E37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Plosker GL Acamprosate: A review of its use in alcohol dependence. Drugs, 2015, 75(11), 1255–68. [DOI] [PubMed] [Google Scholar]
- [217].Abushakra S; Porsteinsson A; Scheltens P; Sadowsky C; Vellas B; Cummings J; Gauthier S; Hey JA; Power A; Wang P; Shen L; Tolar M Clinical effects of tramiprosate in APOE4/4 homozygous patients with mild Alzheimer’s disease suggest disease modification potential. J. Prev. Alzheimers Dis, 2017, 4(3), 149–156. [DOI] [PubMed] [Google Scholar]
- [218].Rumjon A; Coats T; Javaid MM Review of eprodisate for the treatment of renal disease in AA amyloidosis. Int. J. Nephrol. Renovasc. Dis, 2012, 5, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Abdeen S; Salim N; Mammadova N; Summers CM; Goldsmith-Pestana K; McMahon-Pratt D; Schultz PG; Horwich AL; Chapman E; Johnson SM Targeting the HSP60/10 chaperonin systems of Trypanosoma brucei as a strategy for treating African sleeping sickness. Bioorg. Med. Chem. Lett, 2016, 26(21), 5247–5253. [DOI] [PubMed] [Google Scholar]
- [220].Rondanin R; Fochi S; Baruchello R; Bernardi T; Oliva P; Semeraro F; Simoni D; Giannini G Arylamidonaphtalene sulfonate compounds as a novel class of heparanase inhibitors. Bioorg. Med. Chem. Lett, 2017, 27(18), 4421–4425. [DOI] [PubMed] [Google Scholar]
- [221].Ferla S; Netzler NE; Ferla S; Veronese S; Tuipulotu DE; Guccione S; Brancale A; White PA; Bassetto M In silico screening for human norovirus antivirals reveals a novel non-nucleoside inhibitor of the viral polymerase. Sci. Rep, 2018, 8: 4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Naviaux RK; Curtis B; Li K,; Naviaux JC; Bright AT; Reiner GE; Westerfield M; Goh S; Alaynick WA; Wang L; Capparelli EV; Adams C; Sun J; Jain S; He F; Arellano DA; Mash LE; Chukoskie L; Lincoln A; Townsend J Low‐dose suramin in autism spectrum disorder: a small, phase I/II, randomized clinical trial. Ann. Clin. Transl. Neurol, 2017, 4(7), 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Gustafsen C; Olsen D; Vilstrup J; Lund S; Reinhardt A; Wellner N; Larsen T; Andersen CBF; Weyer K; Li J-p.; Seeberger PH; Thirup S; Madsen P; Glerup S Heparan sulfate proteoglycans present PCSK9 to the LDL receptor. Nat. Commun 2017, 8, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Frommherz KJ; Faller B; Bieth JG Heparin strongly decreases the rate of inhibition of neutrophil elastase by alphal-proteinase inhibitor. J. Biol. Chem, 1991, 266(23), 15356–15362. [PubMed] [Google Scholar]
- [225].Jordan RE; Oosta GM; Gardner WT; Rosenberg RD The binding of low molecular weight heparin to hemostatic enzymes. J. Biol. Chem, 1980, 255, 10073–10080. [PubMed] [Google Scholar]
- [226].Krieger E; Geretti E; Brandner B; Goger B; Wells TN; Kungl AJ A structural and dynamic model for the interaction of interleukin-8 and glycosaminoglycans: Support from isothermal fluorescence titrations. Proteins Struct. Funct. Genet, 2004, 54, 768–775. [DOI] [PubMed] [Google Scholar]
- [227].Mayo KH; Ilyina E; Roongta V; Dundas M; Joseph J; Lai CK; Maione T; Daly TJ Heparin binding to platelet factor-4. An NMR and site-directed mutagenesis study: arginine residues are crucial for binding. Biochem. J, 1995, 312 (Pt 2), 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Loscalzo J; Melnick B; Handin RI The interaction of platelet factor 4 and glycosaminoglycans. Arch. Biochem. Biophys, 1985, 240, 446–55. [DOI] [PubMed] [Google Scholar]
- [229].Shukla D; Liu J; Blaiklock P; Shworak NW; Bai X; Esko JD; Cohen GH; Eisenberg RJ; Rosenberg RD; Spear PG A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell, 1999, 99, 13–22. [DOI] [PubMed] [Google Scholar]
- [230].Liu J; Shriver Z; Pope RM; Thorp SC; Duncan MB; Copeland RJ; Raska CS; Yoshida K; Eisenberg RJ; Cohen G; Linhardt RJ; Sasisekharan R Characterization of a heparan sulfate octasaccharide that binds to herpes simplex virus type 1 glycoprotein D. J. Biol. Chem, 2002, 277, 33456–33467. [DOI] [PubMed] [Google Scholar]
- [231].Moulard M; Lortat-Jacob H; Mondor I; Roca G; Wyatt R; Sodroski J; Zhao L; Olson W; Kwong PD; Sattentau QJ Selective interactions of polyanions with basic surfaces on human immunodeficiency virus type 1 gp120. J. Virol, 2000, 74, 1948–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]