

Abstract
Drug delivery to avascular, negatively charged tissues like cartilage remains a challenge. The constant turnover of synovial fluid results in short residence time of administered drugs in the joint space and the dense negatively charged matrix of cartilage hinders their diffusive transport. Drugs are, therefore, unable to reach their cell and matrix targets in sufficient doses, and fail to elicit relevant biological response, which has led to unsuccessful clinical trials. The high negative fixed charge density (FCD) of cartilage, however, can be used to convert cartilage from a barrier to drug entry into a depot by making drugs positively charged. Here we design cartilage penetrating and binding cationic peptide carriers (CPCs) with varying net charge, spatial distribution and hydrophobicity to deliver large-sized therapeutics and investigate their electro-diffusive transport in healthy and arthritic cartilage. We showed that CPC uptake increased with increasing net charge up to +14 but dropped as charge increased further due to stronger binding interactions that hindered CPC penetrability and uptake showing that weak-reversible binding is key to enable their penetration through full tissue thickness. Even after 90% GAG depletion, while CPC +14 uptake reduced by over 50% but still had a significantly high value of 148x showing that intra-tissue long-range charge-based binding is further stabilized by short-range H-bond and hydrophobic interactions. The work presents an approach for rational design of cationic carriers based on tissue FCD and properties of macromolecules to be delivered. These design rules can be extended to drug delivery for other avascular, negatively charged tissues.
Keywords: Intra-cartilage drug delivery, electrostatic interactions, cationic peptide carriers, negatively charged tissues, osteoarthritis, electro-diffusive transport
1. Introduction
Osteoarthritis (OA) is a degenerative joint disease affecting millions of people, yet there is not a cure for it [1]. Current treatment approaches merely provide temporary pain and inflammation relief but do not repair the joint tissues or rescue them from ongoing OA induced degeneration [2–4]. While research over the last two decades has led to discovery of several disease modifying OA drugs (DMOADs) that have shown promise in preclinical studies, none have demonstrated effective recovery of joint function or protection of tissue structure in clinical trials [3], in part due to a lack of effective drug delivery systems that can locally target OA affected joint tissues [5].
Since many of these joint tissues like articular cartilage, meniscus, tendons, ligaments etc. are avascular, systemic drug delivery is ineffective and direct intra-articular (IA) injections are used especially because OA only affects the local joint environment [5, 6]. Injected drugs, however, suffer from short joint residence time as they exit rapidly from the synovium vasculature and lymphatics [6, 7]. Furthermore, drugs are unable to reach their cell and matrix target sites residing inside the deep zones of affected joint tissues like cartilage due to their dense meshwork of collagen and aggrecan proteoglycans containing highly negatively charged glycosaminoglycan (GAG) chains that hinder penetration of most macromolecules [8–11]. These negatively charged groups create a high fixed charge density (FCD) inside the cartilage, which is key to its structure and function and provides the necessary hydration, swelling pressures and compressive stiffness [12–14]. OA onset results in gradual loss of GAGs thus depleting the tissue’s FCD and compromising its matrix integrity.
Recent unsuccessful clinical trials evaluating efficacy of inhibitors of interleukin-1, a cytokine elevated in OA, have attributed their failures to the inability of therapeutics to penetrate the cartilage and reach chondrocytes and other matrix target sites [15, 16]. It is, therefore, critical to develop cartilage penetrating materials that, following IA administration, can enable rapid uptake of drugs through the full thickness of cartilage to reach a therapeutic level before being cleared out and bind within to increase joint residence time. Drug delivery systems under investigation include micron sized polymeric particles [17, 18], micelles [19, 20], liposomes [21, 22] and aggregating hydrogels [23–25] but none of them have been shown to penetrate the cartilage and thus remain ineffective in eliciting a disease modifying biological response. Gene transfer vectors can only penetrate through the intact superficial zone of tissues [26] and suffer from concerns of immunogenicity and safety that make their clinical translation difficult [6, 27].
The high negative FCD of cartilage can be utilized for enhancing transport rate, uptake and retention of drugs or drug carriers by making them positively charged [5]. Previously, a size limit of <10 nm hydrodynamic diameter has been reported for unhindered solute transport into healthy cartilage matrix [28]. We recently showed that glycoprotein avidin, due to its optimal size and charge (7 nm dia, net charge b/w +7 and +20 estimated using Donnan-Boltzmann), to be an effective nanocarrier for delivering small molecule drugs into the deep zones of cartilage in vitro [28, 29] and in vivo using rabbit traumatic injury models of OA [30–32]. To carry proteins and other therapeutics of larger size, smaller sized cationic domains are required such that the carrier-drug size is within the 10 nm limit and the conjugate still has enough electrical driving force to carry macromolecules into the tissue.
With the goal of developing cartilage penetrating and binding materials, we design arginine or lysine rich short-length Cationic Peptide Carriers (CPCs) with varying net charge (between +7 and +20), spatial distribution and hydrophobicity. The work characterizes their intra-cartilage electro-diffusive properties including equilibrium uptake, transport rate, partitioning and binding in healthy and arthritic cartilage with different net FCD, and also investigates relative contributions of long-range charge effects and short-range non-specific interactions such as hydrophobic and H-bonds on intra-tissue binding.
2. Materials and Methods
Protease Inhibitor Mini Tablets were purchased from Thermo Scientific Pierce (Rockford, IL). Proteinase-K was purchased from Roche Diagnostics (Risch-Rotkreuz, Switzerland). Dulbecco’s Modification of Eagle’s Medium (DMEM) was from Cellgro (Manassas, VA). HEPES, Insulin-transferrin-selenium (ITS), trypsin-EDTA phenol red and non-essential amino acids (NEAA) were purchased from Gibco (Carlsbad, CA). Ascorbic acid and L-proline were from Fisher Bioreagents (Pittsburgh, PA). Sulfur-35 radionuclide was obtained from PerkinElmer (Waltham, MA). Propidium Iodide (PI) was obtained from Thermofisher Acros Organics (Geel, Belgium). Fluorescein Diacetate (FDA), Nonfat-dried bovine milk and other salts and reagents were purchased from Sigma (St. Louis, MO).
2.1. Design and synthesis of Cationic Peptide Carriers (CPC):
To study the effect of net positive charge and its spatial distribution on solute transport in cartilage, CPCs were designed by manipulating the number of arginine or lysine residues (Table 1). Alanine residues were used as spacers to symmetrically distribute the charge over the CPC sequence. The net charge (z) of CPCs was varied from +7 to +20; all of them comprised of 20 amino acids (Molecular Weight ~ 2.5–3.5 kDa) except CPC +16 that had 36 amino acid residues (MW ~4 kDa). The sequence of CPC +16 was chosen to provide heparin binding property [33, 34]. Additionally, to study synergistic effects of electrostatic and hydrophobic interactions, hydrophilic tail (ANANAN, asparagine spaced with alanine) or a hydrophobic tail (AFAFAF, phenylalanine spaced with alanine) was added in CPC +7. These are referred as CPC +N and CPC +F respectively throughout the text. CPCs were synthesized by Fmoc solid-phase peptide synthesis using pre-loaded Wang resins (LifeTein, Somerset, NJ). The peptides were then purified by reverse phase C18 HPLC (yielding >95% purity), and their masses were confirmed by electrospray ionization-time-of-light (ESI) mass spectrometry. For fluorescence measurements, CPCs were labeled with 5-FAM at the N-terminus.
Table 1.
Cationic Peptide Carrier (CPC) sequences and net charge
| CPC | CPC Sequence | Net electric charge (z) | MW (Da) |
|---|---|---|---|
| CPC +8 | (RRAAAA)3RR | +8 | 2,478.7 |
| CPC +14 | RRRR(AARRR)3R | +14 | 2,989.3 |
| CPC +16 | (ARRRAARA)4 | +16 | 4,012.4 |
| CPC +20 | (RRRRR)4 | +20 | 3,500.0 |
| CPC +N | (AK)7ANANAN | +7 | 2,326.6 |
| CPC +F | (AK)7AFAFAF | +7 | 2,425.8 |
2.2. Cartilage explant harvest
Cartilage plugs were harvested from 2-week-old bovine knee joints obtained from a local slaughterhouse (Research 87, Boylston, MA). Initially, full-thickness plugs were extracted from the femoropatellar grooves using 3 mm and 6 mm diameter biopsy punches. The plugs were further sliced to obtain 1 mm thick cartilage disks containing the superficial zone as described previously [28]. Upon harvest, the disks were washed and equilibrated in PBS for up to 1 h. For transport experiments, cartilage disks were stored in PBS supplemented with protease inhibitors at −20 °C until the day of experiment.
2.3. Transport measurements for CPC diffusivities in cartilage
A previously described custom designed transport chamber setup [28] consisting of clear poly (methyl methacrylate) chambers was used to study real time one-dimensional diffusion of solutes through cartilage. The inner walls of the compartments were treated with 0.5% nonfat-dried bovine milk solution in PBS for 15 minutes to minimize any non-specific binding of solutes to the chamber walls. The compartments were then rinsed with Deionized (DI) water. 6 mm diameter cartilage disk of 500–800 µm thickness was sealed between the two compartments. 3 µM solution of each CPC type labeled with 5-FAM was added to the upstream chamber and both compartments were kept under constant magnetic stirring during the experiment to avoid formation of any stagnant layers. CPC concentration in the downstream chamber was measured real time using a custom built spectrofluorometer. Once the concentration profile in the downstream chamber achieved a steady state, 20 µl of upstream solution was transferred to the downstream compartment to re-calibrate concentrations for the next repeat run. Non-equilibrium diffusion curves obtained were used to extract steady state (DSS) and effective diffusivities (DEFF) of CPCs in bovine cartilage.
2.4. Equilibrium intra-cartilage uptake and retention of CPCs
2.4.1. Equilibrium uptake of CPCs in normal and glycosaminoglycan (GAG) depleted cartilage explants
3 × 1 mm cartilage disks were individually equilibrated in 300 µl of 30 µM CPC saline solution (supplemented with protease inhibitors) in a 96 well plate at 37 °C for 24 h. The difference in CPC fluorescence of the initial and final equilibration bath was measured using a microplate reader (Synergy H1, Biotek). The standard curve correlating CPC concentration with relative fluorescence intensity was found to be linear. The uptake ratio (RU) was defined as the CPC concentration inside cartilage (per intra-tissue wet weight) normalized by the concentration of remaining CPC in the outside equilibration bath. For uptake experiments in GAG depleted samples, cartilage disks were treated with 0.1 mg/ml trypsin-EDTA phenol red in PBS for 5 h and 14 h at 37 °C to cause ~50% and ~90% GAG depletion respectively, to simulate early to mid-stage and late-stage osteoarthritic conditions. Explants were rinsed four times in PBS and then equilibrated in 1x PBS supplemented with protease inhibitors for 10 h before using them for CPC uptake experiments. Total tissue GAG content and that lost to the media were measured using dimethyl-methylene blue (DMMB) assay [35].
2.4.2. Histology
2.4.3. Native and trypsin digested cartilage explants were fixed in 4% formalin, dehydrated in a graded series of ethanol and xylene and then embedded in paraffin.
Tissue blocks were cut in coronal sections (to obtain full thickness cartilage slices with superficial and deep zones) of 5 µm thickness. The sections were stained with 0.5% Safranin-O and 0.01% fast green for GAGs detection, and immunostained for type II collagen using 1 μg/mL mouse IgG1 (clone II-II6B3; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA, USA) and a VectaStain Elite ABC kit (Vector Laboratories, Burlingame, CA, USA) for detection using 3,3-diaminobenzidine (DAB). Fixed slides of the stained tissue slices were imaged at 4x magnification under brightfield light (Nikon Eclipse TS2R). Intra-cartilage retention of CPCs
Following 24 h absorption in the CPC bath, explants were then desorbed in 1x or 10x PBS (supplemented with protease inhibitors) for a 7-day period. The bath solution was changed every day and the desorbed CPC concentration was measured using a plate reader. Intra-cartilage % retention for CPCs was calculated by normalizing the total CPC moles retained inside the tissue after desorption by the total initial CPC moles uptaken after 24 h absorption.
2.5. Depth of penetration using confocal microscopy
A custom designed transport chamber setup that allows for transport in only one direction through the superficial zone of cartilage was used to study distance penetrated by CPCs into the tissue over time [28]. Briefly, 6 mm cartilage half disks were placed in the center of the chamber well (Supplementary Figure S1). The upstream side, facing the cartilage superficial zone was filled up with 80 µl of 30 µM CPCs, while the downstream side was filled with only 1x PBS supplemented with protease inhibitors. The chamber was then placed in a covered petri dish containing DI water to prevent evaporation and placed on a slow-speed rocker to avoid formation of any stagnant layers for 4 h or 24 h at room temperature. Following 24 h absorption in CPC bath, cartilage half disks were desorbed in 10x PBS for 24 h. A 100–200 µm thick slice was cut from the center of the cartilage half disk and imaged using a confocal microscope (Zeiss LSM 700) at 10x magnification. 488 nm excitation laser line was used to detect the fluorescently labeled CPCs in the slice; laser intensity, gain values and the exposure time were kept constant throughout the experiment. Tile scanning function was used to image full thickness of the slice. The X-Y plane of each slice was imaged across z direction at 10 µm intervals. Consequently, a z-stack of fluorescent images were obtained. Using FIJI [36], concentration profiles were plotted along a straight line drawn across the explant thickness from superficial to deep zone based on the fluorescence intensity levels.
2.6. Cytotoxicity studies
2.6.1. Tissue culture
Cartilage explants were incubated in culture medium containing low glucose DMEM, 1% HEPES buffer, 1% ITS, 1% NEAA, 1% ascorbic acid and 0.4% proline for 48 h at 37 °C and 5% CO2. Subsequently, the explants were treated with increasing concentrations of CPC +14 (20 nM, 200 nM, 2000 nM and 20000 nM) for 48 h. Media was changed every 2 days over a period of 8-day culture. On the 6th day of culture, cartilage explants were incubated in media containing 15 µCi/ml radioisotope 35S-sulfate for 48 h. The explants were then washed 4 times over 1.5 h with PBS to remove free label. Each explant was weighed and then digested using proteinase-K. Radiolabel in each digested sample and the media standard was measured using a liquid scintillation counter (1450 PerkinElmer MicroBeta Trilux); concentration of newly synthesized GAGs was calculated with the help of media standard and normalized to explant wet weight and the time of radiolabel incorporation. Cumulative release of GAGs to the media and residual GAGs in the digested explants were measured using the DMMB dye assay.
2.6.2. Chondrocyte viability in CPC treated explant culture
Cell viability was measured by staining explants on day 8 of culture with 10 mg/ml of PI and 4 mg/ml of FDA for 6 minutes. PI stains non-viable cells red and FDA stains viable cells green. Slices were washed with PBS and imaged at 4x magnification (Nikon Eclipse TS2R). The live and dead images were overlaid using Image J.
2.7. Statistical Analysis
All the data presented here is shown as Mean ± Standard Deviation. Generally, n = 6–8 cartilage explants were used for every condition and experiments were repeated using explants from at least 4 animals yielding consistent results and trends. The general linear mixed effects model with animal as a random variable was used that showed no effect of animal, followed by Tukey’s Honestly Significant Difference (Tukey’s HSD) test to compare multiple treatment conditions. P < 0.05 was considered as statistically significantly different.
3. Results
3.1. CPC +14 resulted in highest intra-cartilage uptake
The equilibrium intra-cartilage uptake (RU) is defined as
| (Eq. 1) |
where is CPC concentration (both free and bound) inside cartilage (per intra-tissue wet weight), and CBath is the CPC concentration remaining in the outside bath at equilibration. Increasing the z of CPCs from +8 to +14 increased the intra-cartilage uptake by 7x (RU ~360 for CPC +14 vs ~50 for CPC +8). Increasing z of CPCs beyond +14, however, did not enhance their uptake (Fig. 1A) but instead reduced it in a charge dependent manner resulting in 2x lower uptake for CPC +20 compared to CPC +14. Desorption studies showed that the majority (~99% of uptaken solute) of higher charged CPCs (z=+14, +16 and +20) remained bound within cartilage after 24 h desorption in PBS with physiological salinity (1x PBS), while about 77% of CPC +8 was retained (Fig. 1B). Desorption over 7 days further reduced its intra-cartilage retention to 35% while ~99% of all the higher charged CPCs continued to remain bound demonstrating stronger binding interactions with intra-cartilage sites compared to CPC +8 (Fig. 1C). To study how much binding can be attributed to electrostatics, we desorbed these explants in 10x PBS to shield and break charge interactions. CPCs desorbed out in a charge dependent manner with about 25% retention measured for CPC +8 and 83% retention for CPC +20 after 24 h desorption (Fig. 1B). A plateau in % intra-cartilage retention was achieved over the 7-day desorption period in 10x PBS (Fig. 1D), which is attributed to interactions other than due to the charge. Of note is that while 50–80% CPCs desorbed in 10x PBS for CPC +20, +14 and +8 indicating that charge interactions are dominant effects, only about 40% CPC +16 desorbed out over 7 days. This is due to the specific heparin binding attribute of this sequence which is known to bind strongly with heparan sulfate GAGs with dissociation constant, KD =21 nM [37].
Figure 1.
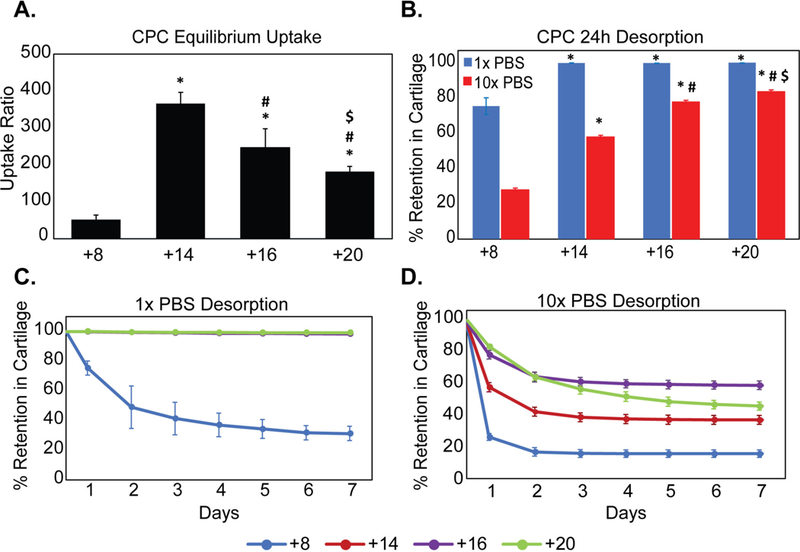
Effect of net positive charge (z) of CPCs (cationic peptide carriers) on equilibrium uptake and retention in bovine cartilage explants. A. Intra-cartilage equilibrium uptake ratios of CPCs B. % CPCs retained within cartilage explants following 24h desorption in 1x and 10x PBS. C. % CPC retained within cartilage with desorption over 7 days in 1x PBS and D. in 10x PBS (* vs corresponding condition in CPC +8, # vs CPC +14, $ vs CPC+16; p<0.05).
3.2. Weak-reversible binding is necessary for full depth penetration into the tissue
Fig. 2 shows distance penetrated by CPCs over time and their concentration profiles from superficial (SZ) to the deep zone (DZ) of cartilage. Within 4 h, CPC +8 diffused through the full thickness of the tissue owing to weak and reversible charge interactions. However, as the net charge of CPCs increased, the diffusion rate was slowed down because of stronger binding interactions within cartilage. CPC +14 and +16 penetrated nearly halfway through the tissue thickness in 4 h, while CPC +20 was stuck within the superficial zone (Fig. 2A). By the 24 h time point, both CPC +14 and +16 penetrated through the full thickness of cartilage while CPC +20 remained stuck within 200 μm of the tissue SZ owing to too strong intra-cartilage binding that hinders transport (Fig 2B). Desorption in 10x PBS shielded charge interactions and released CPCs out from the cartilage (Fig. 2C) in a charge dependent manner, consistent with the trends shown in Figs. 1B and 1D. Interestingly, CPC +20 penetrated deeper into the tissue (Fig. 2C) as the stronger binding interactions were broken by the high salt concentration of 10x PBS
Figure 2.
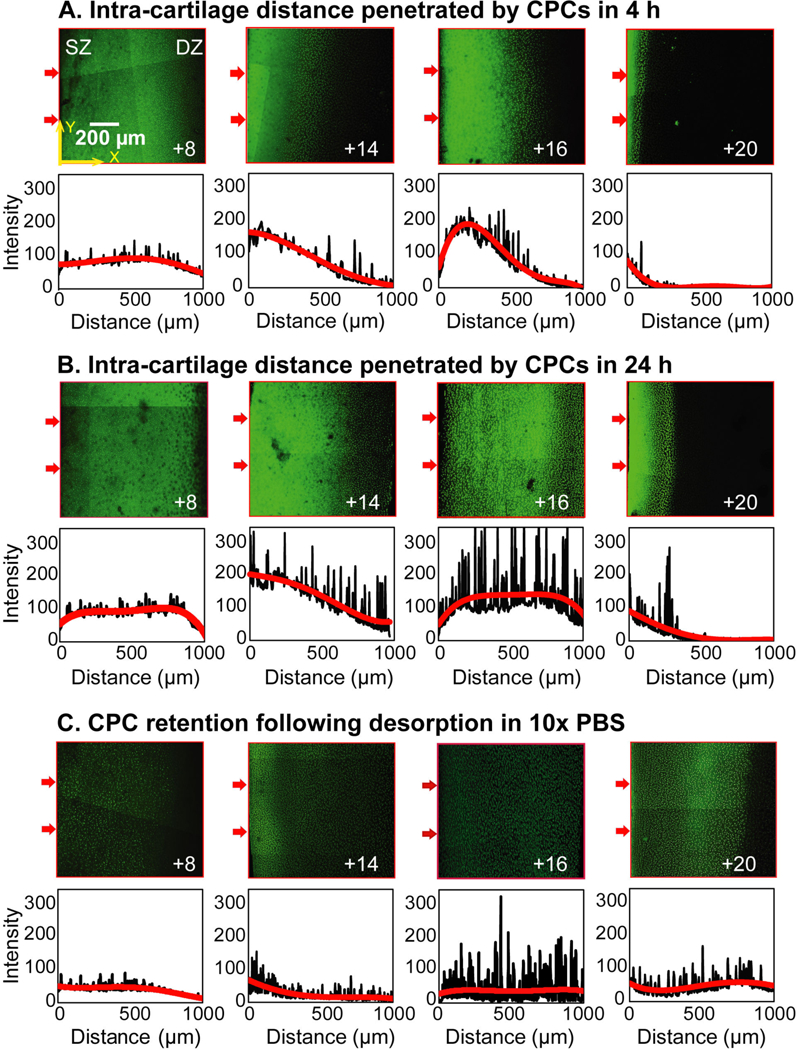
Confocal microscopy images showing intra-cartilage depth of penetration and concentration profiles of CPCs with different net charge (z) from superficial zone (SZ) to deep zone (DZ) in A. 4 h and B. 24 h. C. Confocal microscopy images showing extent of CPC retention and concentration profiles of explants incubated with CPCs for 24 h followed by 24 h desorption in 10x PBS. Arrows on the left side of the images indicate cartilage superficial zone and the direction of CPC penetration. Average fluorescence intensities across the thickness of each slice (X direction) are plotted below the images as a function of distance from the left edge of the images (or cartilage SZ). Scale bar is 200 μm.
3.3. Effect of CPC net charge on non-equilibrium transport in cartilage
The transport chamber setup shown in Fig. 3A was used to study non-equilibrium transport properties of CPCs in cartilage. Fig. 3B shows CPC concentration measured in the downstream chamber (CD) normalized to the applied upstream concentration (CU) plotted against time. The time to reach a steady state flux, τLag, is calculated from the time-axis intercept of the linear slope of normalized concentration versus time. Assuming one-dimensional diffusion of CPCs through the cartilage disk of given thickness, δ (500–800 µm), effective diffusivity that incorporated intra-cartilage effects of binding, DEFF, can be calculated as [28, 38]:
| (Eq. 2) |
Figure 3.
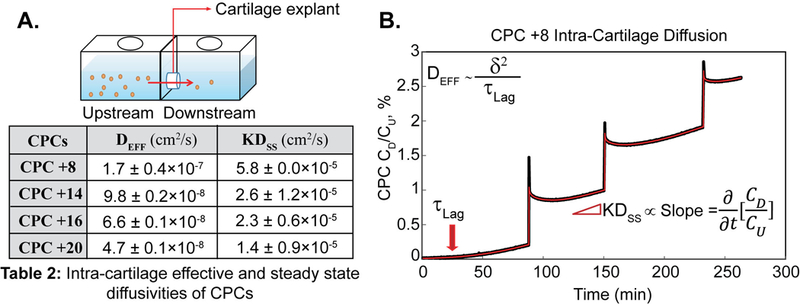
Non-equilibrium diffusion transport of CPCs in cartilage. A. Transport chamber setup to study one-dimensional diffusion of CPCs in cartilage. B. Example diffusion transport curve for CPC +8 plotted as the measured downstream concentration (CD) normalized to the applied upstream concentration (CU), versus time. Intra-cartilage effective diffusivity, DEFF (diffusivity in presence of binding interactions) was calculated using τLag and the steady state diffusivity, Dss (diffusivity when all binding sites are occupied, and a steady state is achieved) was calculated using the steady state slope. K is the Donnan partitioning factor. Each spike shows calibration step where 20 μl of CPC solution from the upstream compartment was added to downstream, and multiple diffusion curves were obtained.
Table 2 shows diffusivity estimates for CPCs in cartilage. Increasing the net charge of CPCs hinders their diffusive transport due to increased binding interactions and reduced effective diffusivities; DEFF of CPC +20 was measured to be an order of magnitude slower than that of CPC +8. These results are consistent with the confocal images showing CPC intra-cartilage depth of penetration as discussed in section 3.2.
Table 2.
Intra-cartilage effective (DEFF) and steady state (Dss) diffusivities of CPCs
| CPCs | DEFF (cm2/s) | KDss (cm2/s) |
|---|---|---|
| CPC +8 | 1.7 ± 0.4 × 10−7 | 5.8 ± 0.0 × 10−5 |
| CPC +14 | 9.8 ± 0.2 × 10−8 | 2.6 ± 1.2 × 10−5 |
| CPC +16 | 6.6 ± 0.1 × 10−8 | 2.3 ± 0.6 × 10−5 |
| CPC +20 | 4.7 ± 0.1 × 10−8 | 1.4 ± 0.9 × 10−5 |
Once the steady-state diffusion of CPCs through cartilage is achieved, the flux (Γ) can be correlated to steady state diffusivity (DSS) and the concentration gradient across tissue thickness by the following equation [28]:
| (Eq. 3) |
where K represents partition coefficient and Φ is cartilage porosity (~0.8) [28]. The time derivative of normalized CPC concentration is related to the steady-state flux described in Eq. 3 as follows:
| (Eq. 4) |
where VD is the volume of CPC solution in the downstream chamber (VD = 2 mL) and A is the cartilage disk surface area exposed to diffusion (A = 0.1257 cm2). Using this, KDSS for all CPCs were estimated (Table 2), which as expected measured to be two to three orders of magnitude faster than their respective effective diffusivities. KDSS were within the same order of magnitude estimate for all CPCs owing to their similar size and shape (stable alpha helical peptide chain); the slight difference in values is attributed to difference in Donnan partitioning factors (K) for CPCs of varying net charge, which is defined as the equilibrium uptake of only free solutes (and does not account for the solute bound to intra-tissue binding sites) [39–42].
3.4. Effect of cartilage negative fixed charge density (FCD) on CPC uptake
The high density of aggrecan GAGs trapped within a dense type II collagen matrix contributes to the high negative FCD of cartilage, which is key to its structure and function. It provides compressive stiffness to cartilage by impeding loading induced interstitial fluid flow; the reduced distance between the negatively charged GAG chains increases electrostatic repulsion that helps the tissue to reswell back to its original shape as the loading is removed [12]. OA progression is accompanied by loss of aggrecan GAGs, which eventually causes joint degradation and loss of functionality and can also significantly affect the uptake and transport of drugs and drug carriers, especially ones that are electrically charged.
Here, mid-stage (~50% GAG depleted) and late-stage (~90% GAG depleted) osteoarthritic cartilage samples were created by varying tissue FCD via time controlled tryptic digestion. As expected, uptake of CPCs reduced as the tissue FCD dropped by 50% (Fig. 4A-B), highlighting the dominant role of charge interactions in their transport; mean uptake values, however, still remained very high (RU ranging between ~30–200). Note that an uncharged small sized solute (with no steric hindrance) is expected to have an equilibrium uptake ratio of one. CPC +14 demonstrated the largest drop in uptake ratio (from ~360 to 192) while uptake of CPC +20 did not change significantly with 50% GAG depletion (mean RU~216 compared to RU~181 in healthy). This can be explained as CPC +20 binding interactions in healthy tissues are too strong and in fact limit their transport beyond the superficial zones but when GAGs are depleted, the reduction in these interactions and increased pore size likely enhance their transport and uptake. This finding is consistent with the effects of charge shielding by 10x PBS that enabled greater intra-cartilage penetration of CPC +20 as described in section 3.2. The change in CPC uptake between 50% and 90% GAG depletion was less significant than the change from healthy to 50% GAG depleted cartilage. Note that CPC +14, +16 and +20 still maintained high uptake ratios of 120–150 even in 90% GAG depleted samples. This suggests the significance of other short-range interactions (like hydrophobic interactions and H-bonds) in stabilizing long-range charge-based binding. As expected, desorption in 10x PBS further reduced CPC retention in 50% GAG depleted samples but not as much in 90% GAG depleted samples (Fig. 4C). CPC +8 desorbed out the most in 1x as well as 10x PBS compared to others suggesting that it had the weakest binding interactions.
Figure 4.
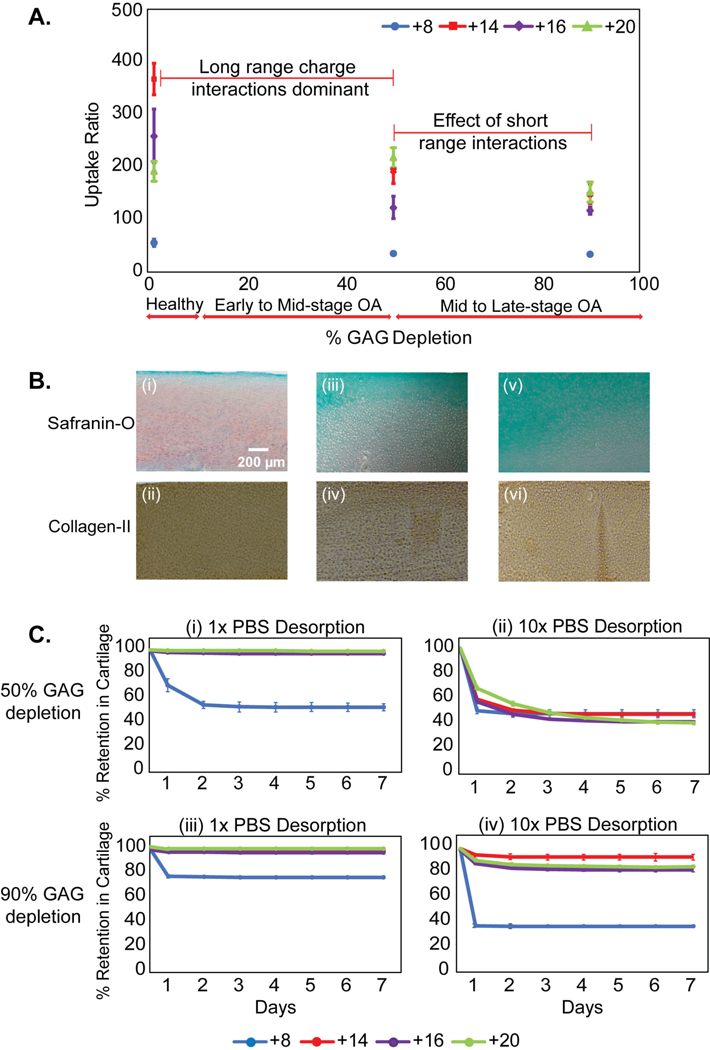
A. Intra-cartilage equilibrium uptake of CPCs in healthy (native) and arthritic (GAG depleted) explants. Cartilage explants were trypsin treated for different durations to cause about 50% GAG depletion, representing early to mid-stage osteoarthritis (OA) and about 90% GAG depletion representing late stage OA. B. Spatial GAG distribution in cartilage slices across thickness using safranin-O staining and type II collagen staining; (i-ii) Healthy, (iii-iv) 50% and (v-vi) 90% GAG depletion. C. % CPC retained after desorption in 1x and 10x PBS over 7 days in (i-ii) 50% and (iii-iv) 90% GAG depleted cartilage explants.
The above results validate the applicability of CPCs as intra-cartilage carriers of therapeutics in healthy as well as in mid-to-late stage arthritic conditions.
3.5. Effect of short-range interactions on transport of CPCs in cartilage
The short-range interactions could arise from guanidinium moieties of arginine that can form stable bidentate hydrogen bonds with GAG sulfates [43, 44]. To separate the effects of weaker charge interactions from other non-specific H-bonds and hydrophobic interactions, we studied the transport of CPC +7 comprising of lysine and alanine along with a hydrophilic (+N) or a hydrophobic tail (+F).
Mean equilibrium uptake ratio of CPC +N in 90% GAG depleted cartilage dropped to ~0.96 compared to RU = 15 in healthy condition, suggesting dominance of electrostatic charge interactions in intra-cartilage uptake of lysine rich CPC +7. In addition, CPC +N completely diffused out of the tissue when desorbed in 10x PBS (Fig. 5A-B), which further validates that the basis of CPC +N uptake and binding are strictly dominated by charge effects, unlike arginine rich CPCs where hydrogen bond and hydrophobic interactions play a role.
Figure 5.
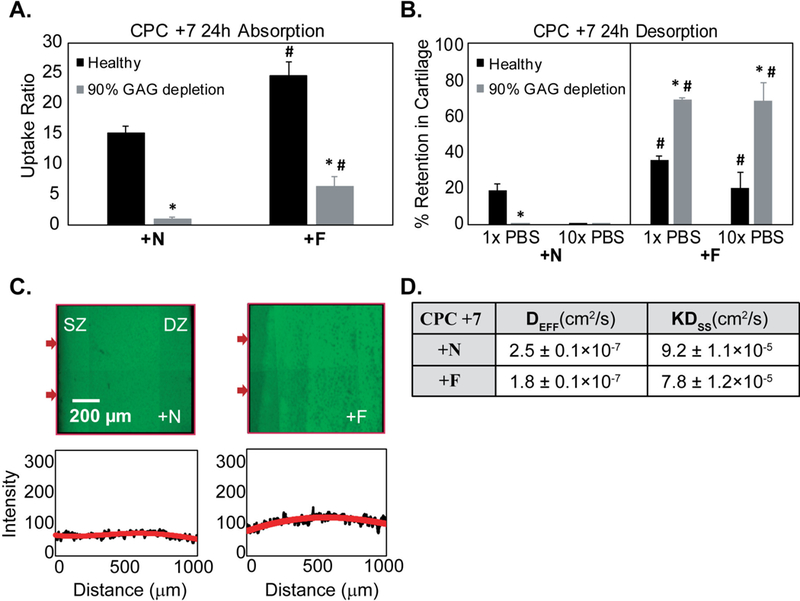
A. Equilibrium uptake of CPC +N (hydrophilic) and +F (hydrophobic) with net charge of +7 in healthy and 90% GAG depleted cartilage explants. B. % Intra-cartilage retention following 24 h desorption in 1x and 10x PBS bath (* vs Healthy, # vs corresponding condition in CPC +N; p<0.05) C. Full thickness penetration of CPC +N and +F in cartilage within 4 h D. DEFF and Dss of CPC +N and CPC +F depicting rapid diffusion within cartilage due to charge interactions.
To study the synergistic effects of hydrophobic interactions with that of charge, equilibrium uptake and intra-cartilage retention of CPC +N was compared with that of CPC +F (Fig. 5A) in both healthy and 90% GAG depleted cartilage. CPC +F (with hydrophobic tail) demonstrated higher uptake (RU ~ 24) compared to its hydrophilic counterpart, CPC +N (RU ~ 15). Similarly, in 90% GAG depleted explants, uptake of CPC +F was 6.5x higher than its hydrophilic counterpart. Therefore, hydrophobic interactions can further enhance intra-cartilage uptake of solutes synergistically with electrostatic charge interactions. Moreover, incorporation of hydrophobic tail significantly improved intra-cartilage retention of CPCs both in healthy and 90% GAG depleted samples (Fig. 5B). CPC +N and +F also rapidly penetrated through the full thickness of cartilage tissues within 4 h (Fig. 5C). This suggests that the lower binding affinity and the absence of short-range interactions in lysine rich CPCs result in faster diffusion rates compared to the arginine rich CPCs. Addition of hydrophobic tail enhanced the intra-cartilage binding affinity of CPC +7 resulting in slightly decreased diffusivities for CPC +F compared to CPC +N (Fig. 5D). In summary, the higher intra-cartilage uptake and longer retention of arginine rich CPCs can be attributed to complex binding mechanisms involving weak-reversible electrostatic charge interactions in combination with shorter range forces of hydrophobic and hydrogen bonds.
To delineate purely Donnan effects (i.e. no intra-tissue binding interactions) from those of binding-based charge and other short-range interactions, we estimated Donnan partitioning factor, K, for CPCs. As described earlier, solute uptake inside cartilage can be free or bound; 24 h desorption in 1x PBS resulted in 80% of the uptaken CPC +N releasing out, which can be approximated as free (and not bound to intra-cartilage sites) and thus can be attributed entirely to Donnan partitioning, K [39–41]. Therefore, KCPC +N is estimated as ~12. The ratio of KDss of CPC +N and +F is 1.17 (Fig. 5D), thus, KCPC +F is estimated as ~10.2. Similarly, K of other CPCs are estimated and reported in Table 3. Higher K implies higher upward partitioning of CPCs at the solution-cartilage interface that results in steeper concentration gradients explaining faster rate of transport for CPCs with higher K values.
Table 3.
Estimates of Donnan Partitioning factor, K, for CPCs
| CPC | K |
|---|---|
| CPC +8 | 7.6 |
| CPC +14 | 3.4 |
| CPC +16 | 3.0 |
| CPC +20 | 1.8 |
| CPC +N | 12.0 |
| CPC +F | 10.2 |
3.6. Dose dependent biological response of CPCs on cartilage health and biosynthesis rates.
We also studied the dose dependent biological response of CPCs in cartilage culture experiments. CPC +14 was chosen as the representative candidate as it resulted in the highest intra-cartilage uptake, full depth penetration and long retention, making it an ideal candidate for intra-cartilage delivery of macromolecules. CPC +14 even at a very high concentration of 20 μM did not affect the cumulative GAG loss to media compared to untreated control. Similarly, the rate of GAG biosynthesis measured using S35 radiolabel incorporation remained unaffected. The chondrocyte viability was not affected either over the 8-day culture period (Fig. 6A-C). Note that some cell death in the superficial zone is typically observed in untreated control explants, depending on the location of harvesting along the joint. Also, excision of tissues from the joint using punches can also lead to cell death at the cut surfaces [45].
Figure 6.
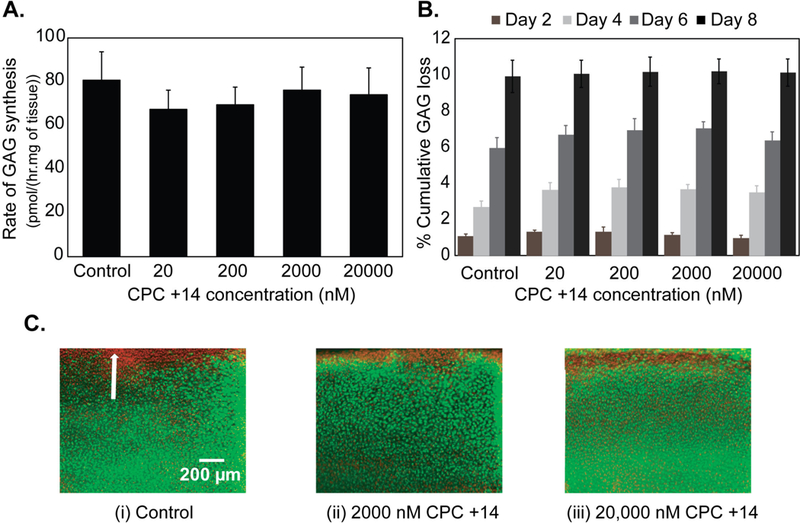
Dose dependent biological response of CPC +14 in cartilage over a period of 8 days in culture. A. Rate of GAG synthesis, B. % cumulative GAG loss in media, and C. Chondrocyte viability. Green indicates viable cells and red indicates non-viable cells. White arrow shows superficial zone of the tissue.
4. Discussion
Despite the existence of promising drugs, OA treatment remains a challenge due to a lack of effective drug delivery systems [6]. Currently, pain and inflammation relievers are administered via direct IA injections but drugs suffer from short residence times clearing out rapidly from the joint via synovium vasculature and lymphatics (e.g., half-lives of NSAIDs reported 1–4 h) [7]. Furthermore, drugs that need to reach their cell and matrix target sites are unable to penetrate into the dense, negatively charged matrix of cartilage. As a result, no DMOADs have passed clinical trials due to concerns of systemic toxicity and lack of cartilage targeting [6].
The high negative FCD of cartilage provides a unique opportunity to enhance the transport rate, uptake, and retention of drugs or drug carriers by making them positively charged [5, 46]. The net positive charge on a solute of given size confers specific transport properties, in a charged tissue of known FCD, which determine its effectiveness as a drug carrier. We show that there exists an optimal charge range for a solute of given size for effective targeting into a tissue of known FCD. Increasing the net charge on a solute does not necessarily imply increased intra-tissue uptake or transport rate; electro-diffusive transport can be slowed down or hindered by the solute binding with intra-tissue sites that can limit its tissue penetrability and uptake. CPC +7 and +8 diffused through the full thickness of cartilage explants within 4 h while CPC +14 and +16 penetrated half-way through and CPC +20 remained stuck within the tissue superficial zone even by 24 h time point due to its stronger binding with the intra-cartilage negatively charged aggrecan-GAGs (Fig. 2). Furthermore, CPC equilibrium uptake increased with increasing net charge from z = +7 to +14 resulting in the highest RU ~360 measured for CPC +14; further increase in net charge to +20 resulted in reduced CPC uptake (Fig. 1). A majority of CPC +7 and +8 desorbed in physiological saline solution while high salt concentration bath (10x PBS) was needed to break charge interactions and release out higher charged CPCs from tissue explants. As a result of this charge shielding, CPC +20 that was stuck in the cartilage superficial zone unbound and penetrated deeper into the cartilage. Also, CPC +20 uptake did not change significantly with 50% GAG depletion as the reduced charge-based binding interactions and increased pore size likely enhanced its transport and uptake into the deeper zones of cartilage (Fig. 4). CPC +14, +16 and +20 remained bound within the cartilage over the 7-day desorption period in 1x PBS. The ability of strong binding interactions to promote transport is therefore paradoxical; while binding enhances retention, it consequently hinders diffusive transport slowing down the penetration of carriers as they would get trapped in the tissue surface before reaching deeper zone targets. This underlines the significance of tuning the net charge of the carrier to enable weak-reversible binding between with oppositely charged tissue that it needs to target.
Charge dependent rates of intra-cartilage transport of CPCs are consistent with our independent estimates of relative effective diffusivities (Fig. 3) and partitioning factors, K (Table 3). Effective diffusivities of CPCs slowed down with increasing net charge. We estimated highest K values for CPC +7 (K=10–12) and CPC +8 (K=7.6) explaining their rapid transport into cartilage (within 4 h). Solute concentration is enhanced by the partition coefficient, K at the bath-cartilage interface that results in steep concentration gradients and flux [5, 28]. K=1.8 for CPC +20 explains its relatively slower transport rate thereby indicating that a majority of the uptaken CPC is bound to intra-cartilage sites (and is not free). CPC +14 measured a higher K=3 enabling its faster intra-cartilage flux. Its structure confers optimal binding affinity to enable highest intra-cartilage uptake as well as long term retention without significantly slowing down its transport.
The work highlights that while the net charge of a carrier can help predict its electro-diffusive behavior, the type of amino acid moiety in the peptide [43, 47, 48] and its spatial distribution [33, 49–51] are also important to consider. The change in uptake of arginine rich CPCs between 50% and 90% GAG depletion, was less significant than the change from healthy to 50% GAG depleted cartilage. Note that these CPCs still maintained high uptake ratios even in 90% GAG depleted samples (Fig. 4C). The decrease in uptake due to 50% reduction in cartilage FCD is largely due to the reduced charge interactions while the high uptake measured even after 90% GAG depletion can be attributed to the presence of other short-range interactions (hydrophobic interactions and H-bonds) that stabilize long-range charge-based binding. This is supported by the observation that in 10x PBS only 50% CPCs desorbed out from 50% GAG depleted samples and there was almost no desorption from 90% GAG depleted samples for all arginine rich CPCs except CPC +8, 60% of which desorbed in 10x PBS. On the other hand, lysine rich CPC +N did not measure as high of an uptake, and 80% of it desorbed in 1x PBS and 100% in 10x PBS confirming the dominance of charge interactions (Fig. 5). Despite the similar net charge in arginine rich CPC +8 and lysine rich CPC +N, the higher uptake and retention of CPC +8 is attributed to the chemistry of the guanidinium head-group on arginine residues that allow formation of stable bidentate hydrogen bonds with polarizable oxo-anions such as the sulfates in aggrecan glycosaminoglycans [44, 52], which further stabilize the long-range electrostatic interactions. Additionally, guanidinium cations can form thermodynamically stable (weakly) like-charge pair in water, where a combination of dispersion and cavitation forces from the medium can overwhelm the coulombic repulsion [53]. Because of this, arginine moieties are capable of binding more strongly (due to synergistic effects of charge, H-bond and hydrophobic interactions) and cooperatively (due to formation of like-charge pair between arginine) with negatively charged GAGs compared to lysine moieties, which instead exhibit mutual coulombic repulsion. To confirm the synergistic effects of charge and hydrophobic interactions on intra-cartilage uptake and retention, a hydrophobic tail was added in lysine rich CPC +F, which resulted in higher uptake and stronger intra-cartilage binding compared to its hydrophilic counterpart in both healthy and GAG depleted samples (Fig. 5A-B).
Polyarginines are known to more effectively penetrate the cell membrane than polylysines due to their stronger binding with the heparan sulfate proteoglycans on the cell surface [43, 54–56] (two orders of higher dissociation constants have been reported for polylysines than for polyarginines) [43]. Arginine rich CPCs, therefore, could also have bound to the cell lipid bilayer, translocated across the membrane and/or internalized via endocytosis [54, 56], a mechanism that was not studied here but one that may partly contribute to the high uptake values measured.
Varying lengths of arginine-based Cell Penetrating Peptides (CPPs) have been used for intra-cellular delivery of drugs and genetic materials [57, 58]. It has been observed that peptides of six or more arginine (or guanidinium groups) were necessary for uptake into the cells. Uptake increased with increasing number of arginine up to 15 (net charge of +15) after which intra-cellular uptake declined. It has been speculated that perhaps a minimum of six arginine residues are required to form stable ionic interactions with either the heparan sulfate proteoglycans on cell surface or specific receptors or to adopt a biologically active conformation that can initiate transcellular transport [59]. Interestingly, our work shows highest intra-cartilage uptake of arginine rich CPC at similar charge range (net charge of +14), which can be explained by strong binding with heparan sulfate GAGs (high affinity for heparan sulfate (KD~109 nM) has been reported for R9) [54].
The high uptake and long-term retention of CPC +14 in both healthy and arthritic cartilage with varying FCD makes it an ideal carrier for delivering larger sized drugs to intra-cartilage cell targets. Additionally, it was found to be safe for cell and cartilage matrix health even at high concentrations (Fig. 6). CPC +8 and +7 have applications in cartilage imaging due to their very fast transport rate and shorter intra-tissue residence time, which can help clear contrast agents from the joint post imaging [60].
Conclusion
Electrostatic interactions can be utilized to significantly advance the field of targeted drug delivery for avascular, negatively charged tissues like cartilage. This way, tissues can be converted from a barrier to drug entry into a drug depot that can provide sustained drug doses over several weeks. The net charge of a carrier determines its electro-diffusive transport behavior in oppositely charged tissues. This work demonstrates that increasing net charge on solute does not necessarily imply increased uptake and that there exists an optimal charge range for a solute of given size to effectively target a tissue of known FCD. Weak and reversible binding interactions are critical for carriers to penetrate through the tissue deep zones, so they can reach their cell and matrix target sites. Arginine rich CPCs bind more strongly with the intra-cartilage negatively charged aggrecan-GAGs compared to the lysine rich CPCs because of short-range H-bond and hydrophobic interactions that further stabilize electrostatic binding. These concepts of charge-based intra-cartilage drug delivery are summarized in Fig. 7.
Figure 7.
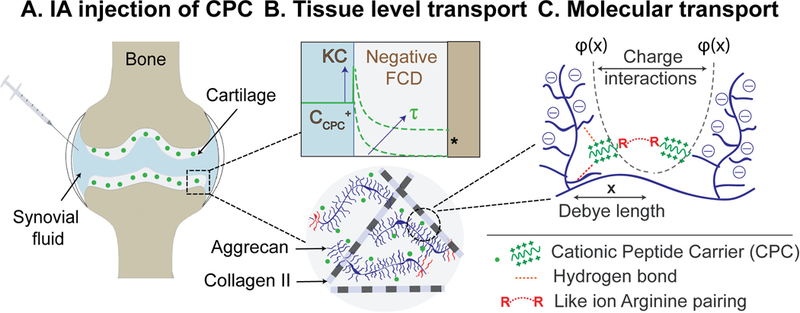
Charge based intra-cartilage delivery of CPCs at multiple length scales. A. Intra-articular (IA) injection of CPCs: Electrostatic interactions enable rapid and full depth penetration of CPCs into negatively charged cartilage. B. Tissue level transport: High upward Donnan partitioning at the synovial fluid-cartilage interface results in steep intra-cartilage concentration gradients for CPCs, thereby reducing the time (τ) required to reach intra-cartilage therapeutic index (*). Weak-reversible binding of CPCs with negatively charged intra-cartilage sites enables their full depth penetration. C. Molecular level transport: Inside cartilage, CPCs bind with aggrecans via long-range charge interactions. Electric potential of negatively charged aggrecans Ф(x) drops exponentially as a function of distance x and defines the debye length or spacing between aggrecan chains. The resulting electrical fields determine intra-cartilage electro-diffusive transport and binding of CPCs. This binding is further stabilized by short-range H-bond and hydrophobic interactions.
Ongoing work includes conjugating CPCs to DMOADs to make them cartilage penetrating and binding in order to enhance their biological efficacy, which can potentially enable their clinical translation. This work outlines an approach to rationally design carriers based on tissue FCD, properties of the molecule to be delivered, and the specific application.
Supplementary Material
Figure S1A. Custom built transport chamber to study one-dimensional (x-direction) diffusion of CPCs in cartilage. B. Schematic of 100 thick slice taken from the middle of the half-disk. C. Cross-section of the slice in X-Y plane imaged using confocal microscopy to estimate intra-cartilage distance penetrated by CPCs as presented in Fig. 2 of the manuscript. (Adapted from Ref. 27).
Acknowledgment
This work was funded by the United States Department of Defense through the Congressionally Directed Medical Research Programs (CDMRP) under contract W81XWH‐17‐1‐0085, NIH R03 EB025903-1 and in part by the Northeastern University Tier 1 Award.
References
- [1].Wieland HA, Michaelis M, Kirschbaum BJ, Rudolphi KA, Osteoarthritis—an untreatable disease?, Nat Rev Drug Discov 4(4) (2005) 331–344. [DOI] [PubMed] [Google Scholar]
- [2].Hellio Le Graverand-Gastineau M-P, Disease modifying osteoarthritis drugs: facing development challenges and choosing molecular targets, Curr. Drug Targets 11(5) (2010) 528–535. [DOI] [PubMed] [Google Scholar]
- [3].Hunter DJ, Pharmacologic therapy for osteoarthritis—the era of disease modification, Nat Rev Rheumatol 7(1) (2011) 13–22. [DOI] [PubMed] [Google Scholar]
- [4].Hellio Le Graverand-Gastineau MP, OA clinical trials: current targets and trials for OA. Choosing molecular targets: what have we learned and where we are headed?, Osteoarthr. Cartil 17(11) (2009) 1393–1401. [DOI] [PubMed] [Google Scholar]
- [5].Bajpayee AG, Grodzinsky AJ, Cartilage-targeting drug delivery: can electrostatic interactions help?, Nat Rev Rheumatol 13(3) (2017) 183–193. [DOI] [PubMed] [Google Scholar]
- [6].Evans CH, Kraus VB, Setton LA, Progress in intra-articular therapy, Nat Rev Rheumatol 10 (2013) 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Larsen C, Østergaard J, Larsen SW, Jensen H, Jacobsen S, Lindegaard C, Andersen PH, Intra‐articular depot formulation principles: Role in the management of postoperative pain and arthritic disorders, J. Pharm. Sci 97(11) (2008) 4622–4654. [DOI] [PubMed] [Google Scholar]
- [8].Maroudas A, Transport of solutes through cartilage: permeability to large molecules, J. Anat 122(Pt 2) (1976) 335–347. [PMC free article] [PubMed] [Google Scholar]
- [9].McK Snowden J, Maroudas A, The distribution of serum albumin in human normal and degenerate articular cartilage, Biochim Biophys Acta Gen Subj 428(3) (1976) 726–740. [DOI] [PubMed] [Google Scholar]
- [10].Byun S, Sinskey YL, Lu YCS, Ort T, Kavalkovich K, Sivakumar P, Hunziker EB, Frank EH, Grodzinsky AJ, Transport of anti-IL-6 antigen binding fragments into cartilage and the effects of injury, Arch. Biochem. Biophys 532(1) (2013) 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].DiDomenico CD, Bonassar LJ, How can 50 years of solute transport data in articular cartilage inform the design of arthritis therapeutics?, Osteoarthr. Cartil (2018). [DOI] [PubMed]
- [12].Mow VC, Wang CC, Hung CT, The extracellular matrix, interstitial fluid and ions as a mechanical signal transducer in articular cartilage, Osteoarthr. Cartil 7(1) (1999) 41–58. [DOI] [PubMed] [Google Scholar]
- [13].Maroudas A, Balance between swelling pressure and collagen tension in normal and degenerate cartilage, Nature 260 (1976) 808–809. [DOI] [PubMed] [Google Scholar]
- [14].Torzilli PA, Influence of cartilage conformation on its equilibrium water partition, J. Orth. Res 3(4) (1985) 473–483. [DOI] [PubMed] [Google Scholar]
- [15].Chevalier X, Goupille P, Beaulieu AD, Burch FX, Bensen WG, Conrozier T, Loeuille D, Kivitz AJ, Silver D, Appleton BE, Intraarticular injection of anakinra in osteoarthritis of the knee: A multicenter, randomized, double-blind, placebo-controlled study, Arthritis Care Res (Hoboken) 61(3) (2009) 344–352. [DOI] [PubMed] [Google Scholar]
- [16].Cohen SB, Proudman S, Kivitz AJ, Burch FX, Donohue JP, Burstein D, Sun Y-N, Banfield C, Vincent MS, Ni L, Zack DJ, A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee, Arthrit. Res. Ther 13(4) (2011) R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bodick N, Lufkin J, Willwerth C, Kumar A, Bolognese J, Schoonmaker C, Ballal R, Hunter D, Clayman M, An intra-articular, extended-release formulation of triamcinolone acetonide prolongs and amplifies analgesic effect in patients with osteoarthritis of the knee: a randomized clinical trial, JBJS 97(11) (2015) 877–888. [DOI] [PubMed] [Google Scholar]
- [18].Butoescu N, Seemayer CA, Foti M, Jordan O, Doelker E, Dexamethasone-containing PLGA superparamagnetic microparticles as carriers for the local treatment of arthritis, Biomaterials 30(9) (2009) 1772–1780. [DOI] [PubMed] [Google Scholar]
- [19].Elsaid K, Ferreira L, Truong T, Liang A, Machan J, D’Souza G, Pharmaceutical nanocarrier association with chondrocytes and cartilage explants: influence of surface modification and extracellular matrix depletion, Osteoarthr. Cartil 21(2) (2013) 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kang M-L, Kim J-E, Im G-I, Thermoresponsive nanospheres with independent dual drug release profiles for the treatment of osteoarthritis, Acta Biomater 39 (2016) 65–78. [DOI] [PubMed] [Google Scholar]
- [21].Dong J, Jiang D, Wang Z, Wu G, Miao L, Huang L, Intra-articular delivery of liposomal celecoxib–hyaluronate combination for the treatment of osteoarthritis in rabbit model, Int. J. Pharm 441(1) (2013) 285–290. [DOI] [PubMed] [Google Scholar]
- [22].Gouveia VM, Lopes-de-Araújo J, Lima SAC, Nunes C, Reis S, Hyaluronic acid-conjugated pH-sensitive liposomes for targeted delivery of prednisolone on rheumatoid arthritis therapy, Nanomedicine 13(9) (2018) 1037–1049. [DOI] [PubMed] [Google Scholar]
- [23].Shamji MF, Betre H, Kraus VB, Chen J, Chilkoti A, Pichika R, Masuda K, Setton LA, Development and characterization of a fusion protein between thermally responsive elastin-like polypeptide and interleukin-1 receptor antagonist: Sustained release of a local antiinflammatory therapeutic, Arthritis Rheum 56(11) (2007) 3650–3661. [DOI] [PubMed] [Google Scholar]
- [24].Vemula PK, Boilard E, Syed A, Campbell NR, Muluneh M, Weitz DA, Lee DM, Karp JM, On-demand drug delivery from self-assembled nanofibrous gels: A new approach for treatment of proteolytic disease, J Biomed Mater Res A 97A(2) (2011) 103–110. [DOI] [PubMed] [Google Scholar]
- [25].Kim KS, Park SJ, Yang JA, Jeon JH, Bhang SH, Kim BS, Hahn SK , Injectable hyaluronic acid–tyramine hydrogels for the treatment of rheumatoid arthritis, Acta Biomater 7(2) (2011) 666–674. [DOI] [PubMed] [Google Scholar]
- [26].Watson RS, Broome TA, Levings PP, Rice BL, Kay JD, Smith AD, Gouze E, Gouze J-N, Dacanay EA, Hauswirth WW, Nickerson DM, Dark MJ, Colahan PT, Ghivizzani SC, scAAV-Mediated Gene Transfer of Interleukin 1-Receptor Antagonist to Synovium and Articular Cartilage in Large Mammalian Joints, Gene Ther 20(6) (2013) 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Evans CH, Drug delivery to chondrocytes, Osteoarthr. Cartil 24(1) (2016) 1–3. [DOI] [PubMed] [Google Scholar]
- [28].Bajpayee AG, Wong CR, Bawendi MG, Frank EH, Grodzinsky AJ, Avidin as a model for charge driven transport into cartilage and drug delivery for treating early stage post-traumatic osteoarthritis, Biomaterials 35(1) (2014) 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bajpayee AG, Quadir MA, Hammond PT, Grodzinsky AJ, Charge based intra-cartilage delivery of single dose dexamethasone using Avidin nano-carriers suppresses cytokine-induced catabolism long term, Osteoarthr. Cartil 24(1) (2016) 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bajpayee AG, Scheu M, Grodzinsky AJ, Porter RM, Electrostatic interactions enable rapid penetration, enhanced uptake and retention of intra-articular injected avidin in rat knee joints, J. Orth. Res 32(8) (2014) 1044–1051. [DOI] [PubMed] [Google Scholar]
- [31].Bajpayee AG, Scheu M, Grodzinsky AJ, Porter RM, A rabbit model demonstrates the influence of cartilage thickness on intra-articular drug delivery and retention within cartilage, J. Orth. Res 33(5) (2015) 660–667. [DOI] [PubMed] [Google Scholar]
- [32].Bajpayee AG, Rodolfo E, Scheu M, Varady NH, Yannatos IA, Brown LA, Krishnan Y, Fitzsimons TJ, Bhattacharya P, Frank EH, Sustained intra-cartilage delivery of low dose dexamethasone using a cationic carrier for treatment of post traumatic osteoarthritis, Eur Cell Mater 34 (2017) 341–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Naik RJ, Chatterjee A, Ganguli M, Different roles of cell surface and exogenous glycosaminoglycans in controlling gene delivery by arginine-rich peptides with varied distribution of arginines, Biochim Biophys Acta Biomembr 1828(6) (2013) 1484–1493. [DOI] [PubMed] [Google Scholar]
- [34].Naik RJ, Sharma R, Nisakar D, Purohit G, Ganguli M, Exogenous chondroitin sulfate glycosaminoglycan associate with arginine-rich peptide–DNA complexes to alter their intracellular processing and gene delivery efficiency, Biochim Biophys Acta Biomembr 1848(4) (2015) 1053–1064. [DOI] [PubMed] [Google Scholar]
- [35].Farndale RW, Sayers CA, Barrett AJ, A Direct Spectrophotometric Microassay for Sulfated Glycosaminoglycans in Cartilage Cultures, Connect. Tissue Res 9(4) (1982) 247–248. [DOI] [PubMed] [Google Scholar]
- [36].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Fiji: an open-source platform for biological-image analysis, Nat. Methods 9(7) (2012) 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Miller RE, Grodzinsky AJ, Cummings K, Plaas AHK, Cole AA, Lee RT, Patwari P, Intra-articular Injection of HB-IGF-1 Sustains Delivery of IGF-1 to Cartilage through Binding to Chondroitin Sulfate, Arthritis Rheum 62(12) (2010) 3686–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Crank J, The Mathematics of Diffusion, second ed., Clarendon Press, 1975. [Google Scholar]
- [39].Bartlett JH, Kromhout RA, The donnan equilibrium, Bull Math Biophys 14(4) (1952) 385–391. [Google Scholar]
- [40].Donnan FG, The Theory of Membrane Equilibria, Chem. Rev 1(1) (1924) 73–90. [Google Scholar]
- [41].Grodzinsky AJ, Fields, forces, and flows in biological systems, Garland Science, London; New York, 2011. [Google Scholar]
- [42].Maroudas A, Physicochemical properties of cartilage in the light of ion exchange theory, Biophys. J 8(5) (1968) 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Robison AD, Sun S, Poyton MF, Johnson GA, Pellois J-P, Jungwirth P, Vazdar M, Cremer PS, Polyarginine interacts more strongly and cooperatively than polylysine with phospholipid bilayers, J Phys Chem B 120(35) (2016) 9287–9296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Åmand HL, Rydberg HA, Fornander LH, Lincoln P, Nordén B, Esbjörner EK, Cell surface binding and uptake of arginine- and lysine-rich penetratin peptides in absence and presence of proteoglycans, Biochim Biophys Acta Biomembr 1818(11) (2012) 2669–2678. [DOI] [PubMed] [Google Scholar]
- [45].Redman S, Dowthwaite G, Thomson B, Archer C, The cellular responses of articular cartilage to sharp and blunt trauma1, Osteoarthr. Cartil 12(2) (2004) 106–116. [DOI] [PubMed] [Google Scholar]
- [46].Krishnan Y, Rees HA, Rossitto CP, Kim S-E, Hung H-HK, Frank EH, Olsen BD, Liu DR, Hammond PT, Grodzinsky AJ, Green fluorescent proteins engineered for cartilage-targeted drug delivery: Insights for transport into highly charged avascular tissues, Biomaterials 183 (2018) 218–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rice A, Wereszczynski J, Probing the disparate effects of arginine and lysine residues on antimicrobial peptide/bilayer association, Biochim Biophys Acta Biomembr 1859(10) (2017) 1941–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Åmand HL, Rydberg HA, Fornander LH, Lincoln P, Nordén B, Esbjörner EK, Cell surface binding and uptake of arginine-and lysine-rich penetratin peptides in absence and presence of proteoglycans, Biochim Biophys Acta Biomembr 1818(11) (2012) 2669–2678. [DOI] [PubMed] [Google Scholar]
- [49].Li LD, Crouzier T, Sarkar A, Dunphy L, Han J, Ribbeck K, Spatial configuration and composition of charge modulates transport into a mucin hydrogel barrier, Biophys. J 105(6) (2013) 1357–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jiang K, Gao X, Shen Q, Zhan C, Zhang Y, Xie C, Wei G, Lu W, Discerning the composition of penetratin for safe penetration from cornea to retina, Acta Biomater 63 (2017) 123–134. [DOI] [PubMed] [Google Scholar]
- [51].Alhakamy NA, Dhar P, Berkland CJ, Charge type, charge spacing, and hydrophobicity of arginine-rich cell-penetrating peptides dictate gene transfection, Mol. Pharm 13(3) (2016) 1047–1057. [DOI] [PubMed] [Google Scholar]
- [52].Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA, Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells, J. Am. Chem. Soc 126(31) (2004) 9506–9507. [DOI] [PubMed] [Google Scholar]
- [53].Vazdar M, Heyda J, Mason PE, Tesei G, Allolio C, Lund M, Jungwirth P, Arginine “Magic”: Guanidinium Like-Charge Ion Pairing from Aqueous Salts to Cell Penetrating Peptides, Acc Chem Res 51(6) (2018) 1455–1464. [DOI] [PubMed] [Google Scholar]
- [54].Fuchs SM, Raines RT, Pathway for polyarginine entry into mammalian cells, Biochemistry 43(9) (2004) 2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tesei G, Vazdar M, Jensen MR, Cragnell C, Mason PE, Heyda J, Skepö M, Jungwirth P, Lund M, Self-association of a highly charged arginine-rich cell-penetrating peptide, PNAS 114(43) (2017) 11428–11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sun M, Zhu Z, Wang H, Han C, Liu D, Tian L, Yang X, Pan W, Surface density of polyarginine influence the size, zeta potential, cellular uptake and tissue distribution of the nanostructured lipid carrier, Drug Deliv 24(1) (2017) 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y, Arginine-rich peptides An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery, J. Biol. Chem 276(8) (2001) 5836–5840. [DOI] [PubMed] [Google Scholar]
- [58].Joseph SC, Blackman BA, Kelly ML, Phillips M, Beaury MW, Martinez I, Parronchi CJ, Bitsaktsis C, Blake AD, Sabatino D, Synthesis, characterization, and biological activity of poly (arginine)‐derived cancer‐targeting peptides in HepG2 liver cancer cells, J. Pept. Sci 20(9) (2014) 736–745. [DOI] [PubMed] [Google Scholar]
- [59].Mitchell DJ, Steinman L, Kim D, Fathman C, Rothbard J, Polyarginine enters cells more efficiently than other polycationic homopolymers, J. Pept. Res 56(5) (2000) 318–325. [DOI] [PubMed] [Google Scholar]
- [60].Pouran B, Arbabi V, Bajpayee AG, van Tiel J, Töyräs J, Jurvelin JS, Malda J, Zadpoor AA, Weinans H, Multi-scale imaging techniques to investigate solute transport across articular cartilage, J. Biomech 78 (2018) 10–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1A. Custom built transport chamber to study one-dimensional (x-direction) diffusion of CPCs in cartilage. B. Schematic of 100 thick slice taken from the middle of the half-disk. C. Cross-section of the slice in X-Y plane imaged using confocal microscopy to estimate intra-cartilage distance penetrated by CPCs as presented in Fig. 2 of the manuscript. (Adapted from Ref. 27).