

Abstract
Background
Experimental studies support a role of complement activation in diabetic nephropathy (DN), yet few clinical correlates exist. We evaluated urinary levels of sC5b-9 membrane attack complex (MAC) in patients with overt DN, and examined its association with the glomerular filtration rate (GFR) decline, proteinuria, and inflammatory biomarkers. We explored different complement pathways and compared our findings to autoimmune glomerulonephritis.
Methods
We prospectively followed 83 patients with DN and obtained repeated measurements of proteinuria, complement fragments (sC5b-9, C4a, C1q, mannose-binding lectin–associated serine protease [MASP]-1, and factor Bb), monocyte chemoattractant protein-1 (MCP-1), and transforming growth factor (TGF)-β1. We assessed independence and interactions using general linear models and repeated measures analyses and compared levels with subjects with active focal and segmental glomerulosclerosis, ANCA-associated vasculitis, and membranous and IgA nephropathies (n = 63).
Results
The diabetic cohort had an initial GFR of 25 ± 9 ml/min per 1.73 m2 and a renal function decline of 2.9 ± 3.0 ml/min per 1.73 m2 per year. All complement biomarkers were strongly intercorrelated and associated with biomarker inflammation and fibrosis, proteinuria, and the rate of renal function decline. There was a significant interaction (P = 0.03) between the level of proteinuria and urinary sC5b-9: in individuals with higher levels of urinary MAC, the relationship between proteinuria and the rate of renal function decline was more pronounced than in those with low urinary MAC. Finally, patients with DN had levels of urinary sC5b-9 comparable to autoimmune glomerulonephritis, when stratified by the level of proteinuria.
Conclusion
Urinary MAC is present in patients with overt DN at levels comparable to autoimmune glomerulonephritis and correlates with the GFR decline, supporting that complement activation and its measurement are clinically relevant in DN.
Keywords: complement activation, diabetic nephropathy, fibrosis, inflammation, proteinuria, glomerulonephritis
Graphical abstract
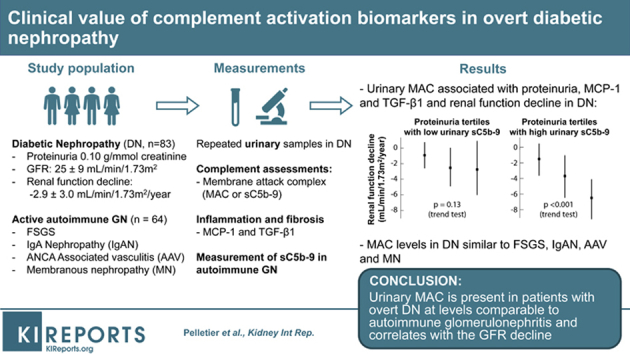
Inflammation is an important pathway of injury in the development of diabetic nephropathy (DN) and increasing evidence points toward a role for the complement system in the pathogenesis of DN.1, 2, 3 Experimental studies have elucidated how hyperglycemia may trigger the complement system and promote inflammation. High levels of circulating mannose-binding lectin exist in diabetes. They activate MASP-1 and MASP-2 of the lectin complement pathway and have been associated with lesions of DN, urinary albumin excretion, and increased mortality in type 1 and type 2 diabetes.3, 4, 5, 6, 7 Sustained hyperglycemia also may induce glycation and inactivation of CD59, a regulatory protein that inhibits the membrane attack complex (MAC or sC5b-9),8 and advanced glycation end-products may bind C1q and activate the classic pathway.9 Furthermore, histologic studies in humans and animal models have shown deposition of C4d and MAC in glomeruli, vessels, and interstitium in correlation with the severity of the lesions of DN.2, 10, 11 Finally, complement inactivation ameliorates animal models of DN.12 These findings support a pathogenic role of complement activation in the progression of DN. Yet, it is unknown how clinically important this is in a condition in which multiple pathways of injury exist13 and whether the measurement of complement biomarkers may identify individuals at risk of progression.
The most predictive clinical risk factor of progression in DN is proteinuria.14 Urinary MCP-1 and TGF-β1, markers of inflammation and fibrosis, have also been shown to predict renal function decline independently and additively to proteinuria.15, 16, 17 To our knowledge, there is no prospective clinical study addressing the association between urinary complement markers and the progression of DN. Our main objective was to assess urinary levels of sC5b-9 (MAC), a measurement of overall complement activation, and examine its association with the GFR decline, proteinuria, inflammatory, and fibrosis biomarkers in overt DN. We also measured C4a, a cleavage product of both classical and lectin pathways activation; MASP-1 and C1q as surrogates of the classical and lectin pathways activity; and factor Bb, cleaved from factor B when the alternative pathway is triggered. We finally compared our findings with autoimmune GN. We hypothesized that higher levels of these urinary biomarkers will show activation of the complement system in the clinical setting mostly by the classical and lectin pathways at levels comparable to active autoimmune GN and will correlate with the severity of DN during follow-up.
Materials and Methods
Study Design
We performed a prospective observational study in subjects with overt DN initiated in 2006 in 4 hospitals affiliated with the Université de Montréal, Canada. Each center’s ethics committee approved this study and all participants gave informed consent before enrollment, including the agreement to biobank urinary specimens for use at a subsequent time to test new hypotheses relevant to their disease. Results pertaining to biomarkers of inflammation and fibrosis from this cohort have previously been published.15 Types 1 and 2 diabetes were defined according to the American Diabetes Association18 and overt DN required a proteinuria of ≥0.5 g/d at least once before enrollment.19 Most patients were enrolled from predialysis clinics and had a long-standing history of diabetes and hypertension.
Our biobank also includes consenting individuals with biopsy-proven focal and segmental glomerulosclerosis, membranous and IgA nephropathies, as well as ANCA-associated vasculitis, participating in a longitudinal study on the clinical value of biomarker measurements. These served as comparison groups.
Patients and Samples
Parameters collected include demographics and serial blood pressure, number of antihypertensive medications, use of renin angiotensin system blockade, serum creatinine, and glycosylated hemoglobin (HbA1C). The mean arterial pressure was the diastolic plus a third of the pulse pressure. The GFR was estimated using the 4-variable Modification of Diet in Renal Disease formula. All patients had at least 3 serum creatinine measurements, and the rate of decline in renal function (GFR slope) was calculated by fitting a straight line through the GFRs using linear regression. Urinary protein excretion was estimated from spot urine collection by computing the protein-to-creatinine ratio (g/mmol of creatinine).
Measurement of serum and urinary creatinine and proteinuria were performed in each center’s laboratory within 24 hours of sampling. For all other biomarkers, urine spot samples taken on site were stored at 4°C, centrifuged at 200g for 10 minutes, aliquoted in multiple 0.4-ml vials, and stored at −80°C until further processing. For each DN participant, at least 2 urine samples were available at separate time points. Proteinuria and complement biomarkers were measured in all samples. The average of all measurements at different time points within an individual was used for analysis. In individuals with focal and segmental glomerulosclerosis, membranous and IgA nephropathies, and ANCA-associated vasculitis, we measured the proteinuria and complement biomarkers from the first available urine sample, corresponding most often to the time of renal biopsy, and before or immediately after starting immunosuppressive therapy.
Urinary Biomarkers
Complement components were measured using human EIA Kits: sC5b-9, C4a, and factor Bb (MicroVue; Quidel Corp., San Diego, CA), MASP-1 (Cloud Clone Corp., Katy, TX), and C1q (AssayPro, St. Charles, MO). Because of their sizes, factor Bb (63 kDa), and the larger C1q, MASP-1, and sC5b-9 cannot in normal conditions pass through the glomerular barrier, unlike the small C4a (9 kDa). In diseased states, they may appear in the urine, either when locally expressed or when filtrated because of higher glomerular permeability. Urine samples were diluted 1:5, except for sC5b-9, which was diluted 1:3. For MASP-1 and C1q measurements, some samples were retested at higher dilutions (1:10 or 1:20) to bring the concentration toward the range of the standard curve. The lower threshold sensitivities of the assays were 15, 18, 15, 0.2, and 0.5 ng/ml for sC5b-9, factor Bb, C4a, MASP-1, and C1q, respectively. Urinary MCP-1 and TGF- β1 were measured using 2 Human Cytokine/Chemokine plex kit (Millipore, St. Charles, MO) on a multiplex platform (Eve Technologies Corp, Calgary, Alberta, Canada). The lower threshold sensitivities of the assays were 2 and 10 ng/l for MCP-1 and TGF- β1. We report the values of urinary complement biomarkers in μg/mmol of creatinine and MCP-1 and TGF- β1 in ng/mmol of creatinine.
Statistical Analyses
Normally distributed variables are presented as means with SDs. Because the distributions of all urinary biomarkers were nonparametric, they are presented as medians with interquartile range, and correlation coefficients were calculated using Spearman’s rho. We also categorized urinary sC5b-9 and proteinuria into tertiles to illustrate our findings and used the Pearson correlation to perform trend tests with the rate of renal function decline.
To further assess the association between the GFR slope (dependent variable) and urinary sC5b-9 separately from the overall proteinuria, we performed general linear models using log-transformation (to respect the assumptions of linear regression) of these 2 biomarkers as covariates with and without an interaction term.
We also verified these analyses using the estimated GFRs, instead of the slopes, using repeated-measures analysis of variance. Because GFR estimations were not done at predefined times, we established GFRs at each 6-month interval based on the available data. We ran mixed repeated-measures analysis of variance adding to the “within-subject variable” (GFRs at different time) a “between-subject variable”: one model using log-transformed proteinuria, another using log-transformed sC5b-9 and finally one using both biomarkers. We present the interactions with the time effect, that is, to test whether the change in GFR over time was influenced by that variable and report the F and partial eta (ηp2) scores. Because the Mauchly's tests of sphericity P values were always <0.001, we used the Greenhouse-Geisser correction for the degree of freedom.
In the subanalyses presented, we also separated individuals with low and high sC5b-9, C4a, and factor Bb by the point on a receiver operating characteristic curve maximizing the sensitivity and specificity product. To perform this analysis, the receiver operating characteristic curve used the GFR slope divided into 2 groups using the median value as the outcome. Two-tailed P values less than 0.05 were considered statistically significant. Analyses were performed using SPSS software (version 24; SPSS Inc., Chicago, IL).
Results
Cohort
The clinical and biochemical characteristics of the study participants are given in Table 1. A total of 83 patients were included in the cohort. All but 2 subjects had type 2 diabetes, 80% were male and 87% white with a mean age of 69 ± 10 years. The initial GFR was 25 ± 9 ml/min per 1.73 m2 and mean arterial pressure was 94 ± 12 mm Hg. Overall, we obtained a median 12 serum creatinine, 6 blood pressure, 3 HbA1C, and 3 urine measurements per patient. Patients were followed for 2.1 (1.6–2.8) years. During the study, patients received on average 4 antihypertensive drugs and renin angiotensin system blockade in 73% of cases. Median HbA1C was 7.3%, 46% of participants had an HbA1C ≤7%, and 12% a mean HbA1C >8.5%. The median protein-to-creatinine ratio was 0.13 (0.05–0.32) g/mmol. The mean decline in renal function was 2.9 ± 3.0 ml/min per 1.73 m2 per year. Only 2 individuals had a renal biopsy, and both demonstrated DN.
Table 1.
Demographics and clinical features of the DN cohort (n = 83)
| At enrollment | |
| Female (%) | 20 |
| Ethnicity (%): white, Asian, African, Middle Eastern | 87, 4, 4, 5 |
| Age (yr) | 69 ± 10 |
| GFR (ml/min per 1.73 m2) | 25 ± 9 |
| Proteinuria (g/mmol creatinine) | 0.10 (0.05–0.32) |
| Urinary MCP-1 (ng/mmol creatinine) | 35 (22–67) |
| Urinary TGF-β1 (ng/mmol creatinine) | 1.4 (0.0–3.0) |
| Urinary sC5b-9 (μg/mmol creatinine) | 1.2 (0.3–6.8) |
| During follow-up | |
| Systolic blood pressure (mm Hg) | 142 ± 18 |
| Diastolic blood pressure (mm Hg) | 69 ± 12 |
| Mean arterial blood pressure (mm Hg) | 94 ± 12 |
| Antihypertensive medication (n) | 4 (3–5) |
| RASB (%) | 73 |
| HbA1C (%) | 7.3 (6.5–7.9) |
| Length of follow-up (yr) | 2.1 (1.6–2.8) |
| Proteinuria (g/mmol creatinine) | 0.13 (0.04–0.32) |
| Urinary MCP-1 (ng/mmol creatinine) | 40 (21–80) |
| Urinary TGF-β1 (ng/mmol creatinine) | 1.7 (0.7–3.4) |
| Urinary sC5b-9 (μg/mmol creatinine) | 1.9 (0.5–10.4) |
| Rate of renal function decline (ml/min per 1.73 m2 per year) | −2.9 ± 3.0 |
HbA1C, glycosylated hemoglobin; MCP, monocyte chemoattractant protein; RASB, renin angiotensin system blockade; TGF, transforming growth factor.
Values are expressed as percent, mean ± SD, or median (interquartile range).
Urinary Complement Biomarker Associations With the Proteinuria, Blood Pressure, Inflammatory, and Fibrosis Biomarkers and the Rate of Renal Function Decline
Urinary measurements of sC5b-9, factor Bb, C4a, and MASP-1 had a skewed distribution, more so compared with proteinuria (Figure 1). The median average sC5b9-to-creatinine ratio in our cohort was 1.9 (0.5–10.4) μg/mmol. All complement biomarkers showed a strong correlation to each other (all P values ≤ 0.001), as well as to proteinuria. Urinary sC5b-9 also showed a correlation with the mean arterial pressure (correlation coefficient 0.29; P = 0.009, Figure 2b), with the number of antihypertensive medications (correlation coefficient 0.42; P < 0.001, Figure 2c), and with MCP-1 and TGF-β1 (Figure 2d and e). sC5b-9 levels did not correlate with the initial GFR. Finally, all complement biomarkers were strongly associated with the rate of renal function decline by univariate analysis (Table 2 and Figure 2f).
Figure 1.

Distribution of urinary sC5b-9, C1q, factor Bb, C4a, mannose-binding lectin–associated serine protease (MASP)-1, and proteinuria based on the average of all measurements per patients.
Figure 2.

Associations between tertiles of urinary sC5b-9 (C1–C3) and proteinuria (a), mean arterial pressure (b), number of blood pressure medications (c), urinary MCP-1 (d), TGF-β1 (e), and the rate of renal function decline (f). Values are expressed either as mean ± SD (full line) or median with 25th and 75th percentile (dashed lines). All trend tests between tertiles of sC5b-9/creatinine and each outcome were statistically significant (all P < 0.01).
Table 2.
Associations between urinary biomarkers and the rate of renal function decline
| Variable | Correlation coefficient | P |
|---|---|---|
| Proteinuria | −0.50 | <0.001 |
| MCP-1 | −0.55 | <0.001 |
| TGF-β1 | −0.39 | <0.001 |
| sC5b-9 | −0.38 | <0.001 |
| C4a | −0.36 | 0.001 |
| MASP-1 | −0.34 | 0.002 |
| C1q | −0.34 | 0.002 |
| Factor Bb | −0.33 | 0.003 |
MASP, mannose-binding lectin–associated serine protease; MCP, monocyte chemoattractant protein; TGF, transforming growth factor.
The distributions of biomarkers were nonparametric and tested using Spearman’s Rho.
Interaction Between Urinary MAC and Proteinuria With GFR Decline
Because all the biomarkers of the complement studied are proteins, we sought to distinguish urinary sC5b-9 from the effects of proteinuria and performed a general linear model addressing independence and interactions. Although a sC5b-9 effect independent of proteinuria was not found, an interaction existed between urinary MAC and proteinuria (P = 0.03) indicating that the relationship of proteinuria with the rate of renal function decline was modified by the level of urinary sC5b-9. To illustrate this, we dichotomized urinary sC5b-9 based into “low” and “high” levels based on the point on the receiver operating characteristic curve maximizing the sensitivity and specificity product (2 μg/mmol). We can see in Figure 3, that the relationship between proteinuria and the rate of renal function was pronounced in the presence of “high” urinary MAC (P < 0.001), whereas it was modest and not statistically significant with “low” urinary MAC (P = 0.13).
Figure 3.
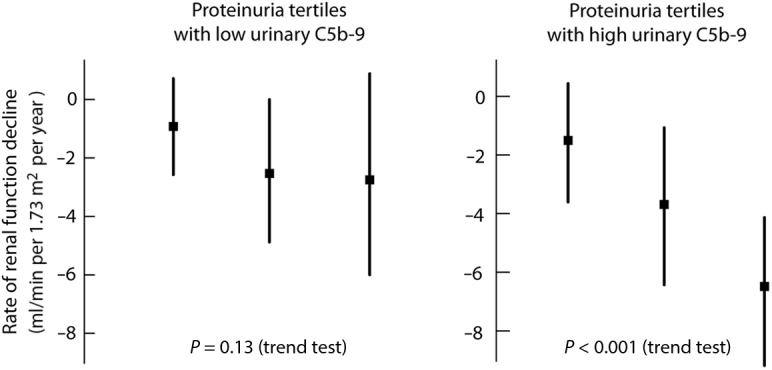
Interaction between proteinuria and the rate of renal function decline according to urinary sC5b-9 levels. Values are expressed as mean ± SD.
We repeated these analyses using a repeated-measures analysis of variance instead of the slope and found similar results. Individually, proteinuria (F2.7,222.0 = 6.4, P = 0.001, ηp2 = 0.07) and urinary sC5b-9 (F2.7,221.2 = 3.8, P = 0.01, ηp2 = 0.05) were associated with the degree of change in GFR over time. When both were entered in the same model, the interaction between them was statistically significant (P = 0.048). We therefore separated into low and high urinary MAC, as we did above when using the slope, and found that proteinuria was not associated with the change in GFR in the low MAC group (F2.7,120.0 = 1.1, P = 0.35, ηp2 = 0.02), whereas it was in the high MAC group (F2.4,85.6 = 6.7, P = 0.001, ηp2 = 0.16).
Comparison of Different Pathways of Activation
To further investigate how the activation of the classical and/or lectin pathways differed clinically from that of the alternative pathway, we separated individuals into categories of C4a and factor Bb levels based on cutoffs determined by receiver operating characteristic curves (1.15 and 2.2 μg/mmol, for C4a and factor Bb, respectively). The slopes were −1.8 ± 2.4, −1.4 ± 1.6, −2.7 ± 2.9, and −5.1 ± 2.9 ml/min per 1.73 m2 per year with “both biomarkers low” (n = 35), “low C4a-high factor Bb” (n = 7), “high C4a-low factor Bb” (n = 16), and “both biomarkers high” (n = 25), respectively (1-way analysis of variance, P < 0.001 with “both high” faster than each of the 3 other groups). The groups “both low,” “high C4a-low factor Bb,” and “low C4a-high factor Bb” were not statistically different.
Comparison With Autoimmune GN
We compared our findings with recently diagnosed autoimmune GN and measured these biomarkers in urine samples from 15 subjects with focal and segmental glomerulosclerosis, 17 with IgA nephropathies, 14 with membranous nephropathies, and 18 with ANCA-associated vasculitis with a median proteinuria of 0.43 (0.19–0.74), 0.15 (0.11–0.43), 0.79 (0.20–1.32), and 0.14 (0.06–0.65) g/mmol of creatinine, respectively. The urinary excretion of sC5b-9 and its corresponding proteinuria varied according to the underlying glomerular disease. Figure 4 illustrates that for equal proteinurias, urinary sC5b-9 in DN are comparable to autoimmune conditions in which complement activation is implicated in the pathogenesis.
Figure 4.

Relation between urinary sC5b-9 and proteinuria in diabetic nephropathy (DN) and autoimmune glomerulonephritis (GN). The purpose of this figure was to simultaneously compare urinary sC5b-9 levels stratified by proteinuria in different GN and demonstrate that the magnitude of sC5b-9 in DN is comparable to autoimmune disorders in which complement activation is implicated in the pathogenesis. AAV, ANCA-associated vasculitis; FSGS, focal segmental glomerulosclerosis; MN, membranous nephropathy.
Discussion
The current study explored renal complement activation in DN in the clinical setting. We found that complement components sC5b-9, C4a, C1q, MASP-1, and factor Bb are present in the urine and strongly correlate with proteinuria, urinary MCP-1 and TGF-β1, blood pressure control, and the rate of renal function decline. In individuals with higher levels of urinary MAC, the relationship between proteinuria and the rate of renal function decline was pronounced, unlike when urinary MAC was low, demonstrating a clinically observable effect. In addition to high levels of urinary MAC, individuals with both elevated C4a and factor Bb had a faster rate of renal function decline. Finally, at comparable levels of proteinuria, the patients with DN had similar or higher levels of urinary sC5b-9 compared with other active autoimmune glomerulonephritis. Taken together, these findings support a meaningful clinical role of complement activation in agreement with experimental findings.
The complement system is an important component of the innate immune system with 3 main pathways: the lectin, classical, and alternative pathways. Each of these has its own triggers, but their activation leads to the production of complement C3 and C5 convertases, ultimately generating the C5b-9 complex, responsible for cell and organ damage. Glomerular proteinuria can activate the complement in the tubules and the kidney has the ability to synthesize most of the components of complement cascade.20, 21 Evidence in DN points toward complement activation via the lectin and classical complement pathways. Recent studies have demonstrated a relationship between high levels of circulating mannose-binding lectin, which initiate the complement lectin pathway, and the development of persistent proteinuria and a more rapid decline in renal function in patients with DN.4, 5, 6, 7 Glomerular C4d and sC5b-9 deposits correlate with the severity of DN and microvascular and interstitial lesions.22 Furthermore, an experimental model of nephrotic syndrome has shown that complement-deficient rats developed significantly milder tubulointerstitial injuries than did complement-sufficient animals, despite equivalent levels of proteinuria.23 Complement activation also participates in the deleterious effect of hypertension.24, 25 Animal models of angiotensin-2–induced hypertension show reduced cardiomyocyte hypertrophy, cardiac inflammation, and perivascular fibrosis using a C5a receptor antagonist.26 Only a few experimental studies have assessed the therapeutic effect of blocking complement signaling in DN. Diabetic mice with increased complement activation had a more severe diabetic nephropathy27 and blockade of complement component C5 diminished the severity of albuminuria.28
To our knowledge, this is the first prospective study with repeatedly measured urinary complement biomarkers in the diabetic population to find a correlation with the rate of renal function decline. Previous studies have addressed sC5b-9 in the clinical setting: Ogrodowski et al.29 reported preliminary data showing that urinary MAC excretion was increased not only in patients with membranous nephropathy but also in those with DN, which was correlated with the degree of proteinuria. A small study in 15 patients with DN with nephrotic proteinuria also reported urinary excretion of sC5b-9 and CD59 (a MAC inhibitor).30 In this study, however, urinary levels of sC5b-9 correlated with CD59, but not with proteinuria or cross-sectional GFR. Morita et al.31 later found that urinary excretion of MAC was elevated in 99 patients (17 with DN) with nephrotic syndrome, and that excretion was correlated with degree of proteinuria. Interestingly, in this study, individuals with minimal change disease, which does not lead to chronic kidney disease, had very low levels of urinary MAC despite a mean proteinuria of 4.89 g/g of creatinine.
It was not possible to highlight the activation of one specific complement pathway in our cohort. MASP-1 and C1q were both correlated with proteinuria and renal function decline. These 2 proteins could thus have led to the formation of C4a fragment, and both the lectin and classical pathways could be implicated. Associations with urinary factor Bb, present in fewer patients, is more surprising, as the alternative complement pathway has not been linked with DN in previous studies. However, deposition of C3b mediated by the classical or lectin pathways can activate the alternative pathway and amplify the global complement response by a feedback loop.32 It can thus be plausible to find some level of alternative pathway fragments in patients with DN who show evidence of complement activation. We found the greatest rate of progression when both factor Bb and C4a were present, supporting this hypothesis.
A possible limitation of our study is that we did not have measurements of plasma complement biomarkers, and it was thus not possible to address if urinary levels simply represent overflow of plasma levels. However, recent studies showed that there was no significant correlation between plasma levels of complement components, proteinuria, and GFR in DN,22 as well as in patients with autoimmune glomerulonephritis.29, 31 Nevertheless, because we know that plasma levels of sC5b-9 are increased in patients with DN, urinary excretion due to change in glomerular permeability cannot be excluded. sC5b-9 is a large molecule (approximately 1000 kDa) that is not filtered by the normal glomerulus29 and healthy individuals have undetectable urinary levels.30, 31 To further address this, we compared urinary levels of sC5b-9 of patients with DN with other immunologic glomerulonephritis, in which a clinically meaningful role of complement activation exists and can be the target of therapy.33 Our results demonstrated that autoimmune GN excretes sC5b-9 at levels comparable to DN. This supports that increased MAC excretion in DN is not solely caused by a passive filtration of plasma sC5b-9 in the setting of higher glomerular permeability, but is locally expressed and implicated in the pathogenesis of DN, at least in a subgroup of patients. It is also possible that certain individuals in the DN cohort unknowingly had an underlying autoimmune GN. Such misdiagnosis of DN is inherent to most diabetic studies in which a renal biopsy is not mandatory.6, 7, 34, 35
Other limitations should be outlined. We only had 2 individuals with type 1 diabetes and 80% of the cohort were male, reducing the generalizability of the study. We must also underline that most individuals in the DN cohort corresponded to the Kidney Disease: Improving Global Outcomes chronic kidney disease categories G3b/G4-A3 and that the results presented here are not necessarily applicable to earlier stages of the disease.36 Finally, the small sample could not allow us to adjust for other important risk factors of progression in DN (e.g., among others, blood pressure control and antihypertensive drugs, HbA1c, type of diabetes treatment).
Future studies are needed to validate that urinary complement fragments are predictive of outcomes in the clinical setting, and not a mere reflection of the level of proteinuria. Of interest, sC5b-9 should be tested in earlier stages of DN with lower proteinuria and preserved GFR to see whether its expression still exists. If such findings are confirmed, we should then contemplate whether inhibition of the complement pathways with newly developed agents should be investigated in DN, as done in animal models. Urinary sC5b-9 measurements could then help us determine which patients might benefit from this approach.
Disclosure
All the authors declared no competing interests. A small part of the results pertaining to measurements of monocyte chemoattractant protein-1 and transforming growth factor-β1 were published in Diabetes Research and Clinical Practice in 2013.15
Acknowledgments
This study was partly supported by grants from the Fonds de la recherche du Québec–Santé (#14395) and from the Fondation de l’Hôpital du Sacré-Coeur de Montréal.
Author Contributions
Conception and design: FM, VP, SB, ST. Manuscript drafting: KP, AB, HC, SB, ST. Analyses: KP, AB, ML, ST. The corresponding author (ST) had full access to the data in the study and final responsibility for the decision to submit for publication.
References
- 1.Wada J., Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond) 2013;124:139–152. doi: 10.1042/CS20120198. [DOI] [PubMed] [Google Scholar]
- 2.Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol. 2017;13:311–318. doi: 10.1038/nrneph.2017.31. [DOI] [PubMed] [Google Scholar]
- 3.Flyvbjerg A. Diabetic angiopathy, the complement system and the tumor necrosis factor superfamily. Nat Rev Endocrinol. 2010;6:94–101. doi: 10.1038/nrendo.2009.266. [DOI] [PubMed] [Google Scholar]
- 4.Hovind P., Hansen T.K., Tarnow L. Mannose-binding lectin as a predictor of microalbuminuria in type 1 diabetes:an inception cohort study. Diabetes. 2005;54:1523–1527. doi: 10.2337/diabetes.54.5.1523. [DOI] [PubMed] [Google Scholar]
- 5.Hansen T.K., Thiel S., Knudsen S.T. Elevated levels of mannan-binding lectin in patients with type 1 diabetes. J Endocrinol Metab. 2003;88:4857–4861. doi: 10.1210/jc.2003-030742. [DOI] [PubMed] [Google Scholar]
- 6.Ostergaard J.A., Thiel S., Lajer M. Increased all-cause mortality in patients with type 1 diabetes and high-expression mannan-binding lectin genotypes:a 12-year follow-up study. Diabetes Care. 2015;38:1898–1903. doi: 10.2337/dc15-0851. [DOI] [PubMed] [Google Scholar]
- 7.Hansen T.K., Gall M.A., Tarnow L. Mannose-binding lectin and mortality in type 2 diabetes. Arch Intern Med. 2006;166:2007–2013. doi: 10.1001/archinte.166.18.2007. [DOI] [PubMed] [Google Scholar]
- 8.Qin X., Goldfine A., Krumrei N. Glycation inactivation of the complement regulatory protein CD59: a possible role in the pathogenesis of the vascular complications of human diabetes. Diabetes. 2004;53:2653–2661. doi: 10.2337/diabetes.53.10.2653. [DOI] [PubMed] [Google Scholar]
- 9.Chikazawa M., Shibata T., Hatasa Y. Identification of C1q as a binding protein for advanced glycation end products. Biochemistry. 2016;55:435–446. doi: 10.1021/acs.biochem.5b00777. [DOI] [PubMed] [Google Scholar]
- 10.Bus P., Chua J.S., Klessens C.Q.F. Complement activation in patients with diabetic nephropathy. Kidney Int Rep. 2018;3:302–313. doi: 10.1016/j.ekir.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tervaert T.W., Mooyaart A.L., Amann K. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 12.Yiu W.H., Li R.X., Wong D.W.L. Complement C5a inhibition moderates lipid metabolism and reduces tubulointerstitial fibrosis in diabetic nephropathy. Nephrol Dial Transplant. 2018;33:1323–1332. doi: 10.1093/ndt/gfx336. [DOI] [PubMed] [Google Scholar]
- 13.Gnudi L., Coward R.J.M., Long D.A. Diabetic nephropathy: perspective on novel molecular mechanisms. Trends Endocrinol Metab. 2016;27:820–830. doi: 10.1016/j.tem.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Nosadini R., Velussi M., Brocco E. Course of renal function in type 2 diabetic patients with abnormalities of albumin excretion rate. Diabetes. 2000;49:476–484. doi: 10.2337/diabetes.49.3.476. [DOI] [PubMed] [Google Scholar]
- 15.Verhave J.C., Bouchard J., Goupil R. Clinical value of inflammatory urinary biomarkers in overt diabetic nephropathy:a prospective study. Diabetes Res Clin Pract. 2013;101:333–340. doi: 10.1016/j.diabres.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Nadkarni G.N., Rao V., Ismail-Beigi F. Association of urinary biomarkers of inflammation, injury, and fibrosis with renal function decline: the ACCORD trial. Clin J Am Soc Nephrol. 2016;11:1343–1352. doi: 10.2215/CJN.12051115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Camilla R., Brachemi S., Pichette V. Urinary monocyte chemotactic protein 1:marker of renal function decline in diabetic and nondiabetic proteinuric renal disease. J Nephrol. 2011;24:60–67. doi: 10.5301/jn.2010.1458. [DOI] [PubMed] [Google Scholar]
- 18.American Diabetes Association 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2018. Diabetes Care. 2018;41:S13–S27. doi: 10.2337/dc18-S002. [DOI] [PubMed] [Google Scholar]
- 19.Gross J.L., de Azevedo M.J., Silveiro S.P. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care. 2005;28:164–176. doi: 10.2337/diacare.28.1.164. [DOI] [PubMed] [Google Scholar]
- 20.Hsu S.I., Couser W.G. Chronic progression of tubulointerstitial damage in proteinuric renal disease is mediated by complement activation:a therapeutic role for complement inhibitors? J Am Soc Nephrol. 2003;14:S186–S191. doi: 10.1097/01.asn.0000070032.58017.20. [DOI] [PubMed] [Google Scholar]
- 21.Zhou W., Marsh J.E., Sacks S.H. Intrarenal synthesis of complement. Kidney Int. 2001;59:1227–1235. doi: 10.1046/j.1523-1755.2001.0590041227.x. [DOI] [PubMed] [Google Scholar]
- 22.Li X.Q., Chang D.Y., Chen M. Complement activation in patients with diabetic nephropathy [e-pub ahead of print]. Diabetes Metab. accessed December 2018. [DOI] [PubMed]
- 23.Nangaku M., Pippin J., Couser W.G. Complement membrane attack complex (C5b-9) mediates interstitial disease in experimental nephrotic syndrome. J Am Soc Nephrol. 1999;10:2323–2331. doi: 10.1681/ASN.V10112323. [DOI] [PubMed] [Google Scholar]
- 24.Wenzel U., Turner J.E., Krebs C. Immune mechanisms in arterial hypertension. J Am Soc Nephrol. 2016;27:677–686. doi: 10.1681/ASN.2015050562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wenzel U.O., Bode M., Kohl J. A pathogenic role of complement in arterial hypertension and hypertensive end organ damage. Am J Physiol Heart Circ Physiol. 2017;312:H349–H354. doi: 10.1152/ajpheart.00759.2016. [DOI] [PubMed] [Google Scholar]
- 26.Zhang C., Li Y., Wang C. Antagonist of C5aR prevents cardiac remodeling in angiotensin II-induced hypertension. Am J Hypertens. 2014;27:857–864. doi: 10.1093/ajh/hpt274. [DOI] [PubMed] [Google Scholar]
- 27.Wang H., Vinnikov I., Shahzad K. The lectin-like domain of thrombomodulin ameliorates diabetic glomerulopathy via complement inhibition. Thromb Haemost. 2012;108:1141–1153. doi: 10.1160/TH12-07-0460. [DOI] [PubMed] [Google Scholar]
- 28.Fujita T., Ohi H., Komatsu K. Complement activation accelerates glomerular injury in diabetic rats. Nephron. 1999;81:208–214. doi: 10.1159/000045278. [DOI] [PubMed] [Google Scholar]
- 29.Ogrodowski J.L., Hebert L.A., Sedmak D. Measurement of SC5b-9 in urine in patients with the nephrotic syndrome. Kidney Int. 1991;40:1141–1147. doi: 10.1038/ki.1991.326. [DOI] [PubMed] [Google Scholar]
- 30.Lehto T., Honkanen E., Teppo A.M. Urinary excretion of protectin (CD59), complement SC5b-9 and cytokines in membranous glomerulonephritis. Kidney Int. 1995;47:1403–1411. doi: 10.1038/ki.1995.197. [DOI] [PubMed] [Google Scholar]
- 31.Morita Y., Ikeguchi H., Nakamura J. Complement activation products in the urine from proteinuric patients. J Am Soc Nephrol. 2000;11:700–707. doi: 10.1681/ASN.V114700. [DOI] [PubMed] [Google Scholar]
- 32.Noris M., Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33:479–492. doi: 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jayne D.R.W., Bruchfeld A.N., Harper L. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J Am Soc Nephrol. 2017;28:2756–2767. doi: 10.1681/ASN.2016111179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wanner C., Heerspink H.J.L., Zinman B. Empagliflozin and kidney function decline in patients with type 2 diabetes: a slope analysis from the EMPA-REG OUTCOME trial. J Am Soc Nephrol. 2018;29:2755–2769. doi: 10.1681/ASN.2018010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saraheimo M., Forsblom C., Hansen T.K. Increased levels of mannan-binding lectin in type 1 diabetic patients with incipient and overt nephropathy. Diabetologia. 2005;48:198–202. doi: 10.1007/s00125-004-1594-1. [DOI] [PubMed] [Google Scholar]
- 36.Levin A., Stevens P.E. Summary of KDIGO. 2012 CKD Guideline:behind the scenes, need for guidance, and a framework for moving forward. Kidney Int. 2014;85:49–61. doi: 10.1038/ki.2013.444. [DOI] [PubMed] [Google Scholar]