

Introduction
CYP24A1 encodes 24-hydroxylase, which metabolizes 1,25-OH vitamin D. Inactivating CYP24A1 pathogenic variants were first identified as causing idiopathic infantile hypercalcemia in 2011.1 More recently, pathogenic variants in this gene have been described in adults with hypercalcemia and nephrolithiasis. Pregnancy appears to exacerbate hypercalcemia due to increased synthesis of 1,25OH Vitamin D. Case reports describe 3 women with CYP24A1 pathogenic variants, who demonstrated worsening hypercalcemia and obstetric complications.2, 3, 4 Management strategies for this condition during pregnancy, particularly the use of calcitonin, have not been described.
Case Presentation
A 32-year-old woman, G2P1, with known homozygous CYP24A1 pathogenic variants and stage 1 chronic kidney disease due to nephrocalcinosis, presented at 14 weeks’ gestation with recurrent hypercalcemia (ionized Ca 1.44 mmol/l [1.13−1.32]) and mild acute on chronic kidney injury (creatinine 82 μmol/l [nonpregnant reference range, 44−80], 0.9 mg/dl, urea 6.8 mmol/l [2.6−6.7]).
Her first pregnancy, 5 years prior, has been previously described5; it was complicated by antenatal diagnosis of fetal renal tract dilatation and polyhydramnios, term preeclampsia, postpartum maternal acute kidney injury, and severe maternal hypercalcemia. Neonatal hypercalcemia (exacerbated by ergocalciferol loading) prompted CYP24A1 sequencing, which identified 2 variants in the infant (child 1): NM_000782.4: c. 427_429del, p.Glu143del, and c.1186C>T, p.Arg396Trp. Subsequent maternal testing demonstrated the c. 1186C>T, p.Arg396Trp pathogenic variant in a homozygous state, consistent with affected status, and paternal testing revealed the c. 427_429del, p.Glu143del mutation, consistent with carrier status. All variants have been previously described in the literature and classified as pathogenic.1 Over months, functional renal tract obstruction in child 1 resolved; however, nephrocalcinosis developed and has persisted.
The mother was lost to endocrine and nephrology follow up in the intergravid period and re-presented without prepregnancy genetic or medical assessment or counseling.
The genetic status of the second fetus was unknown for the duration of the pregnancy, and could not, therefore, influence maternal management.
Investigations during the second pregnancy early in the second trimester demonstrated hypercalcemia with parathyroid hormone suppression (<0.6 pmol/l), elevated serum 1,25-OH vitamin D (304 pmol/l), and low serum phosphate (0.77 mmol/l) and potassium (3.4 mmol/l). Mild hypercalciuria (urinary Ca/Cr ratio 0.76 mmol/mmol) was documented at 23 weeks prior to commencement of calcitonin treatment.
Hypercalcemia was initially managed with dietary calcium restriction, high oral fluid intake, and 2 short admissions for i.v. hydration, oral corticosteroids (prednisolone 10−25 mg/d), and potassium citrate. Cyclical calcitonin was commenced at 23 weeks due to a subsequent consistent increase in serum calcium (ionized calcium 1.5 mmol/l) despite the above interventions. Calcitonin was self-administered subcutaneously 100 iU BD 2 days on and 3 days off, and serum calcium was closely monitored. Doses of calcitonin were not escalated over the pregnancy. The patient developed hypertension in the third trimester, requiring treatment with labetolol. At 38+4 weeks, worsening renal function was noted. A healthy male neonate was delivered at 38+5 weeks gestation by elective repeat caesarean section. The patient elected not to breastfeed.
Worsening of maternal renal function was noted postpartum, associated with an exacerbation of hypercalcemia (serum ionized calcium peaked at 1.71 mmol/l) (Figure 1), requiring increased doses of calcitonin, up to 200 IU 3 times daily for several days. Serum calcium normalized in the absence of breastfeeding by 2 weeks postpartum. A rapid fall in serum 1,25-OH vitamin D postpartum occurred. Renal function slowly improved, but remained above prepregnancy baseline (Cr 87 μmol/l, 0.98 mg/dl at 15 months postpartum). Ongoing follow-up in the 18 months postpartum demonstrated normal serum calcium (corrected Ca 2.39−2.56 mmol/l) and other electrolytes, mild persisting elevation of protein:creatinine ratio (55−106 mg/mmol), and persisting medullary nephrocalcinosis on ultrasound. Hypertension treatment was no longer required by 4 months postpartum. The patient elected not to pursue further pregnancies.
Figure 1.

Maternal serum calcium, parathyroid hormone (PTH), and 1,25-hydroxyvitamin D during the peripartum period. Date of delivery is indicated with red text.
No acute management of neonatal electrolyte disturbance was required in the first week of life (child 2). High normal serum calcium prompted presumptive management with low-calcium formula and avoidance of vitamin D supplementation. Child 2 re-presented at 3 months of age with symptomatic hypocalcemia while on low-calcium formula (iCa 0.76 mmol/l), prompting treatment with ergocalciferol 50,000 IU and normal formula. He was subsequently managed with variable ratios of normal and low-calcium formula, titrated to serum and urinary calcium. At 8 months of age, child 2 remained on 100% low-calcium formula, with normal serum calcium and urine calcium:creatinine ratio. CYP24A1 sequencing of child 2 (result available at 9 months of age) identified the same 2 variants as his sibling: NM_000782.4: c. 427_429del, p.Glu143del, and c.1186C>T, p.Arg396Trp (Figure 2). At the time of writing, at 2 years of age, child 2 was on a low-calcium diet, serum calcium was 2.6 mmol/l, and urine calcium:creatinine ratio was 0.5 (normal range, <1.4). No renal tract abnormalities have been identified at any stage despite regular ultrasound screening.
Figure 2.
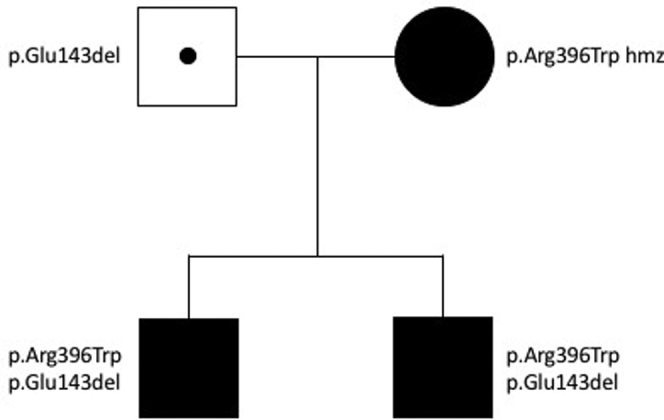
Genogram.
Discussion
This case presentation is the first in the literature to describe the diagnosis of both maternal and neonatal hypercalcemia due to CYP24A1 pathogenic variants, along with differing renal outcomes between 2 genetically identical siblings modified by antenatal treatment of maternal hypercalcemia and neonatal vitamin D and calcium intake.
Child 1 developed fetal polyuria and hydronephrosis, symptomatic hypercalcemia, and hypercalciuria with persisting nephrocalcinosis. This occurred in the context of bolus vitamin D supplementation. His younger brother (child 2) had a normal renal tract at birth and no nephrocalcinosis, with normal serum and urine calcium at 2 years of age.
There were 2 major identifiable differences between the management of child 1 and 2 that we hypothesize account for their differing outcomes. First, gestational hypercalcemia was managed aggressively during the second pregnancy. Second, neonatal vitamin D supplementation was avoided in child 2, and low-calcium formula was used from birth.
During normal pregnancy, maternal calcium metabolism is altered, to ensure adequate calcium delivery to the developing fetus. This is primarily achieved through increased intestinal absorption of calcium. A sharp increase in 1,25-OH vitamin D occurs, as demonstrated with data from normal pregnant women in Saudi Arabia and Denmark.6, 7 This is thought to be mediated via upregulation of renal 1-α-hydroxylase, independent of parathyroid hormone (PTH), as well as possible placental synthesis of 1-α-hydroxylase.8 1,25-OH vitamin D falls rapidly after delivery. Postpartum, an increase in parathyroid hormone−related peptide (PTHrP) mobilizes calcium from bone and increases renal tubular reabsorption to support breastfeeding.8
Maternal deficiency of 24-hydroxylase caused the anticipated increase in 1,25-OH vitamin D to be grossly exaggerated, causing gestational hypercalcemia and PTH suppression. We also documented hypophosphatemia and hypomagnesemia. Hypophosphatemia is likely due to the upregulation of fibroblast growth factor-23 by 1,25-OH vitamin D, increasing renal phosphate wasting.9 Hypomagnesemia is likely secondary to hypercalcemia and PTH suppression. As previously reported, a postpartum rise in serum calcium was seen (Figure 1), which was thought to be due to the abrupt cessation of calcium transport to the fetus.
The treatment of maternal hypercalcemia presents unique challenges. Many medications are relatively contraindicated in pregnancy (Table 1).
Table 1.
Management strategies for hypercalcemia, including safety in pregnancy
| Management strategy | Medications | Pregnancy category |
|---|---|---|
| Forced diuresis | Frusemide | C Theoretical risk hypovolemia, uteroplacental insufficiency |
| Reduced GI absorption | Prednisolone Cellulose phosphate |
A (consider alternative in T1) Not available in Australia. Not in database |
| Reduction of bone turnover | Bisphosphonate, i.e., Pamidronate | B3 Bilateral talipes equinovarus, LBW, shortened GA, spontaneous abortion, transient hypocalcemia in newborn with exposure during pregnancy. Concern regarding interference with fetal bone modeling and development, availability of calcium, and maternal hypocalcemia if used in T3 |
| CYP450 inhibition | Fluconazole Ketoconazole |
D Higher continuous daily doses associated with congenital malformations and increased risk of miscarriage B3 limited data |
| Calcitonin | B2 limited data Does not cross placenta in animal studies |
GA, gestational age; LBW, low birth weight; T, trimester.
The current literature consists of 2 case reports in which the diagnosis of hypercalcemia secondary to maternal CYP24A1 pathogenic variants was known antenatally.3, 4 Shah et al. described conservative measures to manage hypercalcemia.3 Woods et al. described cellulose phosphate to bind intestinal calcium and to prevent absorption.4 Cellulose phosphate was not available in Australia. CYP450 inhibitors have been used in this condition outside of pregnancy. However, the safety of these drugs in pregnancy remains a concern (Table 1). Prednisolone reduces calcium absorption from the gastrointestinal tract and has also been used in this condition. Given that prednisolone is considered safe in pregnancy, this was our first therapeutic option, following the instigation of dietary changes, oral hyperhydration, and cessation of vitamin D and calcium supplements. A decrease in serum calcium was noted. Calcitonin was the next therapeutic option due to documented safety in pregnancy. No tachyphylaxis occurred with our dosing regimen, and dose escalation was not required.
There is no clear consensus on an appropriate target for serum calcium during pregnancy. We targeted a serum ionized calcium of 1.3−1.35 mmol/l to maintain a physiologic intrauterine environment and to avoid complications. Multiple maternal and fetal complications have been described that were associated with maternal CYP24A1 pathogenic variants, including fetal death in utero, intrauterine growth restriction, and hypertensive disorders of pregnancy.2, 4,S1 Although not conclusive, data from the hyperparathyroid population in pregnancy also associated hypercalcemia in pregnancy with adverse outcomes.S2 Neonatal hyper- and hypocalcemia were both possible, as fetal genetic status remained unknown during pregnancy.
The second key difference was the management of child 2 postpartum. Vitamin D supplementation was avoided, and low-calcium formula was prescribed. Children with CYP24A1 pathogenic variants have classically presented following consumption of vitamin D−fortified products,S3 and regulation of vitamin D and calcium intake remain key in managing this condition. Complications of hypercalcemia were avoided in child 2. Similar outcomes have been described in siblings previously.S1 However, in this case, hypocalcemia occurred, demonstrating that close monitoring and titration of intake is essential to maintain calcium homeostasis.
Uniquely in the literature, we were able to closely document the key serum biochemistry of a woman with CYP24A1 pathogenic variants during pregnancy and the postpartum period (Figure 1). However, we found urinary calcium excretion inconsistent with that described in most of the existing literature,S3 including in pregnancy.2, 3 Our patient initially had mildly elevated urinary calcium. Subsequent monitoring of urinary calcium including postpartum revealed normal values, contrary to the anticipated effects of hypercalcemia, hyperfiltration during pregnancy, and increased urinary calcium excretion caused by exogenous calcitonin. We postulate that possible contributing factors to this finding are as follows: (i) underlying chronic kidney disease causing reduced glomerular filtration of calcium, possibly exacerbated by hypercalcemia-induced acute kidney injury and evolving hypertensive disorder of pregnancyS4; (ii) very high levels of 1,25-OH vitamin D increasing renal tubular calcium reabsorption, both directly, in the distal convoluted tubule, and via its upregulation of KlothoS5; and (iii) adherence to a strict low-calcium diet, which increases renal calcium reabsorption, independent of vitamin D.S6 It is also important to note that normocalciuria has been reported in other hypercalcemic patients with CYP24A1 pathogenic variants outside of pregnancy.S7 This finding has not been explained, and there are no data to support the coexistence of CYP24A1 pathogenic variants and inherited or acquired tubular disorders. Testing for renal tubular dysfunction was not formally evaluated in our patient, but serum bicarbonate and chloride levels were within normal limits.
Despite relatively aggressive calcium-lowering therapy, our patient had persisting renal dysfunction, likely due to the renal tubular effects of prolonged hypercalcemia in pregnancy. The potential impact of further future pregnancy is concerning. Prepregnancy planning with consideration of renal biopsy to assist with risk stratification would be considered.
This condition is rare. Current international guidelines recommend universal vitamin D supplementation in pregnant women and infants to avoid complications of deficiency.S8 Should these guidelines be widely adopted, there is concern that more people with CYP24A1 pathogenic variants are likely to become symptomatic. A recent randomized controlled trial of vitamin D supplementation of 1300 Bangladeshi women in pregnancy and postpartumS9 did not demonstrate any significant difference in adverse outcomes; however, genetic and environmental factors may limit generalizability.
In conclusion, this case demonstrates that antenatal hypercalcemia that is resistant to standard therapies can be safely and effectively managed with cyclical calcitonin to achieve a term delivery and to minimize the risk of maternal, fetal, and neonatal complications in the context of CYP24A1 pathogenic variants. Close monitoring and manipulation of calcium and vitamin D intake is also essential in ensuring optimal maternal and neonatal outcomes (see Table 2 for teaching points).
Table 2.
Teaching points
| CYP24A1 pathogenic variants are a cause of vitamin D−mediated hypercalcemia, which is exacerbated by pregnancy, particularly following delivery |
| Maternal and neonatal complications have been described, including renal dysfunction, hypertensive disorders of pregnancy and preeclampsia, preterm delivery, and fetal growth restriction and death |
| Regulation of vitamin D and calcium intake is key |
| Treatment options in pregnancy are limited due to safety concerns Calcitonin was used in this case and appears safe and effective |
| Management of gestational hypercalcemia reduced complications, particularly in the offspring |
Disclosure
All the authors declared no competing interests.
Acknowledgments
The authors thank Dr. Hayden Waterham, Obstetrician, A/Professor Christine Rodda, Paediatric Endocrinologist, and Dr. Niroj Obeyesekere, Nephrologist, for their input into patient care and the writing of this report.
Acknowledgments
Declaration
Ethics approval and consent to participate: Local ethics committee ruled no formal ethics committee approval required. Verbal and written consent was obtained from the patient and/or guardian.
Consent for publication: Verbal and written consent was obtained from the patient and/or guardian for publication.
Availability of data and materials: Further clinical data of this case are available from the corresponding author upon reasonable request.
This project was not supported by any funding.
Author Contributions
LM wrote the manuscript, supervised by AC. LM, AC, and CH were responsible for maternal management. BM, CQ, and ZS followed the pediatric management. All authors had a clinical role in managing the case/providing medical care, reviewed the paper, and revised it critically for important intellectual content. All authors read and approved the final draft.
Footnotes
Supplementary References.
Supplementary material is linked to the online version of the paper at www.kireports.org.
Supplementary Material
References
- 1.Schlingmann K.P., Kaufmann M., Weber S. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med. 2011;365:410–421. doi: 10.1056/NEJMoa1103864. [DOI] [PubMed] [Google Scholar]
- 2.Dinour D., Davidovits M., Aviner S. Maternal and infantile hypercalcemia caused by vitamin-D-hydroxylase mutations and vitamin D intake. Pediatr Nephrol. 2015;30:145–152. doi: 10.1007/s00467-014-2889-1. [DOI] [PubMed] [Google Scholar]
- 3.Shah A.D., Hsiao E.C., O'Donnell B. Maternal hypercalcemia due to failure of 1,25-dihydroxyvitamin-D3 catabolism in a patient with CYP24A1 mutations. J Clin Endocrinol Metab. 2015;100:2832–2836. doi: 10.1210/jc.2015-1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woods G.N., Saitman A., Gao H. A young woman with recurrent gestational hypercalcemia and acute pancreatitis due to CYP24A1 deficiency. J Bone Miner Res. 2016;31:1841–1844. doi: 10.1002/jbmr.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McBride L., Crosthwaite A., Houlihan C. Rare cause of maternal and neonatal hypercalcaemia. J Paediatr Child Health. 2019;55:232–235. doi: 10.1111/jpc.14219. [DOI] [PubMed] [Google Scholar]
- 6.Ardawi M.S., Nasrat H.A., BA'Aqueel H.S. Calcium-regulating hormones and parathyroid hormone-related peptide in normal human pregnancy and postpartum: a longitudinal study. Eur J Endocrinol. 1997;137:402–409. doi: 10.1530/eje.0.1370402. [DOI] [PubMed] [Google Scholar]
- 7.Moller U.K., Streym S., Mosekilde L. Changes in calcitropic hormones, bone markers and insulin-like growth factor I (IGF-I) during pregnancy and postpartum: a controlled cohort study. Osteoporos Int. 2013;24:1307–1320. doi: 10.1007/s00198-012-2062-2. [DOI] [PubMed] [Google Scholar]
- 8.Kovacs C.S. Maternal mineral and bone metabolism during pregnancy, lactation, and post-weaning recovery. Physiol Rev. 2016;96:449–547. doi: 10.1152/physrev.00027.2015. [DOI] [PubMed] [Google Scholar]
- 9.Liu S., Tang W., Zhou J. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17:1305–1315. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.