

Abstract
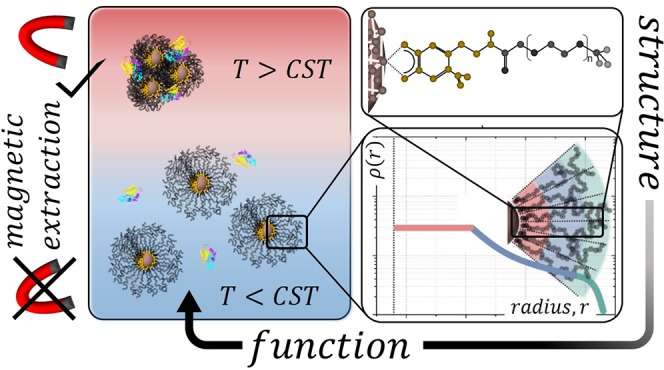
In this feature article, we summarize our recent work on understanding and controlling the thermal behavior of nanoparticles grafted with thermoresponsive polymer shells. Precision synthesis of monodisperse superparamagnetic iron oxide nanocrystals was combined with irreversible dense grafting of nitrodopamide-anchored thermoresponsive polymer chains. We provide an overview of how the dense and stable grafting of biomedically relevant polymers, including poly(ethylene glycol), poly(N-isopropylacrylamide), polysarcosin, and polyoxazolines, can be achieved. This platform has made it possible for us to demonstrate that the polymer brush geometry, as defined by the nanoparticle core and relative polymer brush size, determines the thermal transitions of the polymer brush. We furthermore summarize our work on how the polymer shell transitions and nanoparticle aggregation can be tuned. With the independent variation of the core and the shell, we can optimize and precisely control the thermally controlled solubility of our system. Finally, our feature article gives examples relevant to current and future applications. We show how the thermal response of the shell influences the nanoparticle performance in biological fluids and interactions with proteins and cells, also under purely magnetic actuation of the nanoparticles through the superparamagnetic iron oxide core.
Introduction
Research on so-called smart materials has developed into an important and expanding segment within the field of nanomaterials.1,2 A subgroup of smart materials of special importance for biotechnology and biomedicine includes magnetic nanoparticles with thermoresponsive solubility.3−5 Temperature is an easily applied external stimulus that can be used to change the quality or stability of a dispersion. In the case of superparamagnetic nanoparticles, this means a change in their magnetic properties such that they can be extracted from a dispersion and redispersed depending on the aggregation state. Thermoresponsive magnetic nanoparticles for biomedical applications are individually dispersed at low temperature, while the particles cluster above a certain temperature called the critical solution temperature, which will be further discussed below. Specific and nonspecific interactions of a nanomaterial also rely on the aggregation state of the nanomaterial and the solubility of functional groups on its surface; thus, they can be switched on and off upon temperature change for thermoresponsive dispersions of nanoparticles. Thermoresponsive colloidal smart materials therefore have received increasing attention in recent years.3,4,6 Obvious applications are triggered cell uptake as well as the extraction of biological molecules and cell separation;7 they can also be used for medical applications8,9 such as drug delivery,10−12 hyperthermia,13 and as contrast agents9,14,15 but also for catalysis16 and sensing.14,17 In all of these applications, a key concern should be the control of colloidal stability in a colloidally dense environment such as complex bodily fluids, even if the designed nanoparticle itself is dilute. In more applied work, the need to characterize the basis for this colloidal stability is not always appreciated and is often underestimated. If the application requires a dilute or complex medium and the colloidal stability is provided by polymers and dispersants, then purification to remove excess dispersants is required to correctly characterize the colloidal stability.9,18 Many other colloids, molecules, and ions (e.g., proteins and physiological concentrations of salts) will be present when nanoparticles are dispersed in a biological environment; they will exert a strong influence on the colloidal stability of the engineered nanomaterial, which also must be understood.
The most intuitive architecture that combines an easy and well-controlled variation of structure with tailored function consists of inorganic core nanoparticles with a grafted polymer shell (Figure 1).5,9 They superpose the properties of the core (e.g., magnetic) and the shell (controlling colloidal interactions) in a hierarchical structure with well-defined geometry.5 The core–shell structure not only provides an excellent platform to test ideas regarding functional nanoparticle design and interactions but also provides a straightforward way of linking and controlling the core function with shell colloidal and responsive interactions. In this feature article, we will describe some of our recent work on creating a library of core–shell magnetic iron oxide nanoparticles with reproducible and controlled architecture. We will then focus on our current understanding of how a thermoresponsive polymer shell can and must be tailored to the nanoparticle core to control the interactions of a dispersion of nanoparticles. We see this work as the basis for the further development of any application built on thermoresponsive colloidal dispersions, in particular within biomedicine and biotechnology.
Figure 1.

Schematic representation of a core–shell nanoparticle. The core functionality, such as drug encapsulation or a magnetic or plasmonic contrast enhancer, is protected by the polymer shell that provides colloidal stability. The shell is linked to the nanoparticle core by an anchor forming an irreversible chemical bond between the core and shell. Additional functionalities can be linked to the shell.
Providing a Defined Shell and Colloidal Stability to Iron Oxide Nanoparticles
The shell provides a key function in composite nanoparticles with an inorganic core. It must prevent and control aggregation. A polymer shell does so by providing an osmotically repulsive chemical potential to balance out van der Waals, electric double layer, and other long-range attractive interactions (e.g., magnetic dipolar interactions of magnetic cores).19,20 In particular, the van der Waals attractive interaction is long-ranged and strong enough for nanoparticles to provide a strong driving force for flocculation as well as modulate specific particle interactions even in the presence of a polymer shell.21 In addition to this, the polymer shell can provide a steric shield to prevent direct physical and chemical interactions with the core.20 To fulfill its purpose, the repulsion of the polymer brush must extend over the range of the attractive potentials,9 which requires sufficient solvation, flexibility, dense grafting, and high molecular weight of the polymer chains.20 The relative influence of the long-range van der Waals and electric double-layer interactions, which dominate within the DLVO framework that aids our understanding of colloidal interactions,22 can be estimated and compared to the shell extension by performing experiments that scale their contributions differently. For example, the screening of the electric double-layer interaction is strongly dependent on ion concentration. Whether the electric double-layer interaction is responsible for aggregating polymer-coated particles can therefore be investigated as a function of ionic strength or by direct measurements of particle binding to a charged interface as a function of ionic strength.21,23 Similarly, the van der Waals interaction can be investigated by varying the size of the core particle if the polymer shells of the various particles are identical and the double-layer interaction is screened by counterions.15,21,23 The repulsive interaction of a hydrated polymer shell (e.g., a grafted polymer brush) can either completely neutralize the attractive interactions or can partially suppress them. The latter leads to weak flocculation or increased probability of localization of nanoparticles close to the attracting interface.21,23 In bulk, interactions are studied by measuring the aggregation of nanoparticles using various scattering techniques (see below), while attractive interactions with an interface can be measured quantitatively using surface-sensitive biosensor techniques such as the quartz crystal microbalance23 or, to obtain the full interaction potential, by total-internal reflection microscopy.21
Grafted polymer chains are forced to stretch and overlap in a polymer brush. This is achieved by spacing the polymer grafting sites much closer than the radius of gyration, Rg, which defines the equilibrium size of the free chain.24 This requirement becomes even more challenging on the highly curved surface of a small nanoparticle, comparable in diameter to the radius of gyration of the polymer.25 Dense grafting is thus a high-energy state for the polymer chain, which requires a nonreversible bond to the nanoparticle surface to keep the out-of-equilibrium high concentration of stretched polymer at the surface from dispersing. We exemplified this requirement for iron oxide nanoparticle cores by using different anchors that are known to bind to iron oxide. It was demonstrated that only ligands that strongly coordinate to surface iron ions, such as nitrodopamide-functionalized polymer dispersants, can prevent nanoparticle aggregation upon the continuous removal of excess dispersants or an increase in temperature or ionic strength.18,26 That the stability of the anchor has to be investigated under relevant conditions was demonstrated by the fact that hydroxydopamide-poly(ethylene glycol) (hydroxydopamide-PEG)-stabilized nanoparticles could be dispersed for years at room temperature;27 however, they precipitated if filtered to remove excess dispersants in equilibrium with the weakly surface-bound hydroxydopamide-PEG.18 In contrast, nitrodopamide that provides a much stronger complex with the surface iron withstands not only stringent purification but also thermal actuation, high dilution, and the presence of other dispersants.12,18,28,29 To complicate matters further, the instability of oxide nanoparticle surfaces means that a too strongly complexing anchor can lead to dissolution of the nanoparticles, as was demonstrated by using mimosine-PEG.18
The role of a strongly binding anchor becomes even more important if a ligand, such as oleic acid or oleylamine, is already present on the nanoparticle surface after core synthesis. To produce iron oxide nanoparticles with the highest precision in size, shape, and control, thermal decomposition synthesis methods using organometallic precursors are applied that leave a strongly complexed shell of such ligands.30−32 When the synthesis method already provides a ligand shell, it is important to optimize the ligand replacement protocol to achieve a high grafting density of the preferred dispersant33,34 (i.e., a large number of polymer chains grafted per unit area of the core). This requires especially challenging protocols for nonpolar ligands33,35 but is also a time-consuming process to be optimized for hydrophilic polymer shells, for which the solubilities of the different grafting states vary greatly.28,36 It can also be sensitive to the history of the ligand-coated nanoparticle, such as the aging of oleic acid-capped iron oxide nanoparticles.37
It is imperative to test and achieve the purification of core–shell nanoparticles that correspond to the conditions of stability relevant for biomedical and biotechnological applications. We argue that this means allowing for full purification, dilution, and use under the physiological salt concentration and over a large range of temperature in water. Of these demands, achieving complete purification of excess dispersants after nanoparticle synthesis is also a very demanding and often underestimated task38 because a densely grafted nanoparticle shares almost all of its physicochemical properties with the free polymer. We recently investigated the pros and cons of different methods of purifying PEG-grafted iron oxide nanoparticles.28 Dialysis through high-molecular-weight (MW) dialysis membranes was shown to be an efficient but slow way to remove excess dispersants; it also risks destabilizing densely grafted nanoparticles possibly because of the long purification times and large osmotic stress on dense polymer brushes. When possible, as for PEG-grafted iron oxide nanoparticles, solvent extraction aided by magnetic decantation can be used to completely purify the particle dispersion much faster and gentler in large batches. The magnetic moment of the core is used in combination with the reduced solvent quality for the polymer brush that aggregates them to magnetically extract the nanoparticles faster than the free polymer.28 We have since exploited the effect of the combination of efficient ligand replacement, a strongly complexing anchor, and the purity of the final nanoparticle dispersion to investigate the influence of the polymer grafting density,27,39 polymer molecular weight,40 polymer topology,41 core size,15 and choice of polymer28,29,39,42 on the colloidal stability. This has also enabled the study of their influence on important properties in biomedical and biotechnological applications such as the relaxivity of iron oxide nanoparticle contrast agents,15 protein interactions,23,28,29,39,41,43 membrane interactions,23 and cell uptake.15,23,29 To briefly summarize the result of these numerous studies, a denser grafted shell of higher molecular weight and higher topology with an absolute requirement of stable anchor chemistry, in combination with a larger core (in the superparamagnetic iron oxide size range of ∼4–15 nm in diameter), will result in maximum colloidal stability, resistance to protein adsorption, and functionality for applications such as magnetic resonance imaging (relaxivity).
Thermoresponsive Core–Shell Nanoparticles
Thermoresponsive polymers add an interesting function to the core–shell nanoparticles discussed in the previous section because they allow the modulation of the colloidal stability and nanoparticle function in situ.3−5 This is possible because the solvation of the polymer shell determines the colloidal stability or aggregation state of core–shell particles dispersed in a solvent. The solvation state also determines the polymer shell conformation and thickness (overall particle size). In polar solvents, such as water, dispersible polymers often have a lower critical solution temperature (LCST).44 Above the LCST, the solubility of the polymer decreases drastically, as illustrated in Figure 2. Hydrogen bonding of water to the polymer to form a hydration shell lowers its entropy compared to that of bulk water. Upon heating, the higher entropy of bulk water is favored, leading to the dehydration of the polymer over a short temperature interval, which gives rise to LCST behavior of the polymer in water.
Figure 2.
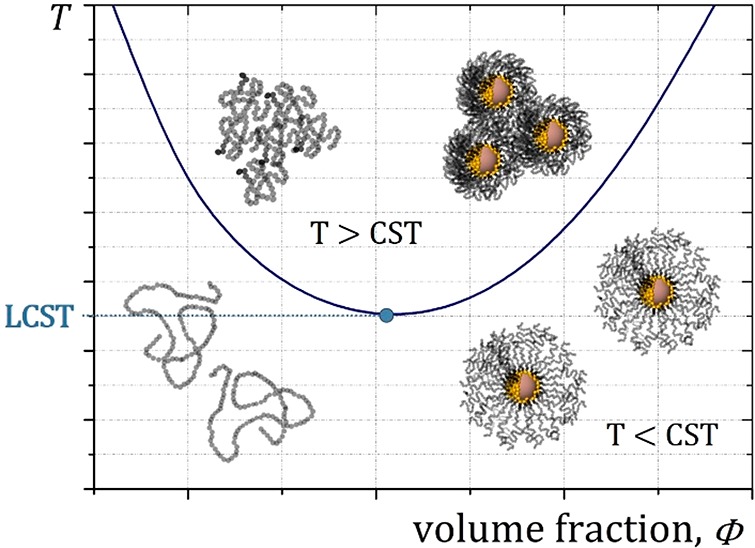
Schematic phase diagram of thermoresponsive polymers in water. Mostly, only a lower critical solution temperature (LCST) is observed for water-soluble polymers. The LCST is the lowest temperature in the phase diagram at which the polymer can lose solubility. At temperatures above the phase boundary (the critical solution temperature (CST)), the polymer loses solubility and can aggregate. This property can be transferred to core–shell nanoparticles grafted with thermoresponsive polymers and used to achieve a reduction in the core–shell nanoparticle size or aggregation of nanoparticles above the CST.
The simple schematic phase diagram for a polymer/solvent dispersion greatly increases in complexity for a nanostructured hybrid material such as core–shell nanoparticles. First, the polymer is already present in locally high density on the nanoparticle surface, and the interdigitation of polymer chains from opposing brushes is low. As the polymer solubility decreases, the brush dimensions and therefore the shell and overall hydrodynamic size of the nanoparticle decrease. Nevertheless, this thinner and denser shell can theoretically still provide colloidal stability, especially in dilute dispersions. However, it is also possible that nanoparticle aggregation occurs because of dehydration and reduction of the shell thickness (Figure 2). Despite the simple morphology of spherical core–shell nanoparticles, the act of confining the polymer at high concentration to a highly curved nanoparticle interface greatly affects the local phase behavior. It is crucial to know the phase diagram of the core–shell system under realistic conditions. The nanoparticles will fail in application if their thermoresponsive properties are optimized under ideal laboratory conditions. Many parameters have an influence on the critical solution temperature (CST) of a polymer, such as the concentration,36 the end group,45 the monomer composition,29 and the ionic strength of the aqueous surrounding.29,36 Intuitively, these parameters are affected locally by the core–shell nanoparticle morphology. In the past few years, we have investigated all of these parameters thoroughly using our platform of monodisperse iron oxide nanoparticles to which polymer brush shells can be grafted in a very controlled way. This has led to a number of insights into how the detailed structure of the polymer shell around the core greatly influences crucial parameters for applications, such as the transition temperature and extent of particle aggregation. Furthermore, these investigations have shown that dispersions of core–shell nanoparticles can be controlled to a much greater extent than previously shown by employing state-of-the-art synthesis methods for nanoparticles grafted with polymer brushes.
Difference between Measuring Polymer Desolvation and Core–Shell Nanoparticle Flocculation
Methodologically, we emphasize that one must distinguish between measuring the critical solution transition of the polymer and the thermally induced flocculation transition of the core–shell nanoparticle dispersion. We have settled on a combination of dynamic light scattering (DLS) and differential scanning calorimetry (DSC) to elucidate the details of how core–shell nanoparticle dispersions respond to an increase in temperature on the individual particle and global dispersion scales. DLS is used to study the flocculation of nanoparticles in a quantitative manner, while DSC is used to study the solvation transitions within the polymer shell (i.e., even on the submolecular level). These methods are much more sensitive to changes in particle size, aggregation state (DLS), and polymer solvation (DSC) than traditional methods for determining the CST of polymers, such as turbidimetry. It is important to have a consistent way of measuring the transitions of thermoresponsive polymers to compare results from different laboratories and experiments, as was discussed recently for the simpler case of turbidimetry in thermoresponsive polymer solutions.46
Dynamic light scattering is the most commonly used method to measure the CST of thermoresponsive core–shell nanoparticles as a result of its role as a standard technique to determine the colloidal size and aggregation in colloidal science.47 DLS determines the hydrodynamic size of colloids based on the Brownian motion of the colloids in suspension. Particles that are part of an aggregate diffuse together and therefore yield an effectively larger hydrodynamic diameter in a DLS measurement than the individual particles. Thermally induced aggregation can therefore be determined by DLS through observing an increase in the average particle size. When used in this way, DLS determines the critical flocculation temperature (CFT) at which the particles aggregate, rather than the solution transition of the polymer in the shell; this distinction is missing in most of the current experimental literature on thermoresponsive colloids. Already without considering the inner complexity of a polymer brush shell, the solution transition of the shell can lead to two different types of outcomes for the core–shell nanoparticle dispersion:
-
(1)
particles with a desolvated shell are still colloidally stable in the dispersion medium; this leads to a reduction in hydrodynamic diameter as the shell becomes thinner as a result of dehydration;
-
(2)
particles with dehydrated and collapsed shells aggregate; this leads to an increase in the measured average hydrodynamic diameter.
DLS can be used to determine the CST/CFT by analogy to turbidity measurements, but it does so more sensitively. The intensity of the scattered light is recorded, from which the scattering intensity correlation function and thereafter from the correlation times the Brownian diffusion coefficient are calculated. According to Rayleigh’s approximation, the intensity of the scattered light follows I ∝ R.6 Mie theory (applicable to very large nanoparticles or nanoparticle aggregates) predicts a more complicated relationship between the scattering intensity and size, which also depends on the scattering direction. However, the dependence of the intensity on the size of the scattering particle is also very strong in Mie theory.48 Thus, small changes in the aggregation state (increase in effective size of the scattering objects) can also lead to a huge increase in the scattering intensity. We therefore use the scattering intensity recorded by the DLS detector to identify more sensitively and precisely the CST/CFT of core–shell nanoparticles than what we can do from observing the change in average size calculated on the basis of the change in the diffusion coefficient.29,43,49
The thermal response of a solvated polymer is from a molecular perspective defined not by macroscopically observable aggregation but by the increased demixing of polymer segments and solvent as the temperature increases to above the CST. The CST phase transition of polymers in water is caused by a cleavage of intermolecular (polymer–water) hydrogen bonds due to the favorable entropy of water in the bulk phase. The breaking of bonds amounts to a change in enthalpy, which is exactly what is directly measured by DSC. DSC allows a much more sensitive observation of the desolvation of the shell than observing it through a change in size through DLS, as is demonstrated by comparing the results in refs (29), (36), and (50). Using DSC, the heat capacity as a function of temperature is recorded, which usually leads to a broader transition than observed by DLS, because the specific heat is sensitive to the local environment of different parts of the polymer shell. The enthalpy of the transition can be obtained by integrating the specific heat with temperature. Therefore, a measure of the number of broken polymer–water hydrogen bonds per monomer can be approximated.29,43 Below, we want to demonstrate an important point for thermoresponsive core–shell nanoparticles, which is that by DSC it is possible to study the CST of the polymer as it varies within the shell without a global change in the aggregation state taking place.
Effect of Grafting Polymers to an Inorganic Core
Grafting a polymer to a surface changes its transition temperature. This is easy to rationalize from the well-known fact that a change in the end group of a polymer has an outsized influence on the CST.51,52 Anchoring one end of a polymer to a surface is a drastic version of that change, leading to a conformational rearrangement from free coil to a so-called mushroom conformation (Figure 3Aii).53,54 Because the anchored end-segment essentially is not soluble anymore, a decrease in CST is observed. This has been demonstrated by many reports on nanoparticles grafted with thermoresponsive polymers for which a high grafting density in the brush regime could not be reached.50 The density of the polymer segments is much higher in a polymer brush than in the free coil (cf. Figure 3Aiii). A polymer brush is therefore also expected to have a lower CST than free chains. We observed a large change in the CST of poly(2-ethyl-2-oxazoline) (PEtOxA)29,55 and poly(2-isopropyl-2-oxazoline) (PiPrOxA)29,36,50,55 grafted to iron oxide nanoparticles. Most likely, this effect is dependent on the grafting density. However, this remains to be conclusively investigated because a dense brush is formed only at grafting densities approaching 1 chain nm–2 for realistic polymer chain sizes, and there are few examples of higher grafting densities being achieved for thermoresponsive polymers. Tentatively, a very large decrease in the CST was observed for PEG grafted to iron oxide cores at grafting densities of ∼3 chains nm–2 through melt grafting39 when investigated by small-angle X-ray scattering.56 How much the CST is expected to shift simply due to grafting as a function of grafting density remains to be investigated. As will be discussed below, on the basis of our research it might not be meaningful to treat this question in isolation without further considering the effect of varying core and polymer chain sizes.
Figure 3.

(A) Schematic comparison of the size and conformation of (i) free polymer chains, (ii) surface-grafted polymer chains in the mushroom conformation, and (iii) polymer chains grafted on a planar surface in the brush conformation, with Rg being the radius of gyration, s the distance between grafting sites, and L the brush height. (B) Schematic of the segment density profile expected for a spherical core grafted with a dense brush of linear polymer chains according to the Daoud–Cotton model.57 (C) Example of the difference in fitting a parabolic or a Daoud–Cotton segment profile to the scattering data of iron oxide nanoparticles densely grafted with poly(ethylene glycol) chains. Reproduced from Grünewald, T. A., et al. Core–Shell Structure of Monodisperse Poly(ethylene glycol)-Grafted Iron Oxide Nanoparticles Studied by Small-Angle X-ray Scattering. Chem. Mater.2015, 27, 4763–4771.56 Copyright 2015 American Chemical Society.
Effect of Core Size on the Thermal Response of a Grafted Polymer Shell
Grafting a polymer chain to a surface matters because of the imposed change in conformation and increased local concentration. Thus, a polymer grafted to a nanoparticle is also sensitive to the geometry imposed by the surface (e.g., a spherical nanoparticle core). The curvature of the surface becomes significant as the sizes of the core and polymer coils approach each other. A polymer brush tethered to a planar surface has a nearly uniform segment density through the brush (i.e., every monomer experiences a similar environment (cf. Figure 3Aiii)). The volume available for the polymer to expand as it stretches into a brush is conical at a curved (e.g., spherical) surface (cf. Figure 3B); therefore, a segment density profile such as a star polymer is expected.57 Careful investigations of the segment density profile have been performed by Zhulina and Borisov.25 Similar to star polymers, one can roughly find three different polymer density decay regimes within such a brush, schematically described in Figure 3B.25,57 Using small-angle X-ray scattering, we demonstrated that for iron oxide nanoparticles with extremely high grafting densities of PEG only a segment density profile with a dense inner core region in accordance with this model fits the scattering curves of the shell well (Figure 3C).56 Thus, the local environment of the monomer segments changes drastically and at different rates within the shell as a function of distance from the core. A concentration- and conformation-dependent property such as the CST should be sensitive to this.
The curvature of the core controls how quickly the segment density decays radially. It therefore also influences the ratio of a polymer that experiences different decay rates within the shell. Thus, we grafted polymer brushes with identical molecular weights and grafting densities to highly monodisperse cores for which the diameter was varied stepwise.50 Interestingly, a pronounced effect of core size was observed by DSC as reproduced in Figure 4.
Figure 4.

Influence of core size (curvature) on the CST of the thermoresponsive PiPrOxA brush shell as measured by DSC. Colors from light orange to red indicate increasing particle concentration from 10–8 to 10–6 g mol–1 for the heating transition, while colors from cyan to dark blue indicate the same increase in particle concentration for the cooling curves. Multiple intra-shell transitions are observed for highly curved particles, while lower curvature yields a more monomodal transition similar to that for free chains. Adapted with permission under the Creative Commons Attribution 4.0 license of M. Schroffenegger and E. Reimhult.50 Copyright 2018 MDPI.
Although free chains show a single broad transition peak with a long tail for the heat capacity in the DSC, small nanoparticles with high curvature show a transition with multiple distinguishable peaks over an even larger temperature interval. Free polymer shows the expected strong dependence on concentration (light orange, low concentration to dark red, high concentration in Figure 4), which results in a shift in the uniform transition to lower temperature with increasing concentration. A shift to lower CST is observed for polymer grafted to nanoparticles as the concentration of core–shell nanoparticles is increased, but only for the transition peaks at higher temperature. This makes it possible to identify the higher CST peaks as belonging to the outer part of the shell. The chemical potential of polymer within the brush is defined with respect to the average bulk potential. The outer parts are at lower segment concentration as a result of the radially nonuniform distribution of polymer in the shell. They are therefore more susceptible to the change in average bulk concentration (chemical potential) that influences the CST of the polymer segments.
Even more interesting is that the same data set shows that the transition becomes more uniform as the core diameter is increased. Increased size means a decrease in curvature and a more uniform brush. Indeed, at a core diameter of 10 nm the transition is again almost described by a single broad peak, especially at high particle concentration. The thermal response measured by DSC is then similar to that of free polymer chains but with the transition at significantly lower temperature.
In summary, we show that nanoparticle geometry, as a result of its influence on polymer brush morphology, must be considered to understand the thermal response of core–shell nanoparticles. Furthermore, the number and order of transitions within the shell seem to roughly correspond to the regimes theoretically and experimentally found by considering the spherical brush to correspond to the Daoud–Cotton model rather than a simple parabolic or homogeneous shell model (Figure 3C).56 Importantly, fitting three Gaussian peaks to the DSC data for each core size showed that the transition temperature defined by each peak does not change with core size despite the large difference in the area of each peak (amount of polymer transitioning at this CST).50 However, the fitted CSTs of each peak changed with concentration. We observed that the fraction belonging to the outermost part of the shell was the one showing the strongest decrease in the fractional enthalpy of the transition, under the influence of particle curvature. The center part of the shell was the second most sensitive, and the innermost part of the shell was largely insensitive to particle concentration.
Because the shell structure is also strongly dependent on the grafting density and molecular weight, one cannot specify a precise core size range where these observations are important. However, it is clear from the presented results that this size range extends over the 5–20 nm range of core sizes that are common in many applications, especially for superparamagnetic nanoparticles, and over the polymer shell thicknesses required to make them stable in biological fluids.29,40
Intriguingly, the difference between the particles was negligible when we investigated the CFT at which the nanoparticles formed small aggregates in the same study (Figure 5). They all showed a distinct formation of small clusters in the same size range at the same transition temperature. The CFT was furthermore distinctly lower than the temperature at which the free polymer chains of the same molecular weight formed much larger aggregates. Interestingly, the CFT correlates well with the CST of the innermost part of the shell, which was the same for all particles regardless of core size and concentration (cf. Figure 4).
Figure 5.
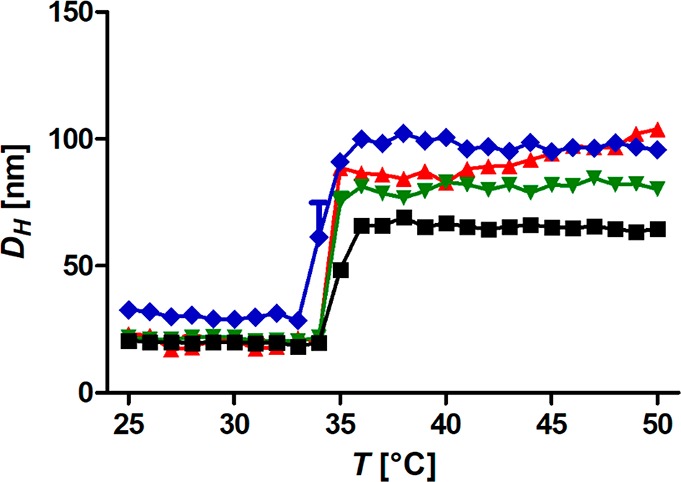
Dynamic light scattering heating curves of iron oxide nanoparticles grafted with 26 kg mol–1 PiPrOxA (1 g L–1 samples) as a function of core size: red triangles, 5 nm cores; green triangles, 7 nm cores; black squares, 10 nm cores; and blue diamonds, 21 nm cores. The step increase in size results from nanoparticle aggregation at the critical flocculation temperature (CFT). The graph was adapted with permission under the Creative Commons Attribution 4.0 license of M. Schroffenegger and E. Reimhult.50 Copyright 2018 MDPI.
Effect of Polymer Chain Length on the Thermal Response of Core–Shell Nanoparticles
The shell extension and density are also affected by the grafted polymer chain size, i.e. degree of polymerization or molecular weight. A larger chain size corresponds to a thicker shell if all other parameters of the polymer brush are kept equal. A higher molecular weight leads to a higher number of polymer segments, and as seen for PiPrOxA grafted to 9.1 nm iron oxide cores in Figure 6, the CFT decreases as a function of the polymer chain molecular weight.36 This is observed at both a constant mass concentration (Figure 6A, green curve) and a constant molar concentration (Figure 6A, pink curve) of core–shell nanoparticles (i.e., the local increase in polymer concentration is sufficient to decrease the CFT without taking into account the global increase in polymer concentration). We also note that the difference between the CFT of grafted and free polymer chains decreases with increased molecular weight (cf. black and green data points in Figure 6) at similar overall polymer concentration. Thus, the effect on the CFT of grafting a polymer to a nanoparticle decreases with increasing degree of polymerization of the grafted chains. Importantly, we note that these results seem independent of polymer chemistry because we obtained the same results when varying the molecular weight of PNiPAAm from 5 to 30 kg mol–1 grafted to ∼10 nm iron oxide cores.58
Figure 6.

(A) Summary of the CFT as a function of PiPrOxA MW for free polymer and 9.1 nm iron oxide nanoparticles grafted with PiPrOxA measured in Milli-Q water by DLS as the temperature at which aggregation starts (the onset of the transition in the count rate curve). Black circles, free PiPrOxA (1 g L–1); pink triangles, PiPrOxA-grafted iron oxide nanoparticles (5 × 1013 particles mL–1); green squares, PiPrOxA-grafted iron oxide nanoparticles (1 g L–1). (B) DSC curves for 21 kg mol–1 MW PiPrOxA at 1 mg mL–1 for free polymer (black) showing one transition and PiPrOxA grafted to nanoparticles (red), showing multiple transitions. Altered and reprinted with permission under the Creative Commons Attribution 4.0 license for Schroffenegger, M., et al.36 Copyright 2018 MDPI. (C) Schematic phase diagram experimentally demonstrated for iron oxide nanoparticles grafted with PNiPAAm. Reprinted from ref (58). Copyright 2017 with permission from Elsevier.
In our investigation on the effect of changing the PiPrOxA molecular weight, it was observed that the shell included at least two CST transitions if the molecular weight was high enough.36 This was true even if only one CFT transition was observed (temperature at which aggregation was observed) for the same particle. These observations agree with the observation from the study of the variation of core diameter because for a low molecular weight the shell should not extend far beyond the region close to the core where the segment density is high and almost uniform. For higher-molecular-weight polymers, the different density regimes within the shells described above43,56 can also be found in the CST transitions measured by DSC.36 Unexpectedly, in our study of varying the molecular weight of PNiPAAm over a similar range, multiple CST transitions were not observed by DSC.58
While all densely PiPrOxA-grafted core–shell nanoparticles formed only small clusters, there was a tendency for shell brushes of high-molecular-weight polymers to lead to slightly larger aggregated cluster sizes.36 Although small, the difference in cluster size was sufficiently large to require more particles on average per aggregate. The effect of grafted polymer molecular weight on aggregate size was observed to be very strong in our earlier study of PNiPAAm-grafted iron oxide nanoparticles.58 Only a decrease in shell (particle hydrodynamic) size was observed by DLS below a critical aggregation concentration of the nanoparticles that had a shell of low molecular weight (5 or 10 kg mol–1) PNiPAAm. However, increasingly large aggregates were observed as either the grafted PNiPAAm molecular weight or the nanoparticle concentration were increased, as schematically described in the phase diagram in Figure 6C.
The grafting of both PNiPAAm58 and PiPrOxA36 at the same grafting density yielded a drastic decrease in the enthalpy per monomer of the CST transition. Interestingly, there is a 6-fold increase in the enthalpy per monomer of the CST transition for grafted PiPrOxA from 6 to 20 kg mol–1, while it less than doubles and mainly stays constant for free polymer chains in this molecular weight range.36 Again, these findings agree with the picture that the inner part of the shell is greatly affected by the grafting onto a nanoparticle, while the outer parts of the shell increasingly show the transition temperature and effect on the aggregate size of free polymer.
An important measure of colloidal stability for applications is how easily and fast core–shell nanoparticles redisperse after being aggregated above the CFT and cooled. In an application, fast and spontaneous redispersion without the need for sonication or other energy-intensive and potentially damaging treatments is mostly preferred. This can be investigated by how fast the original average hydrodynamic size of the (individually dispersed) core–shell nanoparticles is measured again by DLS upon cooling, after the core–shell nanoparticles have been heated to above the CFT. We have shown that particles forming small clusters above the CFT can redisperse instantaneously, at least on the minute time scale, under such conditions.29,49,58 However, it is also clear that several properties of the shell, primarily the molecular weight of the grafted chains, influence if such fast redispersion takes place. A too thin, too sparsely grafted, or too unstably anchored polymer brush shell leads to irreversible aggregation upon heating.36,40,41,49 If the molecular weight is high (i.e., the outer part of the shell behaves similarly to free polymer chains and comprises a major part of the shell), then the aggregates that form are larger and take longer time to redisperse.36,58 A likely explanation for the increased aggregate size is that these clusters form more like those of free polymer chains, which form very large aggregates and turbid suspensions.50 A shell comprising polymers with higher molecular weight (especially at a low core size) means a larger fraction of the polymer shell assuming a low-density conformation similar to that of the free chains. Core–shell nanoparticles forming large aggregates do not redisperse spontaneously and immediately. This contrasts with free polymer chains, which redisperse immediately in the molecular weight range investigated by us. Three major differences that likely contribute to this are the slower diffusion of the cores, the restriction on water distribution that cores pose, and the additional adhesive potential produced by van der Waals attraction of the cores brought close to each other. However, care should be taken to ensure proper preparation of the samples and interpretation of the results. Free polymer in dispersion left over from insufficiently stringent purification can also lead to the formation of large aggregates and a shift to higher CFT.42,58 The adhesive interaction between polymers also becomes stronger as grafted polymer chains and free polymer dehydrate. This drives aggregation to larger aggregates and slows down rehydration.
Effect of Ions and Physiological Buffer Conditions on Thermoresponsive Nanoparticle Aggregation
Many studies investigate the effect of temperature on the colloidal interactions of thermoresponsive nanoparticles in pure water despite the fact that biotechnological and biomedical applications require physiological or close to physiological conditions. Physiological application conditions mean a very high ionic concentration of ∼160 mM, with the concentration of NaCl usually set to ∼150 mM. The presence of ions is expected to change the solubility of polymers in water in accordance with the Hofmeister series. Thus, kosmotropes such as chloride ions are expected to strongly reduce the solubility of the thermoresponsive polymers used to stabilize core–shell nanoparticles, in direct correlation with their hydration.59 By strengthening the hydrogen bond network of bulk water, kosmotropes make water hydrogen bonding to the polymer less favorable and therefore shift the balance between a fully hydrated brush and a dehydrated brush to a lower CST. More specific effects of ions on the thermoresponsive behavior of polymer brushes have also been suggested, such as direct complexing to the polymer by salting out.59
The discrepancy between the ionic conditions in many studies and those of the intended applications is not consequential to investigate fundamental behavior such as the influence of nanoparticle architecture, but it can lead to significant deviations for the evaluation of the performance of a specific nanoparticle design in an application. Despite its CST being lower than human body temperature, most work in the literature has focused on PNiPAAm. For core–shell nanoparticles with PNiPAAm, the difference between performing an experiment in pure water or in physiological buffer is small. However, other polymers, such as poly(2-alkyl-2-oxazolines), have shown greater susceptibility to the presence of ions at high concentration.60
We investigated the effect of the concentration of different salts from the Hofmeister series on the CFT of 9.1 nm diameter iron oxide nanoparticles grafted with PiPrOxA.36 A drop in the CFT of several degrees was observed at salt concentrations in the millimolar range (Figure 7). The temperature drop followed the Hofmeister series for kosmotropes, yielding a larger drop in CFT for stronger kosmotropes and a higher sensitivity to the anion than to the cation. A drop of 10 °C from 36 to 26 °C could be observed for H2PO4– at 160 mM, while NaCl at 160 mM led to a drop in the CFT of 4 °C. Interestingly, the strongest kosmotropes showed a stronger concentration dependence but less influence at low concentrations (Figure 7). In a similar study using 10 nm indiameter cores grafted with 16.5 kg mol–1 PiPrOxA, a drop in the CFT from 38 °C in Milli-Q water to ∼32 °C in physiological buffer (48 °C for the free polymer) was observed.29 That result demonstrated the high reproducibility of the effect of ionic strength across studies if the system parameters are reproduced.
Figure 7.
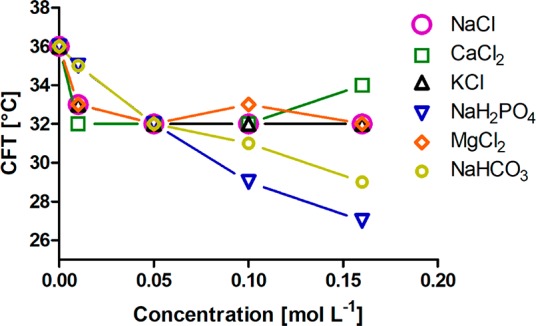
Critical flocculation temperature of dispersions of 10 nm iron oxide nanoparticles densely grafted with 21 kg mol–1 PiPrOxA (1 g L–1) in salt solutions as a function of ionic strength. The data were extracted from the corresponding DLS count rate vs temperature curves. The figure is reproduced with permission under the Creative Commons Attribution 4.0 license for Schroffenegger, M., et al.36 Copyright 2018 MDPI.
Thermoresponsive polymers with very high CST were shown to be more susceptible to a change from pure water to physiological conditions. PEtOxA (14 kg mol–1) does not show a CST in pure water. Nanoparticles (10 nm core) densely grafted with 14 kg mol–1 PEtOxA showed a decrease in the CFT from 74 to 47 °C when the medium was changed from pure Milli-Q water to cell medium based on phosphate-buffered saline.29 The addition of fetal calf serum to the media did not change the CFT further. In the same study, a 15 kg mol–1 random copolymer of 87/13 mol/mol PEtOxA/PiPrOxA was synthesized to have a grafted CFT slightly above body temperature at 45 °C in pure Milli-Q water. In physiological buffer, the CFT dropped to 35 °C (i.e., below body temperature and temperatures used for cell experiments). In summary, the loss of hydration of the polymer shell due to the exposure to anion kosmotropes leads to reduced osmotic repulsion by the shell. Thus, the core–core and polymer–polymer van der Waals attractions can dominate the interparticle interaction and lead to the observed aggregation. Because of the van der Waals attraction between the nanoparticle cores, one can also observe particle aggregation for particles with polymer chains such as poly(ethylene glycol) (PEG) at temperatures much lower than the CFT of the polymer in water.27,56 This is more pronounced as the chains become shorter and the grafting density is decreased, therefore leading to thinner and less hydrated brushes.
Protein Interactions and Cell Uptake of Nanoparticles Controlled by Thermoresponsive Shells
Protein adsorption on core–shell nanoparticles is known to play a crucial role in the clearing cascades of the mononuclear phagocyte system and therefore must be avoided.19,61 This is likely a combined effect of specific interactions of opsonins and a response to the increased size of aggregates. The stability of nanoparticle dispersions in the presence of proteins is affected by temperature. Core–shell nanoparticles with densely grafted polymer brushes are colloidally stable in the presence of protein solutions as long as the polymer brush is irreversibly grafted at a grafting density of at least 0.7–1 chains nm–1; this was shown for our model system in numerous studies using poly(ethylene glycol) and other polymers.18,23,29,39,42 Some protein adsorption seems to inevitably take place with nanoparticles stabilized by linear, high CST, so-called “stealth” polymer brushes such as PEG and PEtOxA as well as for thermoresponsive polymers below their CST such as PNiPAAm and PiPrOxA.43 Increased protein adsorption takes place when the temperature is higher than the CST of the polymer shell,62 as can be seen by the increased cluster size29,39 and sometimes the loss of reversibility of thermally induced aggregation in the presence of (denatured) protein.39 This can lead to precipitation of particles in serum at temperatures and protein concentrations high enough that proteins denature and precipitate.
Furthermore, we recently showed that the cell uptake of core–shell nanoparticles is affected by temperature in relation to the CST/CFT, as shown in our study of PEtOxA, PiPrOxA, and PEtOxA/PiPrOxA brush-stabilized nanoparticles.29 Particles above their CFT lost their stealth properties and were recognized and taken up by HeLa cells.29 Whether this is due to reduced resistance to protein adsorption, due to direct interaction with the cell surface (receptor mediated endocytosis), or simply due to a larger average aggregate size remained unclear from these experiments.
Tuning the Reversible Aggregation of Core–Shell Nanoparticles by Polymer Sequence and Topology
With knowledge of the requirements of grafting density, molecular weight, and CST of a polymer as well as how the CST varies with radial distance from the core through the shell, one can start to design tailor-made sequences that optimize the thermal transitions and colloidal interactions. One could, for example, in the first step design the transition of the inner part of the shell to define the CFT (results in Effect of Core Size on the Thermal Response of a Grafted Polymer Shell) and the outer part of the shell to control the reversibility and size of the formed aggregates (results in Effect of Polymer Chain Length on the Thermal Response of Core–Shell Nanoparticles). This concept was investigated by grafting a block copolymer shell using two blocks that have different CSTs (PEtOxA and PiPrOxA).29 The block closest to the core always saw the largest reduction in CST regardless of the order in which the PEtOxA and PiPrOxA blocks were grafted, as investigated by DSC. Significant cluster formation was observed only for particles with the lower CST block (PiPrOxA) in the outer part of the shell.29 These particles also seemed less stable below the CST/CFT of the PiPrOxA block compared to particles with the PEtOxA block in the outermost part of the shell. It is thus possible to have two well-separated thermal shell transitions for one core–shell nanoparticle by grafting block copolymer chains.
With the inner part of the shell seemingly dominating the colloidal interactions of core–shell nanoparticles, one can also consider changing the type of brush close to the core. An interesting new option is to graft cyclic polymers to the nanoparticle surface. Cyclic brushes have a different topology which makes the brush profile more homogeneous but thinner for a grafted cyclic compared to linear polymer of equivalent molecular weight.63,64 Cyclic polymers, because of their smaller radius of gyration,65 should also be easier to graft densely to the particle surface.64 Changing from a linear to a cyclic topology of the brush was recently shown to lead to superior properties in terms of suppressed protein binding and increased lubrication on planar surfaces.999
Even more recently, we experimentally showed these advantages for highly curved nanoparticle brushes. Iron oxide nanoparticles (9 nm in diameter) grafted with ∼6 and 11 kg mol–1 PEtOxA at grafting densities >1 chain nm–2 for linear chains were compared to the same cores grafted with cyclic PEtOxA of similar MW.41 The grafting of cyclic PEtOxA resulted in almost twice the grafting densities compared to those of their linear equivalents. The nanoparticles grafted with cyclic PEtOxA showed a higher CFT in phosphate-buffered saline than their linear counterparts. They were also redispersed immediate upon cooling, while the linear grafted core–shell nanoparticles redispersed only after cooling and agitation. This was interpreted as that the dense cyclic brushes, which display very low interchain penetration, prevented close van der Waals contact of the cores during aggregation and thereby facilitated rapid reversible redispersion.
It should be noted that this is not the only possible explanation for the observed difference in redispersibility. The reversibility of colloidal aggregation can also be affected by interpolymer bonding. PNiPAAm is not known to form an internal structure upon heating, and there was no effect observed for prolonged heating above the CST for PNiPAAm-grafted core–shell nanoparticles on the reversibility of the aggregates.49,58 However, PiPrOxA is known to be able to adapt crystalline conformations upon dehydration, in which the hydrogen bonding with water is replaced by internal hydrogen bonds.66−68 The internal hydrogen bonding is essentially irreversible, and a polymer brush in which this occurs cannot spontaneously regain its original brush conformation. We frequently observed indications of a reduced ability to rehydrate and redisperse in our studies on the reversible thermal actuation of iron oxide nanoparticles grafted with PiPrOxA or PEtOxA.29,36,41,50 Samples could often no longer be fully redispersed when they were kept at temperatures significantly above the CST for long times. The effect is most clearly demonstrated by the loss of enthalpy of the transition upon cooling compared to heating50 (Figure 4). The nanoparticles with linear PEtOxA brush shells were held for a longer time and were higher above their CFT than the cyclic ones in the investigations described in ref (41). However, crystallization is not known to occur for PEtOxA. It could also be that this observation of time and temperature having an effect on the redispersibiliy of PEtOxA-grafted nanoparticles is only an effect of more complete dehydration leading to more compact clusters with stronger adhesive forces between the polymer shells.
Magnetothermal Reversible Aggregation
Single-crystalline iron oxide nanoparticles in the 3–20 nm diameter range are superparamagnetic. Nanoparticles with individual iron oxide superparamagnetic cores can typically not be extracted from a dispersion by a fixed magnet if they are well-stabilized by an extended polymer shell. They retain full and fast Brownian mobility, which is excellent for maximum binding and capture ability if they are functionalized for specific interactions with biomolecules and cells in separation applications. However, aggregated particles with interacting cores produce a magnetic moment sufficiently high for extraction by a fixed magnet that can be built into biotechnological devices for separation. Using nanoparticles grafted with PNiPAAm,49 polypeptoid (polysarcosin),42 and PiPrOxA29 brush, we showed that heating the nanoparticle dispersion to above the CFT could be used to extract superparamagnetic core–shell nanoparticles (Figure 8A). It was also shown that nanoparticles with dense polymer brush shells can spontaneously redisperse within minutes or redisperse by just providing a light shake. This holds true even after they have been thermally and magnetically aggregated. This ability is not present if the grafting density is low. Magnetic nanoparticles can be heated using alternating magnetic fields that induce losses through Néelian relaxation as the particle magnetic moment is flipped across the boundaries between easy magnetization directions of the nanocrystals.69 Coupling such localized magnetic heating to responsive materials has been shown to be very promising for use of hyperthermia for biomedical applications in a wide sense.6 We demonstrated for PNiPAAm-grafted iron oxide nanoparticles that, indeed, alternating magnetic fields can be used to control the aggregation of the core–shell nanoparticles (Figure 8B).49 With the dispersion in contact with an external magnet, this leads to magnetic extraction, while cooling or removal of the magnet leads to redispersion. The magnetically induced heating and aggregation were shown to work only when the nanoparticle concentration was high (5 g L–1). At low concentration (1 g L–1), the heating is less efficient, and even after passing the CST/CFT, the particles did not aggregate sufficiently to be extracted by the fixed magnet.49 This is in line with the phase diagram in Figure 6C because at a low concentration only small clusters are formed, leading to a lower magnetic moment per aggregate.58
Figure 8.

Dispersion of iron oxide nanoparticles grafted with PNiPAAm (10.7 nm, shell 20 kg mol–1, 5 g L–1) in water placed on a neodym magnet. (Ai) Below the CFT, the dispersion is stable. (Aii) Above the CST, the nanoparticles are extracted but can be redispersed. (B) Dispersion heated exclusively by an alternating magnetic field: (i) clear dispersion before magnetic actuation, solution temperature 24 °C; (ii) aggregation and turbidity after 5 min of actuation, solution temperature 32.4 °C; (iii) precipitation after 10 min of actuation, solution temperature 35.7 °C; and (iv) redispersion of aggregated particles after cooling to below the CST. Reproduced from Kurzhals, S., et al.49 Copyright 2015 American Chemical Society.
Conclusions
With the precise synthesis of both nanoparticle cores and polymer dispersants, it has become possible to investigate in detail the unique properties of core–shell nanoparticles that combine an inorganic (e.g., magnetic) core with a functional (e.g., thermoresponsive) polymer shell. We have therefore in recent years investigated how variations in the core–shell structure influence thermoresponsive properties relevant to applications of nanoparticles in biotechnological and biomedical applications. This was made possible using our toolbox of thermoresponsive polymers synthesized by controlled polymerization, precisely size-controlled iron oxide nanoparticles synthesized by the heat-up method, and optimized nitrodopamide-based protocols for the grafting of dense polymer brushes. The most important of those lessons is that the high curvature of nanoparticles must be matched to the polymer shell morphology and chemical properties. The shell density and thereby thermoresponsive transitions vary with distance from the core surface. Thus, by controlling the monomer composition and density as a function of radial distance from the core (dependent on the curvature), the thermal response can be tailored much more richly than by just accepting uncontrolled nanoparticle aggregation at a single temperature. We foresee that this insight will lead to thermoresponsive polymer shells tailored to applications where multiple transition points and/or controlled cluster size are beneficial. Interesting approaches to accomplish this include gradient and block copolymers as well as combinations with potentially structure-forming thermoresponsive polypeptoids. Furthermore, tailoring the polymer topology and not only the chemistry to match the curvature of the core provides an additional tool that can be exploited to optimize the functional response of core–shell nanoparticles. As we have shown, the thermal colloidal response of core–shell nanoparticles is strongly influenced by moving from pure water to biological salt-, protein-, and cell-containing media. Our demonstrations of the many parameters that can be used to differentiate and tune the shell and colloidal transitions are important with these differences in mind. Applications often put restrictions on changes that can be made to parts of a nanoparticle; having more ways to achieve the same targeted responsiveness (e.g. independently through geometrical dimensions, chemistry, and polymers) is therefore beneficial.
Acknowledgments
We especially acknowledge Dr. Steffen Kurzhals and Dr. Ronald Zirbs for their many contributions to the work summarized in this feature article.
Glossary
Abbreviations
- PEG
poly(ethylene glycol)
- MW
molecular weight
- LCST
lower critical solution temperature
- UCST
upper critical solution temperature
- CST
critical solution temperature
- CFT
critical flocculation temperature
- DLS
dynamic light scattering
- DSC
differential scanning calorimetry
- I
light intensity
- R
radius
- PNiPAAm
poly(N-isopopylacrylamide)
- PEtOxA
poly(2-ethyl-2-oxazoline)
- PiPrOxA
poly(2-isopropyl-2-oxazoline)
Biographies

Erik Reimhult received his Ph.D. in 2004 from Chalmers University of Technology under the supervision of Prof. Bengt Kasemo. Since 2010, he has been full professor of nanobiotechnology at the University of Natural Resources and Life Sciences (BOKU), Vienna, Austria. He was awarded an ERC Consolidator Grant for research on nanoparticle–membrane interactions and was elected member of the Young Academy of the Austrian Academy of Sciences. His research interests include colloidal interactions from smart nanomaterials to bacterial adhesion, with a specific interest in the role of polymer structures at biointerfaces. He serves as an editorial advisory board member for several journals, including ACS Applied Materials & Interfaces and ACS Biomaterials Science & Engineering, as well as on panels for several national science funds.

Martina Schroffenegger is a postdoctoral researcher at the University of Natural Resources and Life Sciences, Vienna, in the Department of Nanobiotechnology. She received her Ph.D. in 2018 from the University of Natural Resources and Life Sciences, Vienna, Austria. She obtained her M.Sc. (2015) and B.Sc. (2012) in technical chemistry from TU Wien with a focus on synthesis. Her research interests are thermoresponsive and multiresponsive core–shell nanoparticles with a focus on medical applications.

Andrea Lassenberger is a postdoctoral researcher at the European neutron research facility Institut Laue-Langevin in Grenoble, France. She received her M.S. in chemistry from the University of Graz in 2011 and her Ph.D. in physical chemistry from the University of Natural Resources and Life Sciences, Vienna, Austria in 2017. Her research focuses on the synthesis, growth mechanisms, and characterization of metal oxide nanoparticles and organic semiconductor nanoparticles and the incorporation of these nanomaterials into silk-derived hydrogels.
The writing of this article and a large part of the research described within were funded by the Austrian Science Fund (FWF, grant number P 28190-N28) and by the European Research Council under the European Union’s Seventh Framework Programme (FP/2007–2013)/ERC grant agreement no. 310034.
The authors declare no competing financial interest.
References
- Lu Y.; Aimetti A. A.; Langer R.; Gu Z. Bioresponsive Materials. Nat. Rev. Mater. 2017, 2 (1), 16075. 10.1038/natrevmats.2016.75. [DOI] [Google Scholar]
- Schattling P.; Jochum F. D.; Theato P. Multi-Stimuli Responsive Polymers – the All-in-One Talents. Polym. Chem. 2014, 5 (1), 25–36. 10.1039/C3PY00880K. [DOI] [Google Scholar]
- Mertz D.; Sandre O.; Bégin-Colin S. Drug Releasing Nanoplatforms Activated by Alternating Magnetic Fields. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861 (6), 1617–1641. 10.1016/j.bbagen.2017.02.025. [DOI] [PubMed] [Google Scholar]
- Schärtl W. Current Directions in Core–shell Nanoparticle Design. Nanoscale 2010, 2 (6), 829. 10.1039/c0nr00028k. [DOI] [PubMed] [Google Scholar]
- Kang T.; Li F.; Baik S.; Shao W.; Ling D.; Hyeon T. Surface Design of Magnetic Nanoparticles for Stimuli-Responsive Cancer Imaging and Therapy. Biomaterials 2017, 136, 98–114. 10.1016/j.biomaterials.2017.05.013. [DOI] [PubMed] [Google Scholar]
- Kumar C. S. S. R.; Mohammad F. Magnetic Nanomaterials for Hyperthermia-Based Therapy and Controlled Drug Delivery. Adv. Drug Delivery Rev. 2011, 63 (9), 789–808. 10.1016/j.addr.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lü T.; Zhang S.; Qi D.; Zhang D.; Zhao H. Thermosensitive Poly(N-Isopropylacrylamide)-Grafted Magnetic Nanoparticles for Efficient Treatment of Emulsified Oily Wastewater. J. Alloys Compd. 2016, 688, 513–520. 10.1016/j.jallcom.2016.07.262. [DOI] [Google Scholar]
- Veiseh O.; Gunn J. W.; Zhang M. Design and Fabrication of Magnetic Nanoparticles for Targeted Drug Delivery and Imaging. Adv. Drug Delivery Rev. 2010, 62 (3), 284–304. 10.1016/j.addr.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amstad E.; Textor M.; Reimhult E. Stabilization and Functionalization of Iron Oxide Nanoparticles for Biomedical Applications. Nanoscale 2011, 3 (7), 2819. 10.1039/c1nr10173k. [DOI] [PubMed] [Google Scholar]
- Shen S.; Ding B.; Zhang S.; Qi X.; Wang K.; Tian J.; Yan Y.; Ge Y.; Wu L. Near-Infrared Light-Responsive Nanoparticles with Thermosensitive Yolk-Shell Structure for Multimodal Imaging and Chemo-Photothermal Therapy of Tumor. Nanomedicine 2017, 13 (5), 1607–1616. 10.1016/j.nano.2017.02.014. [DOI] [PubMed] [Google Scholar]
- Amstad E.; Kohlbrecher J.; MuÜller E.; Schweizer T.; Textor M.; Reimhult E. Triggered Release from Liposomes through Magnetic Actuation of Iron Oxide Nanoparticle Containing Membranes. Nano Lett. 2011, 11 (4), 1664–1670. 10.1021/nl2001499. [DOI] [PubMed] [Google Scholar]
- Shirmardi Shaghasemi B.; Virk M. M.; Reimhult E. Optimization of Magneto-Thermally Controlled Release Kinetics by Tuning of Magnetoliposome Composition and Structure. Sci. Rep. 2017, 7 (1), 7474. 10.1038/s41598-017-06980-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakwere H.; Leal M. P.; Materia M. E.; Curcio A.; Guardia P.; Niculaes D.; Marotta R.; Falqui A.; Pellegrino T. Functionalization of Strongly Interacting Magnetic Nanocubes with (Thermo)Responsive Coating and Their Application in Hyperthermia and Heat-Triggered Drug Delivery. ACS Appl. Mater. Interfaces 2015, 7 (19), 10132–10145. 10.1021/am5088117. [DOI] [PubMed] [Google Scholar]
- Hannecart A.; Stanicki D.; Vander Elst L.; Muller R. N.; Lecommandoux S.; Thévenot J.; Bonduelle C.; Trotier A.; Massot P.; Miraux S.; et al. Nano-Thermometers with Thermo-Sensitive Polymer Grafted USPIOs Behaving as Positive Contrast Agents in Low-Field MRI. Nanoscale 2015, 7 (8), 3754–3767. 10.1039/C4NR07064J. [DOI] [PubMed] [Google Scholar]
- Lassenberger A.; Scheberl A.; Stadlbauer A.; Stiglbauer A.; Helbich T.; Reimhult E. Individually Stabilized, Superparamagnetic Nanoparticles with Controlled Shell and Size Leading to Exceptional Stealth Properties and High Relaxivities. ACS Appl. Mater. Interfaces 2017, 9 (4), 3343–3353. 10.1021/acsami.6b12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G.; Wang D.; Zhou F.; Liu W. Electrostatic Self-Assembly of Au Nanoparticles onto Thermosensitive Magnetic Core-Shell Microgels for Thermally Tunable and Magnetically Recyclable Catalysis. Small 2015, 11 (23), 2807–2816. 10.1002/smll.201403305. [DOI] [PubMed] [Google Scholar]
- Xiao Q.; Li Y.; Li F.; Zhang M.; Zhang Z.; Lin H. Rational Design of a Thermalresponsive-Polymer-Switchable FRET System for Enhancing the Temperature Sensitivity of Upconversion Nanophosphors. Nanoscale 2014, 6 (17), 10179–10186. 10.1039/C4NR02497D. [DOI] [PubMed] [Google Scholar]
- Amstad E.; Gillich T.; Bilecka I.; Textor M.; Reimhult E. Ultrastable Iron Oxide Nanoparticle Colloidal Suspensions Using Dispersants with Catechol-Derived Anchor Groups. Nano Lett. 2009, 9 (12), 4042–4048. 10.1021/nl902212q. [DOI] [PubMed] [Google Scholar]
- Nel A. E.; Mädler L.; Velegol D.; Xia T.; Hoek E. M. V.; Somasundaran P.; Klaessig F.; Castranova V.; Thompson M. Understanding Biophysicochemical Interactions at the Nano–bio Interface. Nat. Mater. 2009, 8 (7), 543–557. 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- Currie E. P. K.; Norde W.; Cohen Stuart M. A. Tethered Polymer Chains: Surface Chemistry and Their Impact on Colloidal and Surface Properties. Adv. Colloid Interface Sci. 2003, 100–102, 205–265. 10.1016/S0001-8686(02)00061-1. [DOI] [PubMed] [Google Scholar]
- Lundgren A.; Agnarsson B.; Zirbs R.; Zhdanov V. P.; Reimhult E.; Höök F. Nonspecific Colloidal-Type Interaction Explains Size-Dependent Specific Binding of Membrane-Targeted Nanoparticles. ACS Nano 2016, 10 (11), 9974–9982. 10.1021/acsnano.6b04160. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Hilal N.; Langston P.; Starov V. Interaction Forces between Colloidal Particles in Liquid: Theory and Experiment. Adv. Colloid Interface Sci. 2007, 134–135, 151–166. 10.1016/j.cis.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Gal N.; Lassenberger A.; Herrero-Nogareda L.; Scheberl A.; Charwat V.; Kasper C.; Reimhult E. Interaction of Size-Tailored PEGylated Iron Oxide Nanoparticles with Lipid Membranes and Cells. ACS Biomater. Sci. Eng. 2017, 3 (3), 249–259. 10.1021/acsbiomaterials.6b00311. [DOI] [PubMed] [Google Scholar]
- Zdyrko B.; Luzinov I. Polymer Brushes by the “Grafting to” Method. Macromol. Rapid Commun. 2011, 32 (12), 859–869. 10.1002/marc.201100162. [DOI] [PubMed] [Google Scholar]
- Zhulina E. B.; Birshtein T. M.; Borisov O. V. Curved Polymer and Polyelectrolyte Brushes beyond the Daoud-Cotton Model. Eur. Phys. J. E: Soft Matter Biol. Phys. 2006, 20 (3), 243–256. 10.1140/epje/i2006-10013-5. [DOI] [PubMed] [Google Scholar]
- Amstad E.; Gehring A. U. U.; Fischer H. H.; Nagaiyanallur V. V. V.; Hähner G.; Textor M.; Reimhult E.; Hähner G.; Textor M.; Reimhult E. Influence of Electronegative Substituents on the Binding Affinity of Catechol-Derived Anchors to Fe 3 O 4 Nanoparticles. J. Phys. Chem. C 2011, 115 (3), 683–691. 10.1021/jp1109306. [DOI] [Google Scholar]
- Amstad E.; Zurcher S.; Mashaghi A.; Wong J. Y.; Textor M.; Reimhult E. Surface Functionalization of Single Superparamagnetic Iron Oxide Nanoparticles for Targeted Magnetic Resonance Imaging. Small 2009, 5 (11), 1334–1342. 10.1002/smll.200801328. [DOI] [PubMed] [Google Scholar]
- Lassenberger A.; Bixner O.; Gruenewald T.; Lichtenegger H.; Zirbs R.; Reimhult E. Evaluation of High-Yield Purification Methods on Monodisperse PEG-Grafted Iron Oxide Nanoparticles. Langmuir 2016, 32 (17), 4259–4269. 10.1021/acs.langmuir.6b00919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzhals S.; Gal N.; Zirbs R.; Reimhult E. Controlled Aggregation and Cell Uptake of Thermoresponsive Polyoxazoline-Grafted Superparamagnetic Iron Oxide Nanoparticles. Nanoscale 2017, 9 (8), 2793–2805. 10.1039/C6NR08654C. [DOI] [PubMed] [Google Scholar]
- Park J.; An K.; Hwang Y.; Park J.-G.; Noh H.-J.; Kim J.-Y.; Park J.-H.; Hwang N.-M.; Hyeon T. Ultra-Large-Scale Syntheses of Monodisperse Nanocrystals. Nat. Mater. 2004, 3 (12), 891–895. 10.1038/nmat1251. [DOI] [PubMed] [Google Scholar]
- Park J.; Lee E.; Hwang N.-M.; Kang M.; Kim S. C.; Hwang Y.; Park J.-G.; Noh H.-J.; Kim J.-Y.; Park J.-H.; et al. One-Nanometer-Scale Size-Controlled Synthesis of Monodisperse Magnetic Iron Oxide Nanoparticles. Angew. Chem., Int. Ed. 2005, 44 (19), 2872–2877. 10.1002/anie.200461665. [DOI] [PubMed] [Google Scholar]
- Lassenberger A.; Grünewald T. A.; Van Oostrum P. D. J.; Rennhofer H.; Amenitsch H.; Zirbs R.; Lichtenegger H. C.; Reimhult E. Monodisperse Iron Oxide Nanoparticles by Thermal Decomposition: Elucidating Particle Formation by Second-Resolved in Situ Small-Angle X-Ray Scattering. Chem. Mater. 2017, 29 (10), 4511–4522. 10.1021/acs.chemmater.7b01207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bixner O.; Lassenberger A.; Baurecht D.; Reimhult E. Complete Exchange of the Hydrophobic Dispersant Shell on Monodisperse Superparamagnetic Iron Oxide Nanoparticles. Langmuir 2015, 31 (33), 9198–9204. 10.1021/acs.langmuir.5b01833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis K.; Qi B.; Witmer M.; Kitchens C. L.; Powell B. A.; Mefford O. T. Quantitative Measurement of Ligand Exchange on Iron Oxides via Radiolabeled Oleic Acid. Langmuir 2014, 30 (36), 10918–10925. 10.1021/la502204g. [DOI] [PubMed] [Google Scholar]
- Shirmardi Shaghasemi B.; Dehghani E. S. S.; Benetti E. M. M.; Reimhult E. Host–guest Driven Ligand Replacement on Monodisperse Inorganic Nanoparticles. Nanoscale 2017, 9 (26), 8925–8929. 10.1039/C7NR02199B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroffenegger M.; Zirbs R.; Kurzhals S.; Reimhult E. The Role of Chain Molecular Weight and Hofmeister Series Ions in Thermal Aggregation of Poly(2-Isopropyl-2-Oxazoline) Grafted Nanoparticles. Polymers (Basel, Switz.) 2018, 10 (4), 451. 10.3390/polym10040451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis K.; Vidmar M.; Khasanov A.; Cole B.; Ghelardini M.; Mayer J.; Kitchens C.; Nath A.; Powell B. A.; Mefford O. T. The Effect of Post-Synthesis Aging on the Ligand Exchange Activity of Iron Oxide Nanoparticles. J. Colloid Interface Sci. 2018, 511, 374–382. 10.1016/j.jcis.2017.09.087. [DOI] [PubMed] [Google Scholar]
- Hühn J.; Carrillo-Carrion C.; Soliman M. G.; Pfeiffer C.; Valdeperez D.; Masood A.; Chakraborty I.; Zhu L.; Gallego M.; Yue Z.; et al. Selected Standard Protocols for the Synthesis, Phase Transfer, and Characterization of Inorganic Colloidal Nanoparticles. Chem. Mater. 2017, 29 (1), 399–461. 10.1021/acs.chemmater.6b04738. [DOI] [Google Scholar]
- Zirbs R.; Lassenberger A.; Vonderhaid I.; Kurzhals S.; Reimhult E. Melt-Grafting for the Synthesis of Core–shell Nanoparticles with Ultra-High Dispersant Density. Nanoscale 2015, 7 (25), 11216–11225. 10.1039/C5NR02313K. [DOI] [PubMed] [Google Scholar]
- Isa L.; Amstad E.; Textor M.; Reimhult E. Self-Assembly of Iron Oxide-Poly(Ethylene Glycol) Core–Shell Nanoparticles at Liquid–Liquid Interfaces. Chimia 2010, 64 (3), 145–149. 10.2533/chimia.2010.145. [DOI] [PubMed] [Google Scholar]
- Morgese G.; Shirmardi Shaghasemi B.; Causin V.; Zenobi-Wong M.; Ramakrishna S. N. S. N.; Reimhult E.; Benetti E. M. Next-Generation Polymer Shells for Inorganic Nanoparticles Are Highly Compact, Ultra-Dense, and Long-Lasting Cyclic Brushes. Angew. Chem., Int. Ed. 2017, 56 (16), 4507–4511. 10.1002/anie.201700196. [DOI] [PubMed] [Google Scholar]
- Kurzhals S.; Pretzner B.; Reimhult E.; Zirbs R. Thermoresponsive Polypeptoid-Coated Superparamagnetic Iron Oxide Nanoparticles by Surface-Initiated Polymerization. Macromol. Chem. Phys. 2017, 218 (13), 1700116. 10.1002/macp.201700116. [DOI] [Google Scholar]
- Gal N.; Schroffenegger M.; Reimhult E. Stealth Nanoparticles Grafted with Dense Polymer Brushes Display Adsorption of Serum Protein Investigated by Isothermal Titration Calorimetry. J. Phys. Chem. B 2018, 122 (22), 5820–5834. 10.1021/acs.jpcb.8b02338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C.; Ma Z.; Zhu X. X. Rational Design of Thermoresponsive Polymers in Aqueous Solutions: A Thermodynamics Map. Prog. Polym. Sci. 2019, 90, 269–291. 10.1016/j.progpolymsci.2019.01.001. [DOI] [Google Scholar]
- Shan J.; Chen J.; Nuopponen M.; Tenhu H. Two Phase Transitions of Poly(N -Isopropylacrylamide) Brushes Bound to Gold Nanoparticles. Langmuir 2004, 20 (11), 4671–4676. 10.1021/la0363938. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Weber C.; Schubert U. S.; Hoogenboom R. Thermoresponsive Polymers with Lower Critical Solution Temperature: From Fundamental Aspects and Measuring Techniques to Recommended Turbidimetry Conditions. Mater. Horiz. 2017, 4 (2), 109–116. 10.1039/C7MH00016B. [DOI] [Google Scholar]
- Zhou W.; Su M.; Cai X. Advances in Nanoparticle Sizing in Suspensions: Dynamic Light Scattering and Ultrasonic Attenuation Spectroscopy. KONA Powder Part. J. 2017, 34 (34), 168–182. 10.14356/kona.2017022. [DOI] [Google Scholar]
- Fan X.; Zheng W.; Singh D. J. Light Scattering and Surface Plasmons on Small Spherical Particles. Light: Sci. Appl. 2014, 3 (6), e179–e179. 10.1038/lsa.2014.60. [DOI] [Google Scholar]
- Kurzhals S.; Zirbs R.; Reimhult E. Synthesis and Magneto-Thermal Actuation of Iron Oxide Core-PNIPAM Shell Nanoparticles. ACS Appl. Mater. Interfaces 2015, 7 (34), 19342–19352. 10.1021/acsami.5b05459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroffenegger M.; Reimhult E. Thermoresponsive Core-Shell Nanoparticles: Does Core Size Matter?. Materials 2018, 11 (9), 1654. 10.3390/ma11091654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber S.; Hutter N.; Jordan R. Effect of End Group Polarity upon the Lower Critical Solution Temperature of Poly(2-Isopropyl-2-Oxazoline). Colloid Polym. Sci. 2008, 286 (14–15), 1653–1661. 10.1007/s00396-008-1942-7. [DOI] [Google Scholar]
- Hoogenboom R.; Schlaad H. Thermoresponsive Poly(2-Oxazoline)s, Polypeptoids, and Polypeptides. Polym. Chem. 2017, 8 (1), 24–40. 10.1039/C6PY01320A. [DOI] [Google Scholar]
- Yim H.; Kent M. S.; Satija S.; Mendez S.; Balamurugan S. S.; Balamurugan S.; Lopez G. P. Evidence for Vertical Phase Separation in Densely Grafted, High-Molecular-Weight Poly(N-Isopropylacrylamide) Brushes in Water. Phys. Rev. 2005, 72 (5), 051801. 10.1103/PhysRevE.72.051801. [DOI] [PubMed] [Google Scholar]
- Goodman D.; Kizhakkedathu J. N.; Brooks D. E. Attractive Bridging Interactions in Dense Polymer Brushes in Good Solvent Measured by Atomic Force Microscopy. Langmuir 2004, 20 (6), 2333–2340. 10.1021/la035843t. [DOI] [PubMed] [Google Scholar]
- Kurzhals S.; Schroffenegger M.; Gal N.; Zirbs R.; Reimhult E. Influence of Grafted Block Copolymer Structure on Thermoresponsiveness of Superparamagnetic Core–Shell Nanoparticles. Biomacromolecules 2018, 19 (5), 1435–1444. 10.1021/acs.biomac.7b01403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald T. A.; Lassenberger A.; Van Oostrum P. D. J.; Rennhofer H.; Zirbs R.; Capone B.; Vonderhaid I.; Amenitsch H.; Lichtenegger H. C.; Reimhult E. Core–Shell Structure of Monodisperse Poly(Ethylene Glycol)-Grafted Iron Oxide Nanoparticles Studied by Small-Angle X-Ray Scattering. Chem. Mater. 2015, 27 (13), 4763–4771. 10.1021/acs.chemmater.5b01488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoud M.; Cotton J. P. Star Shaped Polymers: A Model for the Conformation and Its Concentration Dependence. J. Phys. (Paris) 1982, 43 (3), 531–538. 10.1051/jphys:01982004303053100. [DOI] [Google Scholar]
- Kurzhals S.; Gal N.; Zirbs R.; Reimhult E. Aggregation of Thermoresponsive Core-Shell Nanoparticles: Influence of Particle Concentration, Dispersant Molecular Weight and Grafting. J. Colloid Interface Sci. 2017, 500, 321–332. 10.1016/j.jcis.2017.04.007. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Furyk S.; Bergbreiter D. E.; Cremer P. S. Specific Ion Effects on the Water Solubility of Macromolecules: PNIPAM and the Hofmeister Series. J. Am. Chem. Soc. 2005, 127 (41), 14505–14510. 10.1021/ja0546424. [DOI] [PubMed] [Google Scholar]
- Tatar Güner P.; Demirel A. L. Effect of Anions on the Cloud Point Temperature of Aqueous Poly(2-Ethyl-2-Oxazoline) Solutions. J. Phys. Chem. B 2012, 116 (49), 14510–14514. 10.1021/jp310648d. [DOI] [PubMed] [Google Scholar]
- Karmali P. P.; Simberg D. Interactions of Nanoparticles with Plasma Proteins: Implication on Clearance and Toxicity of Drug Delivery Systems. Expert Opin. Drug Delivery 2011, 8 (3), 343–357. 10.1517/17425247.2011.554818. [DOI] [PubMed] [Google Scholar]
- Cunliffe D.; De las Heras Alarcón C.; Peters V.; Smith J. R.; Alexander C. Thermoresponsive Surface-Grafted Poly(N– Isopropylacrylamide) Copolymers: Effect of Phase Transitions on Protein and Bacterial Attachment. Langmuir 2003, 19 (7), 2888–2899. 10.1021/la026358l. [DOI] [Google Scholar]
- Zhulina E. B.; Leermakers F. A. M.; Borisov O. V. Brushes of Cycled Macromolecules: Structure and Lubricating Properties. Macromolecules 2016, 49 (22), 8758–8767. 10.1021/acs.macromol.6b01275. [DOI] [Google Scholar]
- He S.-Z.; Holger M.; Su C.-F.; Wu C.-X. Static and Dynamic Properties of Grafted Ring Polymer: Molecular Dynamics Simulation. Chin. Phys. B 2013, 22 (1), 016101. 10.1088/1674-1056/22/1/016101. [DOI] [Google Scholar]
- Higgins J. S.; Dodgson K.; Semlyen J. A. Studies of Cyclic and Linear Poly(Dimethyl Siloxanes): 3. Neutron Scattering Measurements of the Dimensions of Ring and Chain Polymers. Polymer 1979, 20 (5), 553–558. 10.1016/0032-3861(79)90164-2. [DOI] [Google Scholar]
- Morgese G.; Trachsel L.; Romio M.; Divandari M.; Ramakrishna S. N.; Benetti E. M. Topological Polymer Chemistry Enters Surface Science: Linear versus Cyclic Polymer Brushes. Angew. Chem., Int. Ed. 2016, 55 (50), 15583–15588. 10.1002/anie.201607309. [DOI] [PubMed] [Google Scholar]
- Katsumoto Y.; Tsuchiizu A.; Qiu X.; Winnik F. M. Dissecting the Mechanism of the Heat-Induced Phase Separation and Crystallization of Poly(2-Isopropyl-2-Oxazoline) in Water through Vibrational Spectroscopy and Molecular Orbital Calculations. Macromolecules 2012, 45 (8), 3531–3541. 10.1021/ma300252e. [DOI] [Google Scholar]
- Sun S.; Wu P. From Globules to Crystals: A Spectral Study of Poly(2-Isopropyl-2-Oxazoline) Crystallization in Hot Water. Phys. Chem. Chem. Phys. 2015, 17 (48), 32232–32240. 10.1039/C5CP05626H. [DOI] [PubMed] [Google Scholar]
- Li T.; Tang H.; Wu P. Molecular Evolution of Poly(2-Isopropyl-2-Oxazoline) Aqueous Solution during the Liquid-Liquid Phase Separation and Phase Transition Process. Langmuir 2015, 31 (24), 6870–6878. 10.1021/acs.langmuir.5b01009. [DOI] [PubMed] [Google Scholar]
- Laurent S.; Dutz S.; Häfeli U. O.; Mahmoudi M. Magnetic Fluid Hyperthermia: Focus on Superparamagnetic Iron Oxide Nanoparticles. Adv. Colloid Interface Sci. 2011, 166 (1–2), 8–23. 10.1016/j.cis.2011.04.003. [DOI] [PubMed] [Google Scholar]