

Abstract
The union of photoredox and nickel catalysis has resulted in a renaissance in radical chemistry as well as in the use of nickel-catalyzed transformations, specifically for carbon-carbon bond formation. Collectively, these advances address the longstanding challenge of late-stage cross-coupling of functionalized alkyl fragments. Empowered by the notion that photocatalytically-generated alkyl radicals readily undergo capture by Ni complexes, wholly new feedstocks for cross-coupling have been realized. Herein, we highlight recent developments in several types of alkyl cross-couplings that are accessible exclusively through this approach.
Keywords: photocatalysis, cross-coupling, alkyl radicals, nickel, single-electron transmetalation
Graphical Abstract
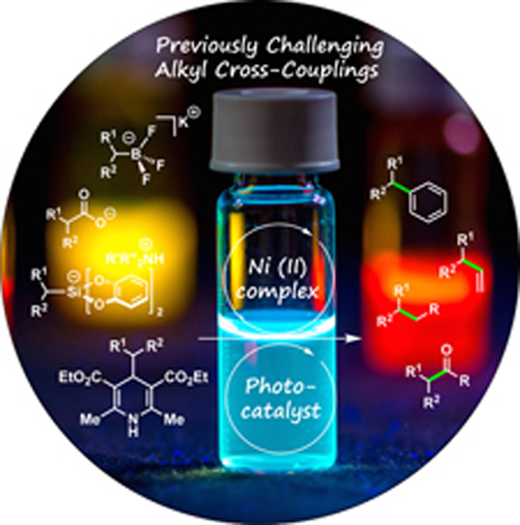
Nickel in the limelight:
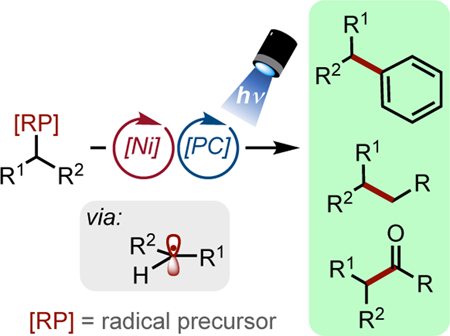
Nickel/photoredox dual cross-coupling is a rapidly developing method that enables alkyl carbon-carbon bond formation. This Minireview discusses the progress of this field by highlighting various types of bond formations that have recently been developed.
1. Introduction
Reactions for the controlled, catalytic formation of carbon-carbon bonds are crucial for modern organic synthesis. In an idealized sense, they enable a rapid, convergent assembly of molecular complexity.[1] Toward this goal, numerous elegant and robust methods for C-C coupling, such as Pd-catalyzed cross couplings,[2] Ru-mediated olefin-metathesis,[3] and C–H functionalization,[4] have been developed. Among such transformations, the formation of C–C bonds at sp3-hybridized centers is a particularly desirable construct because of its potential to provide rapid access to 3D-rich architectures and, akin to the Suzuki sp2-sp2 coupling, impact the way that novel chemical space is accessed.[5] A significant limitation of many state-of-the-art cross-coupling methods is the inability to access sp3-hybridized carbon centers, particularly in complex molecular environments.[6] Although some success has been achieved,[7] the use of forcing conditions and/or the pre-formation of more reactive coupling partners are needed for productive reactivity, thereby limiting functional group compatibility when attempting to forge this type of bond.[8] The challenges encountered when attempting sp2–sp3 cross-coupling prompted the exploration of alternate mechanistic paradigms.
Photoredox catalysis converts visible light into chemical energy under benign conditions by using a photoexcitable catalyst, which is typically a transition metal complex (1–3, Figure 1) or a highly conjugated organic molecule such as 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN, 4).[9] Upon absorption of light and interconversion to a triplet excited state, these catalysts engage in sequential single electron transfer (SET) events to return to their ground state by either an oxidative or reductive quenching cycle (Figure 2). In stark contrast to classical means of free radical generation, photoredox catalysis enables the mild, catalytic generation of reactive C(sp3)-hybridized radicals in a regulated manner. These high-energy intermediates can thus be tamed and utilized for targeted transformations.
Figure 1.
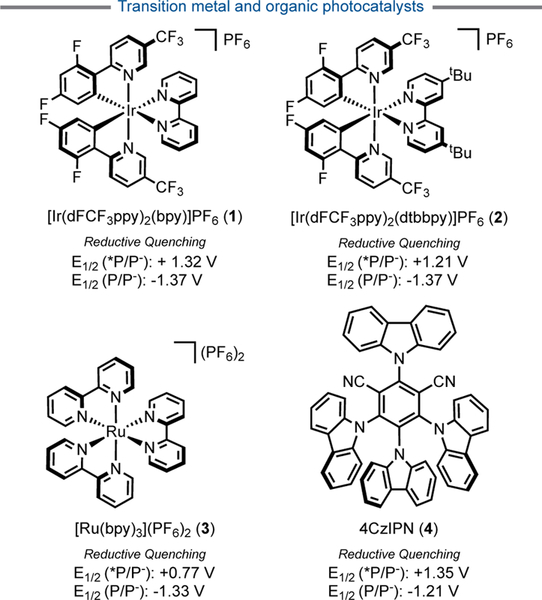
Common photocatalysts for Ni/photoredox dual cross-coupling
Figure 2.
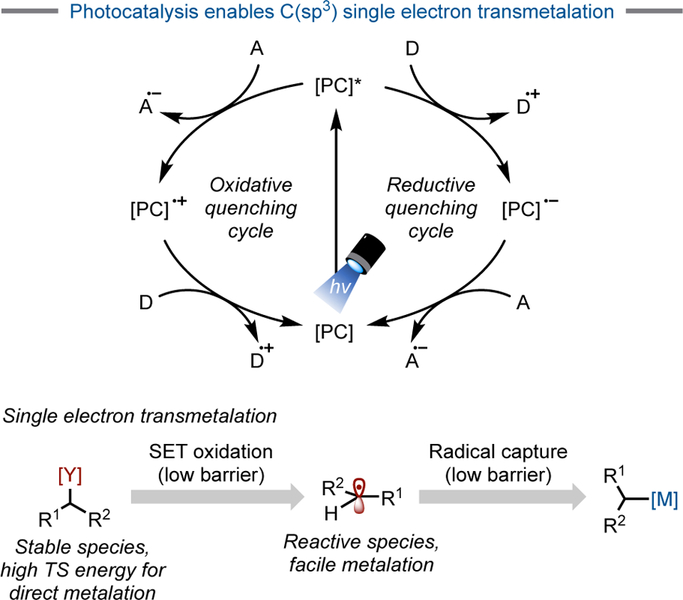
Photocatalyst excitation and quenching cycles. [PC] = photocatalyst.
In 2014, alkyl radicals were first shown to be viable partners in Ni-catalyzed cross-coupling, funneling into a cycle to assemble new C(sp3)–C(sp2) bonds (Figure 3).[10] The development of this paradigm was the culmination of several concepts. First, the intermediacy of carbon-centered C(sp3)-hybridized radicals in Ni-catalyzed coupling processes such as reductive cross-electrophile couplings had been well-documented.[11] Second, the pioneering reports on palladium- and copper photoredox dual catalysis suggested transition-metals could readily be accommodated in photocatalytic cycles.[12] Finally, the rich chemistry of Ni-catalyzed cross-couplings[13] implied that an array of ligands and electrophiles would be compatible with such a dual catalytic reaction.
Figure 3.
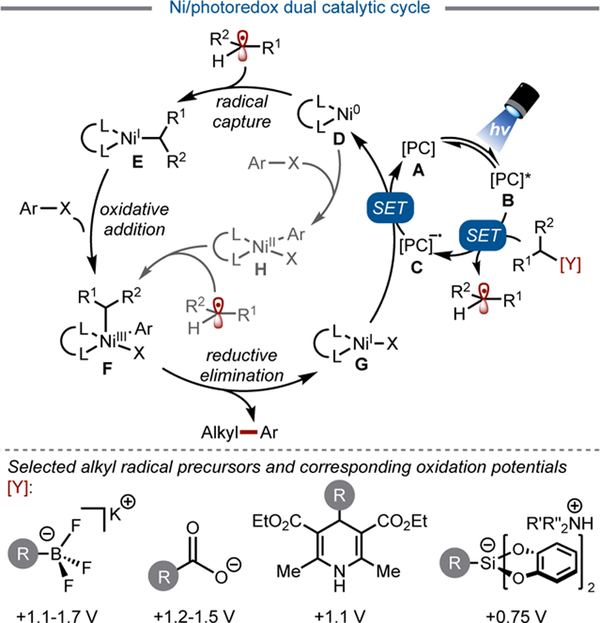
General mechanism of Ni/photoredox dual catalytic cycle
This new coupling paradigm offered a solution to the challenge of conducting two-electron alkyl cross-couplings by subdividing the process into multiple, lower barrier single electron steps (Figure 2). These steps can be partitioned into two distinct, yet interconnected, catalytic cycles – a photoredox cycle and a cross-coupling cycle. In the cross-coupling cycle, capture of a photoredox-generated radical species by ligated Ni0 (D) to generate a NiI intermediate (E) and subsequent oxidative addition of an aryl halide onto this species provides a NiIII intermediate (F) (Figure 3).[14] This NiIII intermediate (F) could also be accessed through the reverse order of events (oxidative addition followed by radical capture), depending on the nature of the electrophile. In any event, reductive elimination of the carbon fragments provides the desired product along with a NiI halide species (G), which is reduced back to Ni0 (D) by the reduced photocatalyst in the photoredox cycle.
The inherently mild nature of this reaction has permitted unprecedented retrosynthetic disconnections to be established. Even more impressive than its mild conditions, this dual catalytic system is extraordinarily modular. An array of radical precursors originating from feedstock chemicals such as organoboron reagents, carboxylic acids, aldehydes, and organosilanes has been employed in these transformations (Figure 3, bottom). Since the initial disclosures in this area, the general pathway depicted in Figure 3 has been adapted for the arylation, vinylation, acylation, and alkylation of these alkyl radical precursors. Because of the relative “blindness” of the reaction pathway to the origin of the radical, even alkyl radicals arising from hydrogen atom transfer (HAT) processes can be employed in these cross-couplings.
Owing to the sheer volume of recent literature on transition metal/photoredox dual catalysis and the existence of several relevant reviews,[9,15] we have chosen to focus the scope of this Minireview on photoredox reactions that use C(sp3) radicals to forge carbon-carbon bonds through a Ni-catalyzed cross-coupling cycle. Photoredox reactions that accomplish carbon-heteroatom coupling or that employ other metals are therefore excluded. Processes that form carbon-carbon bonds through non-metal catalyzed photoredox mechanisms such as the Giese addition,[16] proton-coupled electron transfer (PCET),[17] radical/polar crossover processes,[18] the Minisci reaction,[19] or cycloadditions,[20] although useful in their own right, are also not discussed.
2. Alkyl-aryl cross-couplings
A longstanding challenge for transition-metal cross-coupling has been the construction of alkyl-aryl linkages under mild conditions and with broad functional group tolerance. The seminal work in the field of Ni/photoredox dual catalysis reported methods for the cross-coupling of alkyl radical precursors with aryl halides.[10] Subsequently, multiple strategies for C(sp3) radical generation have been developed. Some approaches rely on redox-active groups, such as alkyltrifluoroborates, carboxylates, bis(catecholato)silicates, and 1,4-dihydropyridines, to achieve programmed reactivity (Figure 3), and others utilize the innate reactivity of substrates (via hydrogen or halogen atom transfer) to generate alkyl radicals. Radicals originating from the oxidation of sulfinate salts,[21] xanthates,[22] and α-silylamines[23] have also been employed in this cross-coupling strategy, although they are not discussed in this review.
2.1. Couplings with alkyltrifluoroborates
Alkyltrifluoroborates are a class of readily prepared, bench-stable reagents that have gained prominence over the past two decades.[24] The enhanced polarity of the carbon-boron bond in these salts allows the controlled, in situ hydrolytic generation of boronic acid derivatives that would be difficult or impossible to isolate. Despite the advantages of these reagents, the palladium-mediated cross-coupling of alkyltrifluoroborates is beset by the characteristic limitations of C(sp3) cross-coupling; namely, the forcing conditions required (elevated temperatures, stoichiometric base) and the susceptibility of these species to undergo β-hydride elimination.
To bypass the challenges associated with alkyl cross-couplings using traditional methods, a seminal report described a visible light-enabled cross-coupling of benzyltrifluoroborates with a variety of aryl- and heteroaryl bromides (Table 1, Entry 1).[10a] Oxidative fragmentation of these trifluoroborates (Ered = +1.10 V vs SCE, on average) gives rise to a C-centered radical, which subsequently combines with the Ni catalyst through an energetically barrierless, single-electron metalation process. Density functional theory (DFT) calculations suggest that although either radical capture or oxidative addition could occur first to arrive ultimately at the same NiIII complex (F), the lower energy pathway proceeds via radical capture followed by oxidative addition (Figure 3). Furthermore, DFT calculations indicate that the stereodetermining step in the catalytic cycle is reductive elimination.[14] Because of the stereoablative nature of the radical generation, stereoconvergent cross-coupling reactivity is possible through dynamic kinetic resolution of the NiIII intermediates (F) with reversibility to H (Figure 3). Modest enantioselectivity (65% ee) has been achieved with secondary benzylic trifluoroborates and a chiral nickel bis(oxazoline) catalyst,[14] but a general solution to accomplish enantioselective cross-coupling remains challenging.
Table 1.
Alkyl-aryl dual cross-couplings using alkyltrifluoroborates. Pin = 2,3-dimethylbutane-2,3-diol. Boc = tert-butyloxycarbonyl.
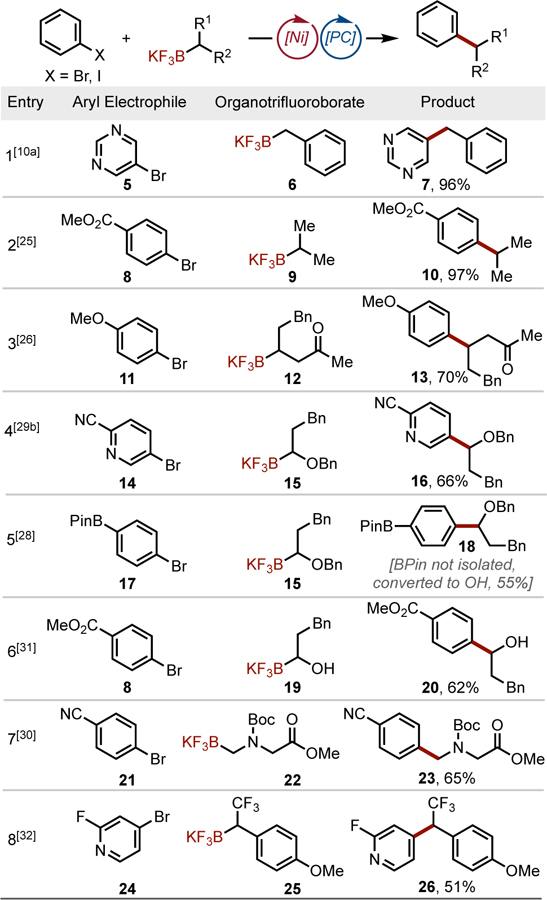 |
A wide array of alkyltrifluoroborates can be cross-coupled with aryl- and heteroaryl bromides using the dual catalytic manifold, including secondary alkyltrifluoroborates, which have relatively high reduction potentials (Ered = +1.50 V vs SCE).[25] Based on this outcome, a unified approach toward the arylation of secondary alkyl β-trifluoroborato carbonyl substrates was designed as a complementary approach to existing synthetic routes (Table 1, Entry 3).[26]
A major feature of this Ni/photoredox dual catalytic cross-coupling method is the orthogonality of the single electron oxidation of alkyltrifluoroborates to the palladium-mediated activation of arylboron reagents.[27] This was leveraged in an iterative cross-coupling approach, wherein rapid diversification of borylated arenes such as 17 was realized (Table 1, Entry 5).[28]
In an effort to target pharmaceutically relevant structural motifs, conditions for coupling α-alkoxy-,[29] α-amino-,[30] α-hydroxyalkyl-,[31] and α-trifluoromethyltrifluoroborates[32] were developed (Table 1, Entries 4–8). In the latter case, the reaction represents the first general route toward unsymmetrical 1,1-diaryl-2,2,2-trifluoroethanes such as 26. The intrinsically low nucleophilicity of α-CF3 organoboron reagents and the propensity for β-fluoride elimination rendered conventional Pd-catalyzed cross-coupling protocols unfeasible.[33] Other organoboron derivatives can be employed in these couplings, as illustrated by a protocol for the cross-coupling of benzylboronic pinacol esters in continuous flow by Ley and coworkers.[34]
More recently, a strategy was reported for the installation of challenging arylated quaternary carbon centers using tertiary alkyltrifluoroborates (Scheme 1).[35] High-throughput screening[36] was critical for identifying a unique ligand, 2,2,6,6-tetramethylheptanedione (TMHD), that enabled the cross-coupling of these sterically-hindered radical species. Although the aryl halide scope was limited to electron-withdrawing and electron-neutral arenes (likely because of poor oxidative addition rates), the types of 3o alkyl fragments that could be installed relevant precedents for the coupling of tertiary pinacol boronates with electron-rich arenes.[37]
Scheme 1.
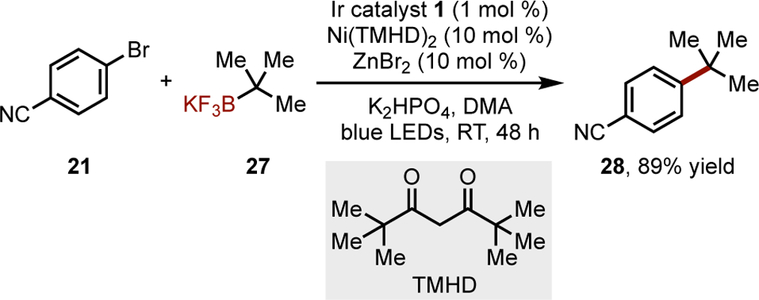
Cross-coupling of tertiary alkyl fragments.[35] DMA = N,N-dimethylacetamide
2.2. Couplings with carboxylic acid radical precursors
Concurrent with the efforts to activate alkyltrifluoroborates using Ni/photoredox dual catalysis, carboxylic acids were demonstrated to undergo single-electron oxidative decarboxylation to generate C(sp3)-centered radicals under a parallel mechanistic manifold.[10b] Carboxylic acids are highly attractive as radical precursors because of their synthetic accessibility and widespread commercial availability. Although transition-metal-catalyzed decarboxylative cross-couplings using aryl carboxylic acids are well-documented,[38] engaging aliphatic carboxylic acids in these two-electron pathways can be challenging. To overcome this limitation, a single electron approach was undertaken by using NiCl2(dme)/dtbbpy in combination with the photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) 2 (Figure 1) to forge the desired C(sp2)-C(sp3) bond. This protocol is effective with secondary-, benzylic-, α-amino-, and α-oxy carboxylic acids with aryl iodides, -bromides, and -chlorides (Scheme 2A). An enantioselective arylation of α-amino acids was subsequently reported.[39] By employing ligand 37 (Scheme 2B), the stereoconvergent synthesis of benzylamine 36 was accomplished.
Scheme 2.
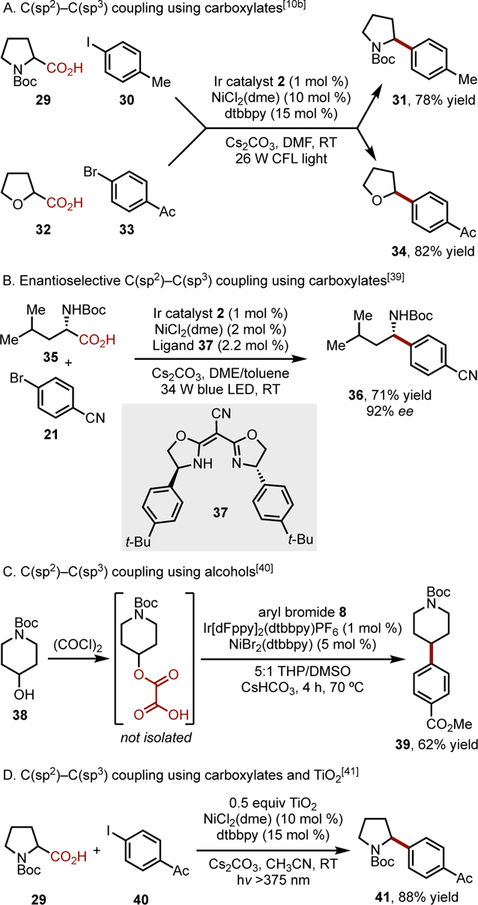
Alkyl-aryl cross-coupling using carboxylic acids. DME = 1,2-dimethoxyethane. dtbbpy = 4,4′-di-tert-butyl-2,2′-dipyridyl. DMF = N,N-dimethylformamide. CFL = compact fluorescent light. LED = light emitting diode. dFppy = 2-(2,4-difluorophenyl)pyridine. THP = tetrahydropyran. DMSO = dimethyl sulfoxide.
Alcohols, activated in situ with oxalyl chloride, have also been used in alkyl-aryl cross-coupling (Scheme 2C).[40] The oxalic acid redox handle functions analogously to carboxylic acids. After deprotonation and single-electron oxidation, two successive decarboxylations occur to form an alkyl radical that is engaged in cross-coupling. More recently, innovative approaches to engage alkyl carboxylic acids in Ni/photoredox cross-coupling have been developed, including those that use a TiO2 photocatalyst (Scheme 2D)[41] or flow chemistry.[42]
2.3. Couplings with alkyl bis(catecholato)silicates
Alkyltrifluoroborates and carboxylic acids that result in primary, non-stabilized radicals are challenging to oxidize, and consequently, their use in Ni/photoredox cross-coupling is sometimes challenging.[9a] New classes of radical precursors with lower oxidation potentials were therefore sought. Specifically, molecules were designed in which a redox-active group would provide an “antenna” for SET processes to occur, facilitating a homolytic scission of a C–C bond. One class of precursor that was readily identified were alkyl bis(catecholato)silicates. Although encumbered by suboptimal atom-economy, these reagents are bench-stable, crystalline solids or powders that possess low oxidation potentials (Ered = +0.75 V vs SCE), allowing the use of less oxidizing (and inexpensive) photocatalysts.[43]
In 2015 a Ni/photoredox-catalyzed C(sp2)-C(sp3) cross-coupling between 4-bromobenzonitrile and a series of alkyl bis(catecholato)silicates possessing potassium 18-crown-6 counterions was reported.[44] These silicate coupling partners incorporated a variety of functional groups, including esters, nitriles, oxiranes, and halides (Scheme 3A).[45] Concurrent with these studies, cross-coupling protocols with bis(catecholato)silicates bearing more practical and less expensive (albeit more acidic) alkylammonium counterions (analogous to the previously reported aryl variants,[46] Scheme 3B) was developed.[47] Couplings with these reagents display exquisite chemoselectivity when dihalogenated arenes were used as coupling partners (Scheme 3C).[48] This class of radical precursors has also been cross-coupled with borylated aryl bromides[49] as well as aryl triflates, tosylates, and mesylates.[50]
Scheme 3.
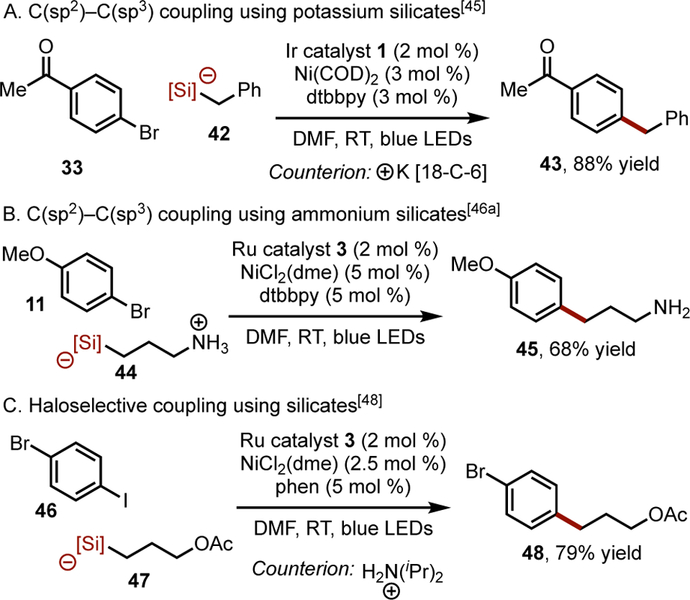
Alkyl-aryl cross-coupling using potassium and ammonium silicates. [Si] = bis(catecholato)silicate. COD = 1,5-cyclooctadiene. phen = 1,10-phenanthroline.
2.4. Couplings with dihydropyridine radical precursors
Another type of radical precursor that has been investigated are 4-alkyl-1,4-dihydropyridines (DHPs). DHPs are typically bench-stable solids that can be prepared in a single step from the corresponding aliphatic aldehyde, the widespread commercial availability of which make DHPs highly accessible radical feedstocks.[51] These heterocyclic species can be thought of as residing at a thermodynamic local minimum, primed to become fully aromatic pyridines through a facile photoredox-catalyzed oxidation (Eox = +1.05 V vs SCE, on average).[52,53] Indeed, many long-utilized methods for the synthesis of substituted pyridines pass through DHP intermediates and require stoichiometric oxidants to achieve aromaticity. It therefore comes as no surprise that photocatalytic SET oxidation occurs with ease.[52c] DHPs bearing 4-alkyl substituents readily undergo oxidative fragmentation to extrude alkyl radicals,[53] and these radicals have been shown to participate in transformations such as aromatic substitution.[54]
Two groups, [55,56] have reported the cross-coupling of aryl halides and alkyl DHPs. One employed a basic additive to deprotonate the DHP and form a more easily oxidized anionic species (Scheme 4A). The second was able to omit the basic additive by employing the more oxidizing 4CzIPN photocatalyst 4 (Figure 1). Both sets of conditions allow the cross-coupling of various alkyl radicals with either aryl bromides or -iodides. Saccharide-derived DHPs can also be used in the Ni/photoredox cross-coupling cycle to afford reversed C-aryl glycosides such as 53 (Scheme 4B).[57] The latter substrate would be prone to β-elimination using traditional cross-coupling methods. A recent report indicated the possibility of direct photoexcitation of DHPs to trigger the formation of alkyl radicals in the absence of a photocatalyst (Scheme 4C).[58]
Scheme 4.
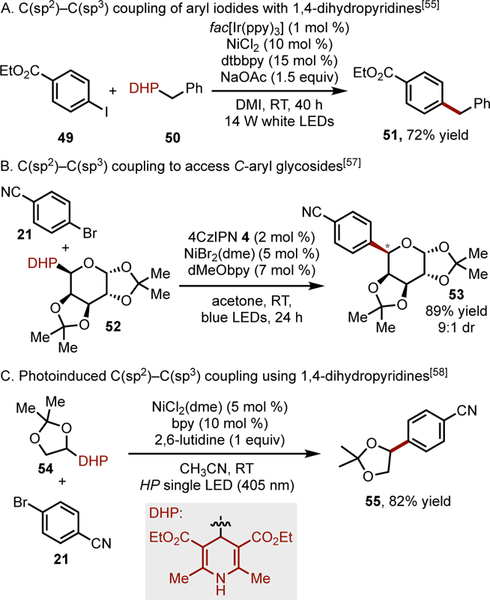
Alkyl-aryl cross-coupling using 1,4-dihydropyridines. ppy = 2-phenylpyridine. DMI = 1,3-dimethyl-2-imidazolidinone. bpy = 2,2’-bipyridine dMeObpy = 4–4′-dimethoxy-2–2′-bipyridine.
2.5. Hydrogen atom transfer in the dual catalytic manifold
A key feature of Ni/photoredox dual catalysis is the relative “blindness” of the Ni cross-coupling cycle to the origin of the alkyl radical coupling partner. This allows compatibility with unique methods for alkyl radical generation, such as hydrogen atom transfer (HAT).[17b,59] HAT processes exploit the innate reactivity of saturated heterocycles possessing homolytically labile C–H bonds adjacent to heteroatoms. Regioselective hydrogen atom abstraction provides a heteroatom-stabilized radical that enters into a Ni cross-coupling cycle. The major significance of this approach is that radical generation via HAT obviates the need for a pre-functionalized alkyl radical precursor, thus enabling highly atom economical cross-coupling processes.
In 2016, two groups reported that Ni/photoredox cross-couplings can be initiated by hydrogen atom abstraction of ethers (Table 2, Entries 1–2).[60,61] Experimental evidence suggests that the couplings proceed through stable and isolable NiII aryl halide oxidative addition complexes (I, Figure 4A). Photoexcitation of these complexes in the presence of ethers or amines, typically as the solvent, promotes C–H cleavage to form the coupled product. Dioxolane 59 is also amenable to HAT/cross-coupling, which provides a mild and redox-neutral method to access masked aldehydes (Table 2, Entry 3).[62]
Table 2.
Alkyl-aryl dual cross-couplings mediated by hydrogen atom transfer. TBADT = tetrabutylammonium decatungstate
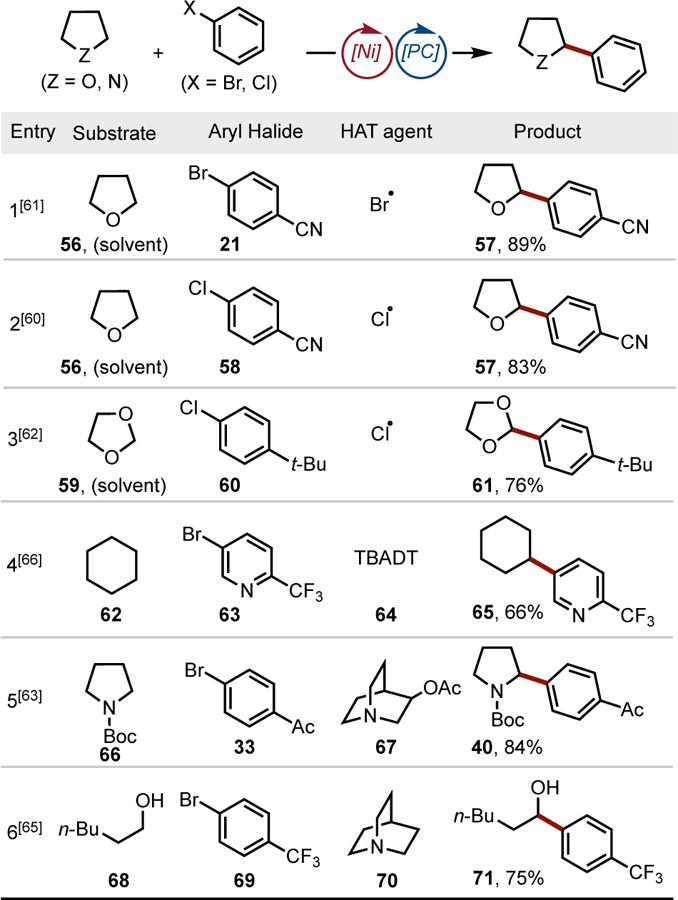 |
Figure 4.
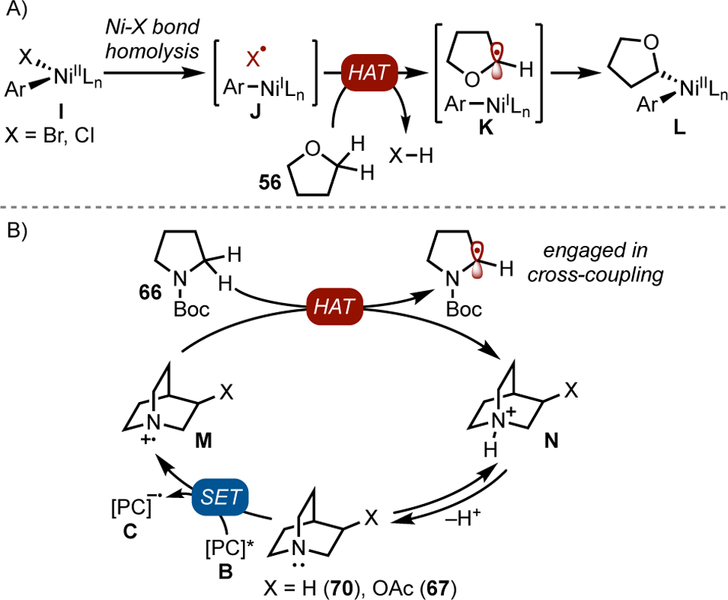
Pathways for hydrogen atom transfer.
An alternative approach to accomplish HAT-initiated dual catalytic cross-coupling is the use of an amine reagent to conduct targeted H-atom abstraction (Figure 4B). Thus, quinuclidine derivatives were used to accomplish C-H arylation of pyrrolidine derivatives (Table 2, Entry 5).[63,64] This strategy has been extended to the C-H arylation of free alcohols. The latter transformation required the use of ZnCl2 as a Lewis acid activator to achieve efficient hydrogen atom transfer (Table 2, Entry 6).[65] Alternative HAT agents, such as photoexcitable polyoxometalates, have been recently described.[66]
2.6. Net reductive alkyl-aryl couplings
Although most Ni/photoredox dual catalytic reactions occur through a redox neutral pathway, several net reductive transformations have been reported. These transformations are conceptually similar to Ni-catalyzed reductive cross-electrophile couplings.[11] By using a photoredox catalyst as an electron shuttle, the stoichiometric manganese and zinc reductants that are typically used can be replaced by stoichiometric amines, Hantzsch esters, or silanes as terminal reductants. The process proceeds through a similar series of mechanistic steps as the redox-neutral variants (Figure 5). The major distinction is that the photocatalyst cycle is turned over by a stoichiometric reductant and that the alkyl bromide is reduced by a Ni0 intermediate to generate the alkyl radical for metalation.
Figure 5.

Net reductive Ni/photoredox cross-coupling cycle.
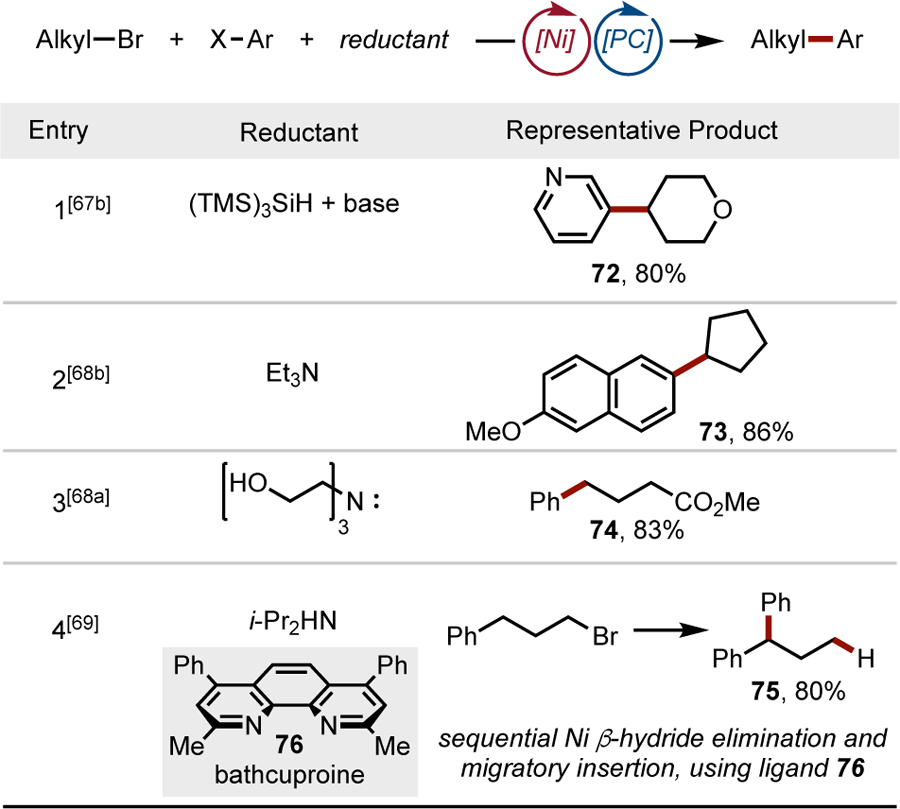
To effect the photocatalytic, net-reductive cross-electrophile coupling of alkyl bromides and aryl halides, silanes and amines have been used as terminal reductants (Table 3). In the first entry, oxidation of tris(trimethylsilyl)silane was employed as an electron source to enable the reductive generation of radicals from alkyl bromides. The reaction could proceed by either direct reduction of the alkyl halide or, more likely, halogen atom abstraction. Overall, both pathways result in a net reductive catalytic cycle. This method was used to couple heteroaryl bromides with various functionalized 1°, 2°, and even 3° alkyl bromides.[67] Other reductive systems employ amines as terminal reductants to achieve couplings similar to those of alkyl bromides, although with a considerably more narrow heteroaryl halide scope.[68] Notably, in Entry 4 the use of bathocuproine 76 as a Ni ligand promotes sequential β-hydride elimination and migratory insertion events to form a more stabilized benzylic NiII complex that generates diarylmethine 75 upon reductive elimination.[69] In addition to alkyl-aryl cross-couplings, reductive Ni/photoredox conditions have been developed for the homocoupling of aryl halides[70] and alkyl halides,[68a] as well as the carboxylation of both aryl- and alkyl bromides.[71]
Table 3.
Net reductive Ni/photoredox cross-couplings of alkyl bromides and aryl halides.
 |
3. Alkyl-alkenyl cross-couplings
Although Pd-mediated cross-coupling with alkenyl halides is a well-established synthetic method, couplings of this type have only been sparsely studied using the Ni/photoredox dual catalytic paradigm. In 2015, an alkyl-vinyl cross coupling of this type was disclosed.[72] The procedure involved the generation of alkyl radicals from carboxylic acids using iridium photocatalyst 2. Many of the substrates employed in the alkenylation benefitted from α-heteroatom stabilization (as in proline derivative 29), but alkenylation of secondary alkyl and benzylic carboxylic acids was also demonstrated (Table 4, Entry 1). Related strategies for the hydroalkylation of alkynes have also been developed,[73] whereby unactivated alkynes undergo Ni-mediated coupling with alkyl radical precursors (Table 4, Entries 2,3). When differentially substituted alkynes were employed in the transformation, the additions took place with good regio- and E:Z selectivity.
Table 4.
Ni/photoredox dual-catalytic alkenylation methods.
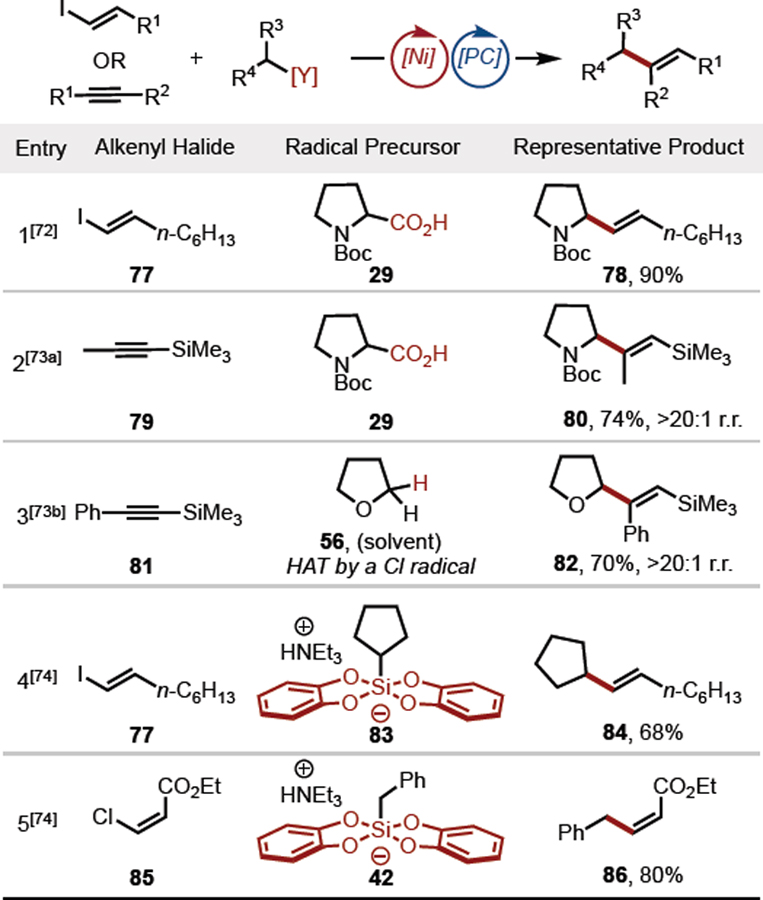 |
Alkyl bis(catecholato)silicate salts such as 83 have also been coupled to alkenyl halides using the dual catalytic method.[74] The characteristic lower oxidation potential of silicate radical precursors enabled the use of the less expensive Ru(bpy)3(PF6)2 3 (Figure 1) in these transformations. The reported protocol is effective with various alkenyl coupling partners, including less reactive alkenyl chlorides (Table 4, Entry 5).
4. Alkyl-acyl cross-couplings
Ketones are a versatile functional group, capable of accessing myriad chemical motifs. One convergent strategy for ketone synthesis is through direct acylation of alkyl fragments. Typically, such disconnections employ reactive, nucleophilic organometallic reagents (as in the Weinreb ketone synthesis), inherently limiting both the scope and functional group compatibility of the transformation. Conversely, a catalytic method for the acylation of otherwise unreactive acyl and alkyl fragments would provide rapid access to complex molecules and mark a clear improvement over the traditional nucleophilic acyl substitution chemistry.
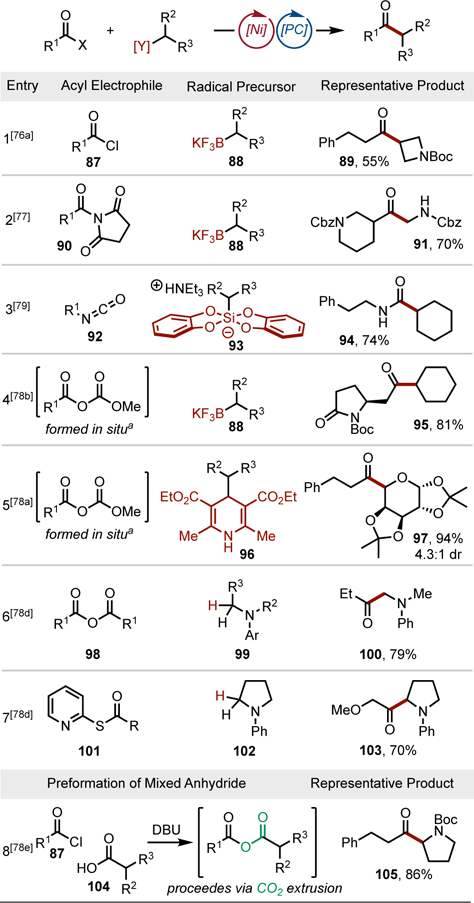
The ability of organonickel intermediates to engage acyl electrophiles in oxidative addition has been reported.[75] Building on this work, Ni/photoredox dual catalysis was employed to couple numerous acyl electrophiles with alkyl radical precursors. As a testament to the modularity of the catalytic system, multiple classes of both acyl electrophiles (acyl chlorides,[76] acyl imides,[77] anhydrides,[78] isocyanates,[79] and thioesters[78d]) and radical precursors have been engaged within the reaction manifold. Achieving selective acyl transfer when using mixed anhydrides can prove challenging because of the presence of two C(sp2)–O bonds that must be differentiated by the Ni-catalyst. One strategy relies on the in situ formation of carbonic anhydrides (Table 5, Entries 4 and 5). In another approach, reactivity reminiscent of Tsuji-Trost chemistry is exploited to effect CO2 extrusion from a pre-formed mixed anhydride (Table 5, Entry 8). HAT strategies for acylation have also been reported, including the coupling of aldehydes with alkyl bromides[80] and the synthesis of alkyl thioesters.[81]
Table 5.
Acyl-alkyl cross-couplings. aFormed through the reaction of the corresponding acid with dimethyl dicarbonate. Cbz = carboxybenzyl.
 |
A Ni/photoredox-catalyzed cross-coupling of meso-anhydrides was reported to access phenylacetone derivatives such as 107 (Scheme 5).[78c] The key stereodetermining step is the oxidative addition of the chiral Ni species onto the meso-anhydride. Although the transformation was limited to aliphatic anhydrides and benzylic trifluoroborates as radical precursors, it is notable as one of the few enantioselective Ni/photoredox dual catalytic transformations.
Scheme 5.
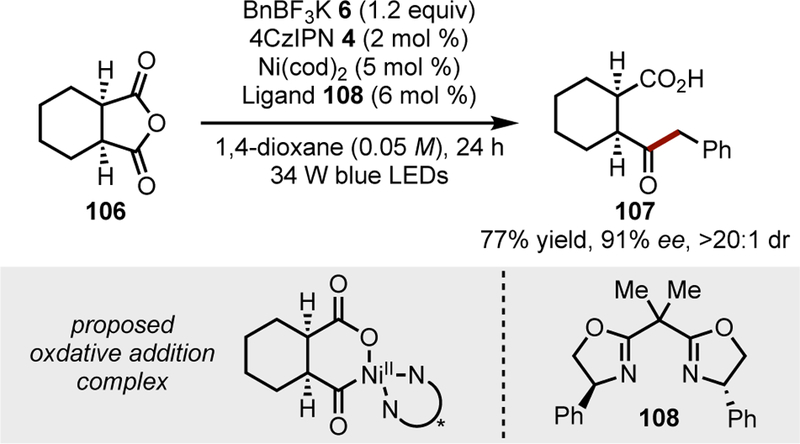
Desymmetrization of meso-anhydrides to effect acyl-alkyl cross-coupling.[78c]
5. Alkyl-alkyl cross-couplings
The ability to join two C(sp3)-hybridized alkyl fragments selectively is a highly desirable and longstanding challenge in C–C bond formation. Such couplings enable the rapid assembly of complex fragments that would otherwise require multiple oxidation/reduction manipulations to access. From the standpoint of Ni-catalyzed cross-coupling, alkyl-alkyl unions are particularly difficult because of the relative weakness of he Ni–C bond and propensity for radical rebound. Further, pairing these challenges with the problem of selective radical generation under photoredox conditions (to differentiate two alkyl radicals so as not to obtain a statistical mixture of products) suggested initially that C(sp3)–C(sp3) coupling under Ni/photoredox crosscoupling would be intractable.[82] To overcome these intrinsic barriers, the innate stabilization of radicals to differentiate two odd-electron species was exploited.[83] Coupling of a more persistent, stabilized a-heteroatom (N, O, S) radical with a nonstabilized alkyl bromide resulted in good selectivity for the crosscoupled product. Stabilized radicals derived from carboxylates (Scheme 6A) or HAT processes (Figure 6B) were used in the cross-coupling. For select cases, non-stabilized secondary alkyl carboxylates could be used, with the quinuclidine catalyst preferentially abstracting the C(sp3)–H bonds a to the nitrogen in preference to those a to the oxygen or at benzylic positions.
Scheme 6.
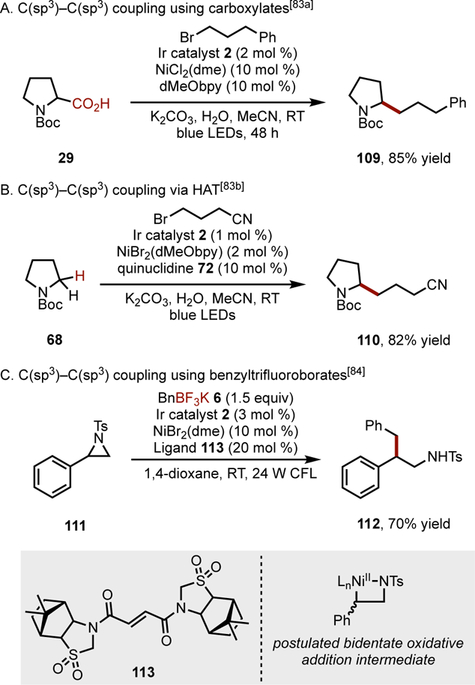
C(sp3)–C(sp3) cross-couplings.
An alternative approach to alkyl-alkyl coupling is to activate the strained C–N bond in aziridines, across which Ni can perform oxidative addition (Scheme 6C).[84] The reaction was postulated to proceed through a bidentate oxidative addition complex, which could impart the desired selectivity for cross-coupling. These reports, along with other recent work in alkyl-alkyl transition-metal-catalyzed cross-coupling,[5] promises to have a profound impact in organic synthesis. However, further development in this area is required to form a broad range of alkyl-alkyl bonds selectively.
6. Conclusions and outlook
In the span of approximately four years, Ni/photoredox dual catalysis has become a workhorse for the construction of an array of carbon-carbon bonds. Many of the bond disconnections achievable by this method could not be plausibly accomplished using traditional cross-coupling approaches. This, combined with the mild and modular nature of these reactions, enables the rapid generation of diverse chemotypes that possess a range of unprotected functional groups. In this manner, Ni/photoredox dual catalysis is a major contribution to the modern vision of “ideal” organic synthesis,[85] wherein highly functionalized molecules can be prepared with minimal protection/deprotection or redox fluctuation steps. The active and near immediate uptake of Ni/photoredox cross-coupling in the industrial sector serves as a testament to both the utility and potential of this reaction paradigm.[41,86] Further opportunities for expansion include the development of additional radical precursors, general protocols for coupling at tertiary centers, enantioselective crosscoupling, and alkyl-alkyl couplings. Continued development in these areas will serve to augment further the impact of this rapidly growing field of research.
Acknowledgements
The authors acknowledge the ongoing financial support provided by the NIGMS (R01 GM 113878). J.P.P. is grateful for an NIH NRSA postdoctoral fellowship (F32 GM125241). We thank Prof. Chris Kelly (Virginia Commonwealth University) for stimulating discussions and Mr. Borna Saeednia (University of Pennsylvania) for photographic assistance.
Biography

John Milligan earned his B.S. in chemistry from Allegheny College in 2012. He then joined the laboratory of Professor Peter Wipf at the University of Pittsburgh, where he earned his Ph.D. in 2018. He is currently a postdoctoral researcher in the laboratory of Professor Gary Molander.

James Phelan received his B.A. in Chemistry from Grinnell College in 2011. He pursued his doctoral studies at Yale University under the supervision of Professor Jonathan A. Ellman. After obtaining his Ph.D. in 2016, he joined Professor Gary Molander’s group at the University of Pennsylvania, where he is a National Institutes of Health postdoctoral fellow.

Shorouk Badir earned her A.B. in Chemistry from Bryn Mawr College in 2016. She then joined the laboratory of Professor Gary A. Molander at the University of Pennsylvania, where she is currently pursuing her Ph.D. She is actively engaged in developing new transformations using photoredox catalysis.

Gary Molander completed his undergraduate studies at Iowa State University under the tutelage of Professor Richard C. Larock. He earned his Ph.D. at Purdue University under the direction of Professor Herbert C. Brown and undertook postdoctoral training with Professor Barry M. Trost at the University of Wisconsin, Madison. He began his academic career at the University of Colorado, Boulder, moving to the University of Pennsylvania in 1999, where he is currently the Hirschmann–Makineni Professor of Chemistry.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Seechurn CCCJ, Kitching MO, Colacot TJ, Snieckus V Angew. Chem., Int. Ed 2012, 51, 5062–5085. [DOI] [PubMed] [Google Scholar]
- [2].a) Wu X-F, Anbarasan P, Neumann H, Beller M, Angew. Chem., Int. Ed 2010, 49, 9047–9050. [DOI] [PubMed] [Google Scholar]; (b) Nobel Prizes 2010: Heck Richard F./Negishi Ei-ichi/Suzuki Akira Angew Chem. Int. Ed 2010, 49, 8300–8300. [DOI] [PubMed] [Google Scholar]
- [3].Casey CP, J. Chem. Educ 2006, 83, 192–195. [Google Scholar]
- [4].Davies HML, Morton D, J. Org. Chem 2016, 81, 343–350. [DOI] [PubMed] [Google Scholar]
- [5].a) Brown DG, Bostrom J, J. Med. Chem 2016, 59, 4443–4458; [DOI] [PubMed] [Google Scholar]; b) Lovering F, Med. Chem. Commun 2013, 4, 515–519; [Google Scholar]; c) Roughley SD, Jordan AM, J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- [6].a) Choi J, Fu GC, Science 2017, 356, 152–160; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rudolph A, Lautens M, Angew. Chem., Int. Ed 2009, 48, 2656–2670. [DOI] [PubMed] [Google Scholar]
- [7].Leonori D, Aggarwal VK, Angew. Chem., Int. Ed 2015, 54, 1082–1096; [DOI] [PubMed] [Google Scholar]; b) Li L, Zhao S, Joshi-Pangu A, Diane M, Briscoe MR, J. Am. Chem. Soc 2014, 136, 14027–14030; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) He A, Falck JR, J. Am. Chem. Soc 2010, 132, 2524–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jana R, Pathak TP, Sigman MS, Chem. Rev 2011, 111, 1417–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Luo J, Zhang J, ACS Catal 2016, 6, 873–877; [Google Scholar]; b) Romero NA, Nicewicz DA, Chem. Rev 2016, 116, 10075–10166; [DOI] [PubMed] [Google Scholar]; c) Shaw MH, Twilton J, MacMillan DWC, J. Org. Chem 2016, 81, 6898–6926; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Prier CK, Rankic DA, MacMillan DWC, Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Tellis JC, Primer DN, Molander GA, Science 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC, Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Weix DJ, Acc. Chem. Res 2015, 48, 1767–1775; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Biswas S, Weix DJ, J. Am. Chem. Soc 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) For Palladium examples, see: Neufeldt SR, Sanford MS, Adv. Synth. Catal 2012, 354, 3517–3522; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kalyani D, McMurtrey KB, Sanford MS J. Am. Chem. Soc 2011, 133, 18566–18569; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) For copper example, see: Yingda Y, Sanford MS, J. Am. Chem. Soc 2012, 134, 9034–9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Ananikov VP, ACS Catal 2015, 5, 1964–1971; [Google Scholar]; b) Tasker SZ, Standley EA, Jamison TF, Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC, J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Twilton J, Le C, Zhang P, Shaw MH, Evans RW, MacMillan DWC, Nat. Rev. Chem 2017, 0052;; b) Matsui JK, Lang SB, Heitz DR, Molander GA, ACS Catal 2017, 7, 2563–2575; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tellis JC, Kelly CB, Primer DN, Jouffroy M, Patel NR, Molander GA, Acc. Chem. Res 2016, 49, 1429–1439; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Skubi KL, Blum TR, Yoon TP, Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].For a recent example, see: Ma J, Lin J, Zhao L, Harms K, Marsch M, Xie X, Meggers E, Angew. Chem., Int. Ed 2018, 57, 11193–11197. [DOI] [PubMed] [Google Scholar]
- [17].a) For recent reviews, see: Hoffmann N, Eur. J. Org. Chem 2017, 1982–1992;; b) Gentry EC, Knowles RR, Acc. Chem. Res 2016, 49, 1546–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) For select recent examples, see: Phelan JP, Lang SB, Compton JS, Kelly CB, Dykstra R, Gutierrez O, Molander GA, J. Am. Chem. Soc 2018, 140, 8037–8047; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cavanaugh CL, Nicewicz DA Org. Lett 2015, 17, 6082–6085; [DOI] [PubMed] [Google Scholar]; c) Hollister KA, Conner ES, Spell ML, Deveaux K, Maneval L, Beal MW, Ragains JR, Angew. Chem., Int. Ed 2015, 54, 7837–7841. [DOI] [PubMed] [Google Scholar]
- [19].For a recent review, see: Wang C-S, Dixneuf PH, Soulé J-F, Chem. Rev 2018, 118, 7532–7585. [DOI] [PubMed] [Google Scholar]
- [20].For a review, see: Yoon TP, ACS Catal 2013, 3, 895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Knauber T, Chandrasekaran R, Tucker JW, Chen JM, Reese M, Rankic DA, Sach N, Helal C, Org. Lett 2017, 19, 6566–6569. [DOI] [PubMed] [Google Scholar]
- [22].Vara BA, Patel NR, Molander GA, ACS Catal 2017, 7, 3955–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Remeur C, Kelly CB, Patel NR, Molander GA, ACS Catal 2017, 7, 6065–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Molander GA, J. Org. Chem 2015, 80, 7837–7848. [DOI] [PubMed] [Google Scholar]
- [25].Primer DN, Karakaya I, Tellis JC, Molander GA, J. Am. Chem. Soc 2015, 137, 2195–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tellis JC, Amani J, Molander GA, Org. Lett 2016, 18, 2994–2997. [DOI] [PubMed] [Google Scholar]
- [27].a) For Palladium examples, see: Sun CR, Potter B, Morken JP, J. Am. Chem. Soc 2014, 136, 6534–6537; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mlynarski SN, Schuster CH, Morken JP, Nature 2014, 505, 386–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yamashita Y, Tellis JC, Molander GA, Proc. Natl. Acad. Sci. U. S. A 2015, 112, 12026–12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Matsui JK, Molander GA, Org. Lett 2017, 19, 436–439; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Karimi-Nami R, Tellis JC, Molander GA, Org. Lett 2016, 18, 2572–2575; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Karakaya I, Primer DN, Molander GA, Org. Lett 2015, 17, 3294–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].El Khatib M, Serafim RAM, Molander GA, Angew. Chem., Int. Ed 2016, 55, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Alam R, Molander GA, J. Org. Chem 2017, 82, 13728–13734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ryu D, Primer DN, Tellis JC, Molander GA, Chem. Eur. J 2016, 22, 120–123. [DOI] [PubMed] [Google Scholar]
- [33].Furuya T, Kamlet AS, Ritter T, Nature 2011, 473, 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lima F, Kabeshow MA, Tran DN, Battilocchio C, Sedelmeier J, Sedelmeier G, Schenkel B, Ley SV, Angew. Chem., Int. Ed 2016, 55, 14085–14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Primer DN, Molander GA, J. Am. Chem. Soc 2017, 139, 9847–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schmink JR, Bellomo A, Berritt S, Aldrichimica Acta 2013, 46, 71–80. [Google Scholar]
- [37].Sandford C, Aggarwal VK, Chem. Commun 2017, 53, 5481–5494 [DOI] [PubMed] [Google Scholar]
- [38].Rodriguez N, Goossen LJ, Chem. Soc. Rev 2011, 40, 5030–5048. [DOI] [PubMed] [Google Scholar]
- [39].Zuo Z, Cong H, Li W, Choi J, Fu GC, MacMillan DWC, J. Am. Chem. Soc 2016, 138, 1832–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang X, MacMillan DWC, J. Am. Chem. Soc 2016, 138, 13862–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].McTiernan CD, Leblanc X, Scaiano JC, ACS Catal 2017, 7, 2171–2175. [Google Scholar]
- [42].Hsieh H-W, Coley CW, Baumgartner LM, Jensen KF, Robinson RI, Org. Process Res. Dev, 2018, 22, 542–550. [Google Scholar]
- [43].a) Matsuoka D, Nishigaichi Y, Chem. Lett 2015, 44, 163–165; [Google Scholar]; b) Matsuoka D, Nishigaichi Y, Chem. Lett 2014, 43, 559–561; [Google Scholar]; c) Nishigaichi Y, Suzuki A, Takuwa A, Tetrahedron Lett 2007, 48, 211–214. [Google Scholar]
- [44].Corce V, Chamoreau LM, Derat E, Goddard JP, Ollivier C, Fensterbank L, Angew. Chem., Int. Ed 2015, 54, 11414–11418. [DOI] [PubMed] [Google Scholar]
- [45].Lévèque C, Chenneberg L, Corcé V, Goddard J-P, Ollivier C, Fensterbank L, Org. Chem. Front 2016, 3, 462–465. [Google Scholar]
- [46].Seganish WM, DeShong P, J. Org. Chem 2004, 69, 1137–1143. [DOI] [PubMed] [Google Scholar]
- [47].a) Jouffroy M, Primer DN, Molander GA, J. Am. Chem. Soc 2016, 138, 475–478; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) For preparation of ammonium silicates, see: Lin K, Kelly C, Jouffroy M, Molander GA, Org. Synth 2017, 94, 16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lin K, Wiles RJ, Kelly CB, Davies GHM, Molander GA, ACS Catal 2017, 7, 5129–5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Vara BA, Jouffroy M, Molander GA, Chem. Sci 2017, 8, 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Patel NR, Molander GA, J. Org. Chem 2016, 81, 7271–7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].a) For examples, see: Heydari A, Khaksar S, Tajbakhsh M, Bijanzadeh HR, J. Fluorine Chem 2009, 130, 609–614; [Google Scholar]; b) Adibi H Samimi HA, Beygzadeh M, Catal. Commun 2007, 8, 2119–2124; [Google Scholar]; c) Tewari N, Dwivedi N, Tripathi R, Tetrahedron Lett 2004, 45, 9011–9014. [Google Scholar]
- [52].a) Wei X, Wang L, Jia W, Du S, Wu L, Liu Q, Chin. J. Chem 2014, 32, 1245–1250; [Google Scholar]; b) Wang, Liu Q, Chen B, Zhang L, Tung C, Wu L, Chin. Sci. Bull 2010, 55, 2855–2858; [Google Scholar]; c) Zhang D, Wu L-Z, Zhou L, Han X, Yang Q-Z, Zhang L-P, Tung C-H, J. Am. Chem. Soc 2004, 126, 3440–3441; [DOI] [PubMed] [Google Scholar]; d) Fukuzumi S, Suenobu T, Patz M, Hirasaka T, Itoh S, Fujitsuka D, M.; Ito O, J. Am. Chem. Soc 1998, 120, 8060–8068. [Google Scholar]
- [53].Cheng JP, Lu Y, Zhu XQ, Sun Y, Bi F, He J, J. Org. Chem 2000, 65, 3853–3857. [DOI] [PubMed] [Google Scholar]
- [54].Nakajima K, Nojima S, Sakata K, Nishibayashi Y, ChemCatChem 2016, 8, 1028–1032. [Google Scholar]
- [55].Nakajima K, Nojima S, Nishibayashi Y, Angew. Chem., Int. Ed 2016, 55, 14106–14110. [DOI] [PubMed] [Google Scholar]
- [56].Gutierrez-Bonet A, Tellis JC, Matsui JK, Vara BA, Molander GA, ACS Catal 2016, 6, 8004–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dumoulin A, Matsui JK, Gutierrez-Bonet A, Molander GA, Angew. Chem., Int. Ed 2018, 57, 6614–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Buzzetti L, Prieto A, Roy SR, Melchiorre P, Angew. Chem., Int. Ed 56, 15039–15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].a) For a review on PCET, see: Warren JJ, Tronic TA, Mayer JM, Chem. Rev 2010, 110, 6961–7001; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) For a review on photocatalyzed HAT reactions, see: Capaldo L, Ravelli Eur D J. Org. Chem 2017, 2056–2071. [DOI] [PMC free article] [PubMed]
- [60].Shields BJ, Doyle AG, J. Am. Chem. Soc 2016, 138, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Heitz DR, Tellis JC, Molander GA, J. Am. Chem. Soc 2016, 138, 12715–12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Nielsen MK, Shields BJ, Liu J, Williams MJ, Zacuto MJ, Doyle AG, Angew. Chem., Int. Ed 2017, 56, 7191–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DW, Science 2016, 352, 1304–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].For a reaction that provides similar products via a non-HAT mechanism, see: Ahneman DT, Doyle AG Chem. Sci 2016, 7, 7002–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Twilton J, Christensen M, DiRocco DA, Ruck RT, Davies IW, MacMillan DWC, Angew. Chem., Int. Ed 2018, 57, 5369–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Perry IB, Brewer TF, Sarver PJ, Schultz DM, DiRocco DA, MacMillan DWC, Nature 2018, 560, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].a) Bacauanu V, Cardinal S, Yamauchi M, Kondo M, Fernandez F, Remu R, MacMillan DWC, Angew. Chem. Int. Ed 2018, 10.1002/anie.201807629; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang P, Le CC, MacMillan DW, J. Am. Chem. Soc 2016, 138, 8084–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].a) Paul A, Smith MD, Vannucci AK, J. Org. Chem 2017, 82, 1996–2003; [DOI] [PubMed] [Google Scholar]; b) Duan Z, Li W, Lei A, Org. Lett 2016, 18, 4012–4015. [DOI] [PubMed] [Google Scholar]
- [69].Peng L, Li Z, Yin G, Org. Lett 2018, 20, 1880–1883. [DOI] [PubMed] [Google Scholar]
- [70].Masuda Y, Ishida N, Murakami M, Eur. J. Org. Chem 2016, 5822–5825.
- [71].Meng QY, Wang S, Konig B, Angew. Chem., Int. Ed 2017, 56, 13426–13430. [DOI] [PubMed] [Google Scholar]
- [72].Noble A, McCarver SJ, MacMillan DWC, J. Am. Chem. Soc 2015, 137, 624–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].a) Till NA, Smith RT, MacMillan DWC, J. Am. Chem. Soc 2018, 140, 5701–5705; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Deng H-P, Fan X-Z, Chen Z-H, Xu Q-H, Wu J, J. Am. Chem. Soc 2017, 139, 13579–13584. [DOI] [PubMed] [Google Scholar]
- [74].Patel NR, Kelly CB, Jouffroy M, Molander GA, Org. Lett 2016, 18, 764–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Guo L, Rueping M, Chem. Eur. J 2018, 24, 7794–7809. [DOI] [PubMed] [Google Scholar]
- [76].a) Amani J, Molander GA, J. Org. Chem 2017, 82, 1856–1863; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Amani J, Sodagar E, Molander GA, Org. Lett 2016, 18, 732–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Amani J, Alam R, Badir S, Molander GA, Org. Lett 2017, 19, 2426–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].a) Badir SO, Dumoulin A, Matsui JK, Molander GA, Angew. Chem., Int. Ed 2018, 57, 6610–6613; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Amani J, Molander GA, Org. Lett 2017, 19, 3612–3615; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Stache EE, Rovis T, Doyle AG, Angew. Chem., Int. Ed 2017, 56, 3679–3683; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Joe CL, Doyle AG, Angew. Chem., Int. Ed 2016, 55, 4040–4043; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Le CC, MacMillan DW, J. Am. Chem. Soc 2015, 137, 11938–11941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Zheng S, Primer DN, Molander GA, ACS Catal 2017, 7, 7957–7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhang X, MacMillan DWC, J. Am. Chem. Soc 2017, 139, 11353–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Kang B, Hong SH, Chem. Sci 2017, 8, 6613–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].For a report on the challenge of obtaining non-statistical mixtures, see: Lévèque C, Corcé V, Chenneberg L, Ollivier C, Fensterbank L, Eur. J. Org. Chem 2017, 2118–2121.
- [83].a) Johnston CP, Smith RT, Allmendinger S, MacMillan DWC, Nature 2016, 536, 322–325; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Le C, Liang Y, Evans RW, Li X, MacMillan DWC, Nature 2017, 547, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Yu XY, Zhou QQ, Wang PZ, Liao CM, Chen JR, Xiao WJ, Org. Lett 2018, 20, 421–424. [DOI] [PubMed] [Google Scholar]
- [85].Gaich T, Baran PS, J. Org. Chem 2010, 75, 4657–4673. [DOI] [PubMed] [Google Scholar]
- [86].a) For reports on Ni/photoredox chemistry from companies, see: Zhang R, Li G, Wismer M, Vachal P, Colletti SL, Shi Z-C, ACS Med. Chem. Lett 2018, 9, 773–777; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Le CC, Wismer MK, Shi ZC, Zhang R, Conway DV, Li G, Vachal P, Davies IW, MacMillan DWC, ACS Cent. Sci 2017, 3, 647–653; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) DeLano TJ, Bandarage UK, Palaychuk N, Green J, Boyd MJ, J. Org. Chem 2016, 81, 12525–12531. [DOI] [PubMed] [Google Scholar]; d) Palaychuk N, DeLano TJ, Boyde MJ, Green J, Bandarage UK, Org. Lett 2016, 18, 6180–6183. [DOI] [PubMed] [Google Scholar]; e) Raynor KD, May GD, Bandarage UK, Boyd MJ, J. Org. Chem 2018, 83, 1551–1557. [DOI] [PubMed] [Google Scholar]