

Abstract
The large and transparent cells of cleavage-stage zebrafish embryos provide unique opportunities to study cell division and cytoskeletal dynamics in very large animal cells. Here, we summarize recent progress, from our laboratories and others, on live imaging of the microtubule and actin cytoskeletons during zebrafish embryonic cleavage. First, we present simple protocols for extending the breeding competence of zebrafish mating ensembles throughout the day, which ensures a steady supply of embryos in early cleavage, and for mounting these embryos for imaging. Second, we describe a transgenic zebrafish line [Tg(bactin2:HsENSCONSIN17–282-3xEGFP)hm1] that expresses the green fluorescent protein (GFP)-labeled microtubule-binding part of ensconsin (EMTB-3GFP). We demonstrate that the microtubule-based structures of the early cell cycles can be imaged live, with single microtubule resolution and with high contrast, in this line. Microtubules are much more easily visualized using this tagged binding protein rather than directly labeled tubulin (injected Alexa-647-labeled tubulin), presumably due to lower background from probe molecules not attached to microtubules. Third, we illustrate live imaging of the actin cytoskeleton by injection of the actin-binding fragment of utrophin fused to GFP. Fourth, we compare epifluorescence-, spinning-disc-, laser-scanning-, and two-photon-microscopic modalities for live imaging of the microtubule cytoskeleton in early embryos of our EMTB-3GFP-expressing transgenic line. Finally, we discuss future applications and extensions of our methods.
I. Introduction
The zebrafish embryo has long been recognized as an excellent model system for molecular-genetic analysis of vertebrate embryonic development (Detrich et al., 1999), one whose advantages complement, and perhaps exceed, those of the mouse (Orkin and Zon, 1997). Forward genetic screens using large-scale zygotic (Driever et al., 1996; Haffter et al., 1996), maternal (Pelegri and Mullins, 2004), and numerous targeted strategies have generated thousands of mutations in the zebrafish that affect all levels of development. Systematic identification and cloning of the mutated genes, whether by candidate (Skromne and Prince, 2008), positional (Bahary et al., 2004), or insertional (Amsterdam and Hopkins, 2004) approaches, has greatly enhanced our understanding of the signaling pathways that regulate expression of the vertebrate body plan. Modern deep sequencing methods will make gene identification even faster.
The advantages of the zebrafish for mechanistic studies of developmental processes in vivo at the cellular level have been less well appreciated although the tide is clearly turning (Beis and Stainier, 2006). The remarkable optical clarity of the large blastomeres of the pre-pigmentation embryo facilitates the microscopic examination of cellular processes that underlie morphogenesis. The reduced pigmentation mutant lines nacre (Lister et al., 1999) and casper (White et al., 2008) extend tissue and organ transparency to juvenile and adult animals. As researchers apply transgenic approaches to tag proteins of interest with a fluorescent protein (FP), we foresee a major shift of cellular research to the context of the living fish. Zebrafish excel over amphibian models for live imaging of early development because their meroblastic cleavage separates the transparent blastodisc from the opaque yolk, whereas the holoblastic cleavage of amphibian embryos renders cells nontransparent at early stages due to distributed yolk particles. The high fecundity of the zebrafish and its low maintenance costs are also major advantages, particularly in comparison to the mouse.
Characterization of the cytoskeleton of zebrafish eggs and embryos and its role in morphogenesis of the zygote began in the early 1990s. These studies, which had been stimulated by the pioneering work of J. P. Trinkaus on epiboly and gastrulation in embryos of Fundulus heteroclitus (Betchaku and Trinkaus, 1978; Trinkaus, 1949, 1951), focused initially on microtubules and microfilaments. Using ultraviolet irradiation and antimitotic drugs, Strahle and Jesuthasan (1993) and Solnica-Krezel and Driever (1994) demonstrated that microtubules participate either directly or indirectly in epibolic cell movements, and Jesuthasan and Strahle (1997) concluded that specification of the zebrafish dorsoventral axis required the microtubule-dependent transport of dorsal determinants from the vegetal pole to marginal blastomeres. In recent years, numerous studies have shown that maternal products of the zebrafish oocyte and early embryo are organized, and reorganized, by microtubules and microfilaments during oogenesis and embryogenesis (Dekens et al., 2003; Knaut et al., 2000; Strasser et al., 2008; Theusch et al., 2006; Yabe et al., 2009; reviewed by Lindeman and Pelegri, 2010).
To date, the cytoskeletal components of zebrafish oocytes and embryos have generally been analyzed by the application of immunofluorescence light microscopy and/or electron microscopy to fixed preparations. Although methods of fixation to optimize cytoskeletal preservation in embryos have been developed (reviewed by Topczewski and Solnica-Krezel, 2009) and their use has led to important discoveries (reviewed by Lindeman and Pelegri, 2010), research on the function of the cytoskeleton in zebrafish development would benefit enormously from live-cell imaging of fluorescent cytoskeletal proteins. Such studies have revolutionized our understanding of cytoskeleton organization and dynamics in somatic cells, where essentially all cutting-edge cytoskeletal work is now performed using live imaging. Various laboratories have embarked on live-imaging strategies to study cytoskeletal dynamics in zebrafish; examples include microtubule imaging by injection of rhodamine-labeled tubulin into zebrafish zygotes (Li et al., 2006, 2008), the labeling of microfilaments by injection of plasmids that drive the transient expression of the F-actin-binding domain of utrophin fused to mCherry (Andersen et al., 2010), and the creation of a transgenic zebrafish line that expresses a GFP-tagged α-tubulin (Asakawa and Kawakami, 2010).
For any experiment aimed at live visualization of the cytoskeleton, the key question is, “What probe to use?”‘ Useful probes must fulfill multiple criteria: they must not perturb the biology, they must report faithfully on the organization and dynamics of the filament system, they must emit as many photons as possible for as long as possible, and they must provide optimal contrast in the face of background signal from the cytoplasm. The last consideration is often under-appreciated. For all cytoskeleton filaments and their associated binding proteins, there exist at least two protein pools: (1) molecules that are in filaments or binding to filaments and (2) molecules that are free in the cytoplasm and often exchange rapidly with the filament-associated pool. In the thick cells of an early embryo, the majority of signal may come from the soluble pool, which lowers the contrast for imaging the filament. For this reason, the best probes for filament visualization in embryos are often not tagged versions of the primary polymer subunits themselves (e.g. tubulin, actin), but rather probes that bind selectively to the polymeric form of the subunit and thus have a lower pool of free proteins. Such polymer-binding probes must be critically evaluated for unwanted interactions; they may tend to stabilize or bundle the polymer if their levels are too high, and they may also bind selectively to certain subsets of the filaments. Despite these caveats, this strategy has been quite successful, and here we discuss its application to microtubule and actin visualization in zebrafish.
In this chapter, we describe methods for live imaging of microtubules and microfilaments in cleaving zebrafish embryos, the former by use of a transgenic zebrafish line (Wühr et al., 2010) that expresses the GFP-tagged microtubule-binding domain of ensconsin (Faire et al., 1999) and the latter by injection of the actin-binding domain of utrophin bearing a GFP tag (Burkel et al., 2007), respectively. We also compare the quality of images obtained by various optical platforms.
II. Maintaining the Breeding Competence of Zebrafish throughout the Day
In the wild, zebrafish spawn at the onset of light in the morning (Detrich et al., 1999). In the lab, this behavior potentially limits the time frame for experimentation on cleavage-stage embryos. Several procedures exist for circumventing this restriction: (1) use of isolation cabinets on light cycles that shift “morning”‘ for zebrafish mating ensembles to suit the investigator or (2) use of in vitro fertilization, in which females are squeezed and their eggs collected in defined medium or salmon ovarian fluid to prevent activation (Corley-Smith et al., 1999; Sakai et al., 1997). The former technique requires considerable cabinetry, whereas the latter can delay egg activation by at most 6 h (Siripattarapravat et al., 2009) and females require significant time to recover from egg donation.
We have found that it is possible to obtain newly fertilized embryos over a large portion of the day (6–8 h) with a simpler routine. We maintain males and females in separate tanks in our zebrafish facility. One day before the experiment, two females and one male are placed together in a crossing cage and allowed to acclimate, which appears to predispose them to mate. The following morning (as defined by the light cycle), males and females are separated immediately after they have spawned sufficient embryos for initiation of experimentation. When more embryos are required, the connubial trio is reunited. We are careful not to separate a threesome for more than 2 h, because they become refractory to further mating that day. Using this method, we typically obtain sufficient embryos to conduct three to four experiments at 2-h intervals in a single day, provided that the fish are at optimal age (~4–12 months), well fed, and husbanded.
III. Mounting Zebrafish Embryos for Live Imaging
A. Rationale
Proper mounting of cleaving embryos is one of the most important steps for live imaging. To obtain images of high quality, one must immobilize the embryos and place them within the working distance of moderate to high numerical aperture (NA) objectives. In this context, upright and inverted microscopes have different experimental advantages and disadvantages. Mounting of dechorionated embryos on an upright microscope with a water-immersion or air objective is comparatively easy, but one is limited to using objectives of modest NA. This restriction will be reduced as vendors build immersion lenses with increasingly high numerical NA and long working distance. For example, Nikon has introduced a 25 × 1.1 NA water objective with a 2-mm working distance (Nikon Inc.), which delivers an extraordinary increase in performance at low magnification. Unfortunately, such lenses are extremely expensive and may require special adapters. In contrast, mounting embryos for inverted microscopy is more difficult but permits the use of oil-immersion, high NA objectives.
In our experience, the best method to immobilize a dechorionated embryo is to place it in the pocket of an agarose specimen chamber cast on a Petri dish. The lateral dimensions of the pockets are slightly smaller than the diameter of an embryo (Fig. 1A) so that friction between the gently compressed embryo and the walls of the pocket resists embryo movement. To cast a chamber with pockets that can accommodate embryos of differing size, we use a plastic mold that creates squares with sides of 550–700 μm; the depth of each pocket is 400 μm. Mounting of the embryo is performed on a dissecting microscope after which the Petri dish is transferred to the microscope used for imaging.
Fig.1.

Mounting of cleavage-stage zebrafish embryos. (A) Mold used to prepare agarose mounting pockets for cleavage-stage embryos. Pockets of differing dimensions provide flexibility in mounting of embryos of heterogeneous size. (B) Configuration of embryos for observation using an upright microscope equipped with a water-immersion (dipping) objective (left panel) or for imaging via inverted microscopy and an oil-immersion objective (right panel).
The increased difficulty of mounting an embryo for inverted microscopy arises from its natural tendency to rotate so that the heavy yolk faces downward, that is, opposite to the desired yolk-up orientation in the mounting pocket. In addition, the specimen chamber must be cast on the glass cover slip of a suitable culture dish (Fig. 1B). The key, in our experience, is to use an agarose pocket of optimal dimensions, so the embryo is prevented from rotating but not overly compressed.
B. Methods
Machine the embryo mounting mold from a suitable plastic or metal (e.g. plexiglass, aluminum) to the dimensions shown in Fig. 1A.
Pour melted 2% agarose in egg water (Westerfield, 2007) into a Petri dish. Insert the mold into the agarose solution and put a weight on top. Allow the agarose to set at 4° C. If mounting for inverted microscopy, use a coverslip-bottom culture dish (e.g. MatTek Corp., Ashland, MA, USA).
Remove the mold and add egg water and embryos into the dish.
Using the dissecting microscope for visualization, dechorionate embryos with two sharp No. 5 forceps.
With a blunt glass rod, maneuver an embryo into a square chamber whose dimensions are slightly smaller than the embryo’s diameter. Orient the embryo as desired and, if necessary, add low-melting-point agarose (0.8%) to fix the embryo in position. Position additional embryos in remaining pockets as desired.
To reduce swaying when transferring, remove most of the egg water from the dish, leaving just enough to cover the embryo. Transfer the dish to the microscope stage and add egg water to a level sufficient for immersing the objective (Fig. 1B).
For inverted microscopy, carefully transfer the culture dish to the microscope stage and bring the objective into oil contact with the dish’s coverslip (Fig. 1B). Add egg water to cover the embryos so that they do not dehydrate during imaging.
IV. Live Imaging of Microtubules in Cleaving Zebrafish Embryos
A. Rationale
Li et al. (2006, 2008) demonstrated real-time imaging of microtubules in cleaving zebrafish embryos by injection of rhodamine-labeled tubulin at the one-cell stage. Due to the thickness of early zebrafish blastomeres and the large proportion of rhodamine-tubulin that remains monomeric, the signal-to-noise ratio of fluorescent microtubule polymer relative to the fluorescent subunit pool is substantially lower than that achieved by comparable injection of thin, adherent tissue culture cells (Zhai et al., 1996). Fig. 2A (right panel) shows an image from our laboratory of a cleaving zebrafish embryo whose microtubules are labeled by incorporation of injected Alexa-647-labeled tubulin. Although the contrast is unusually high, astral microtubules are barely visible over the background from soluble tubulin. To obtain higher contrast images of microtubule dynamics and organization in cleaving zebra-fish blastomeres and to circumvent the injection step, we (Wühr et al., 2010) generated a transgenic fish line, Tg(bactin2:HsENSCONSINI7−282-3xEGFP)hml, that expresses the microtubule-binding domain of ensconsin fused to three sequential GFP moieties (EMTB-3GFP). This probe was first tested in tissue culture cells, where it was shown to associate tightly but dynamically with microtubules without perturbing either microtubule organization or dynamics when expressed at levels appropriate for imaging (Faire et al., 1999). Later, EMTB-3GFP was adapted for imaging microtubules in echinoderm embryos, where its increased contrast relative to directly labeled tubulin was a compelling advantage (von Dassow et al., 2009). In that work, the probe was introduced by mRNA injection at the one-cell stage, which precluded live imaging of the first division. We introduced this probe into zebrafish by making a transgenic line. In addition to permitting visualization immediately after egg spawning and fertilization due to maternal expression of the transgene, the transgenic approach has the advantage that we know the probe has not perturbed embryonic development since the fish line is fully fertile. Below we compare the two methods directly by injection of Alexa-647-labeled tubulin into the transgenic zebrafish line. We have also used EMTB fused to a single GFP and expressed in bacteria to visualize microtubules live in Xenopus egg extracts, demonstrating the versatility of ensconsin-based probes.
Fig. 2.

Microtubule imaging in cleaving embryos from the Tg(bactin2:HsENSCONSIN17−282-3xEGFP)hml zebrafish line. (A) Transgenic, EMTB-3GFP-expressing embryos were injected with Alexa-647-labeled tubulin. The GFP and Alexa-647 signals were imaged simultaneously as described in Section IV.B.3. (B) Time lapse images of microtubules labeled by EMTB-3GFP (enlargements of the boxed region of panel A, left side). The dynamic instability of the ends of individual microtubules can be followed; the green arrows delineate a growing microtubule end, whereas the red arrows show a shortening microtubule. (C) Ratiometric image generated as the composite of the two images shown in panel A. The image has been pseudocolored to differentiate regions labeled preferentially by EMTB-3GFP (red) or by Alexa-647-labeled tubulin (blue). See Section IV.C for further details. (D) Simultaneous labeling of microtubules and chromatin in embryos expressing EMTB-3GFP and H2B-mCherry. See Section IV.C for details. (See Plate no. 1 in the Color Plate Section.)
B. Methods
1. Generation of Transgenic Zebrafish Lines
The transgenic line, Tg(bactin2:HsENSCONSIN17−282-3xEGFP)hml, was created in an unspecified wild-type strain by use of the Tol2Kit (Kawakami, 2004; Kwan et al., 2007; Urasaki et al., 2006; Wühr et al., 2010). EMTB-3GFP expression is driven by the beta actin promoter, chosen for its high expression levels. Beginning with eight founders, we selected progeny that gave the highest expression levels without detectable developmental toxicity. The line is now in its third generation, the transgene is mostly stably transmitted, and expression of EMTB-3GFP remains robust. EMTB-3GFP expression levels do tend to decline with generational passage of the transgene, and we compensate for this by selecting adult females that express the brightest eggs for propagation. Using identical methods, we have created a transgenic zebrafish line in wild-type strain AB that expresses human histone H2B fused in frame to mCherry2 for visualization of chromatin dynamics.
2. Preparation of Alexa-647-Labeled Tubulin and Embryo Microinjection
Tubulin was purified from calf brain and labeled with Alexa647-succinimide-ester (Invitrogen) as described by Hyman et al. (1991). The ratio of fluorophore to tubulin dimer was 0.7. Tg(bactin2:HsENSCONSIN17−282-3xEGFP)hm1 zebrafish were mated, and, shortly after fertilization, ~5 nL of labeled tubulin (11 mg/ml) were injected through the yolk into the blastodiscs of embryos.
3. Laser-Scanning Confocal Microscopy
Images were recorded using a Zeiss LSM 710 inverted microscope equipped with a 63 × plan-apochromat objective (NA = 1.4). The pinhole was set at 63 μm, the pixel size was 0.11 μm, and pixel dwell time was 0.79 μs. Specimens were illuminated simultaneously by argon (488 nm, 25 mW) and helium-neon (633 nm, 5 mW) lasers. The emission spectra of GFP and Alexa 647 were recorded from 492 to 598 nm and from 637 to 755 nm, respectively. Images of a single focal plane were collected at 7.7-s intervals.
C. Results
Fig. 2A compares the labeling of spindle microtubules by EMTB-3GFP and by Alexa-647-tubulin in early anaphase of the second mitosis in an injected, transgenic embryo: the left panel shows the EMTB-3GFP signal, whereas the right panel shows the Alexa-647 signal. Movie S1 shows the same embryo in both imaging modalities as anaphase commences. Spindle microtubules were brightly labeled in the green channel, and their contrast with respect to the background was high. Neither spindle morphology nor function appears to be perturbed by binding of EMTB-3GFP to spindle microtubules (see Movie S1), as expected since the transgenic line is fertile. Furthermore, the dynamic instability (Mitchison and Kirschner, 1984) of individual microtubules at the front of asters as they expanded in telophase was readily detected (see time-lapse imagery of Fig. 2B, which shows enlargements of the boxed region of 2A). In contrast, the same spindle observed in the Alexa-647 channel was less clearly visualized; the central spindle microtubules were bright, but the background fluorescence of the cytoplasm was high and individual microtubule ends in the asters could not be identified with confidence.
The clarity of microtubule labeling by the transgenic zebrafish line is striking, but one may ask whether EMTB-3GFP, which interacts with microtubules noncovalently, faithfully delineates all of the microtubules throughout the spindle.1 The ensconsin (GFP) and tubulin (Alexa-647) signals correlate well, but subtle differences are apparent that cannot be explained by lower background fluorescence. To compare microtubule labeling by the two approaches, we generated a composite, ratiometric image from the two images of Fig. 2A. Fig. 2C shows that EMTB-3GFP preferentially labels certain microtubule populations, which are shown in red in the pseudocolored ratio image. These include the distal ends (distal with respect to the centrosome) of astral microtubules and microtubules of the furrow microtubule array [microtubules to the left of the spindle (Danilchik et al., 2003)]. Astral microtubules proximal to the centrosomes label equivalently with the two probes (green pseudocolor). EMTB-3GFP staining of the aster interaction zone (Wühr et al., 2010), where the two asters meet, is very low (blue pseudocolor) compared to the signal in the tubulin channel, suggesting the probe is selectively excluded from these microtubules. Possible non-mutually exclusive explanations for the differential labeling of microtubules by EMTB-3GFP could be: (1) EMTB-3GFP’s local concentration within the asters and the aster interaction zones might not be sufficiently high to saturate its binding sites on the polymer, whereas this condition is met for the dispersed microtubule ends at the astral peripheries; (2) EMTB-3GFP may compete with other microtubule-associated proteins for binding to specific subsets or subregions of spindle microtubules; and/or (3) the affinity of EMTB-3GFP may be altered by regional regulation of posttranslational modification. We are currently working on evaluating these hypotheses, with the aim of engineering a modified probe with less differential binding. However, for most purposes the current probe provides excellent microtubule imaging and is clearly superior to directly labeled tubulin. For visualization of the advancing front of astral microtubules, selective binding of the probe is even advantageous.
To observe microtubule and chromatin dynamics simultaneously during embryonic cleavage, we crossed the EMTB-3GFP line (hml) with a beta-actin:H2B-mCherry2 line [Tg(ba:h2b-mCherry2)hm13] (Fig. 2D, Movie S2). Fig. 2D shows that the mCherry2-tagged histone labels the chromosomes at the metaphase plate (red signal), and Movie S2 shows a blastomere undergoing a complete mitotic cycle of chromosome condensation, spindle assembly, and chromosome partition. The high signal-to-noise ratios of the EMTB-3GFP-labeled microtubules and of the mCherry2-H2B-labeled chromosomes in these movies should facilitate quantitative analysis of cleavage in the large blastomeres of the meroblastic zebrafish embryo.
V. Live Imaging of Microfilaments in Cleaving Zebrafish Embryos
A. Rationale
Live imaging of microfilaments in the large blastomeres of the zebrafish embryo is even more problematic than live imaging of microtubules, most likely because the concentration of soluble, unpolymerized actin is very high compared to polymerized actin in fibers. In a comparable embryo (Xenopus laevis), actin is present at ~20 μM, and most is bound to sequestering proteins (Rosenblatt et al., 1995). Sequestered monomer probably contributes to very high background staining if actin is imaged via immunofluorescence of labeled actin monomers. Rhodamine-labeled phalloidin, which binds only to F-actin with extremely high selectivity, has been used to study microfilaments during cleavage of zebrafish embryos (Li et al., 2008; Theusch et al., 2006), but this approach typically requires fixation and restricts fixation methods to those that preserve filament structure (aldehyde fixation works, organic solvent fixation does not). When labeled phalloidin has been used to image microfilaments in living zebrafish embryos (Li et al., 2008), the probe must be restricted to very low concentrations and thus low signal since phalloidin is in fact a toxin derived from the death cap mushroom. Bement and co-workers developed several FP-tagged probes for microfilaments based on the actin-binding calponin homology domain of utrophin (Utr-CH)(Burkel et al., 2007). They showed that transient expression of GFP-Utr-CH in Xenopus oocytes by injection of synthetic mRNA or plasmid constructs reports on the distribution of F-actin without perturbing actin dynamics and is less toxic than phalloidin. It is possible, even likely, that this probe binds selectively to a certain population of filaments, but this is difficult to determine when we lack alternative methods for filament visualization. We attempted to generate a transgenic fish line that would express Utr-CH-GFP, but we were unable to establish founder fish, most likely due to probe toxicity during development. As an alternative, we developed a protocol for injection of bacterially expressed Utr-CH-GFP (kindly provided by David Burgess, Boston College) into the one-cell stage of the zebrafish embryo. With this probe we could visualize cortical microfilaments in living embryos with excellent contrast.
B. Method
His-tagged Utr-CH-GFP was expressed in Escherichia coli, purified via chromatography on nickel columns, and flash frozen in 150 mM aspartic acid and 10 mM HEPES solution (pH 7.2–7.3) in the laboratory of David Burgess. Shortly after fertilization, zebrafish embryos were injected through the yolk into the blastodisc with ~2 nL of utrophin-GFP (1 mg/ml) and then mounted for upright microscopy as described in Section III.B. Images were recorded using a Zeiss LSM 710 upright microscope equipped with a 20 × water-immersion objective (plan-apochromat DIC, NA = 1.0). The pinhole was set at 32 μm, the pixel size was 0.59 μm, and pixel dwell time was 1.58 μs. Specimens were illuminated with a 25-mW Argon laser at 488 nm. Emission spectra were recorded from 492 to 598 nm. Images of a single focal plane were collected at 41-s intervals.
C. Results
Figure 3 shows the lateral views of a one-cell zebrafish embryo undergoing cytokinesis; the boxed regions are enlarged and shown at higher contrast below. The cortical actin filaments are brightly labeled by Utr-CH-GFP as the cleavage furrow develops. Cleavage appears to be unaffected by the utrophin probe (Movie S3, the same embryo). At t = 0 min, the cell is in late telophase, and Utr-CH-GFP fluorescence marks the aster-aster interaction zone (Wühr et al., 2010), where cytokinesis will cleave the cell. By t = 26 min, the daughter cells have re-entered mitosis, and Utr-CH-GFP stains comet tails behind rapidly moving vesicles (see Movie S3 beginning at t =18 min). These comet tails in mitotic cells presumably represent Arp2/3 nucleated assemblies akin to Listeria comet tails, which have been seen before in live embryos (Taunton et al., 2000; Velarde et al., 2007). Comet tail assembly and vesicle movement were abolished by treatment of embryos with the actin-depolymerizing agent cytochalasin B but were insensitive to the anti-microtubule drug nocodazole (data not shown). Although further validation of our utrophin-based labeling strategy is required, we consider it likely to be a useful, non-perturbing method for live imaging of the structure and function of the actin cytoskeleton in zebrafish embryos. One important question for future work is to what extent this probe reports on localization of all actin filaments versus a subset with particular structure or biochemistry.
Fig. 3.

F-actin imaging in an embryo injected with Utr-CH-GFP at the one-cell stage. Viewed from the side, the cortex is brightly labeled. The enlargements (shown at higher contrast below the top panels) illustrate the dynamics of the microfilaments. At t = 0 min, the cell is in late telophase, and Utr-CH-GFP staining marks the aster-aster interaction zone (yellow arrows). As the daughter cells from the first division re-enter mitosis (t = 26 min), Utr-CH-GFP-labeled comets propel vesicles (e.g. red arrow) whose movement is actin-dependent (see Section V.C for details). (See Plate no. 2 in the Color Plate Section.)
VI. Comparison of Microscopic Techniques for Imaging the Cytoskeleton of Cleaving Zebrafish Embryos
Table I summarizes the advantages and disadvantages of four fluorescence-imaging modalities we tested and provides representative micrographs obtained by each. We note that these comments apply to the instrument we used and may not represent fundamental limitations. For example, new gallium arsenide and avalanche photodiode detectors for scanning microscopes may increase sensitivity and lower noise and photobleaching, albeit at additional cost. In the two-cell embryo imaged by conventional epifluorescence, the spindles of each blastomere are quite blurry due to high background signal from out-of-focus fluorescence. Although basic measurements of mitosis, such as rate of spindle assembly, spindle size, and spindle orientation, could be made (see Movie S4), the dynamics of single microtubules, or even bundles, cannot be resolved. Spinning disc confocal microscopy provides greater clarity and single microtubule resolution of early mitotic spindles (Table I, Movie S5) provided that they are relatively close to the surface of the embryo. Spinning disc confocal often seems to provide an advantage over scanning laser confocal for live imaging of the cytoskeleton in tissue culture cells due to lower photobleaching and in some cases superior signal to noise. However, the lack of depth of penetration is a problem for application of current Yokogawa spinning discs to zebrafish embryos. We look forward to development of new spinning disc units with smaller pinholes that are optimized for lower magnification work at depth. For the large, cleaving blastomeres of the zebrafish embryo, laser-scanning confocal microscopy and two-photon microscopy were clearly superior because of their greater depth of penetration. With care to limit exposure, photobleaching and phototoxicity were not a problem with the one-photon modality. Both produced images of mitotic spindles with very high spatial resolution and contrast (Table I, Movie S6). The dynamics of individual microtubules were easily observed. Our current method of choice is laserscanning microscopy with one-photon excitation. This is partly due to the faster bleaching of GFP caused by two-photon excitation, but of greater importance is the higher signal-to-noise that was obtained with one-photon excitation (Movie S6). We do not understand to what extent these factors represent fundamental advantages of one-photon excitation versus limitations of the particular instruments we used.
Table I.
Imaging the zebrafish microtubule cytoskeleton during cleavage: Advantages and disadvantages of four fluorescence-imaging modalities
| Type | Advantages | Disadvantages | Example |
|---|---|---|---|
| Epifluorescence microscopy | - Least expensive - Fast - Efficient collection of photons (low bleaching and phototoxicity) - CCD camera (low noise, high sensitivity) |
- Blurry, especially in thick specimens at high magnification (low contrast) - Low depth penetration - Low contrast of microtubules |
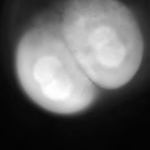 Movie S4 |
| Spinning disc confocal microscopy (SDCM) | - Fast - Less expensive than LSCM or 2PM - Faster than LSCM and 2PM - Fairly efficient collection of photons (low bleaching and phototoxicity) - Reasonable background suppression - CCD camera - Can generate optical sections |
- Fixed pinhole size, only optimal for certain objectives - Cross-talk between pinholes - Higher background than LSM especially in a thick specimen, leading to lower contrast images |
 Movie S5 |
| Laser-scanning confocal microscopy (LSCM) | - Can generate optical sections - Good depth penetration - Good background suppression |
- Slow scan rate - PMT is noisy (8-bit) - Expensive - Very few photons collected (bright signal required, fast bleaching, phototoxicity is likely without care to limit exposure) |
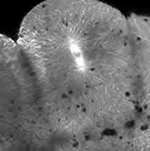 Movie S6 |
| Two-photon microscopy (2PM) | - Can generate optical sections - Excellent depth penetration - Very good background suppression |
- Slow scan rate - PMT is noisy (8-bit) - Expensive - Low brightness, fast bleaching, phototoxicity is likely (probably because common fluorophores are optimized for one-photon excitation) |
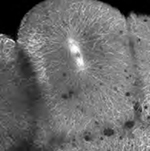 Movie S6 |
VII. Discussion and Future Directions
In this chapter, we describe methods to image microtubules and actin filaments in the thick cells of living cleavage-stage zebrafish embryos. Our methods make use of FP-tagged filament-binding proteins, EMTB-3GFP for microtubules and Utr-CH-GFP for microfilaments, that appear not to affect the dynamics or organization of the respective polymers. These probes yield superior contrast during live imaging when compared to filament labeling by fluorescently derivatized polymer subunits themselves (i.e. tubulin, actin), presumably because the free pools of the binding proteins are much lower than those of the filament subunits. Furthermore, we have successfully developed a transgenic zebrafish line that expresses EMTB-3GFP and shown that it yields valuable information about microtubule organization and function in cleavage-stage zebrafish embryos (Wühr et al., 2010). For analysis of microtubule function at later stages of development, we suggest that the EMTB-3GFP probe be introduced into strains lacking pigmentation [e.g. nacre (Lister etal., 1999) and casper (White et al., 2008)]. A disadvantage of EMTB-3GFP is that the probe turns over rapidly on microtubules (Bulinski et al., 2001), which prevents its use to measure microtubule turnover or sliding by photoactivation experiments (Mitchison, 1989). To enable such measurements, we suggest the creation of a zebrafish line that expresses tubulin linked to a photoconvertible FP (McKinney et al, 2009). Although we have not been able to generate a transgenic line that constitutively expresses Utr-CH-GFP, suggesting subtle toxic effects of this probe expressed at high levels, we consider it probable that such a line can be created, perhaps by using a weaker, or inducible, promoter. Indeed, we envision a bright future for live analysis of cellular dynamics of all kinds by use of transgenic zebrafish that express appropriate FP-tagged probes.
We chose to study the zebrafish embryo not only for its potential in understanding cytoskeletal function during development, but also because the blastomeres created during cleavage are among the largest of vertebrate cells. One of our goals is to understand how the cytoskeleton scales with cell size to solve the physical challenges of organizing large cells (Wühr et al., 2008). To this end, a combination of the unique experimental advantages of Xenopus egg extracts and transgenic zebrafish embryos is likely to yield important experimental synergisms. Xenopus egg extracts provide the opportunity to observe cytoskeletal function in vitro with single molecule resolution (Needleman et al., 2010), and the system can be easily titrated with proteins and drugs. Conversely, the zebrafish embryo provides experimental read out from a truly in vivo system. The two systems, used in combination, are likely to lead to rapid advances in our knowledge of cytoskeletal function and other cellular processes.
Supplementary Material
Acknowledgments
We thank David Burgess for generous gift of purified Utr-CH-GFP. H.W.D. was supported by NSF grant ANT-0635470. N.D.O and S.G.M. were supported by NHGRI P50 HG004071 and NIDCD R01 DC010791. This work was supported by the National Institutes of Health (NIH) grant GM39565.
Footnotes
In this discussion we make the explicit assumption that labeling of microtubules by Alexa-647-tubulin is uniform throughout the spindle.
Appendix A. Supplementary Movies
Supplementary data associated with this chapter can be found, in the online version, at http://www.elsevierdirect.com/companions/9780123870360.
References
- Amsterdam A, and Hopkins N (2004). Retroviral-mediated insertional mutagenesis in zebrafish In “Methods in Cell Biology,” (Detrich HW III, Westerfield M, and Zon LI, eds.), Vol. 77, pp. 3–20. Elsevier, San Diego. [DOI] [PubMed] [Google Scholar]
- Andersen E, Asuri N, Clay M, and Halloran M (2010). Live imaging of cell motility and actin cytoskeleton of individual neurons and neural crest cells in zebrafish embryos. J. Vis. Ex 36 DOI: 10.3791/1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakawa K, and Kawakami K (2010). A transgenic zebrafish for monitoring in vivo microtubule structures. Dev. Dyn 239, 2695–2699. [DOI] [PubMed] [Google Scholar]
- Bahary N, Davidson A, Ransom D, Shepard J, Stern H, Trede N, Zhou Y, Barut B, Zon LI (2004). The Zon laboratory guide to positional cloning in zebrafish In “Methods in Cell Biology,” (Detrich HW III, Westerfield M, and Zon LI, eds.), Vol. 77, pp. 305–329. Elsevier, San Diego. [DOI] [PubMed] [Google Scholar]
- Beis D, and Stainier DY (2006). In vivo cell biology: following the zebrafish trend. Trends Cell. Biol 16, 105–112. [DOI] [PubMed] [Google Scholar]
- Betchaku T, and Trinkaus JP (1978). Contactrelations, surface activity, and cortical microfilaments of marginal cells of the enveloping layer and of the yolk syncytial and yolk cytoplasmic layers of Fundulus before and during epiboly. J. Exp. Zool 206, 381–426. [DOI] [PubMed] [Google Scholar]
- Bulinski JC, Odde DJ, Howell BJ, Salmon TD, and Waterman-Storer CM (2001). Rapid dynamics of the microtubule binding of ensconsin in vivo. J. Cell Sci 114, 3885–3897. [DOI] [PubMed] [Google Scholar]
- Burkel BM,vonDassow G, andBement WM (2007). Versatile fluorescentprobes for actin filaments based on the actin-binding domain of utrophin. Cell Motil. Cytoskeleton 64, 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corley-Smith GE, Brandhorst BP, Walker C, and Postlethwait JH (1999). Production ofhaploid and diploid androgenetic zebrafish (including methodology for delayed in vitro fertilization) In “Methods in Cell Biology,” (Detrich HW III, Westerfield M, and Zon LI, eds.), Vol. 59, pp. 45–60. Elsevier, San Diego. [DOI] [PubMed] [Google Scholar]
- Danilchik MV, Bedrick SD, Brown EE, and Ray K (2003). Furrow microtubules and localized exocytosis in cleaving Xenopus laevis embryos. J. Cell Sci 116, 273–283. [DOI] [PubMed] [Google Scholar]
- Dekens MP, Pelegri FJ, Maischein HM, andNusslein-Volhard C (2003). The maternal-effect gene futile cycle is essential for pronuclear congression and mitotic spindle assembly in the zebrafish zygote. Development 130, 3907–3916. [DOI] [PubMed] [Google Scholar]
- Detrich HW, Westerfield M, and Zon LI (1999). Overview of the zebrafish system In “Methods in Cell Biology,” (Detrich HW III, Westerfield M, and Zon LI, eds.), Vol. 59, pp. 3–10. Elsevier, San Diego. [DOI] [PubMed] [Google Scholar]
- Driever W, Solnica-Krezel L, Schier AF, Neuhauss SCF, Malicki J, Stemple DL, Stainier DYR, Zwartkruis F, Abdelilah S, Rangini Z, et al. (1996). A genetic screen for mutations affecting embryogenesis in zebrafish. Development 123, 37–46. [DOI] [PubMed] [Google Scholar]
- Dumont S, and Mitchison TJ (2009). Compression regulates mitotic spindle length by a mechano- chemical switch at the poles. Curr Biol. 19, 1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faire K, Waterman-Storer CM, Gruber D, Masson D, Salmon ED, Bulinski JC (1999). E-MAP- 115 (ensconsin) associates dynamically with microtubules in vivo and is not a physiological modulator of microtubule dynamics. J. Cell Sci 112, 4243–4255. [DOI] [PubMed] [Google Scholar]
- Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, Kane DC, Odenthal J, van Eeden FJM, Jiang Y-J, Heisenberg C-P, et al. (1996). The identificationof geneswith unique and essential functions in the development of the zebrafish, Danio rerio. Development 123, 1–36. [DOI] [PubMed] [Google Scholar]
- Hyman A, Drechsel D, Kellogg D, Salser S, Sawin K, Steffen P, Wordeman L, Mitchison T (1991). Preparation of modified tubulins. Methods Enzymol. 196, 478–85. [DOI] [PubMed] [Google Scholar]
- Jesuthasan S, and Stahle U (1997). Dynamic microtubules and specification of the zebrafish embryonic axis. Curr. Biol 7, 31–42. [DOI] [PubMed] [Google Scholar]
- Kawakami K (2004). Transgenesis and gene trap methods in zebrafish by using the Tol2 transposable element In “Methods in Cell Biology,” (Detrich HW III, Westerfield M, and Zon LI, eds.), Vol. 77, pp. 201–222. Elsevier, San Diego. [DOI] [PubMed] [Google Scholar]
- Knaut H, Pelegri F, Bohmann K, Schwarz H, and Nusslein-Volhard C (2000). Zebrafish vasa RNA but not its protein is a component of the germ plasm and segregates asymmetrically before germline specification. J. Cell Biol 149, 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM,Yost HJ, Kanki JP, and Chien CB (2007). The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn 236, 3088–3099. [DOI] [PubMed] [Google Scholar]
- Li WM, Webb SE, Chan CM, and Miller AL (2008). Multiple roles of the furrow deepening Ca2+ transient during cytokinesis in zebrafish embryos. Dev. Biol 316, 228–248. [DOI] [PubMed] [Google Scholar]
- Li WM, Webb SE, Lee KW, and Miller AL (2006). Recruitment and SNARE-mediated fusion of vesicles in furrow membrane remodeling during cytokinesis in zebrafish embryos. Exp. Cell Res 312, 3260–3275. [DOI] [PubMed] [Google Scholar]
- Lindeman RE, and Pelegri F (2010). Vertebrate maternal-effect genes: Insights into fertilization, early cleavage divisions, and germ cell determinant localization from studies in the zebrafish. Mol. Reprod. Dev 77, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister JA, Robertson CP, Lepage T, Johnson SL, and Raible DW (1999). nacre encodes a zebrafish microphthalmia-related protein that regulates neural-crest-derived pigment cell fate. Development 126, 3757–3767. [DOI] [PubMed] [Google Scholar]
- McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, and Looger LL (2009). A bright and photostable photoconvertible fluorescent protein. Nat. Methods 6, 131–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ (1989). Polewards microtubule flux in the mitotic spindle: evidence from photoactivation of fluorescence. J Cell Biol 109, 637–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison T, and Kirschner M (1984). Dynamic instability of microtubule growth. Nature 312, 237–242. [DOI] [PubMed] [Google Scholar]
- Needleman DJ, Groen A, Ohi R, Maresca T, Mirny L, Mitchison T (2010). Fast microtubule dynamics in meiotic spindles measured by single molecule imaging: evidence that the spindle environment does not stabilize microtubules. Mol. Biol. Cell 21, 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, and Zon LI (1997). Genetics of erythropoiesis: induced mutations in mice and zebrafish. Annu. Rev. Genet 31, 33–60. [DOI] [PubMed] [Google Scholar]
- Pelegri F, and Mullins MC (2004). Genetic screens for maternal-effect mutations In “Methods in Cell Biology,” (Detrich HW III, Westerfield M, andZon LI, eds.),Vol. 77, pp. 21–51. Elsevier, San Diego. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Peluso P, and Mitchison TJ (1995). The bulk of unpolymerized actin in Xenopus egg extracts is ATP-bound. Mol. Biol. Cell 6, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N, Burgess S, and Hopkins N (1997). Delayed in vitro fertilization of zebrafish eggs in Hank’s saline containing bovine serum albumin. Mol. Mar. Biol. Biotechnol 6, 84–87. [PubMed] [Google Scholar]
- Siripattarapravat K, Busta A, Steibel JP, and Cibelli J (2009). Characterization and in vitro control of MPF activity in zebrafish eggs. Zebrafish 6, 97–105. [DOI] [PubMed] [Google Scholar]
- Skromne I, and Prince VE (2008). Current perspectives in zebrafish reverse genetics: moving forward. Dev. Dyn 237, 861–882. [DOI] [PubMed] [Google Scholar]
- Solnica-Krezel L, and Driever W (1994). Microtubule arrays of the zebrafishyolk cell: organization and function during epiboly. Development 120, 2443–2455. [DOI] [PubMed] [Google Scholar]
- Stiähle U, and Jesuthasan S (1993). Ultraviolet irradiation impairs epiboly in zebrafish embryos: evidence for a microtubule-dependent mechanism of epiboly. Development 119, 909–919. [DOI] [PubMed] [Google Scholar]
- Strasser MJ, Mackenzie NC, Dumstrei K, Nakkrasae LI, Stebler J, Raz E (2008). Control over the morphology and segregation ofzebrafish germ cell granules during embryonic development. BMC. Dev. Biol 8, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taunton J, Rowning BA, Coughlin ML, Wu M, Moon RT, Mitchison TJ, Larabell CA (2000). Actin-dependent propulsion of endosomes and lysosomes by recruitment of N-WASP. J. Cell Biol 148, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theusch EV, Brown KJ, and Pelegri F (2006). Separate pathways of RNA recruitment lead to the compartmentalization of the zebrafish germ plasm. Dev Biol 292, 129–141. [DOI] [PubMed] [Google Scholar]
- Topczewski J, and Solnica-Krezel L (2009). Cytoskeletal dynamics of the zebrafish embryo. In “Essential Zebrafish Methods, Part A: Cell and Developmental Biology,” (Detrich HW III, Westerfield M, and Zon LI, eds.), pp. 133–157. Elsevier, San Diego. [Google Scholar]
- Trinkaus JP (1949). The surface gel layer of Fundulus eggs in relation to epiboly. Proc. Natl. Acad. Sci. U. S.A 35, 218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkaus JP(1951).A study of mechanisms of epiboly inthe egg of Fundulus heteroclitus. J. Exp. Zool 118, 269–319. [Google Scholar]
- Urasaki A, Morvan G, and Kawakami K (2006). Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics 174, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velarde N, Gunsalus KC, and Piano F (2007). Diverse roles of actin in C. elegans early embryogenesis. BMC Dev. Biol 7, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Dassow G, Verbrugghe KJ, Miller AL, Sider JR, and Bement WM (2009). Action at a distance during cytokinesis. J. Cell Biol 187, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M (2007). The zebrafish book: A guide for the laboratory use of zebrafish (Danio rerio) University of Oregon Press, Eugene, Oregon. [Google Scholar]
- White RM, Sessa A, Burke C, Bowman T, LeBlanc J, Ceol C, Bourque C, Dovey M, Goessling W, Burns CE, et al. (2008). Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2, 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wühr M, Chen Y, Dumont S, Groen AC, Needleman DJ, Salic A, Mitchison TJ (2008). Evidence for an upper limit to mitotic spindle length. Curr Biol. 18, 1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wühr M, Tan ES, Parker SK, Detrich HW III, and Mitchison TJ (2010). A model for cleavage plane determination in early amphibian and fish embryos. Curr. Biol 20, 2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe T, Ge X, Lindeman R, Nair S, Runke G, Mullins MC, Pelegri F (2009). The maternal-effect gene cellular island encodes aurora B kinase and is essential for furrow formation in the early zebrafish embryo. PLoS. Genet 5, e1000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y, Kronebusch PJ, Simon PM, and Borisy GG (1996). Microtubule dynamics at the G2/M transition: abrupt breakdown of cytoplasmic microtubules at nuclear envelope breakdown and implications for spindle morphogenesis. J. Cell Biol 135, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.