

Key Points
Question
Is monoamine oxidase B (MAO-B) distribution volume, an index of MAO-B density, greater in the prefrontal cortex during major depressive episodes of MDD?
Findings
This case-control study demonstrated that MAO-B distribution volume was significantly elevated in the prefrontal cortex of patients with major depressive episodes (n = 20) compared with healthy controls (n = 20) (mean, 26%). Duration of illness was significantly and positively correlated with MAO-B distribution volume in the prefrontal cortex.
Meaning
Results suggest that there is a novel phenotype of elevated MAO-B level in the prefrontal cortex of patients with major depressive episodes secondary to major depressive disorder for which low-cost identifiers and selective therapeutics compatible with serotonin reuptake inhibitors should be developed.
This case-control study investigates whether monoamine oxidase B distribution volume is elevated in the prefrontal cortex in major depressive episodes in patients with major depressive disorder.
Abstract
Importance
Monoamine oxidase B (MAO-B) is an important, high-density enzyme in the brain that generates oxidative stress by hydrogen peroxide production, alters mitochondrial function, and metabolizes nonserotonergic monoamines. Recent advances in positron emission tomography radioligand development for MAO-B in humans enable highly quantitative measurement of MAO-B distribution volume (MAO-B VT), an index of MAO-B density. To date, this is the first investigation of MAO-B in the brain of major depressive disorder that evaluates regions beyond the raphe and amygdala.
Objective
To investigate whether MAO-B VT is elevated in the prefrontal cortex in major depressive episodes (MDEs) of major depressive disorder.
Design, Setting, and Participants
This case-control study was performed at a tertiary care psychiatric hospital from April 1, 2014, to August 30, 2018. Twenty patients with MDEs without current psychiatric comorbidities and 20 age-matched controls underwent carbon 11–labeled [11C]SL25.1188 positron emission tomography scanning to measure MAO-B VT. All participants were drug and medication free, nonsmoking, and otherwise healthy.
Main Outcomes and Measures
The MAO-B VT in the prefrontal cortex (PFC). The second main outcome was to evaluate the association between MAO-B VT in the PFC and duration of major depressive disorder illness.
Results
Twenty patients with MDEs (mean [SD] age, 34.2 [13.2] years; 11 women) and 20 healthy controls (mean [SD] age, 33.7 [13.1] years; 10 women) were recruited. Patients with MDEs had significantly greater MAO-B VT in the PFC (mean, 26%; analysis of variance, F1,38 = 19.6, P < .001). In individuals with MDEs, duration of illness covaried positively with MAO-B VT in the PFC (analysis of covariance, F1,18 = 15.2, P = .001), as well as most other cortex regions and the thalamus.
Conclusions and Relevance
Fifty percent (10 of 20) of patients with MDEs had MAO-B VT values in the PFC exceeding those of healthy controls. Greater MAO-B VT is an index of MAO-B overexpression, which may contribute to pathologies of mitochondrial dysfunction, elevated synthesis of neurotoxic products, and increased metabolism of nonserotonergic monoamines. Hence, this study identifies a common pathological marker associated with downstream consequences poorly targeted by the common selective serotonin reuptake inhibitor treatments. It is also recommended that the highly selective MAO-B inhibitor medications that are compatible for use with other antidepressants and have low risk for hypertensive crisis should be developed or repurposed as adjunctive treatment for MDEs.
Introduction
Major depressive disorder (MDD) is the leading cause of death and disability across moderate- to high-income nations as a result of a high lifetime prevalence of 15% and rates of treatment resistance of 50%.1,2 Although dysfunction of several key systems in MDD have been identified, such as signal transduction, neuroplasticity, hypothalamic-pituitary-adrenal axis function, glutamate cycling, inflammation, hippocampal volume, and monoamine availability, a plausible reason for the lack of progress in creating new therapeutics for MDD is that it is heterogeneous and some fundamental pathologies remain elusive.3,4,5,6
Monoamine oxidase B (MAO-B), a protein of 520 amino acids, is an important enzyme largely overlooked in the pathophysiology of MDD. In the brain, MAO-B is a high-density protein mainly located on the outer mitochondrial membrane within astrocytes and serotonin-releasing neurons that has key roles in generating oxidative signaling by hydrogen peroxide production and metabolizing monoamines, such as dopamine, norepinephrine, benzylamine, and phenylethylamine.7,8,9 In brain tissue, the density of MAO-B is highly positively correlated with its activity.9,10 There are only 2 previous studies of MAO-B in postmortem brain tissue of MDD (by the same group of researchers) measuring [3H]lazabemide binding in overlapping samples of individuals with MDD, with the first study11 evaluating the dorsal raphe nucleus in 12 individuals with MDD and the second study12 investigating the amygdala in 15 individuals with MDD. Both investigations reported negative results, but the sensitivity may have been reduced by concurrent cigarette smoking in 40% of the individuals because cigarette smoke contains MAO-B binding chemicals and is associated with lower [3H]lazabemide binding.12 Moreover, because these 2 studies sampled a combination of early- and late-onset MDD, the question of whether MAO-B level or activity is altered in early-onset MDD is unknown.
There are several compelling reasons why MAO-B level may be elevated in the brain of early-onset MDD, particularly in the prefrontal cortex (PFC), a region not previously studied in MDD to date. Many biological abnormalities of MDD are consequent to increased glucocorticoid exposure, and greater MAO-B expression is inducible by paradigms increasing glucocorticoid exposure. Glucocorticoids increase transcription of MAO-B through diverse mechanisms, including (1) glucocorticoid binding to the glucocorticoid response element 4 (GRE4) site of the MAO-B promoter and (2) reduction of transcription factors EAPP and R1, which are 2 transcription factors that repress MAO-B transcription through binding to Sp1 sites in the central core promoter region of MAO-B. With less binding of EAPP and R1, as well as greater binding of glucocorticoids to GRE4, endogenous Sp1 is able to cooperatively increase transcription of MAO-B.13 Also, glucocorticoids promote the expression of transforming growth factor-β-inducible early gene (TIEG2), which bind to proximal Sp1 binding sites on the core promoter region of MAO-B,14 leading to greater MAO-B transcription. These mechanisms, identified in glioblastoma and neuroblastoma cell lines, are particularly relevant to the PFC because brain-penetrant doses of dexamethasone and chronic unpredictable stress are consistently associated with greater MAO-B messenger RNA when this region is prioritized in the analyses.13,15,16,17,18,19 Furthermore, in the PFC of major depressive episodes (MDEs), R1 (also known as RAM2/CDCA7L/JPO2), which inhibits transcription of MAO-B in cell culture,13 is low, while TIEG2, which increases transcription of MAO-B in cell culture,13,18 is elevated. Hence, both transcription factors (R1 and TIEG2) are altered in the PFC of MDEs in directions consistent with glucocorticoid agonism that increase MAO-B transcription.18,20
Recent advances in positron emission tomography (PET) radioligand development for MAO-B in humans enable robust quantitation of MAO-B distribution volume (VT), an index of MAO-B density. Carbon 11–labeled [11C]deprenyl, the first radiotracer for MAO-B imaging with PET,21 had poor reversibility and radioactive metabolites found in both brain and periphery. To improve reversibility, newer analogues were created, such as deuterium-labeled [11C]deprenyl and then deuterium-labeled [18F]deprenyl, the latter of which has been modeled in monkeys, although there may be some bias from brain-penetrant metabolites of these compounds.22,23 Bramoullé et al24 discovered a radiotracer with a different structure, [11C]SL25.1188, then modeled it in baboons.25 Unfortunately, the first production method for [11C]SL25.1188 required the esoteric carbon 11–labeled phosgene, making it difficult to use for clinical translation studies,24 so a new synthesis method was discovered by Vasdev and colleagues, and then the radiotracer was modeled in humans.26,27,28 [11C]SL25.1188 has outstanding properties, including high reversibility, brain uptake, and selectivity for MAO-B, as well as no brain-penetrant metabolites and a very reproducible total VT measure that is highly correlated with the known concentration of MAO-B in postmortem human brain (r2 > 0.9).25,26,27,28
Given that dexamethasone and chronic stress increase MAO-B gene expression and activity transcription and that 2 nuclear transcription factors (R1 and TIEG2) are dysregulated in the PFC of MDEs in a manner associated with increasing MAO-B transcription, our primary hypothesis was that MAO-B VT, measured with [11C]SL25.1188 PET, is elevated in the PFC of MDEs secondary to early-onset MDD. The second hypothesis was that greater MAO-B VT in the PFC is associated with longer duration of illness because increased expression of MAO-B also occurs in glial fibrillary acidic protein–positive (GFAP) reactive astrocytes during reactive astrogliosis and because age-associated increase in GFAP immunoreactivity and protein level is greater in MDD.29,30 Moreover, astrogliosis, a common response to injury in central nervous system disease, may also be associated with greater microglial activation,10,31 and later-stage MDD is associated with the highest level of microglial activation.32 The third objective, which is more exploratory, was to investigate differences in MAO-B VT between MDEs and health in other gray matter brain regions, including those with roles associated with symptoms of MDD and/or which have high MAO-B density, such as the anterior cingulate cortex (ACC), ventral striatum, and dorsal putamen.
Methods
Participants
This case-control study was performed from April 1, 2014, to August 30, 2018. Twenty patients with unmedicated MDEs secondary to MDD without current psychiatric comorbidities and 20 age-matched healthy controls completed the study (Table 1). Participants were recruited from the community (Toronto, Ontario, Canada) and a tertiary care psychiatric hospital (Centre for Addiction and Mental Health, Toronto). Participants provided written informed consent after all procedures were fully explained. The protocol and informed consent forms were approved by the Research Ethics Board of the Centre for Addiction and Mental Health.
Table 1. Characteristics of Participants.
| Variable | Patients With MDEs (n = 20) | Healthy Controls (n = 20) | P value |
|---|---|---|---|
| Age, mean (SD), y | 34.2 (13.2) | 33.7 (13.1) | .91a |
| Age at first MDE, mean (SD), y | 23.7 (8.9) | NA | NA |
| Female sex, No. (%) | 11 (55) | 10 (50) | .75b |
| BMI, mean (SD) | 23.2 (4.6) | 23.7 (3.4) | .71a |
| Alcohol consumption, mean (SD), standard drinks per week | 0.81 (1.21) | 1.55 (2.18) | .21a |
| Education, mean (SD), y | 15.4 (2.2) | 15.3 (1.8) | .88a |
| Racial/ethnic background, No. | |||
| European/white | 11 | 11 | .71c |
| East Asian | 3 | 6 | |
| East Indian | 1 | 1 | |
| African American/Caribbean | 2 | 1 | |
| Hispanic/Latino/Spanish | 1 | 1 | |
| Mixed | 2 | 0 | |
| HDRS score, mean (SD) | 19.1 (3.0) | NA | NA |
| No. of previous MDEs, mean (SD) | 4 (2) | NA | NA |
| Antidepressant free, No. (%) | 20 (100) | NA | NA |
| Previous antidepressant trial, No. (%) | 9 (45) | NA | NA |
| Duration of illness, mean (SD), y | 10.6 (10.4) | 0 | NA |
Abbreviations: BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); HDRS, 17-item Hamilton Depression Rating Scale; MDEs, major depressive episodes; NA, not applicable.
Statistical analysis was performed using independent-samples t test.
Statistical analysis was performed using χ2 test.
Statistical analysis was performed using Fisher exact test.
Participants ranged in age from 19 to 66 years, were nonsmoking, and had good physical health. None had concurrent active Axis I disorders, and none had a history of neurological illness, cerebrovascular disease, autoimmune disease, Axis II disorders, psychotic symptoms, or substance abuse. None had used herbal remedies in the previous month. All were drug and medication free within the past month except for oral contraceptives. Healthy controls were age matched within 5 years to patients with MDEs.
Diagnosis was verified by the Structured Clinical Interview for DSM-IV33 and confirmed by consultation with a psychiatrist (J.H.M.). Patients had early-onset type MDEs (before age 45 years), were antidepressant free for at least 1 month (6 weeks if previously taking fluoxetine), and had a minimum score of 16 on the 17-item Hamilton Depression Rating Scale at screening.34 None had active comorbid Axis I disorders or a history of psychotic symptoms or mania. All participants were requested to abstain from alcohol for 2 days before the PET scan and to consume no grapefruit juice or caffeinated products on the PET scan day.
PET and Magnetic Resonance Imaging Acquisition and Image Analysis
A 3-dimensional high-resolution research tomograph (HRRT; CPS/Siemens) PET scanner system, which provides radioactivity in 207 slices with an interslice distance of 1.22 mm, was used for all patients and healthy controls. After the transmission scan, [11C]SL25.1188 was infused intravenously over a 30-second period at a constant rate using a Harvard infusion pump (Harvard Apparatus). Data were acquired as previously described.28 For the anatomic delineation of regions of interest, a brain magnetic resonance image was acquired for each individual (eAppendix in the Supplement). The MAO-B VT was calculated using a 2-tissue compartment model previously shown to be optimal for [11C]SL25.1188 PET quantification.28
Statistical Analysis
For the first main hypothesis, the PFC MAO-B VT was compared across groups (patients with MDEs vs healthy controls) using analysis of variance (ANOVA) to assess the effect of group. Also, to compare MAO-B VT between MDEs and health across a broader range of regions, including the PFC, ACC, ventral striatum, and dorsal putamen, a repeated-measures ANOVA was applied with regional MAO-B VT as the repeated measure, assessing the effect of group and region. For the second main hypothesis, to assess the association of illness duration with MAO-B VT in the PFC, an analysis of covariance (ANCOVA) was applied with duration of illness as the covariate. Duration of illness is calculated as the date of onset of first MDE subtracted from the date of the PET scan. Then, the association of duration of illness with MAO-B VT was explored in each brain region applying a repeated-measures ANCOVA with duration of illness as the covariate and region as the repeated measure. For the 2 main hypotheses, a corrected threshold of 2-sided P = .025 was required, and remaining analyses were exploratory. Exploratory analyses of the association of MAO-B VT in each brain region with other clinical variables, such as age, Hamilton Depression Rating Scale severity, and the number of previous MDEs, were also investigated applying an ANCOVA with MAO-B VT in the PFC and repeated-measures ANCOVA with regional MAO-B VT as the repeated dependent variable.
Results
Twenty patients with MDEs (mean [SD] age, 34.2 [13.2] years; 11 women) and 20 healthy controls (mean [SD] age, 33.7 [13.1] years; 10 women) were recruited. The PFC MAO-B VT was a mean of 26% greater in patients with MDEs (ANOVA effect of group with MAO-B VT, F1,38 = 19.6; P < .001; Cohen d = 1.4). Given that the lowest VT value in the PFC among healthy controls was notably lower than the others, we investigated the sensitivity of our model to removing this value, and the statistical significance remained (ANOVA effect of group with MAO-B VT, F1,37 = 19.7; P < .001). The highest VT values in each region for patients with MDEs were not from the same individual. A broader comparison of MAO-B VT between MDEs and health across subregions of the PFC and a number of additional gray matter brain regions found a significant effect of diagnosis (repeated-measures ANOVA, F1,38 = 8.7; P = .005) and a significant interaction with region (F7,32 = 6.5; P < .001). The interaction with region reflected a relatively greater difference between groups in the ventrolateral PFC (mean, 26%; ANOVA, F1,38 = 12.2; P = .001), dorsolateral PFC (mean, 16%; ANOVA, F1,38 = 7.4; P = .01), orbitofrontal cortex (mean, 13%; ANOVA, F1,38 = 4.7; P = .04), thalamus (mean, 17%; ANOVA, F1,38 = 8.8; P = .005), and inferior parietal cortex (mean, 16%; ANOVA, F1,38 = 9.0; P = .005) (Figure 1, Figure 2, Table 2, and eFigure 1 and eFigure 2 in the Supplement).
Figure 1. Elevated MAO-B Total Distribution Volume During Major Depressive Episodes (MDEs) in the Prefrontal Cortex.

The MAO-B VT was significantly greater in the 20 patients with MDEs compared with the 20 healthy controls. Calculation of analysis of variance resulted in the following associations of diagnosis with region: prefrontal cortex (F1,38 = 19.6; P < .001), ventrolateral prefrontal cortex (F1,38 = 12.2; P = .001), dorsolateral prefrontal cortex (F1,38 = 7.4; P = .01), orbitofrontal cortex (F1,38 = 4.7; P = .04), and medial prefrontal cortex (F1,38 = 2.6; P = .12). MAO-B indicates monoamine oxidase B; MAO-B VT, MAO-B density measured by distribution volume.
Figure 2. Elevated MAO-B Total Distribution Volume During Major Depressive Episodes (MDEs).
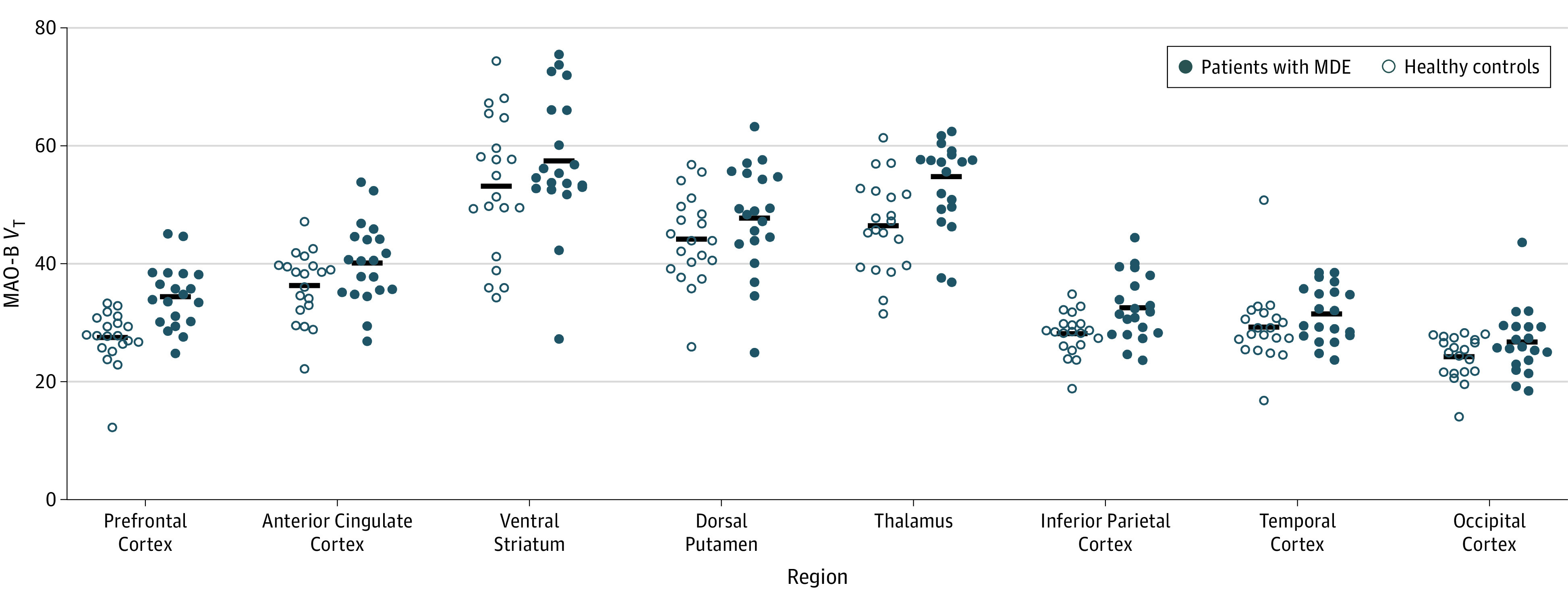
Calculation of analysis of variance resulted in the following interactions of diagnosis with region: prefrontal cortex (F1,38 = 19.6; P < .001), thalamus (F1,38 = 8.8; P = .005), and inferior parietal cortex (F1,38 = 9.0; P = .005). MAO-B indicates monoamine oxidase B; MAO-B VT, MAO-B total distribution volume.
Table 2. Analysis of Variance Comparing Regional MAO-B VT Between Patients With MDEs and Healthy Controlsa.
| Variable | MAO-B VT, Mean (SD) | Difference, % | F1,38 Score | P Value | |
|---|---|---|---|---|---|
| Patients With MDEs | Healthy Controls | ||||
| Prefrontal cortex | 34.5 (5.3) | 27.5 (4.6) | 26 | 19.6 | <.001 |
| Ventrolateral prefrontal cortex | 32.2 (7.6) | 25.6 (3.8) | 26 | 12.2 | .001 |
| Dorsolateral prefrontal cortex | 32.8 (5.5) | 28.2 (5.4) | 16 | 7.4 | .01 |
| Orbitofrontal cortex | 29.1 (4.6) | 25.7 (5.3) | 13 | 4.7 | .04 |
| Medial prefrontal cortex | 35.4 (6.2) | 32.6 (4.8) | 9 | 2.6 | .12 |
| Anterior cingulate cortex | 40.2 (6.9) | 36.3 (5.9) | 11 | 3.7 | .06 |
| Ventral striatum | 57.5 (11.5) | 53.2 (11.8) | 8 | 1.4 | .25 |
| Dorsal putamen | 47.7 (9.0) | 44.2 (7.5) | 8 | 1.8 | .19 |
| Thalamus | 54.6 (9.4) | 46.5 (7.9) | 17 | 8.8 | .005 |
| Inferior parietal cortex | 32.6 (5.5) | 28.2 (3.6) | 16 | 9.0 | .005 |
| Temporal cortex | 31.6 (4.7) | 29.3 (6.3) | 8 | 1.7 | .20 |
| Occipital cortex | 26.7 (5.5) | 24.2 (3.7) | 10 | 2.8 | .10 |
Abbreviations: MAO-B, monoamine oxidase B; MAO-B VT, MAO-B density measured by distribution volume; MDEs, major depressive episodes.
Analysis of variance evaluated MAO-B VT as the dependent variable and group as the predictor variable.
In the PFC in MDEs, longer duration of illness was associated with greater MAO-B VT (ANCOVA, r = 0.68, F1,18 = 15.2; P = .001) (Figure 3). Given that 95% of our sample was aged 18 to 55 years and associations of age with MAO-B density are not observed before age 55 years,9,35 as expected there was no association herein between MAO-B VT and age in the PFC (F1,38 = 0.7; P = .40).
Figure 3. Association Between the Prefrontal Cortex MAO-B Total Distribution Volume and Duration of Illness.

Analysis of covariance evaluated MAO-B VT as the dependent variable and duration of illness as the covariate (analysis of covariance, r = 0.68; F1,18 = 15.2; P = .001). After removing the highest MAO-B VT value in the prefrontal cortex, the statistical significance remained (analysis of covariance, r = 0.65; F1,17 = 12.3; P = .003). MAO-B indicates monoamine oxidase B; MAO-B VT, MAO-B total distribution volume.
In MDEs, longer duration of illness was also associated with greater MAO-B VT in other brain regions. A general regional evaluation of the association of MAO-B VT with duration of illness applying a repeated-measures ANCOVA found a significant association of duration of illness (F1,18 = 18.1; P < .001) and a significant interaction with region (F7,12 = 9.1; P = .02). In MDEs, longer duration of illness was associated with greater MAO-B VT in the ventrolateral PFC, dorsolateral PFC, orbitofrontal cortex, ACC, thalamus, inferior parietal cortex, temporal cortex, and occipital cortex (eTable in the Supplement).
Discussion
Our primary finding is that MAO-B VT is robustly elevated in the PFC during MDEs of MDD (Cohen d = 1.4). The differences between MDEs and health were more prominent in cortical regions proximal to the ventrolateral PFC and in the thalamus. The secondary finding is that longer duration of MDD illness is associated with greater PFC MAO-B VT. This latter finding was prominent across cortical regions and the thalamus. These results have important implications for MAO-B in the pathophysiology of MDD, neuroprogression in MDD, and therapeutic development.
Robustly elevated MAO-B VT in the PFC of MDD is important because elevated MAO-B level is implicated in impairment of mitochondrial function, synthesis of neurotoxic products, and dysregulation of nonserotonergic monoamines. In cell lines, rodent transgenic models of overexpression, and postmortem human brain, greater level of MAO-B is associated with increased MAO-B activity.9,10,36,37 Overexpression of MAO-B is also associated with greater production of hydrogen peroxide, which may adversely influence mitochondrial function and reserve capacity as implicated by reductions in pyruvate dehydrogenase, succinate dehydrogenase, and mitochondrial aconitase, as well as downstream inhibition of mitochondrial complex 1 activity through formation of dopaminochrome from dopamine.36,37 Reduced mitochondrial complex 1 occurs in the PFC of MDD.38 A pathological role for elevated MAO-B in neurodegeneration was proposed through its function in metabolizing rare endogenous neurochemicals, leading to neurotoxic metabolic products, such as aldehydes,36,39,40 and it is known that MAO-B metabolizes exogenously administered 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine, leading to neurotoxic and behavioral sequelae of reduced and slowed movement.41,42 In a transgenic mouse model of globally increased MAO-B in astrocytes, abnormal behaviors compatible with several psychiatric syndromes, including those in MDD, were found during open field assessment, such as reduced total movement, less distance traveled, lower movement speed, and decreased duration of time spent moving.36,43 In humans, when MAO-B substrates dopamine and norepinephrine are depleted with the tyrosine hydroxylase inhibitor α-methylparatyrosine, depressed mood occurs with concurrent activation networks of structures that generate depressed mood, including subregions of the PFC.44,45 For these reasons, it is widely believed that in neuropsychiatric diseases elevated MAO-B level is a pathological target.
With 50% (10 of 20) of MDE cases herein having PFC MAO-B VT values exceeding the highest MAO-B VT values of healthy controls, there is an opportunity to develop and repurpose selective MAO-B inhibitors for MDD. Treatment of early-onset MDD most commonly involves selective serotonin reuptake inhibitors (SSRIs) and serotonin norepinephrine reuptake inhibitors (SNRIs), which are not well matched to the aforementioned potentially harmful sequelae of elevated MAO-B level, such as greater oxidative stress and reduced mitochondrial function, increased metabolism of nonserotonergic monoamines, and production of toxic metabolic products. While traditional nonselective and partially selective MAO-B inhibitors are not frequently used because they inhibit MAO-A, which is incompatible with SSRI and SNRI use due to the risk of serotonin syndrome, and because their reduction of peripheral tissue metabolism of tyramine creates risk for a hypertensive crisis, MAO-B inhibitors with high selectivity for MAO-B over MAO-A, such as sembragiline (completed phase 2 studies for Alzheimer disease46) and pioglitazone (approved for type 2 diabetes47) demonstrate compatibility with SSRI use. Although a recent meta-analysis of double-blind placebo-controlled studies favors pioglitazone,47 neither sembragiline nor pioglitazone has been developed for MDD.
Longer duration of illness was associated with greater MAO-B VT in the PFC, as well as most other cortical regions and the thalamus. The association between longer duration of illness and greater regional MAO-B VT is not accounted for by age because the association of age with MAO-B VT is negligible in this sample, with age-related influences on MAO-B density not beginning until ages 55 to 70 years.9,35 Greater MAO-B density may occur during reactive astrogliosis and may be associated with progression of neuropsychiatric diseases, such as Alzheimer disease, amyotrophic lateral sclerosis, multisystem atrophy, and progressive supranuclear palsy.10,48,49 In these conditions, GFAP and MAO-B levels are often highly correlated, which led to the argument that greater MAO-B density may be an in vivo marker of reactive astrogliosis49,50; however, development of in vivo PET markers of astrogliosis is an active area of ongoing investigation.51 Astrogliosis would not account for differences between MDD and health because GFAP is reduced in the orbitofrontal, dorsolateral, and subgenual PFC in MDE samples inclusive of younger individuals.52,53,54 However, such differences are not established for later stages of MDD because reduced GFAP was not present in late-life MDEs.55 Moreover, 2 studies29,30 that investigated GFAP in relation to age and duration of illness found a much greater rise in GFAP with age in MDD than health in the region sampled, the dorsolateral PFC. Hence, a plausible explanation for the association between greater MAO-B and duration of illness is gradually increasing astrogliosis.
Limitations
Some limitations of the present study should be addressed. First, as is standard with PET imaging studies, the measure of MAO-B VT reflects binding of the radiotracer to MAO-B plus nonspecific binding. However, our latest estimate of nonspecific binding based on blocking studies in humans is low in the PFC at 7% (J.H.M., unpublished data, 2018), so it is unlikely to account for a mean 26% elevation in MAO-B VT because this would require extremely robust elevations in free and nonspecific binding, plausibly exceeding 300%. Second, the specific binding compartment of the MAO-B VT reflects both density and affinity of the radiotracer to MAO-B, although empirically MAO-B VT quantitated with [11C]SL25.1188 PET is highly correlated with MAO-B density in human brain.28 Third, the association between duration of illness and MAO-B VT is based on cross-sectional data, so it is possible that the association with long histories of MDEs could reflect another closely related phenomenon of illness persistence, such as treatment resistance, a direction that may be investigated further in the future. Fourth, while a strength of our study is that we restricted comorbidity to assess the associations of MDEs vs health, future study will need to ascertain the extent to which our results also apply to MDEs with specific comorbid illnesses.
Conclusions
We found higher MAO-B VT in MDEs secondary to MDD, primarily in the PFC. Differences were largest in the ventrolateral PFC and nearby cortical regions, as well as in the thalamus. In the PFC, but also throughout the cortex and in the thalamus, MAO-B VT was more elevated in those with longer duration of illness, a common finding in neuroprogressive illnesses. The collective findings argue that MAO-B, which when overexpressed is implicated in mitochondrial dysfunction, excessive generation of hydrogen peroxide, nonserotonergic monoamine metabolism, and production of toxic metabolites, should be considered a distinct target in MDD and receive greater attention in therapeutic development. This is especially recommended given that well-tolerated MAO-B inhibitors with preferential selectivity compared with MAO-A inhibitors are compatible with use of commonly prescribed SSRI and SNRI antidepressants.
eAppendix. Methods, Results, and Discussion
eFigure 1. Relationship Between Prefrontal Cortex MAO-B Distribution Volume and Severity of Major Depressive Episode
eFigure 2. Total Distribution Volume in Healthy and Major Depressive Episode Subjects
eTable. Analysis of Variance Comparing Regional MAO-B Density and Duration of Major Depressive Disorder
References
- 1.Mathers C, Fat DM, Boerma JT. The Global Burden of Disease: 2004 Update. Geneva, Switzerland: World Health Organization; 2008. doi: 10.1016/B978-012373960-5.00335-X [DOI] [Google Scholar]
- 2.Trivedi MH, Rush AJ, Wisniewski SR, et al. ; STAR*D Study Team . Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163(1):28-40. doi: 10.1176/appi.ajp.163.1.28 [DOI] [PubMed] [Google Scholar]
- 3.Dwivedi Y. Pathogenetic and therapeutic applications of microRNAs in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:341-348. doi: 10.1016/j.pnpbp.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Setiawan E, Wilson AA, Mizrahi R, et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry. 2015;72(3):268-275. doi: 10.1001/jamapsychiatry.2014.2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyer JH. Neuroprogression and immune activation in major depressive disorder. Mod Trends Pharmacopsychiatry. 2017;31:27-36. doi: 10.1159/000470804 [DOI] [PubMed] [Google Scholar]
- 6.Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. 2016;22(3):238-249. doi: 10.1038/nm.4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saura J, Bleuel Z, Ulrich J, et al. Molecular neuroanatomy of human monoamine oxidases A and B revealed by quantitative enzyme radioautography and in situ hybridization histochemistry. Neuroscience. 1996;70(3):755-774. doi: 10.1016/S0306-4522(96)83013-2 [DOI] [PubMed] [Google Scholar]
- 8.Saura J, Kettler R, Da Prada M, Richards JG. Quantitative enzyme radioautography with 3H-Ro 41-1049 and 3H-Ro 19-6327 in vitro: localization and abundance of MAO-A and MAO-B in rat CNS, peripheral organs, and human brain. J Neurosci. 1992;12(5):1977-1999. doi: 10.1523/JNEUROSCI.12-05-01977.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tong J, Meyer JH, Furukawa Y, et al. Distribution of monoamine oxidase proteins in human brain: implications for brain imaging studies. J Cereb Blood Flow Metab. 2013;33(6):863-871. doi: 10.1038/jcbfm.2013.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saura J, Luque JM, Cesura AM, et al. Increased monoamine oxidase B activity in plaque-associated astrocytes of Alzheimer brains revealed by quantitative enzyme radioautography. Neuroscience. 1994;62(1):15-30. doi: 10.1016/0306-4522(94)90311-5 [DOI] [PubMed] [Google Scholar]
- 11.Klimek V, Roberson G, Stockmeier CA, Ordway GA. Serotonin transporter and MAO-B levels in monoamine nuclei of the human brainstem are normal in major depression. J Psychiatr Res. 2003;37(5):387-397. doi: 10.1016/S0022-3956(03)00045-1 [DOI] [PubMed] [Google Scholar]
- 12.Karolewicz B, Klimek V, Zhu H, et al. Effects of depression, cigarette smoking, and age on monoamine oxidase B in amygdaloid nuclei. Brain Res. 2005;1043(1-2):57-64. doi: 10.1016/j.brainres.2005.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen K, Ou XM, Wu JB, Shih JC. Transcription factor E2F-associated phosphoprotein (EAPP), RAM2/CDCA7L/JPO2 (R1), and simian virus 40 promoter factor 1 (Sp1) cooperatively regulate glucocorticoid activation of monoamine oxidase B. Mol Pharmacol. 2011;79(2):308-317. doi: 10.1124/mol.110.067439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ou XM, Chen K, Shih JC. Dual functions of transcription factors, transforming growth factor-β-inducible early gene (TIEG)2 and Sp3, are mediated by CACCC element and Sp1 sites of human monoamine oxidase (MAO) B gene. J Biol Chem. 2004;279(20):21021-21028. doi: 10.1074/jbc.M312638200 [DOI] [PubMed] [Google Scholar]
- 15.Slotkin TA, Seidler FJ, Ritchie JC. Effects of aging and glucocorticoid treatment on monoamine oxidase subtypes in rat cerebral cortex: therapeutic implications. Brain Res Bull. 1998;47(4):345-348. doi: 10.1016/S0361-9230(98)00111-7 [DOI] [PubMed] [Google Scholar]
- 16.Lin YH, Liu AH, Xu Y, Tie L, Yu HM, Li XJ. Effect of chronic unpredictable mild stress on brain–pancreas relative protein in rat brain and pancreas. Behav Brain Res. 2005;165(1):63-71. doi: 10.1016/j.bbr.2005.06.034 [DOI] [PubMed] [Google Scholar]
- 17.Kumar B, Kuhad A, Chopra K. Neuropsychopharmacological effect of sesamol in unpredictable chronic mild stress model of depression: behavioral and biochemical evidences. Psychopharmacology (Berl). 2011;214(4):819-828. doi: 10.1007/s00213-010-2094-2 [DOI] [PubMed] [Google Scholar]
- 18.Harris S, Johnson S, Duncan JW, et al. Evidence revealing deregulation of the KLF11-MAO A pathway in association with chronic stress and depressive disorders. Neuropsychopharmacology. 2015;40(6):1373-1382. doi: 10.1038/npp.2014.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edelstein SB, Breakefield XO. Monoamine oxidases A and B are differentially regulated by glucocorticoids and “aging” in human skin fibroblasts. Cell Mol Neurobiol. 1986;6(2):121-150. doi: 10.1007/BF00711066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson S, Stockmeier CA, Meyer JH, et al. The reduction of R1, a novel repressor protein for monoamine oxidase A, in major depressive disorder. Neuropsychopharmacology. 2011;36(10):2139-2148. doi: 10.1038/npp.2011.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fowler JS, MacGregor RR, Wolf AP, et al. Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science. 1987;235(4787):481-485. doi: 10.1126/science.3099392 [DOI] [PubMed] [Google Scholar]
- 22.Fowler JS, Wang GJ, Logan J, et al. Selective reduction of radiotracer trapping by deuterium substitution: comparison of carbon-11-l-deprenyl and carbon-11-deprenyl-D2 for MAO B mapping. J Nucl Med. 1995;36(7):1255-1262. [PubMed] [Google Scholar]
- 23.Nag S, Fazio P, Lehmann L, et al. In vivo and in vitro characterization of a novel MAO-B inhibitor radioligand, 18F-labeled deuterated fluorodeprenyl. J Nucl Med. 2016;57(2):315-320. doi: 10.2967/jnumed.115.161083 [DOI] [PubMed] [Google Scholar]
- 24.Bramoullé Y, Puech F, Saba W, et al. Radiosynthesis of (S)-5-methoxymethyl-3-[6-(4,4,4-trifluorobutoxy)benzo[d]isoxazol-3-yl] oxazolidin-2-[11C]one ([11C]SL25.1188), a novel radioligand for imaging monoamine oxidase-B with PET. J Labelled Comp Radiopharm. 2008;51(3):153-158. doi: 10.1002/jlcr.1492 [DOI] [Google Scholar]
- 25.Saba W, Valette H, Peyronneau MA, et al. [(11)C]SL25.1188, a new reversible radioligand to study the monoamine oxidase type B with PET: preclinical characterisation in nonhuman primate. Synapse. 2010;64(1):61-69. doi: 10.1002/syn.20703 [DOI] [PubMed] [Google Scholar]
- 26.Vasdev N, Sadovski O, Garcia A, et al. Radiosynthesis of [11C]SL25.1188 via [11C]CO2 fixation for imaging monoamine oxidase B. J Labelled Comp Radiopharm. 2011;54(10):678-680. doi: 10.1002/jlcr.1908 [DOI] [Google Scholar]
- 27.Vasdev N, Sadovski O, Moran MD, et al. Development of new radiopharmaceuticals for imaging monoamine oxidase B. Nucl Med Biol. 2011;38(7):933-943. doi: 10.1016/j.nucmedbio.2011.03.003 [DOI] [PubMed] [Google Scholar]
- 28.Rusjan PM, Wilson AA, Miler L, et al. Kinetic modeling of the monoamine oxidase B radioligand [11C]SL25.1188 in human brain with high-resolution positron emission tomography. J Cereb Blood Flow Metab. 2014;34(5):883-889. doi: 10.1038/jcbfm.2014.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miguel-Hidalgo JJ, Baucom C, Dilley G, et al. Glial fibrillary acidic protein immunoreactivity in the prefrontal cortex distinguishes younger from older adults in major depressive disorder. Biol Psychiatry. 2000;48(8):861-873. doi: 10.1016/S0006-3223(00)00999-9 [DOI] [PubMed] [Google Scholar]
- 30.Si X, Miguel-Hidalgo JJ, O’Dwyer G, Stockmeier CA, Rajkowska G. Age-dependent reductions in the level of glial fibrillary acidic protein in the prefrontal cortex in major depression. Neuropsychopharmacology. 2004;29(11):2088-2096. doi: 10.1038/sj.npp.1300525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ekblom J, Jossan SS, Bergström M, Oreland L, Walum E, Aquilonius SM. Monoamine oxidase-B in astrocytes. Glia. 1993;8(2):122-132. doi: 10.1002/glia.440080208 [DOI] [PubMed] [Google Scholar]
- 32.Setiawan E, Attwells S, Wilson AA, et al. Association of translocator protein total distribution volume with duration of untreated major depressive disorder: a cross-sectional study. Lancet Psychiatry. 2018;5(4):339-347. doi: 10.1016/S2215-0366(18)30048-8 [DOI] [PubMed] [Google Scholar]
- 33.Ventura J, Liberman RP, Green MF, Shaner A, Mintz J. Training and quality assurance with the Structured Clinical Interview for DSM-IV (SCID-I/P). Psychiatry Res. 1998;79(2):163-173. doi: 10.1016/S0165-1781(98)00038-9 [DOI] [PubMed] [Google Scholar]
- 34.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56-62. doi: 10.1136/jnnp.23.1.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saura J, Andrés N, Andrade C, Ojuel J, Eriksson K, Mahy N. Biphasic and region-specific MAO-B response to aging in normal human brain. Neurobiol Aging. 1997;18(5):497-507. doi: 10.1016/S0197-4580(97)00113-9 [DOI] [PubMed] [Google Scholar]
- 36.Mallajosyula JK, Kaur D, Chinta SJ, et al. MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. PLoS One. 2008;3(2):e1616. doi: 10.1371/journal.pone.0001616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mallajosyula JK, Chinta SJ, Rajagopalan S, Nicholls DG, Andersen JK. Metabolic control analysis in a cellular model of elevated MAO-B: relevance to Parkinson’s disease. Neurotox Res. 2009;16(3):186-193. doi: 10.1007/s12640-009-9032-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andreazza AC, Shao L, Wang JF, Young LT. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen Psychiatry. 2010;67(4):360-368. doi: 10.1001/archgenpsychiatry.2010.22 [DOI] [PubMed] [Google Scholar]
- 39.Riederer P, Danielczyk W, Grünblatt E. Monoamine oxidase-B inhibition in Alzheimer’s disease. Neurotoxicology. 2004;25(1-2):271-277. doi: 10.1016/S0161-813X(03)00106-2 [DOI] [PubMed] [Google Scholar]
- 40.Youdim MB, Edmondson D, Tipton KF. The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci. 2006;7(4):295-309. doi: 10.1038/nrn1883 [DOI] [PubMed] [Google Scholar]
- 41.Heikkila RE, Manzino L, Cabbat FS, Duvoisin RC. Protection against the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine by monoamine oxidase inhibitors. Nature. 1984;311(5985):467-469. doi: 10.1038/311467a0 [DOI] [PubMed] [Google Scholar]
- 42.Kupsch A, Sautter J, Götz ME, et al. Monoamine oxidase-inhibition and MPTP-induced neurotoxicity in the non-human primate: comparison of rasagiline (TVP 1012) with selegiline. J Neural Transm (Vienna). 2001;108(8-9):985-1009. doi: 10.1007/s007020170018 [DOI] [PubMed] [Google Scholar]
- 43.Siddiqui A, Mallajosyula JK, Rane A, Andersen JK. Ability to delay neuropathological events associated with astrocytic MAO-B increase in a Parkinsonian mouse model: implications for early intervention on disease progression. Neurobiol Dis. 2011;43(2):527-532. doi: 10.1016/j.nbd.2010.12.014 [DOI] [PubMed] [Google Scholar]
- 44.Hasler G, Fromm S, Carlson PJ, et al. Neural response to catecholamine depletion in unmedicated subjects with major depressive disorder in remission and healthy subjects. Arch Gen Psychiatry. 2008;65(5):521-531. doi: 10.1001/archpsyc.65.5.521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bremner JD, Vythilingam M, Ng CK, et al. Regional brain metabolic correlates of α-methylparatyrosine–induced depressive symptoms: implications for the neural circuitry of depression. JAMA. 2003;289(23):3125-3134. doi: 10.1001/jama.289.23.3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nave S, Doody RS, Boada M, et al. Sembragiline in moderate Alzheimer’s disease: results of a randomized, double-blind, placebo-controlled phase II trial (MAyflOwer RoAD). J Alzheimers Dis. 2017;58(4):1217-1228. doi: 10.3233/JAD-161309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colle R, de Larminat D, Rotenberg S, et al. Pioglitazone could induce remission in major depression: a meta-analysis. Neuropsychiatr Dis Treat. 2016;13:9-16. doi: 10.2147/NDT.S121149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ekblom J, Jossan SS, Oreland L, Walum E, Aquilonius SM. Reactive gliosis and monoamine oxidase B. J Neural Transm Suppl. 1994;41:253-258. [DOI] [PubMed] [Google Scholar]
- 49.Tong J, Rathitharan G, Meyer JH, et al. Brain monoamine oxidase B and A in human parkinsonian dopamine deficiency disorders. Brain. 2017;140(9):2460-2474. doi: 10.1093/brain/awx172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gulyás B, Pavlova E, Kása P, et al. Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-l-deprenyl using whole hemisphere autoradiography. Neurochem Int. 2011;58(1):60-68. doi: 10.1016/j.neuint.2010.10.013 [DOI] [PubMed] [Google Scholar]
- 51.Tyacke RJ, Myers JFM, Venkataraman A, et al. Evaluation of 11C-BU99008, a PET ligand for the imidazoline2 binding site in human brain. J Nucl Med. 2018;59(10):1597-1602. doi: 10.2967/jnumed.118.208009 [DOI] [PubMed] [Google Scholar]
- 52.Rajkowska G, Miguel-Hidalgo JJ, Wei J, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45(9):1085-1098. doi: 10.1016/S0006-3223(99)00041-4 [DOI] [PubMed] [Google Scholar]
- 53.Ongür D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A. 1998;95(22):13290-13295. doi: 10.1073/pnas.95.22.13290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rajkowska G, Stockmeier CA. Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr Drug Targets. 2013;14(11):1225-1236. doi: 10.2174/13894501113149990156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khundakar A, Morris C, Oakley A, Thomas AJ. A morphometric examination of neuronal and glial cell pathology in the orbitofrontal cortex in late-life depression. Int Psychogeriatr. 2011;23(1):132-140. doi: 10.1017/S1041610210000700 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eAppendix. Methods, Results, and Discussion
eFigure 1. Relationship Between Prefrontal Cortex MAO-B Distribution Volume and Severity of Major Depressive Episode
eFigure 2. Total Distribution Volume in Healthy and Major Depressive Episode Subjects
eTable. Analysis of Variance Comparing Regional MAO-B Density and Duration of Major Depressive Disorder