

Abstract
Traumatic brain injury (TBI) is associated with psychiatric dysfunction—including pain, cognitive impairment, anxiety, and increased alcohol use. We previously demonstrated that inhibiting endocannabinoid degradation post-TBI with JZL184 attenuates neuroinflammation and neuronal hyperexcitability at the site of injury and improves neurobehavioral recovery. This study aimed to determine the effect of JZL184 on post-TBI behavioral changes related to psychiatric dysfunction and post-TBI neuroadaptations in brain regions associated with these behaviors. We hypothesized that JZL184 would attenuate post-TBI behavioral and neural changes in alcohol-drinking rats. Adult male Wistar rats were trained to operantly self-administer alcohol before receiving lateral fluid percussion injury. Thirty minutes post-TBI, rats received JZL184 (16 mg/kg, i.p.) or vehicle. Spatial memory (Y-maze), anxiety-like behavior (open field), alcohol motivation (progressive ratio responding), and mechanosensitivity (Von Frey) were measured 3–10 days post-injury, and ventral striatum (VS) and central amygdala (CeA) tissue were collected for western blot analysis of phosphorylated glutamate receptor subunit 1 (GluR1) and glucocorticoid receptor (GR). TBI impaired spatial memory, increased anxiety-like behavior, and increased motivated alcohol drinking. JZL184 prevented these changes. TBI also increased phosphorylated GluR1 and GR in the CeA (but not the VS) compared with sham controls. JZL184 attenuated post-TBI GR phosphorylation in the CeA. These findings suggest that TBI produces comorbid cognitive dysfunction, increased alcohol motivation, and anxiety-like behavior, possibly related to amygdala dysfunction, and these changes are prevented by systemic post-TBI endocannabinoid degradation inhibition. Thus, boosting endocannabinoid tone post-TBI may represent a viable therapeutic strategy for TBI-related psychiatric comorbidities such as alcohol use disorder and anxiety.
Keywords: alcohol motivation, anxiety, central amygdala, glucocorticoid receptor, JZL184
Introduction
Traumatic brain injury (TBI) is a major public health concern in the United States, accounting for over 2.5 million emergency department visits every year and over 50,000 deaths.1 Mild TBI (mTBI), or “concussion,” is most common and may result from falls, motor vehicle accidents, or contact sports such as football or boxing, and as such this injury typically affects otherwise healthy individuals.2 Football players, for example, might sustain multiple concussive or subconcussive hits over the course of a single season.3,4
mTBI is underreported due to the transient nature of clinical symptoms such as unconsciousness and the false sense, particularly in athletics, that the injury does not produce long-term consequences.2 Even after the clinical symptoms dissipate, less obvious changes persist at the cellular and molecular level including neuroinflammation and synaptic hyperexcitability.5–8 Because there is no effective treatment for TBI, these less-obvious cellular and molecular pathologies can contribute to the development of short- and long-term behavioral changes such as pain, anxiety, and neurodegenerative disease.9–14
Among the most detrimental behavioral changes post-TBI is the tendency of patients to increase alcohol drinking. Former professional athletes with a history of concussions often increase drug and alcohol use.15 Individuals with a history of high alcohol consumption prior to sustaining a TBI are particularly susceptible to developing alcohol use disorder post-TBI.16–19 Escalated alcohol use following TBI can be especially problematic because excessive drinking is associated with poor executive function, memory impairment, neuroinflammation, excitotoxicity, and increased long-term risk for neurodegenerative disease.20–23 Thus, treatments that can attenuate post-TBI psychopathologies are greatly needed, and likewise a better understanding of the cellular and molecular mechanisms contributing to such behavioral changes could inform novel therapies for TBI.
There is mounting evidence that indicates a protective role of cannabinoids in reducing inflammation and promoting synaptic homeostasis following injury.24 The two main endocannabinoids that are released in response to injury are 2-arachidonoyl glycerol (2-AG) and N-arachidonoylethanolamine (anandamide, AEA). These molecules are synthesized on demand and are rapidly broken down by monoacylglycerol lipase (MAGL) and fatty acid amide hydrolase (FAAH) respectively.25,26 The experimental drug JZL184 is a potent and long-lasting irreversible MAGL inhibitor, with >80% MAGL inhibition seen for 24 h following a single 16-mg/kg i.p. injection in mice.27 We previously showed that a single JZL184 administration (16 mg/kg i.p. 30 min post-TBI) was sufficient to attenuate increased neuroinflammation and neuronal hyperexcitability at the site of injury as well as improve neurobehavioral and neurological severity scores up to 14 days after mTBI to the sensorimotor cortex (SMC) in male rats.28,29
Our results demonstrated an escalation in alcohol drinking following SMC-mTBI in rats trained to operantly self-administer alcohol.30 Whether mTBI produces behavioral changes related to cognition, anxiety, or pain in alcohol-drinking rats is unknown. Moreover, whether similar protective effects of JZL are observed in terms of motivation for alcohol and associated behavioral comorbidities remains to be explored. Because the endocannabinoid system buffers stress-related glucocorticoid signaling, we speculated this would be an additional mechanism that needed to be explored, particularly given the evidence supporting a potentiation of central brain glucocorticoid signaling in alcohol dependence-related behaviors.31,32
This study tested the hypothesis that mTBI to the SMC in alcohol-drinking male Wistar rats would produce behavioral changes including impaired spatial memory, increased anxiety-like behavior, increased alcohol motivation, and mechanical hypersensitivity. We further hypothesized that JZL184 administration post-TBI would improve behavioral outcomes. Finally, because JZL administration was sufficient to attenuate cortical hyperexcitability at the site of injury 10 days post-TBI, we examined whether JZL administration attenuated subcortical excitatory and glucocorticoid system neuroadaptations in brain regions associated with reward (ventral striatum; VS) and stress/anxiety/negative reinforcement (central amygdala; CeA).
Methods
Animals
Male Wistar rats (175–200 g; Charles River Laboratories, Wilmington, MA) were pair-housed in a temperature- and humidity-controlled room with a 12-h light/dark cycle and ad libitum access to food and water. All animal procedures and experiments were approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center and were in accordance with the guidelines of the National Institutes of Health.
Operant self-administration
Rats were allowed to acclimate to housing conditions for one week before operant alcohol self-administration training, conducted as described previously.33 Briefly, rats were placed in operant self-administration chambers 5 days/week in limited access sessions of 30 min that began 6 h into the dark cycle. Rats had access to two levers (water vs. alcohol) on a fixed ratio (FR1) schedule, in which one lever press resulted in delivery of 0.1 mL of either water or 10% w/v ethanol. Once consistent baseline drinking levels were achieved (i.e., three consecutive sessions during which the variance of the number of alcohol lever presses was no more than ±20%), animals were counterbalanced into experimental groups based on baseline alcohol drinking levels, calculated as mean alcohol lever presses for the last five 30-min operant sessions.
Traumatic brain injury via lateral fluid percussion
Forty-eight hours after the last operant self-administration training session, animals received a 5 mm in diameter craniotomy above the left SMC (from bregma: AP: −2 mm, ML: −3 mm) before undergoing TBI via lateral fluid percussion (Fluid Percussion Injury [FPI], Model 01-B, Custom Design and Fabrication, Virginia Commonwealth University) as previously described.34 Animals in the sham group were anesthetized and received craniotomy but were not subjected to TBI (surgical controls). Only animals with an injury of at least 2 atm of pressure, which produces an mTBI, were included for analysis. Following surgery, topical lidocaine was applied to the incision site, and animals were allowed to recover in their home cages for 48 h with ad libitum food and water prior to resuming operant drinking post-TBI.
MAGL inhibition
JZL184 (Item #13158, Cayman Chemical, Ann Arbor, MI) was used to selectively inhibit MAGL, the enzyme that degrades 2-AG. Systemic JZL184 administration results in rapid and potent MAGL inhibition, with maximal inhibition within 30 min resulting in a 7- to 9-fold increase in brain 2-AG levels.27 JZL184 (16 mg/kg, i.p.) or vehicle (1:1:18 solution of alcohol, emulphor, and saline) was administered 30 min after TBI procedures. Behavioral testing resumed 48 h after injection. To determine whether JZL184 treatment alone improves spatial memory, anxiety-like behavior, or mechanosensitivity, a separate cohort of non-drinking animals received JZL184 (16 mg/kg, i.p.) or vehicle 30 min after sham procedures and then underwent behavioral testing.
Post-TBI operant drinking
Animals resumed 30-min limited access drinking sessions every other day 48 h after TBI or sham procedures (days 2, 4, 6, and 8 post-TBI). To test for motivated alcohol drinking, animals completed a progressive ratio (PR) task 9 days post-TBI. In this task, the work required to receive 0.1 mL of 10% w/v alcohol progressively increases (i.e., initially one press delivers one reward of alcohol, then two presses is required for one reward, then three presses for one reward, etc.). The experimental session ends when the subject fails to achieve an alcohol reward for 15 consecutive min. The dependent measure is the breakpoint, defined as the value of the last completed (reinforced) ratio. The breakpoint under a PR schedule is considered to reflect the motivation of the animal to self-administer a drug.35–38
Post-TBI behavioral assessments
Animals underwent behavioral testing to assess spatial memory, anxiety-like behavior, and mechanosensitivity on non-drinking days post-TBI. These behavioral assessments were conducted at least 24 h after the last alcohol self-administration session to minimize the effects of alcohol on behavioral measures.
Spatial memory was assessed 3 days post-TBI using the Y-maze as previously described.39 Novelty was guided by distinct spatial cues set up around the testing room. The Y-maze had three arms (61 × 14 × 35 cm) that extended from a central platform at a 120-degree angle. Each rat was placed in the middle of the maze and allowed to move freely in the three arms of the maze during a 10-min testing period. An arm entry was defined as the entry of four paws into one arm. The sequence of the arm entries was recorded using video tracking software (ANY-maze, Stoelting Co.). Alternation was defined as entry into each of the three different arms per overlapping triplet set. The percentage of spontaneous alternation was calculated as the ratio of the actual to possible alternations (defined as the total number of arm entries minus 2) multiplied by 100:
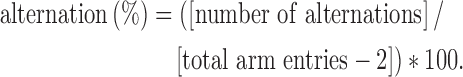 |
Data were analyzed via one-sample t test against chance performance (50% spontaneous alternation) using Prism 5 (GraphPad Software).
Anxiety-like behavior was assessed 7 days post-TBI using the open field test. Each animal had 5 min to explore a black open field. A video camera mounted on the ceiling directly above the open field box wirelessly transmitted video to a computer in the lab and recorded each test. Behavior was later scored by an observer blind to the treatment. The amount of time each rat spent exploring the center (defined by leaving the peripheral layer of boxes on the checkerboard patterned floor) versus the periphery (the layer of boxes along the wall) was quantified. Data are expressed as percent time in center (seconds in center divided by 300). Decreased time spent in the center of the open field is interpreted as anxiety-like behavior.
Mechanosensitivity testing was conducted once 24 h before TBI (baseline) and again 10 days post-injury. Evaluation of paw withdrawal thresholds was performed according to methods described previously.40 Briefly, rats were acclimated for 15 min in elevated cages with a wire mesh floor. A series of Von Frey filaments were applied perpendicularly to the plantar surface of the hindpaw for 3 sec. A sharp withdrawal of the hindpaw indicated a positive response. The mechanical intensity of the stimulus was incrementally increased until a positive response was obtained, then decreased until a negative result was observed to determine a pattern of responses to apply to the statistical method of Dixon.41 The 50% paw withdrawal threshold was determined by the formula Xf + kδ, where Xf = last Von Frey filament employed, k = Dixon value corresponding to response pattern, and δ = mean difference between stimuli. Baseline mechanical nociceptive thresholds were similar to those reported for the ages of rats employed in this study.42
Western blot analysis
Animals were sacrificed by decapitation under isoflurane anesthesia 2 weeks post-TBI, and brains were excised, flash frozen, and analyzed as previously described.43 Ventral striatum and central amygdala punches were obtained from 0.5-mm frozen coronal brain slices and homogenized by sonication in lysis buffer (320 mM sucrose, 5 mM 4(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 1 mM egtazic acid [EGTA], 1 mM ethylenediaminetetraacetic acid [EDTA], and 1% sodium dodecyl sulphate [SDS], with protease inhibitor cocktail and phosphatase inhibitor cocktails II and III diluted 1:100; Sigma, St. Louis, MO). Protein concentration was determined by the Lowry method (Bio-Rad, Hercules, CA), and 20-μg protein samples were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) on 4–15% gradient acrylamide gels before transfer to polyvinylidene difluoride membranes (GE Healthcare, Piscataway, NJ). Membranes were blocked then incubated in primary antibody overnight against phosphorylated glutamate receptor subunit 1 at Ser845 (pGluR1S845, diluted 1:1000 in 5% non-fat milk; Cell Signaling, Danvers, MA), or phosphorylated glucocorticoid receptor at Ser211/232 (pGRS211 [human]/pGR232 [rat], diluted 1:1000; Cell Signaling).
Membranes were incubated with peroxidase-conjugated secondary antibody (1:10,000; Bio-Rad) for 1 h at room temperature and chemiluminescence was detected (SuperSignal West Pico; Thermo Scientific, Rockford, IL). Blots were stripped for 20 min (Restore; Thermo Scientific) and re-probed for total protein levels of GluR1 (1:2500; Cell Signaling) and GR (1:1000; Thermo Fisher). Immunoreactivity was quantified using ImageJ, and the ratio of phosphoprotein to total protein for each sample was calculated for statistical comparison.
Statistical analysis
All data are expressed as mean ± standard error of the mean (SEM). Statistical differences were determined by either Student's t test, one-sample t test (for Y-maze), one-way analysis of variance (ANOVA), or two-way repeated measures ANOVA (for Von Frey) using GraphPad Prism 5.0 statistical software (Graphpad Software Inc., La Jolla, CA), as indicated in each figure legend. Post hoc tests for multiple comparisons were utilized when appropriate, as indicated in the text. Statistical significance was set at p < 0.05. Behavioral studies were conducted (using n = 8–10 animals per experimental group), and western blot analysis was completed with a subset of those experimental groups (n = 4–6 per group).
Results
JZL184 treatment improved spatial memory 3 days after SMC-TBI
Three days post-TBI, all experimental groups were tested for spatial memory in the Y-maze (Fig. 1). One-way ANOVA revealed no significant difference between groups (F[2,27] = 1.708, p > 0.05). In a one-sample t test against 50% (random chance), sham-vehicle animals performed significantly better than 50% (t[9] = 4.447, p < 0.01). There was no significant difference between the mean performance of the TBI-vehicle animals and 50% (random chance) (t[9] = 2.085, p > 0.05). However, TBI animals treated with JZL184 performed significantly better than 50% (random chance), indicating restored cognitive performance post-TBI (t[9] = 5.308, p < 0.01).
FIG. 1.

Spontaneous alternation behavior in the Y-maze 3 days post-TBI. All animals were tested in the Y-maze for spatial memory 3 days post-TBI. Percent spontaneous alternations by sham vehicle-treated animals (open bar), TBI vehicle-treated animals (black bar), and TBI JZL184-treated animals (gray bar) are shown. One-way ANOVA revealed no significant difference between groups. *p < 0.01 for both sham-vehicle and TBI-JZL groups compared with 50% alternation (random chance). Data analyzed by one-sample t test versus 50%, n = 10/group. Data are expressed as mean ± SEM. ANOVA, analysis of variance; SEM, standard error of the mean; TBI, traumatic brain injury.
To determine whether JZL184 treatment alone improves spatial memory, a separate cohort of vehicle- or JZL-treated sham animals was tested in the Y-maze 3 days post-sham procedure. Both sham-vehicle and sham-JZL groups performed better than 50% (t[7] = 7.345 and 8.867, respectively; p < 0.001), and performance between the two groups was not significantly different (unpaired t test: t[14] = 0.7890, p > 0.05; data not shown).
JZL184 treatment attenuated anxiety-like behavior 7 days after SMC-TBI
One week post-TBI, all experimental groups were tested for anxiety-like behavior using the open field test (Fig. 2). In this test, an animal exhibiting greater anxiety-like behavior will spend less time exploring the center of the box.44 One-way ANOVA revealed a significant difference between groups (F[2,27] = 4.651, p < 0.05). Tukey's test for multiple comparisons revealed that TBI-vehicle animals spent significantly less time exploring the center when compared with sham-vehicle animals (p < 0.05), whereas TBI-JZL animals' percent time in the center was not significantly different from that of sham-vehicle controls (p > 0.05).
FIG. 2.

Anxiety-like behavior in the open field test 7 days post-TBI. All animals were assessed in the open field test for anxiety-like behavior 7 days post-TBI. Percent times in center spent by sham-vehicle animals (open bar), TBI-vehicle animals (black bar), and TBI-JZL184 animals (gray bar) are shown. *p < 0.05 for TBI-vehicle group compared with sham-vehicle group. Data analyzed via one-way ANOVA with Tukey's multiple comparisons test, n = 10/group. Data are expressed as mean ± SEM. ANOVA, analysis of variance; SEM, standard error of the mean; TBI, traumatic brain injury.
To determine whether JZL184 treatment alone reduces anxiety-like behavior, a separate cohort of vehicle- or JZL-treated sham animals was tested in the open field test 7 days post-sham procedures. Two-tailed unpaired t test revealed that percent time in the center between the two groups was not significantly different (t[14] = 1.373, p > 0.05; data not shown) indicating a lack of effect of JZL184 alone on anxiety-like behavior.
JZL184 treatment attenuated increased motivation for alcohol 9 days after SMC-TBI
All animals were tested on a progressive ratio schedule to determine motivation for alcohol drinking 9 days post-TBI (Fig. 3). The dependent measure was breakpoint, with higher values on the y-axis indicating a greater motivation to drink alcohol. One-way ANOVA revealed a significant difference in breakpoints across groups (F[2,27] = 4.237, p < 0.05). Tukey's test for multiple comparisons revealed TBI-vehicle animals showed significantly more motivated behavior for alcohol compared with sham-vehicle animals (p < 0.05), and that JZL successfully attenuated the TBI-induced increase in breakpoint (p < 0.05 TBI-JZL compared with TBI-vehicle).
FIG. 3.

Breakpoint in a progressive ratio task measuring motivated operant alcohol responding 9 days post-TBI. All animals were tested for motivation to drink alcohol on a progressive ratio schedule 9 days post-TBI. Number of lever presses for an alcohol reward prior to termination of the session (breakpoint) by sham-vehicle animals (open bar), TBI-vehicle animals (black bar), and TBI-JZL184 animals (gray bar) are shown. *p < 0.05 for TBI-vehicle group compared with sham-vehicle group. #p < 0.05 for TBI-JZL184 group compared with TBI-vehicle group. Data analyzed via one-way ANOVA with Tukey's multiple comparisons test, n = 10/group. Data are expressed as mean ± SEM. ANOVA, analysis of variance; SEM, standard error of the mean; TBI, traumatic brain injury.
Mechanical sensitivity is not significantly altered 10 days after SMC-TBI
Animals were tested for mechanical sensitivity 24 h before TBI (baseline) and 10 days post-TBI using the Von Frey test (Fig. 4). Two-way repeated-measures ANOVA revealed no interaction (F[2,27] = 4.97, p > 0.05) and no main effect of time (F[1,27] = 0.49, p > 0.05) or treatment (F[2,27] = 3.47, p > 0.05).
FIG. 4.

Mechanical sensitivity in the Von Frey test 10 days post-TBI. All animals were tested for mechanical sensitivity 24 h before TBI (pre-TBI baseline) and 10 days post-TBI using the Von Frey test. Hindpaw withdrawal thresholds (in grams) for sham-vehicle animals (open circles), TBI-vehicle animals (black circles), and TBI-JZL184 animals (gray circles) are shown. Data analyzed via two-way repeated measures ANOVA, n = 10/group. Data are expressed as mean ± SEM. ANOVA, analysis of variance; SEM, standard error of the mean; TBI, traumatic brain injury.
To determine whether JZL184 treatment alone affects mechanical sensitivity, a separate cohort of vehicle- or JZL-treated sham animals was tested in the Von Frey test 24 h before and 10 days post-sham procedures. Two-way repeated measures ANOVA revealed no interaction (F[1,14] = 3.50, p > 0.05) and no main effect of time (F[1,14] = 6.91, p > 0.05) or treatment (F[1,14] = 4.66, p > 0.05; data not shown).
JZL184 treatment attenuated post-SMC TBI glucocorticoid signaling in the central amygdala
Western blot analysis of ventral striatum tissue revealed no significant changes in pGluR1 (F[2,14] = 0.7588, p > 0.05) or pGR (F[2,14] = 1.342, p > 0.05). In contrast, one-way ANOVA analysis of pGluR1 expression in the central amygdala revealed significant changes (F[2,12] = 19.66, p < 0.01, Fig. 5). Tukey's test for multiple comparisons revealed that TBI-vehicle animals had increased pGluR1S845 when compared with sham-vehicle animals (p < 0.01). Tukey's test also revealed that TBI-JZL animals had significantly elevated pGluR1 levels as well when compared with sham-vehicle controls (p < 0.01), and these levels were not significantly decreased when compared with TBI-vehicle animals (p > 0.05). In comparison, one-way ANOVA of pGR expression in the central amygdala revealed significant differences (F[2,12] = 6.115, p < 0.05), and Tukey's test for multiple comparisons revealed that TBI-vehicle animals had increased pGRS232 when compared with sham-vehicle animals (p < 0.05) and that JZL successfully attenuated the TBI-induced increase in pGRS232 (p < 0.05 TBI-JZL compared with TBI-vehicle).
FIG. 5.

Phosphorylation of GluR1 and GR in the CeA 2 weeks post-TBI. A subset of animals was sacrificed after behavioral testing (2 weeks post-TBI) for western blot analysis of phosphorylated GluR1 at Ser845 (pGluR1S845) and phosphorylated GR at Ser232 (pGRS232) in CeA tissue of sham-vehicle (open bars), TBI-vehicle (black bars), and TBI-JZL184 animals (gray bars). (A) Ratio of pGluR1S845 to total GluR1 in the CeA. (B) Ratio of pGRS232 to total GR in the CeA. *p < 0.05 compared with sham-vehicle group. #p < 0.05 compared with TBI-vehicle group. Data in all panels analyzed by one-way ANOVA with Tukey's multiple comparisons test, n = 4–6/group. Data in all panels are expressed as mean ± SEM. ANOVA, analysis of variance; CeA, central amygdala; GluR1, glutamate receptor subunit 1; GR, glucocorticoid receptor; SEM, standard error of the mean; TBI, traumatic brain injury.
Discussion
The results from this study show that mTBI to the SMC in alcohol self-administering rats concurrently increases anxiety-like behavior and motivated alcohol drinking and may impair spatial memory, modeling TBI-related psychiatric comorbidities reported in the clinical literature.9,45,46 Systemic JZL184 administration 30 min after injury prevented these post-TBI behavioral changes, suggesting that enhanced endocannabinoid tone, specifically 2-AG levels, may be sufficient to prevent the development of TBI-induced psychopathologies such as anxiety and increased alcohol use. Additionally, a trend for a decrease in paw withdrawal threshold in the TBI-vehicle group 10 days post-TBI when compared with baseline that was not seen in the TBI-JZL group may indicate TBI could produce some mechanical hypersensitivity that may be reduced by JZL treatment following TBI, although further testing at additional time-points post-TBI is required. Further, in the same animals that exhibited behavioral changes, TBI also increased levels of pGluR1S845 and pGRS232 in the CeA, but not the VS, suggesting that increased glutamatergic and glucocorticoid signaling occurs subcortically in a brain area related to stress/anxiety/negative reinforcement and, interestingly, not in a brain region thought to mediate the initial, positive reinforcing effects of alcohol.
Although JZL184 did not significantly attenuate the increased levels of pGluR1 seen in the CeA after TBI, JZL treatment did prevent the TBI-induced increase in pGR in the CeA, which may highlight a role for GR signaling in the pathology and treatment of TBI-related anxiety and alcohol use. Together, these findings demonstrate a model of TBI-related psychiatric comorbidities, including increased motivation for alcohol drinking, and implicate endocannabinoid-modulated glucocorticoid signaling in the CeA as a potential therapeutic target for these long-lasting behavioral changes.
Several pre-clinical studies have shown the important role of the endocannabinoid system in regulating anxiety-like behavior in rodents following auditory fear conditioning tasks.47,48 Studies in either CB1 deficent mice or mice treated with a CB1 receptor antagonist found that subjects with blunted endocannabinoid system function exhibited greater anxiety-like behavior up to 10 days post-fear conditioning.47,48 These studies are relevant to the present work because Reger and colleagues showed that rats display increased anxiety-like behavior in response to similar fear conditioning tasks post-TBI, which are in line with our results demonstrating a post-TBI increase in anxiety-like behavior in the open field test.49 Some pre-clinical studies have attempted to manipulate the endocannabinoid system to treat anxiety-like behavior as well. Administration of the CB1 agonist ACPA (5 ng/rat) into the CeA of rats resulted in reduced anxiety-like behavior on the elevated plus-maze.50
Our results show that MAGL inhibition results in reduced TBI-related anxiety-like behavior, and because JZL was not administered intra-CeA but systemically (i.p.), our results show effectiveness in targeting the endocannabinoid system for post-TBI anxiety-like behavior using a more clinically relevant route of administration. This clinical relevance is important considering clinical studies show that almost half of people who sustain mTBI exhibit anxiety-like behavior up to a year post-injury, and anxiety is often comorbid with excessive alcohol use in humans.9,51–55
Although JZL treatment was effective in reducing anxiety-like behavior, there was no statistical difference between TBI-JZL animals and vehicle-treated TBI animals in terms of mechanical sensitivity. These results are somewhat limited because we only collected post-TBI mechanical sensitivity data at one time-point (10 days post-TBI) and there did appear to be a trend toward a decrease in paw withdrawal thresholds with TBI that may have been prevented with JZL treatment. Measuring mechanical sensitivity post-TBI is important because approximately 20% of patients will develop chronic pain after TBI, a condition that is characterized as allodynia (hypersensitivity to otherwise innocuous stimuli).12 Also, heavy drinking is associated with the development of neuropathy that may further promote escalated drinking and the transition to alcohol use disorder (AUD).56 Moreover, there is pre-clinical evidence that the endocannabinoid system may play a role in antinociception. Hasanein and colleagues showed that bilateral microinjection of the CB1 agonist WIN52,212-2 into the basolateral amygdala (BLA) produced antinociceptive effects in the tail flick assay, and that these effects were blocked by a CB1 antagonist.57 Future studies are required to assess mechanical sensitivity at different time-points post-TBI to further contribute to the field, and potentially provide evidence that a more clinically relevant route of treatment (i.p.) is effective in attenuating post-TBI mechanical hypersensitivity.
It is believed that one consequence of post-TBI anxiety and chronic pain in humans is the tendency to increase alcohol drinking.51–55,58,59 We showed that animals that received TBI in our model demonstrated increased anxiety-like behavior and modest (yet not significant) increased mechanical sensitivity that associated with increased motivation to drink alcohol as measured by the progressive ratio task. The endocannabinoid system, in addition to reducing anxiety-like behavior and allodynia (see above), can control motivational behavior for drugs of abuse. The endocannabinoid degradation inhibitor phenylmethylsulphonyl fluoride (PMSF, a FAAH inhibitor) administered i.p. significantly reduces both drug priming- and cue-induced nicotine-seeking behavior in rats, and URB597 (FAAH inhibitor) administered i.p. significantly attenuates both priming- and cue-induced cocaine-seeking behavior in rats.60–62 In our model, we were able to show that the MAGL inhibitor JZL184 administered i.p. significantly reduced the breakpoint for alcohol self-administration on the progressive ratio task following TBI in rats.
Although these results are in accordance with other studies suggesting a beneficial role for endocannabinoid degradation inhibition in ameliorating drug-seeking behavior, our results extend those observations to the context of TBI, demonstrating the efficacy of a single dose of JZL184 administered 30 min post-TBI, inceasing the clinical relevance of these findings. Previous studies demonstrate the long-lasting inhibition of MAGL achieved by the dose of the irreversible inhibitor JZL184 used in our studies (16 mg/kg).27 Additional studies are warranted to determine the critical time period post-TBI when endocannabinoid degradation is most effective in preventing or attenuating the neuropathological behaviors seen post-TBI.
We previously showed that mTBI to the SMC produced synaptic hyperexcitability at the site of injury as measured by increased pGluR1S845 and increased miniature excitatory post-synaptic current (mEPSC) frequency and amplitude 10 days post-injury, changes that were prevented by JZL184 administration.28 However, it is unclear how hyperexcitability in the SMC, a brain area involved in controlling movement, would impact affective behaviors such as anxiety and alcohol motivation. We thus examined subcortical areas involved in anxiety (CeA) and reward (VS) for evidence of TBI-related hyperexcitability and discovered that whereas no significant changes in pGluR1S845 were seen in the VS, elevated pGluR1S845 was seen in the CeA of TBI animals relative to surgical controls.
In addition to mediating anxiety, the amygdala also mediates learning that gives motivational salience to alcohol cues and underlies craving. Thus, it is possible that TBI-induced hyperexcitability in the CeA may give rise to the TBI-induced increases in anxiety-like behavior and alcohol motivation seen in our model.63–65 Indeed, altered amygdala glutamate homeostasis is associated with anxiety-like behavior. Overall reduction in BLA gamma-aminobutyric-acid (GABA)ergic tone (reduction in GABAa-dependent inhibitory post-synaptic currents or IPSCs) 30 days after controlled cortical impact (CCI) TBI in rats results in amygdala hyperexcitability and the development of behavioral abnormalities including anxiety-like behavior.66 Another study of CCI TBI in C57BL/6J mice showed a significant increase in spontaneous excitatory post-synaptic currents and membrane excitability 3 months post-TBI in the lateral amygdala indicating a persistent change in amygdala synaptic plasticity post-TBI.67
Several mechanisms exist by which endocannabinoids may counter hyperexcitability. For instance, recent evidence clearly demonstrates the existence of endocannabinoid-mediated depolarization-induced suppression of excitation (DSE) in cortical inputs to the lateral and central amygdala.68,69 The endocannabinoid system may also reduce hyperexcitability through its suppression of inflammation, thus attenuating hyperexcitability by improving glutamate recycling or preventing pro-inflammatory cytokine release.70 Previous studies from our group have shown attenuated neuroinflammation at the site of injury in JZL184-treated TBI rats.29 Moreover, we have previously reported attenuation of TBI-induced elevations in pGluR1S845 at the site of injury with JZL184 treatment, corresponding to changes in SMC mEPSCs.28 However, results from this study did not show a significant attenuation of pGluR1S845 in the CeA with JZL treatment. Further investigation is needed, however, to determine the excitability of the CeA in our model after JZL treatment, particularly at time-points corresponding to changes in amygdala-related behaviors (days 7–9 post-injury), as well as excitability of other nuclei of the amygdala with JZL treatment (i.e., BLA). Otherwise, these data might suggest that increasing endocannabinoid tone with JZL184 may prevent the development of TBI-related anxiety and enhanced alcohol motivation through a mechanism independent of CeA hyperexcitability.
Our results did demonstratethat JZL treatment prevented the significant increase in pGRS232 seen in the CeA of TBI-vehicle rats. The stress response to TBI may involve significantly increased exposure to glucocorticoids, which could explain the increase in pGR seen in our TBI rats. This increase in amygdalar GR activity could underlie the increases in anxiety-like behavior seen in our TBI animals. A recent study has shown that rats pre-treated with mifepristone, a GR antagonist, prior to social defeat stress followed by mild weight-drop TBI demonstrated attenuated anxiety-like behavior 9 days post-injury.71 Additionally, increases in pGR are well-documented in the CeA of alcohol-dependent rats, and this increase is associated with escalated alcohol drinking behavior that can be reduced with GR antagonism.72 Thus, the attenuation of TBI-induced pGR seen in the CeA with JZL treatment could be the mechanism underlying the improvements in anxiety-like behavior and motivated alcohol drinking seen in this study.
Despite our finding that CeA pGluR1 levels were not attenuated with JZL treatment in the present study, it is not unlikely that the changes in amygdalar GR phosphorylation are related to changes in amygdala excitability. It is possible that post-TBI GR activity sensitizes the amygdala to hyperexcitability, for instance by increasing N-methyl-D-aspartate (NMDA) receptor expression in the amygdala.73 Although our results present an apparent disconnect between excitatory signaling and stress system in the CeA, it is worth noting that most studies that examine amygdala hyperexcitability following trauma or stress focus on the BLA. Future studies are warranted to examine BLA changes in our model to better understand the link between hyperexcitability and GR activity. Importantly, our results do expand previous examinations of post-TBI neuroadaptations to the CeA, suggesting that similar changes in amygdalar glucocorticoid signaling occur after TBI as they do in alcohol dependence. Moreover, our approach of treating after injury is clinically relevant in contrast to other studies that pre-treat animals before TBI.72,71
Overall, these results show that TBI produces comorbid cognitive dysfunction, anxiety-like behavior, and increased alcohol motivation in alcohol-drinking animals, and that these behaviors are possibly related to TBI-induced amygdala dysfunction. Further, these findings show that these TBI-related behaviors are prevented by systemic post-TBI inhibition of endocannabinoid degradation, highlighting an important role for MAGL inhibition as a potential therapeutic target to improve post-TBI outcomes. Moreover, our results point to GR signaling in the CeA as a possible additional mechanism for these therapeutic effects. Future studies should continue to evaluate the effectiveness of endocannabinoid system modulation in providing benefits following TBI, and further investigation of amygdalar mechanisms underlying TBI-induced behavioral changes could help identify treatments for TBI-related psychiatric comorbidities such as AUD and anxiety.
Acknowledgments
Support for this study was provided by NIAAA training grant T32 AA007577, NIAAA fellowship award F31 AA025812-01A1, NIAAA research grant R00 AA020839, DOD grant W81XWH-11-2-0011, and funds from the LSUHSC-NO Department of Physiology.
Author Disclosure Statement
NWG holds shares in Glauser Life Sciences, Inc. JPM is currently employed by and holds shares in Halyard Health. SE is on the Scientific Advisory Board of Halyard Health. These activities have no relation to any of the work presented in this article. No competing financial interests exist for EAF, MAM, and PEM.
References
- 1. Taylor C., Bell J., Breiding M., and Xu L. (2017). Traumatic brain injury-related emergency department visits, hospitalizations, and deaths—United States, 2007 and 2013. MMWR Surveill. Summ. 66, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Corrigan J., Selassie A., and Orman J. (2010). The epidemiology of traumatic brain injury. J.Head Trauma Rehabil. 25, 72–80 [DOI] [PubMed] [Google Scholar]
- 3. Crisco J., Fiore R., Beckwith J., Chu J., Brolinson P., Duma S., McAllister T., Duhaime A., and Greenwald R. (2010). Frequency and location of head impact exposures in individual collegiate football players. J. Athl. Train. 45, 549–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greenwald R., Gwin J., Chu J., and Crisco J. (2008). Head impact severity measures for evaluating mild traumatic brain injury risk exposure. Neurosurgery 62, 789–798; discussion 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Al Nimer F., Lindblom R., Ström M., Guerreiro-Cacais A., Parsa R., Aeinehband S., Mathiesen T., Lidman O., and Piehl F. (2013). Strain influences on inflammatory pathway activation, cell infiltration and complement cascade after traumatic brain injury in the rat. Brain Behav. Immun. 27, 109–122 [DOI] [PubMed] [Google Scholar]
- 6. Kumar A. and Loane D. (2012). Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav. Immun. 26, 1191–1201 [DOI] [PubMed] [Google Scholar]
- 7. Loane D. and Faden A. (2010). Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 31, 596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McIntosh T., Smith D., Meaney D., Kotapka M., Gennarelli T., and Graham D. (1996). Neuropathological sequelae of traumatic brain injury: relationship to neurochemical and biomechanical mechanisms. Lab. Invest. 74, 315–342 [PubMed] [Google Scholar]
- 9. Hibbard M., Uysal S., Kepler K., Bogdany J., and Silver J. (1998). Axis I psychopathology in individuals with traumatic brain injury. J. Head Trauma Rehabil. 13, 24–39 [DOI] [PubMed] [Google Scholar]
- 10. Bryant R., O'Donnell M., Creamer M., McFarlane A., Clark C., and Silove D. (2010). The psychiatric sequelae of traumatic injury. Am. J. Psychiatry 167, 312–320 [DOI] [PubMed] [Google Scholar]
- 11. Nampiaparampil D. (2008). Prevalence of chronic pain after traumatic brain injury: a systematic review. JAMA 300, 711–719 [DOI] [PubMed] [Google Scholar]
- 12. Ofek H., and Defrin R. (2007). The characteristics of chronic central pain after traumatic brain injury. Pain 131, 330–340 [DOI] [PubMed] [Google Scholar]
- 13. Eikelenboom P., van Exel E., Hoozemans J., Veerhuis R., Rozemuller A., and van Gool W. (2010). Neuroinflammation–an early event in both the history and pathogenesis of Alzheimer's disease. Neurodegener. Dis. 7, 38–41 [DOI] [PubMed] [Google Scholar]
- 14. Brettschneider J., Toledo J., Van Deerlin V., Elman L., McCluskey L., Lee V., and Trojanowski J. (2012). Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One 7, e39216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKee A., Cantu R., Nowinski C., Hedley-Whyte E., Gavett B., Budson A., Santini V., Lee H., Kubilus C., and Stern R. (2009). Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J.Neuropathol. Exp. Neurol. 68, 709–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bombardier C., Temkin N., Machamer J., and Dikmen S. (2003). The natural history of drinking and alcohol-related problems after traumatic brain injury. Arch. Phys. Med. Rehabil. 84, 185–191 [DOI] [PubMed] [Google Scholar]
- 17. Hall K., and Johnston M. (1994). Outcomes evaluation in TBI Rehabilitation. Part II: measurement tools for a nationwide data system. Arch. Phys. Med. Rehabil. 75, SC10–SC18; discussion SC 27–SC18. [PubMed] [Google Scholar]
- 18. Horner M., Ferguson P., Selassie A., Labbate L., Kniele K., and Corrigan J. (2005). Patterns of alcohol use 1 year after traumatic brain injury: a population-based, epidemiological study. J. Int. Neuropsychol. Soc. 11, 322–330 [DOI] [PubMed] [Google Scholar]
- 19. Kreutzer J., Witol A., and Marwitz J. (1996). Alcohol and drug use among young persons with traumatic brain injury. J. Learn. Disabil. 29, 643–651 [DOI] [PubMed] [Google Scholar]
- 20. Washington P., Villapol S., andBurns M. (2015). Polypathology and dementia after brain trauma: does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp. Neurol. 275, 381–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Crews F., Qin L., Sheedy D., Vetreno R., and Zou J. (2013). High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol. Psychiatry 73, 602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu J., Lewohl J., Harris R., Iyer V., Dodd P., Randall P., and Mayfield R. (2006). Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology 31, 1574–1582 [DOI] [PubMed] [Google Scholar]
- 23. He J., Overstreet D., and Crews F. (2009). Abstinence from moderate alcohol self-administration alters progenitor cell proliferation and differentiation in multiple brain regions of male and female P rats. Alcohol Clin. Exp. Res. 33, 129–138 [DOI] [PubMed] [Google Scholar]
- 24. Xu J., and Chen C. (2015). Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist 21, 152–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cravatt B., Giang D., Mayfield S., Boger D., Lerner R., and Gilula N. (1996). Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384, 83–87 [DOI] [PubMed] [Google Scholar]
- 26. Dinh T., Freund T., and Piomelli D. (2002). A role for monoglyceride lipase in 2-arachidonoylglycerol inactivation. Chem. Phys. Lipids 121, 149–158 [DOI] [PubMed] [Google Scholar]
- 27. Long J., Li W., Booker L., Burston J., Kinsey S., Schlosburg J., Pavón F., Serrano A., Selley D., Parsons L., Lichtman A., and Cravatt B. (2009). Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 5, 37–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mayeux J., Katz P., Edwards S., Middleton J., and Molina P. (2017). Inhibition of endocannabinoid degradation improves outcomes from mild traumatic brain injury: a mechanistic role for synaptic hyperexcitability. J.Neurotrauma 34, 436–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katz P., Sulzer J., Impastato R., Teng S., Rogers E., and Molina P. (2015). Endocannabinoid degradation inhibition improves neurobehavioral function, blood–brain barrier integrity, and neuroinflammation following mild traumatic brain injury. J. Neurotrauma 32, 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mayeux J., Teng S., Katz P., Gilpin N., and Molina P. (2015). Traumatic brain injury induces neuroinflammation and neuronal degeneration that is associated with escalated alcohol self-administration in rats. Behav. Brain Res. 279, 22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roltsch E., Baynes B., Mayeux J., Whitaker A., Baiamonte B., and Gilpin N. (2014). Predator odor stress alters corticotropin-releasing factor-1 receptor (CRF1R)-dependent behaviors in rats. Neuropharmacology 79, 83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Teng S., and Molina P. (2014). Acute alcohol intoxication prolongs neuroinflammation without exacerbating neurobehavioral dysfunction following mild traumatic brain injury. J. Neurotrauma 31, 378–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Depoortere R., Li D., Lane J., and Emmett-Oglesby M. (1993). Parameters of self-administration of cocaine in rats under a progressive-ratio schedule. Pharmacol. Biochem.Behav. 45, 539–548 [DOI] [PubMed] [Google Scholar]
- 36. Hodos W. (1961). Progressive ratio as a measure of reward strength. Science 134, 943–944 [DOI] [PubMed] [Google Scholar]
- 37. Hoffmeister F. (1979). Progressive-ratio performance in the rhesus monkey maintained by opiate infusions. Psychopharmacology (Berl) 62, 181–186 [DOI] [PubMed] [Google Scholar]
- 38. Mello N., Lukas S., Bree M., and Mendelson J. (1988). Progressive ratio performance maintained by buprenorphine, heroin and methadone in Macaque monkeys. Drug Alcohol Depend. 21, 81–97 [DOI] [PubMed] [Google Scholar]
- 39. Recinto P., Samant A., Chavez G., Kim A., Yuan C., Soleiman M., Grant Y., Edwards S., Wee S., Koob G., George O., and Mandyam C. (2012). Levels of neural progenitors in the hippocampus predict memory impairment and relapse to drug seeking as a function of excessive methamphetamine self-administration. Neuropsychopharmacology 37, 1275–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Edwards S., Vendruscolo L., Schlosburg J., Misra K., Wee S., Park P., Schulteis G., and Koob G. (2012). Development of mechanical hypersensitivity in rats during heroin and ethanol dependence: alleviation by CRF1 receptor antagonism. Neuropharmacology 62, 1142–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dixon W. (1980). Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 20, 441–462 [DOI] [PubMed] [Google Scholar]
- 42. Ririe D., and Eisenach J. (2006). Age-dependent responses to nerve injury–induced mechanical allodynia. Anesthesiology 104, 344–350 [DOI] [PubMed] [Google Scholar]
- 43. McGinn M., Paulsen R., Itoga C., Farooq M., Reppel J., Edwards K., Whitaker A., Gilpin N., and Edwards S. (2016). Withdrawal from chronic nicotine exposure produces region‐specific tolerance to alcohol‐stimulated GluA1 phosphorylation. Alcohol Clin. Exp. Res. 40, 2537–2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prut L., and Belzung C. (2003). The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur. J. Pharmacol. 463, 3–33 [DOI] [PubMed] [Google Scholar]
- 45. Whelan-Goodinson R., Ponsford J., Johnston L., and Grant F. (2009). Psychiatric disorders following traumatic brain injury: their nature and frequency. J. Head Trauma Rehabil. 24, 324–332 [DOI] [PubMed] [Google Scholar]
- 46. Koponen S., Taiminen T., Portin R., Himanen L., Isoniemi H., Heinonen H., Hinkka S., and Tenovuo O. (2002). Axis I and II psychiatric disorders after traumatic brain injury: a 30-year follow-up study. Am. J. Psychiatry 159, 1315–1321 [DOI] [PubMed] [Google Scholar]
- 47. Marsicano G., Wotjak C., Azad S., Bisogno T., Rammes G., Cascio M., Hermann H., Tang J., Hofmann C., Zieglgänsberger W., Di Marzo V., and Lutz B. (2002). The endogenous cannabinoid system controls extinction of aversive memories. Nature 418, 530–534 [DOI] [PubMed] [Google Scholar]
- 48. Rahimi A., Hajizadeh Moghaddam A., and Roohbakhsh A. (2015). Central administration of GPR55 receptor agonist and antagonist modulates anxiety-related behaviors in rats. Fundam. Clin. Pharmacol. 29, 185–190 [DOI] [PubMed] [Google Scholar]
- 49. Reger M., Poulos A., Buen F., Giza C., Hovda D., and Fanselow M. (2012). Concussive brain injury enhances fear learning and excitatory processes in the amygdala. Biol. Psychiatry 71, 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zarrindast M., Sarahroodi S., Arzi A., Khodayar M., Taheri-Shalmani S., and Rezayof A. (2008). Cannabinoid CB1 receptors of the rat central amygdala mediate anxiety-like behavior: interaction with the opioid system. Behav. Pharmacol. 19, 716–723 [DOI] [PubMed] [Google Scholar]
- 51. Kushner M., Sher K., and Erickson D. (1999). Prospective analysis of the relation between DSM-III anxiety disorders and alcohol use disorders. Am. J. Psychiatry 156, 723–732 [DOI] [PubMed] [Google Scholar]
- 52. Möller C., Wiklund L., Thorsell A., Hyytiä P., and Heilig M. (1997). Decreased measures of experimental anxiety in rats bred for high alcohol preference. Alcohol Clin. Exp. Res. 21, 656–660 [PubMed] [Google Scholar]
- 53. Primeaux S., Wilson S., Bray G., York D., and Wilson M. (2006). Overexpression of neuropeptide Y in the central nucleus of the amygdala decreases ethanol self-administration in “anxious” rats. Alcohol Clin. Exp. Res. 30, 791–801 [DOI] [PubMed] [Google Scholar]
- 54. Spanagel R., Montkowski A., Allingham K., Stöhr T., Shoaib M., Holsboer F., and Landgraf R. (1995). Anxiety: a potential predictor of vulnerability to the initiation of ethanol self-administration in rats. Psychopharmacology (Berl) 122, 369–373 [DOI] [PubMed] [Google Scholar]
- 55. Stewart R., Gatto G., Lumeng L., Li T., and Murphy J. (1993). Comparison of alcohol-preferring (P) and nonpreferring (NP) rats on tests of anxiety and for the anxiolytic effects of ethanol. Alcohol 10, 1–10 [DOI] [PubMed] [Google Scholar]
- 56. Egli M., Koob G., and Edwards S. (2012). Alcohol dependence as a chronic pain disorder. Neurosci. Biobehav. Rev. 36, 2179–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hasanein P., Parviz M., Keshavarz M., and Javanmardi K. (2007). CB1 receptor activation in the basolateral amygdala produces antinociception in animal models of acute and tonic nociception. Clin. Exp. Pharmacol. Physiol. 34, 439–449 [DOI] [PubMed] [Google Scholar]
- 58. Kim C., Vincent A., Clauw D., Luedtke C., Thompson J., Schneekloth T., and Oh T. (2013). Association between alcohol consumption and symptom severity and quality of life in patients with fibromyalgia. Arthritis Res. Ther. 15, R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lawton J., and Simpson J. (2009). Predictors of alcohol use among people experiencing chronic pain. Psychol. Health Med. 14, 487–501 [DOI] [PubMed] [Google Scholar]
- 60. Forget B., Coen K., and Le Foll B. (2009). Inhibition of fatty acid amide hydrolase reduces reinstatement of nicotine seeking but not break point for nicotine self-administration–comparison with CB(1) receptor blockade. Psychopharmacology (Berl) 205, 613–624 [DOI] [PubMed] [Google Scholar]
- 61. Scherma M., Medalie J., Fratta W., Vadivel S., Makriyannis A., Piomelli D., Mikics E., Haller J., Yasar S., Tanda G., and Goldberg S. (2008). The endogenous cannabinoid anandamide has effects on motivation and anxiety that are revealed by fatty acid amide hydrolase (FAAH) inhibition. Neuropharmacology 54, 129–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Adamczyk P., McCreary A., Przegalinski E., Mierzejewski P., Bienkowski P., and Filip M. (2009). The effects of fatty acid amide hydrolase inhibitors on maintenance of cocaine and food self-administration and on reinstatement of cocaine-seeking and food-taking behavior in rats. J. Physiol.Pharmacol. 60, 119–125 [PubMed] [Google Scholar]
- 63. Volkow N., Fowler J., and Wang G. (2003). The addicted human brain: insights from imaging studies. J.Clin. Invest. 111, 1444–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. White N. (1996). Addictive drugs as reinforcers: multiple partial actions on memory systems. Addiction 91, 921–949; discussion 951–965. [PubMed] [Google Scholar]
- 65. Davidson R. (2002). Anxiety and affective style: role of prefrontal cortex and amygdala. Biol. Psychiatry 51, 68–80 [DOI] [PubMed] [Google Scholar]
- 66. Almeida-Suhett C., Prager E., Pidoplichko V., Figueiredo T., Marini A., Li Z., Eiden L., and Braga M. (2014). Reduced GABAergic inhibition in the basolateral amygdala and the development of anxiety-like behaviors after mild traumatic brain injury. PLoS One 9, e102627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Klein R., Acheson S., Qadri L., Dawson A., Rodriguiz R., Wetsel W., Moore S., Laskowitz D., and Dawson H. (2016). Opposing effects of traumatic brain injury on excitatory synaptic function in the lateral amygdala in the absence and presence of preinjury stress. J. Neurosci. Res. 94, 579–589 [DOI] [PubMed] [Google Scholar]
- 68. Kamprath K., Romo-Parra H., Häring M., Gaburro S., Doengi M., Lutz B., and Pape H. (2011). Short-term adaptation of conditioned fear responses through endocannabinoid signaling in the central amygdala. Neuropsychopharmacology 36, 652–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kodirov S., Jasiewicz J., Amirmahani P., Psyrakis D., Bonni K., Wehrmeister M., and Lutz B. (2010). Endogenous cannabinoids trigger the depolarization-induced suppression of excitation in the lateral amygdala. Learn. Mem. 17, 43–49 [DOI] [PubMed] [Google Scholar]
- 70. Bisogno T., and Di Marzo V. (2010). Cannabinoid receptors and endocannabinoids: role in neuroinflammatory and neurodegenerative disorders. CNS Neurol. Disord. Drug Targets 9, 564–573 [DOI] [PubMed] [Google Scholar]
- 71. Fox L., Davies D., Scholl J., Watt M., and Forster G. (2016). Differential effects of glucocorticoid and mineralocorticoid antagonism on anxiety behavior in mild traumatic brain injury. Behav. Brain Res. 312, 362–365 [DOI] [PubMed] [Google Scholar]
- 72. Vendruscolo L., Estey D., Goodell V., Macshane L., Logrip M., Schlosburg J., McGinn M., Zamora-Martinez E., Belanoff J., Hunt H., Sanna P., George O., Koob G., Edwards S., and Mason B. (2015). Glucocorticoid receptor antagonism decreases alcohol seeking in alcohol-dependent individuals. J.Clin. Invest. 125, 3193–3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Prendergast M., and Mulholland P. (2012). Glucocorticoid and polyamine interactions in the plasticity of glutamatergic synapses that contribute to ethanol-associated dependence and neuronal injury. Addict. Biol. 17, 209–223 [DOI] [PMC free article] [PubMed] [Google Scholar]