

Abstract
Proteinases are essential drivers of allergic airway disease and innate antifungal immunity in part through their ability cleave the clotting factor fibrinogen (FBG) into fibrinogen cleavage products (FCPs) that signal through Toll-like receptor 4 (TLR4). However, the mechanism by which FCPs engage TLR4 remains unknown. Here, we show that the proteinases from Aspergillus melleus (PAM) and other allergenic organisms rapidly hydrolyze FBG to yield relatively few FCPs that drive distinct antifungal mechanisms through TLR4. Functional FCPs, termed cryptokines, were characterized by rapid loss of the FBG α chain with substantial preservation of the β and γ chains, including a γ chain sequence (Fibγ390–396) that binds the integrin Mac-1 (CD11b/CD18). PAM-derived cryptokines could be generated from multiple FBG domains, and the ability of cryptokines to induce fungistasis in vitro and innate allergic airway disease in vivo strongly depended on both Mac-1 and the Mac-1–binding domain of FBG (Fibγ390–396). Our findings illustrate the essential concept of proteinase-activated immune responses and for the first time link Mac-1, cryptokines, and TLR4 to innate antifungal immunity and allergic airway disease.
Keywords: allergy, proteinase, fibrinogen, fungi, integrin, innate immunity, Toll-like receptor 4 (TLR4)
Introduction
Allergic airway disease, composed of the strongly associated conditions asthma and chronic rhinosinusitis, represents a rapidly increasing morbidity and healthcare burden across the world. More than 1 in 12 persons in the United States have asthma, with higher rates seen among children, the impoverished, and women (1). Despite the epidemic nature of allergic airway disease and an intensive international effort to understand its causes, modern standard management remains entirely palliative, offering no chance of long-term remission or cure (2). Nonetheless, important advances in our understanding of allergic airway disease pathogenesis have been made.
Environmental proteinases are increasingly recognized to be critical drivers of both allergies, the subset of allergic diseases that are driven by IgE-dependent type 1 hypersensitivity reactions, and allergic airway disease that is mediated by both lymphocytes and IgE-dependent immune reactions (3). Canonical environmental allergens such as pollens, dust mites, cockroaches and other arthropods, and pet danders are all important sources of active proteinases that alone or combined as part of whole-organism extracts readily induce experimental allergic airway disease in rodents (4). A particularly important source of exogenous, allergenic proteinases, however, is the fungi, which are linked to asthma through remarkably diverse environmental contexts that include thunderstorms, mold-contaminated domestic and work environments, and moldy automobiles (5–10).
The pathophysiology of fungus-related allergic airway disease is likely related to several factors, including fungal hypersensitivity, which does not involve actual infection, and true airway infection by fungus that involves the noninvasive growth of fungi in either the upper or lower airways, a unique type of superficial fungal infection termed airway mycosis (11). We and others have previously established that airway mycosis can be readily established in mice (6, 12–14) and that airway mycosis-dependent allergic airway disease requires the secretion of active proteinases by the fungi (6). Moreover, an identical disease phenotype can be induced simply by challenging mice with single proteinases derived from diverse species (5, 15–17).
Several proposed, nonmutually exclusive mechanisms potentially explain how the airway immune system recognizes allergenic proteinases from fungi and potentially other sources to initiate allergic airway disease. These mechanisms include disruption of tight junction proteins of airway epithelial cells to facilitate antigen presentation by dendritic cells (18, 19), induction of signaling from protease-activated receptor 2 (PAR2) (20–22), and activation of innate pattern recognition receptors such as TLR4 (23–27). We showed previously that proteinase-dependent induction of allergic airway disease requires in part the activation of the coagulation factor fibrinogen, from which fibrinogen cleavage products (FCPs)4 initiate both allergic airway disease and activate innate immune cells such as macrophages to limit the growth of fungi in vitro, termed fungistasis.
Both innate fungistatic responses and allergic airway disease are driven by FCPs obligatorily through TLR4 (23), but it remains unclear what structural features of FCPs determine their ability to engage this immune receptor. Moreover, multiple ligands, including LPS, interact with TLR4 indirectly through binding partners, raising the possibility that FCPs engage TLR4 through a similar indirect mechanism. The leukocyte integrin Mac-1 (CD18/CD11b) has previously been shown to bind to fibrinogen and regulate macrophage-dependent innate immunity (28). Moreover, Mac-1 interacts with TLR4 through an unknown mechanism to promote LPS-dependent inflammation and T cell activation (29). Considering these prior observations with our current findings, we reasoned that Mac-1 could facilitate the FCP–TLR4 interaction that promotes antifungal immunity and allergic airway disease.
In this study, we have combined proteinase cleavage assays with MS to demonstrate the fundamental structure of FCPs that determines their ability to interact with TLR4. We further demonstrate that FCPs most likely engage TLR4 indirectly through a mechanism involving the integrin Mac-1 (CD18/CD11b).
Results
Fungal proteinase-derived FCPs promote fungistatic immunity in vitro through diverse mechanisms
Our prior studies demonstrated that FBG can be cleaved by the endogenous proteinase thrombin to produce FCPs that stimulate fungistatic immunity (23). We reasoned that a more clinically relevant source of fibrinogen cleavage could be fungi that, through their secreted proteinases, are likely causes of human asthma and sinusitis (6). To determine whether FCPs can be generated from exogenous fungal proteinases, we first determined how FBG is degraded in the presence of the allergenic proteinase from Aspergillus melleus (PAM) using SDS-PAGE. This analysis confirmed that PAM yields numerous FCPs ranging in size from a few kDa to nearly 250 kDa (Fig. 1A).
Figure 1.

Proteolytic cleavage of fibrinogen enhances the fungistatic activity of diverse cells in vitro. A, SDS-polyacrylamide gel of fibrinogen, PAM (concentrated), and PAM–FCPs. B and C, SDS-PAGE showing PAM-mediated degradation of fibrinogen (B) over time and with increasing concentrations of PAM (C), but limited to 30-min digests. D, to mouse splenocytes cultured in serum-containing and serum-free conditions were added (200) A. niger conidia after which FGEs were enumerated after 18 h. E, same fungistasis assay was performed on splenocytes treated with sham (PBS), whole fibrinogen (100 nm; 34 μg/ml), or FCPs produced using the proteinase of A. melleus (PAM–FCPs; 100 nm cleaved fibrinogen). Percent (%) of FGE inhibition was calculated as the (no. of FGE in wells containing no cells − no. of FGE in wells containing cells/no. of FGE in wells containing no cells) × 100%. F–H, fungistasis assays measuring the ability of FCPs (100 nm) to prime fungal growth inhibition compared with PAM (FCP-equivalent; 40.8 ng/ml) added to splenocytes (F), BMDMs (G), and splenocytes (H) from WT and TLR4−/− mice. n = 3 biological replicates per group. *, p < 0.05; ***, p < 0.001 by Student's t test or one-way ANOVA with Bonferroni's multiple comparisons test. Data are representative of at least two independent experiments.
When added at a constant concentration of 6 μg/ml to FBG diluted to 5 mg/ml, PAM gradually cleaved FBG from 340 to ∼200 kDa, yielding FCPs of many sizes. However, even after 2 h of incubation in these strongly hydrolyzing conditions, PAM yielded only three major FCPs of ∼75, 150, and 200 kDa in size that persisted out to at least 6 h (data not shown). After 24 h, PAM had reduced fibrinogen to fragments no larger than ∼75 kDa (Fig. 1B). We found that similar trends in FBG fragmentation could be observed by modulating the concentration of PAM while keeping the incubation time constant (30 min; Fig. 1C). Thus, an allergenic fungal proteinase rapidly cleaves fibrinogen into FCPs of discrete size that remain relatively stable once formed.
We next determined the functional importance of PAM-derived FCPs using a modification of an in vitro assay that quantifies the fungistatic ability of immune cells, i.e. their ability to inhibit fungal growth in vitro (23). First, we verified that addition of bovine serum enhances the ability of mouse splenocytes to inhibit the growth of Aspergillus niger in vitro as indicated by a reduction in the number of fungal germination events (Fig. 1D).5 We further confirmed that supplementing serum-free cultures of murine splenocytes with 100 nm whole FBG or 100 nm PAM–FCPs resulted in similar fungal inhibition (Fig. 1E). We conducted additional functional experiments to confirm that the ability of PAM–FCPs to promote innate antifungal immunity cannot be accounted for by the residual proteinase remaining in the FCP mixture following their preparation (Fig. 1F). PAM–FCPs were further capable of promoting fungistasis in both murine bone marrow–derived macrophages (BMDMs) and splenocytes in a manner that absolutely depended on expression of TLR4 (Fig. 1, G and H) (23). Thus, PAM-derived FCPs are functionally equivalent to thrombin-derived FCPs with respect to induction of in vitro fungistatic activity from diverse immune cells.
We further evaluated the difference between whole FBG and FCPs in this assay and whether FCP-dependent fungistasis was cell contact–mediated or the result of secreted anti-microbial soluble products. Naïve mouse splenocytes were primed with fibrinogen, FCPs, or sham, and after 18 h the conditioned supernatants were transferred to separate wells, and fresh media were added to the remaining splenocytes. The ability of primed splenocytes, conditioned media, or sham to inhibit A. niger growth was then determined (Fig. 2A). We conducted additional experiments in which splenocytes were incubated with A. niger spores but were kept separated by a semi-permeable transwell membrane (Fig. 2B). These studies demonstrated that only FCP and not FBG-primed splenocytes mediate fungistasis and that splenocytes inhibit fungal growth through a mechanism that requires contact with the fungus and not secretion of a soluble antifungal substance (Fig. 2, A and B). We further conclude from these experiments that the ability of intact FBG to mediate fungistasis when added to cultures that simultaneously contain splenocytes and fungi (Fig. 1E) is most likely due to fungus-dependent cleavage of FBG to produce FCPs.
Figure 2.
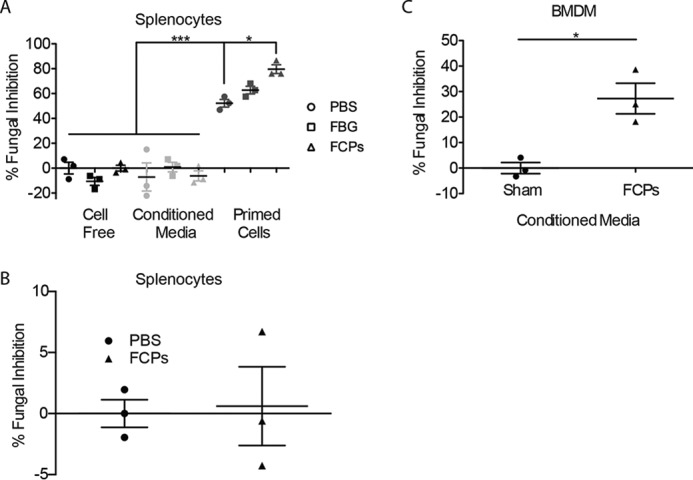
Splenocyte-mediated fungistasis is contact-dependent, whereas BMDMs secrete soluble antifungal factors. A, fungistasis assay comparing the fungistatic activity of PBS, FBG, and FCPs either alone, in treatment-primed cell-conditioned media, or in cultures containing splenocytes primed with each indicated treatment but in fresh media. B, fungistasis assay in which splenocytes were primed with PBS or FCPs in a transwell plate that precluded direct contact between fungi and mouse cells. C, fungistasis assay using conditioned media from bone marrow–derived macrophages primed with sham or FCPs. n = 3 biological replicates per group. *, p < 0.05; ***, p < 0.001 by Student's t test or one-way ANOVA with Bonferroni's multiple comparisons test. Data are representative of at least two independent experiments.
In contrast to splenocytes, although PAM–FCPs enhanced BMDM fungistatic immunity, these cells secreted a soluble factor that accounted for most of this fungistatic activity (Fig. 2C). Thus, fungal proteinase-derived FCPs, but not fibrinogen, induce fungistatic immunity from diverse cell types through a variety of mechanisms involving either cell contact or the secretion of soluble antifungal factors.
PAM is an alkaline serine proteinase
We have previously demonstrated that Aspergillus spp. proteinases are potent allergens that strongly induce allergic airway disease in mice, including profound airway hyper-responsiveness and eosinophilia when delivered intranasally (5, 6, 23). Although long available from commercial sources, the identity of PAM as a proteinase has not previously been characterized. By SDS-PAGE, PAM consists of at least three dominant protein bands of ∼11, 22, and 33 kDa (Fig. 3, A and B). Mass spectrometry (MS) performed on all of these protein bands revealed nearly identical sequence information, indicating that the different bands represented multimers of a single gene product. We observed similar multimerization occurring with the functionally similar allergenic proteinase derived from Aspergillus oryzae (5). MS-based sequencing indicated that the A. melleus proteinase is an extracellular alkaline serine proteinase that is substantially similar to the proteinases produced by other Aspergillus species (Fig. 3, C and D). We further conducted proteinase inhibition experiments involving PAM and multiple class-specific proteinase inhibitors that confirmed that PAM is only susceptible to inhibition by the serine class-specific inhibitor AEBSF (Fig. S1). Therefore, PAM is an alkaline serine proteinase, similar to other fungal proteinases (e.g. Asp f13) that have previously been linked to allergic airway disease in mice (18, 31).
Figure 3.

PAM is an alkaline serine proteinase. A and B, protein electrophoreses of PAM indicating the prevalence of 33-, 22-, and 11-kDa bands performed using a Novex NuPAGE 4–12% gel (A) and by the Baylor College of Medicine Mass Spectrometry Proteomics core facility (B). C and D, protein sequence alignments of PAM in tabular format (C) and in graphical format for the (D) 33-kDa electrophoresis band.
Proteinases from diverse species produce FCPs by preferentially cleaving the fibrinogen α chain
In addition to fungi, allergic airway diseases such as asthma and allergic rhinitis are linked to many other organisms, including plants (especially pollens), arthropods (e.g. environmental mites), and bacteria, all of which produce proteinases. Therefore, we next examined whether allergenic proteinases derived from nonfungal sources could induce similar fibrinogen-dependent fungistatic immunity in vitro. We found that FCPs produced from PAM, Pronase from the bacterium Streptomyces griseus, an extract derived from the dust mite Dermatophagoides farinae, and ragweed pollen from Ambrosia artemisiifolia (ragweed) induced similar fungistatic activity from splenocytes (Fig. 4A).
Figure 4.

Diverse proteinases preferentially cleave the fibrinogen α chain to induce antifungal responses in vitro. A, mouse splenocyte fungistasis assays using FCPs (100 nm) derived from the proteolytic cleavage of human fibrinogen using PAM (6 μg/ml), Pronase from S. griseus (PRO; 6 μg/ml), D. farinae dust mite extract (DF; 600 μg/ml), and A. artemisiifolia (Ragweed) extract (RW; 180 μg/ml). B, schematic diagram of fibrinogen demonstrating the α, β, and γ polypeptide chains as well as the D and E domain, based on crystallography data (30, 47, 66). C, SDS-PAGE analysis of β-mercaptoethanol-reduced human fibrinogen before addition of proteinases (2nd lane from left) and following cleavage by PAM (0.6 μg/ml), Pronase (Pro, 0.6 μg/ml), D. farinae extract (DF; 600 μg/ml), D. pteronyssinus extract (DP; 600 μg/ml), G. domesticus extract (GD; 180 μg/ml), T. putrescentiae extract (TP; 180 μg/ml), and A. artemisiifolia extract (180 μg/ml). D, reducing SDS-PAGE analysis of 6 μg/ml PAM-mediated degradation of fibrinogen over the indicated times. n = 3 biologic replicates per group. *, p < 0.05; **, p < 0.01 by Student's t test or one-way ANOVA with Bonferroni's multiple comparisons test. Data are representative of at least two similar, independent experiments.
After normalizing for proteinase activity, FCPs produced from these sources were analyzed by SDS-PAGE under reducing conditions to reveal the individual FBG polypeptide chains (α, β, and γ chains; Fig. 4B). We further included in this analysis additional FCPs derived from the dust mite Dermatophagoides pteronyssinus and the storage mites Glyciphagus domesticus and Tyrophagus putrescentiae. This analysis revealed that each of the tested proteinases preferentially degraded the fibrinogen α chain (Fig. 4C). Additional analyses of PAM-derived FCPs revealed that whereas the α chain was almost completely degraded immediately upon addition of proteinase, the β and γ chains remained substantially resistant to hydrolysis even after 2 h (Fig. 4D). Thus, although we cannot exclude the possibility that β- or γ-chain cleavage is also essential, our findings indicate that proteinases derived from diverse organisms cleave FBG in a highly stereotypical manner, with predominantly the α chain being rapidly degraded to reveal the relatively proteinase-resistant β and γ chains.
Cleavage products produced from the fibrinogen D and E fragments also promote fungistasis
We next sought to determine the FBG region from which functional FCPs derive. Our initial attempts to identify functionally active FCPs from PAM-digested whole FBG were unsuccessful because standard isolation techniques (molecular sieving; column chromatography) inevitably resulted in denatured and inactive FCPs (data not shown). We therefore determined the ability of distinct FBG regions (e.g. D and E domains, Fig. 4B) to yield active FCPs. At a constant 100 nm, PAM-derived FCPs derived from whole fibrinogen and D and E domains all induced significant fungistatic responses from murine splenocytes as compared with sham-activated cells (Fig. 5A). As noted previously, fibrinogen readily induced fungistasis independent of the addition of PAM, likely due to the cleavage induced by fungal proteinases secreted into the culture media.
Figure 5.

Fibrinogen D and E fragments are sufficient substrates through which PAM can generate functional FCPs. A, fungistasis assays were performed that compared PAM-induced FCPs derived from whole-human fibrinogen, the fibrinogen D fragment, and the fibrinogen E fragment as indicated using murine splenocytes. All fibrinogen moieties were added at the same concentration (100 nm). B, nonreducing SDS-PAGE analysis of native proteins and PAM–FCPs derived from whole FBG, and the D and E fragments (Frag) of fibrinogen. n = 3 biologic replicates per group. *, p < 0.05; ***, p < 0.001 by one-way ANOVA with Bonferroni's multiple comparisons test of fragments and FCPs compared with blank and PAM controls. Statistical data are representative of at least two similar, independent experiments.
In contrast, whole D and E domains behaved distinctly under the same conditions. Whereas D domains had no intrinsic ability to induce fungistasis, E domains showed a nonsignificant trend to promote fungistasis prior to the addition of PAM, suggesting that E domains might be either partially competent or more readily induced by ambient fungal proteinases to become competent to induce fungistatic responses (Fig. 5A). Thus, multiple domains of FBG, including D and E domains, are capable of inducing fungistatic responses from immune cells following proteolytic activation. Whole FBG is more potent in inducing fungistasis under these conditions that control for equivalent molar addition of FBG product as whole FBG contains stoichiometrically more fungistasis-inducing regions than the individual domains.
In contrast to whole FBG, SDS-PAGE analysis of the D and E domains indicated that PAM-directed proteolysis resulted in relatively minor structural changes (Fig. 5B). Mass spectrometry was subsequently performed on the ∼85-kDa fragment of the cleaved D domain that again indicated predominant loss of the α chain and substantial preservation of the β and γ chains as was observed with whole FBG (Fig. 4D). Moreover, the 85-kDa fragment retained a region in the fibrinogen γ chain (amino acids 390–396) that is known to be critical for binding to Mac-1 (data not shown) (28, 32).
CD11b critically regulates FCP-dependent fungistatic immunity and allergic airway disease
To determine whether FCPs could interact with Mac-1, we first compared the ability of WT and CD11b-deficient mouse (itgam−/−) splenocytes to control fungal growth in response to FCP challenge in vitro. We found that although FCPs induced significant fungistatic responses from itgam−/− splenocytes, these responses were markedly attenuated as compared with WT cells (Fig. 6A).
Figure 6.

CD11b is required for the full expression of fungus-induced allergic airway disease. A, fungistasis assay comparing splenocytes from WT and CD11b−/− mice. B, anesthetized C57BL6/J, Fibγ390–396A, and CD11b−/− mice were intranasally challenged with 4 × 105 live conidia of A. niger (AN) or PBS on alternating days for 15 days for a total of eight treatments with data collected 24 h after the final challenge. C, respiratory system resistance (RRS) as assessed by increasing intravenous acetylcholine challenge. D, quantification of cells from bronchoalveolar lavage fluid samples (M, macrophages; E, eosinophils; N, neutrophils; L, lymphocytes). E and F, quantitative real-time PCR measurements of cDNA derived from mouse whole-lung mRNA for Muc5AC and IL-13. G, IL-4 as measured by ELISA from supernatants derived from whole-lung homogenates. n = 3 (A) or n = 4–5 (B–G) biologic replicates per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 by Student's t test or one- or two-way ANOVA with Bonferroni's multiple comparisons test. Data are representative of at least two similar, independent experiments.
We next determined the ability of CD11b-deficient mice to develop allergic airway disease, a model of asthma in which key disease features such as airway hyper-responsiveness, airway eosinophil recruitment, mucus hyper-production, and type 2 cytokine secretion are induced by airway fungi in part through TLR4 (5, 23). Wildtype (WT) and itgam−/− mice were challenged with 400,000 conidia of A. niger or sham every other day for 2 weeks and compared with identically treated Fibγ390–396A mice in which the amino acids of the FBG-binding site have been converted to alanines (33). All mice were assessed for key allergic airway disease features 24 h following the final challenge. Compared with WT mice, both Fibγ390–396A and itgam−/− mice challenged with fungus demonstrated significantly less airway hyper-responsiveness as assessed by increases in respiratory system resistance (RRS) following provocative challenge with escalating doses of acetylcholine (Fig. 6C).
The itgam−/− mice challenged with fungi also displayed a significant reduction in total cell and eosinophil infiltration into the airways as compared with WT mice, but Fibγ390–396A mice showed no defects in cell trafficking (Fig. 6D). Quantitative real-time PCR analysis of RNA isolated from mouse lung homogenates further revealed no significant differences between WT, Fibγ390–396A, and itgam−/− mice with regard to Muc5AC or IL-13 expression (Fig. 6, E and F). Finally, IL-4 secretion from whole-lung homogenates was not impaired by the absence of Mac-1 or in the presence of mutant fibrinogen (Fig. 6G).
Together, these observations demonstrate that a cognate interaction between the FBG γ chain and Mac-1 is essential for the expression of antifungal responses from splenocytes and airway hyper-responsiveness. Our findings further confirm the independent role played by Mac-1 in eosinophil recruitment in diverse contexts (34–36).
FCPs can induce “innate” asthma in Fibγ390–396A mice, but not itgam−/− mice
Full expression of proteinase or fungus-induced allergic airway disease in mice requires generation of both FCPs and TH2 cells, the latter of which drive the strong expression of airway hyper-responsiveness, marked eosinophilia, mucus hypersecretion, and IgE antibodies that are markers of advanced disease (23). However, although they only trigger innate immune responses, FCPs can alone drive an attenuated or “innate” form of allergic airway disease that includes modest induction of airway hyper-responsiveness and nonallergic airway inflammation through a mechanism that involves the activation of signal transducer and activator of transcription 6 (STAT6).5
We therefore hypothesized that providing exogenous FCPs would overcome the inability of Fibγ390–396A mice, but not itgam−/− mice, to express the innate allergic airway disease. To test this, we challenged syngeneic Fibγ390–396A mice, itgam−/−, and WT mice for 5 consecutive days with FCPs or a vehicle control intranasally and quantified key features of the allergic airway disease phenotype 1 day after the final challenge (Fig. 7A). We found that both WT and Fibγ390–396A mice developed modest but significant airway hyper-responsiveness, whereas itgam−/− mice did not (Fig. 7, B–D). Moreover, we found that itgam−/− mice challenged with FCPs yielded fewer total leukocytes from the airways as compared with Fibγ390–396A mice, indicating that homing defects were restricted to the itgam−/− mice (Fig. 7E). Mucus gene induction as assessed by quantifying muc5ac transcripts was similarly enhanced in all three genotypes of mice after FCP challenge (Fig. 7F). Together, these findings demonstrate that “innate” allergic airway disease, similar to leukocyte fungistatic responses, is in part mediated by FCPs through a cognate interaction with CD11b.
Figure 7.

FCP-induced inflammation promotes allergic airway disease in WT and Fibγ390–396A mice but not CD11b (Mac-1) knockouts. A, anesthetized C57BL6/J, Fibγ390–396A, and itgam−/− mice were challenged intranasally with 0.25 mg of PAM–FCPs or sham (PBS) containing 0.3 μg of PAM (FCP dose equivalent) for 5 consecutive days. 24 h after the final challenge, mice were assessed for development of airway hyper-responsiveness over 5 half-log increments of acetylcholine (B). The same data are shown for the 4th and 5th doses only (C and D). E, bronchoalveolar lavage fluid inflammatory cells. F, muc5ac mRNA expression for the same mouse groups. n = 4–7 biologic replicate mice per group. *, p < 0.05; **, p < 0.01; ***, p < 0.001 by one- or two-way ANOVA with Bonferroni's multiple comparisons test. Statistical tests for C and D are carried over from those in B. Data represent two combined independent experiments.
Discussion
Increasing evidence implicates coagulation factors in the development and perpetuation of allergic airway disease (23, 37–40). These studies underscore specifically the importance of the antimicrobial role of clotting factors that can now be seen as a primitive immune response that is orthologous to the coagulogen–Spaetzle–Toll system of arthropods (41, 42). In distinct contexts, fibrinogen has been linked to immunity against bacteria and fungi through its interactions with CD11b/Mac-1 (28, 43, 44) and TLR4 (23, 45, 46). Proteinases are also known to initiate inflammation in the settings of allergic asthma (2, 6, 18, 48) and innate antifungal immunity (23, 49), but it is also now clear that the distinct and complex inflammatory pathways that comprise allergic inflammation also serve complex antifungal roles (12). Thus, fibrinogen activation by proteinases leading to allergic and antifungal responses through TLR4 and CD11b as shown here unifies major elements of this ancient immune paradigm and provides new insight into the pathogenesis of allergic airway diseases.
Our findings recapitulate and extend our previous findings (23) that the formation of fibrinogen cleavage products from proteinases promotes antifungal immunity in vitro in a manner that requires TLR4. Using protein gel electrophoresis, we have characterized the ordered degradation of fibrinogen by the allergenic fungal proteinase PAM, which was determined to be an alkaline serine proteinase. FCPs were found to induce antifungal immunity by both contact-dependent and contact-independent means. Splenocytes, which represent a diverse cell population consisting predominantly of B and T cells and to a lesser extent monocytes, granulocytes, and dendritic cells, were found to require direct contact with fungi to inhibit fungal growth. In contrast, fully mature BMDM secreted soluble antimicrobial factors in response to FCPs that accounted for most of their antifungal activity, although we cannot rule out the possibility that these cells can inhibit fungal growth through additional mechanisms that require contact. Whereas FCPs could prime splenocytes to become fungistatically active, intact fibrinogen required contact with the fungus to initiate fungistatic activity, likely representing the production of FCPs in situ from whole fibrinogen. These observations suggest that surface-bound FBG may bind leukocytes with low affinity, perhaps through Mac-1 or TLR4, but with much higher affinity to Mac-1 in the presence of proteinases produced by germinating fungi that convert FBG to FCPs. These newly formed FCPs would already be present at the cell surface for rapid transfer to other critical receptors such as TLR4 to complete a highly efficient signaling complex (Fig. 8).
Figure 8.

FCPs act through Mac-1 and TLR4 to promote allergic and antifungal inflammation. Schematic depiction of the proposed interactions between FBG, FCPs, Mac-1, and TLR4 to trigger allergic and antifungal responses in the airway. Intact FBG may interact weakly with CD11b and TLR4, but high-affinity binding to CD11b occurs after proteolytic cleavage of especially the α chain to yield FCPs (cryptokines). Subsequent signaling initiated through the Mac-1/TLR4/FCP complex initiates both innate antifungal and allergic inflammatory programs through distinct transcription factors, NF-κB and STAT6 (30).
Once formed, the putative Mac-1/TLR4/FCP complex then initiates a signaling cascade that coordinates two effector functions, innate fungistatic immunity as illustrated by splenocytes and BMDM and innate allergic airway disease that includes modest airway hyper-responsiveness and predominant neutrophilia (Fig. 7), but also limited eosinophilia.5 The latter events likely also serve an antifungal role (12), but despite their similar functions, we have shown in parallel studies that leukocyte-based fungistatic immunity and innate allergic airway disease are coordinated through distinct transcription factors, NF-κB and signal transducer and activator of transcription 6 (STAT6), respectively5 (Fig. 8).
Our combined findings thus reveal unexpected complexity with regard to the antimicrobial programs that are deployed in response to airway mycosis and that trace back to a unique and cleaved form of fibrinogen that results in the presence of fungi and many other exogenous proteinases derived from four kingdoms of life. Given the pleiotropic, cytokine-like properties of FCPs that extend well beyond the clotting function of FBG, we propose the new term cryptokine (“hidden cytokine”) to more functionally describe this unique form of an immunologically active fibrinogen fragment (Fig. 8). Our findings further illustrate the broader concept of proteinase-activated immune responses (PAIRs) in which endogenous proteinases cleave a variety of pro-cytokines to yield their fully active forms (e.g. IL-1 family (50) and IL-25 (51)), i.e. enPAIRs, which presumably are formed downstream of exogenous proteinase-activated immune responses, i.e. exPAIRs, exemplified through fibrinogen cryptokines. Of special interest is the IL-1 family member IL-33, which may be proteolytically activated by both endogenous and exogenous proteinases (52). A more complete understanding of exPAIRs and enPAIRS and their molecular relationships is likely to clarify our understanding of how innate and adaptive immune responses are coordinately regulated.
A limitation of this study is that we were unable to directly demonstrate the binding of FCPs to Mac-1. This is due to the marked affinity that fibrinogen and FCPs have for immunoglobulin, especially IgG (53, 54). This issue of nonspecificity precludes use of immunological assays that are routinely used to demonstrate specific binding between two molecules, including Western blotting and flow cytometry. Thus, although the model presented in Fig. 8 is strongly supported through the indirect evidence presented herein, it remains speculative pending additional studies that overcome this issue.
Lack of Mac-1 did not completely abrogate leukocyte fungistatic responses or airway hyper-responsiveness, in contrast to the more severe such defects seen in TLR4−/− mice (Fig. 1H) (23). It is thus possible that closely-related integrins that also bind FBG such as CD11a/CD18 (leukocyte function–associated antigen 1; LFA-1) may be in part compensating for the lack of CD11b in these experiments. Moreover, ICAM-1 (CD54), an endogenous ligand for LFA-1 and Mac-1 that is also expressed on leukocytes, also recognizes FBG, albeit through a site distinct from that recognized by CD11b (55). Cognate interactions between cryptokines, LFA-1, and ICAM-1 may therefore suffice to support cellular infiltration seen in itgam−/− mice, yet be insufficient for the induction of airway hyper-responsiveness. We previously showed that allergic airway disease develops poorly in mice deficient in all CD18 integrins and specifically LFA-1 and ICAM-1 due to a defect in TH2 cell homing to lung, but we cannot exclude the possibility that perturbations in cryptokine signaling related to lack of these adhesion molecules also contributed to this phenotype (56, 57).
Additional studies have examined the role of Mac-1 in allergic airway disease. Kanwar et al. (58) found that compared with WT, Mac-1-deficient mice developed increased airway hyper-responsiveness and eosinophilic inflammation in an ovalbumin-based model after 28 days. However, because this protocol involves no exPAIR-based mechanism for the creation of cryptokines (ovalbumin is not proteolytically active), it is possible that the function of Mac-1 in this context is entirely distinct, resulting in a substantially different phenotype.
Our findings here and elsewhere5 suggest that cryptokines coordinate a physical relationship between TLR4 and CD11b to yield a highly-efficient signaling complex that coordinates the development of allergic airway disease and innate antifungal immunity (Fig. 8). Additional studies that document the association of fibrinogen with the CD11b I domain and Mac-1 with TLR4 in remarkably distinct contexts that include endotoxemia and malaria strongly support this model (29, 59–61). Thus, although our work has emphasized the importance of this pathway to fungus-related allergic inflammation and disease, it should be noted that cryptokine–TLR4 signaling broadly shapes the immune response to very different pathogens. Additional studies are required to understand how a fundamentally similar ligand–receptor interaction translates into diverse immune responses that are tailored to distinct pathogens.
As the healthcare costs associated with allergic airway disease continue to rise (62), the need for understanding the mechanisms that initiate and propagate these ailments increases commensurately. Future research will further clarify the mechanisms by which fungi and proteinases coordinate the expression of allergic inflammation and diseases at innate and adaptive levels and ultimately translate into more effective and durable therapies.
Experimental procedures
Mice
C57Bl6J (JAX no. 000664), TLR4−/− (B6.B10ScN-Tlr4lps-del/JthJ; JAX no. 007227) (63, 64), and CD11b−/− (B6.129S4-Itgamtm1Myd/J; JAX no. 003991) (65) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Fibγ390–396A mice developed as described previously (28). Mice were housed and bred under specific pathogen-free conditions at the Baylor College of Medicine Transgenic Mouse Facility. Female mice 4–10 weeks old were used for all experiments and matched for age and sex. Animal studies were conducted under Institutional Animal Care and Use Committee–approved protocols and conformed to all Federal and institutional guidelines.
Genotyping of mice
TLR4−/− (B6.B10ScN-Tlr4lps-del/JthJ), CD11b−/− (B6.129S4-Itgamtm1Myd/J), and Fibγ390–396A mice were routinely genotyped using the following primers (all 5′ → 3′): for TLR4−/−, mutant forward (GCAAGTTTCTATATGCATTCTC), and mutant reverse (CCTCCATTTCCAATAGGTAG); WT forward (ATATGCATGATCAACACCACAG), and WT reverse (TTTCCATTGCTGCCCTATAG); for CD11b−/−, mutant reverse (TGATTCCCACTTTGTGGTTC); common (TGTTTTTACCCCTCCCTCCT); WT reverse (CCTTTGATCTCTCCCCACCT); for Fibγ390–396A, γF(+) (ATTGACATGATCACCAAAATTGCTTATTG), and γE (−) (CCATTTAAGGCTAGGTATCATGTTAAGAAAG), followed by the addition of 1 μl of PvuII for 2 h at 37 °C to target the novel mutant restriction site, as described previously (28). PCR products were electrophoresed on 1% agarose gels for genotype determination.
SDS-PAGE
Protein electrophoresis was performed using NuPAGE 4–12% BisTris protein gels, MES SDS Running Buffer, and SimplyBlue SafeStain (all from Invitrogen). Samples for protein electrophoresis were diluted at least 1:2 in Tricine sample buffer and Precision Plus Protein Kaleidoscope prestained protein standards were used as a protein electrophoresis molecular weight marker (both from Bio-Rad).
Isolation of murine splenocytes and generation of murine BMDMs
Murine splenocytes were isolated using the ACK lysing buffer treatment of homogenized mouse spleens in RPMI 1640 cell culture media (without serum) passed through a 40-μm nylon cell strainer (Corning, Corning, NY). BMDMs were isolated by suspending syringe-flushed marrow from murine femurs and tibias in RPMI 1640 cell culture media supplemented with 10% fetal bovine serum and passing the suspension through a 40-μm cell strainer. The marrow was then cultured in RPMI 1640 cell culture media supplemented with 10% fetal bovine serum and 20 ng/ml recombinant murine GM-CSF (R&D Systems, Minneapolis, MN) for 1 week with one media wash and replenishment on day 4. All cell culture media contained 100 units/ml penicillin/streptomycin (Invitrogen).
Proteinases
Proteinase from A. melleus (PAM; P4032; Sigma) and Pronase from S. griseus (PRON-RO; Sigma) were purchased, aliquoted in PBS, and stored at −80 °C. Ragweed extract (short ragweed, A. artemisiifolia) was obtained from Greer (Cambridge, MA), aliquoted, suspended in PBS, and stored at −80 °C. Lyophilized mite samples of D. farinae, D. pteronyssinus, G. domesticus, and T. putrescentiae were generously provided by Fabrizio Ottoboni (Life Science Knowledge Consulting, Milan, Italy). Mite samples were suspended in cold PBS, and protein was extracted using zirconium beads in a Retsch PM 100 Planetary Ball Mill for three 5-min cycles at 550 rpm, each followed by 5 min on ice. Sample slurries were then centrifuged to remove undissolved solids at 2200 × g for 30 min at 4 °C, followed by a second centrifugation of the supernatant at 10,000 × g for 30 min at 4 °C. The remaining supernatants were aliquoted and stored at −80 °C.
Mass spectrometry protein sequencing
Samples were excised from SDS-PAGE preparations, and MS protein sequencing was performed by the Baylor College of Medicine Mass Spectrometry Proteomics Core Facility. Protein identity was confirmed by aligning detected protein in the samples to known sequences.
Generation of FCPs
FCPs were made by suspending human fibrinogen (HCI-0150R; Hematologic Technologies, Essex Junction, VT) at 5 mg/ml in PBS. PAM was added to the fibrinogen solution at a concentration of 6 μg/ml for 30 min at 37 °C. Fibrinogen fragments D (HCI-0150D) and E (HCI-0150E) were also acquired from Hematologic Technologies and processed by the same methods. For in vivo studies, fibrinogen was incubated with PAM for 6 h. Other proteinases, concentrations, and incubation durations were performed using similar methods as indicated in the figure legends.
Fungistasis assay
Splenocytes or BMDMs were cultured in 24-well flat-bottom tissue culture plates (353047, Corning) or 24-well transwell plates (351152, Corning) as indicated and incubated for 24 h with FCPs or other described preparations and controls in a 37 °C, 5% CO2 incubator using RPMI 1640 cell culture media without serum. 200 conidia of A. niger were then added to each well. After 18–22 h, allowing for sufficient growth of the spores, fungal germination events (FGEs) were enumerated. The percentage of fungal growth inhibition was calculated by the difference in the number of FGEs in control no murine cell wells versus the experimental wells, divided by the number of FGEs in no cell control wells, multiplied by 100%: 100%·(FGENC − FGEExp)/FGENC.
Murine model of fungus-induced allergic airway disease
Anesthetized mice were intranasally challenged with 400,000 conidia of A. niger in 50 μl of PBS or PBS only control every other day for 15 days for a total of eight treatments as described previously (6, 23). 24 h following the final challenge, allergic airway disease was assessed.
Murine model of FCP-induced acute allergic airway inflammation
Anesthetized mice were intranasally challenged with 0.25 mg of PAM–FCPs (6 h degradation) diluted in 50 μl of PBS or vehicle control (0.3 μg PAM) in 50 μl of PBS every day for 5 days. 24 h following the final challenge, allergic airway disease was assessed.
Assessment of allergic airway disease
24 h following the final challenge of the allergic airway disease models described above, mice were anesthetized with etomidate, and increasing concentrations of acetylcholine were administered intravenously via the tail vein to determine airway hyper-responsiveness using airway resistance as described previously (6, 23). After these data were collected, bronchoalveolar lavage fluid (BALF) and whole lung for RNA and cell cultures were obtained. Total cells were counted in the BALF, and cell differential analysis was performed using modified Giemsa-stained cytospin preparations. Homogenized whole lung was prepared for RNA extraction in TRIzol (Thermo Fisher Scientific, Waltham, MA) or for cell culture for detection of cytokines. Cell culture supernatants were analyzed for IL-4 concentration by ELISA using anti-IL-4 (Clone 11B11, 1:100) and anti-IL-4-Biotin (BD Biosciences, 554390, 1:200) antibodies.
Proteinase inhibition
PAM (50 ng/sample) was incubated with FITC-labeled casein overnight; whole protein was precipitated by trichloroacetic acid, and sample fluorescence was quantified using the SpectraMax i3x (Molecular Devices) in accordance to the protease fluorescent detection kit protocol (PF0100, Sigma). Inhibition of proteolytic activity by serial dilutions of the broad spectrum, class-specific inhibitors E-64 (Sigma, E3132), pepstatin A (Sigma, P5318), Ilomastat (Selleckchem, S7157), and AEBSF hydrochloride (Santa Cruz Biotechnology, sc-202041) at a starting concentration above the manufacturer's recommended working concentration. Repeated experiments were performed at select concentrations to verify the initial analysis.
mRNA isolation, conversion to cDNA, and quantitative PCR
Homogenized murine lung tissue dissolved in TRIzol was extracted with chloroform (Sigma), precipitated in isopropyl alcohol (Sigma), washed in ethanol, and then resuspended in nuclease-free water. cDNA was generated from RNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). Quantitative PCR was then performed using the ABI 7500 real-time PCR system (Applied Biosystems) with standard techniques with 18S rRNA (Applied Biosystems 4318839) as the normalization standard. Muc5ac (Mm01276718_m1) and IL-13 (Mm00434204_m1) TaqMan probes (Thermo Fisher Scientific) were used in quantitative PCR.
Statistical analysis
Data were analyzed using GraphPad Prism 5 and are presented as mean ± S.E. of the mean. Significant differences relative to PBS-challenged mice or appropriate controls are expressed by p values of <0.05, as measured by the two-tailed Student's t test or one-way or two-way ANOVA followed by Bonferroni's test for multiple comparison using datasets involving similar distributions of variance. Sample sizes, both in vitro and in vivo, were determined empirically. No in vitro data were excluded unless the entire experiment or assay failed due to technical reasons. No mouse data were excluded unless animals died prior to data collection. A process for mouse randomization was not used as all animals within a given genotype were genetically identical, and all mice appeared identical regardless of genotype.
Data availability
Data will be made available by the authors upon reasonable request.
Author contributions
C. T. L. and D. B. C. data curation; C. T. L., Z. Z., F. K., and D. B. C. formal analysis; C. T. L., Z. Z., P. C. P., and D. B. C. investigation; C. T. L., F. K., and D. B. C. writing-original draft; H.-Y. T., J. M. K., M. C. M., Y. W., Z. Z., P. C. P., A. R., M. J. F., F. K., and D. B. C. methodology; H.-Y. T., J. M. K., M. C. M., Y. W., A. R., M. J. F., F. K., and D. B. C. writing-review and editing; M. J. F. resources; F. K. and D. B. C. conceptualization; F. K. and D. B. C. supervision; F. K. and D. B. C. funding acquisition.
Supplementary Material
Acknowledgments
We thank Li-Zhen Song and Nima Baghaei for technical assistance and Daniela Zelaschi (Life Science Knowledge Consulting, Milan, Italy) for providing mite samples.
This work was supported by National Institutes of Health Grants T32AI053831, R01HL117181, R01AI135803, and R41AI124997 and Veterans Affairs Office of Research and Development Grant 5I01BX002221. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
H. Y. Tung, C. T. Landers, J. M. Knight, Y. Wu, W. Lu, P. Porter, S. C. Sun, A. Rodriguez, F. Kheradmand, and D. B. Corry, submitted for publication.
- FCP
- fibrinogen cleavage product
- FBG
- fibrinogen
- PAM
- proteinase from A. melleus
- PAIR
- proteinase-activated immune response
- ANOVA
- analysis of variance
- BALF
- bronchoalveolar lavage fluid
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- BMDM
- bone marrow–derived macrophage
- AEBSF
- 4-(2-aminoethyl)benzenesulfonyl fluoride
- FGE
- fungal germination event
- LPS
- lipopolysaccharide
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1. Akinbami L. J., Bailey C. M., Johnson C. A., King M. E., Liu X., Moorman J. E., and Zahran H. S. (2012) Trends in asthma prevalence, health care use, and mortality in the United States, 2001–2010, 2012, 1–8 [PubMed] [Google Scholar]
- 2. Tung H. Y., Li E., Landers C., Nguyen A., Kheradmand F., Knight J. M., and Corry D. B. (2018) Advances and evolving concepts in allergic asthma. Semin. Respir. Crit. Care Med. 39, 64–81 10.1055/s-0037-1607981 [DOI] [PubMed] [Google Scholar]
- 3. Tung H. Y., Landers C., Li E., Porter P., Kheradmand F., and Corry D. B. (2016) Allergen-encoded signals that control allergic responses. Curr. Opin. Allergy Clin. Immunol. 16, 51–58 10.1097/ACI.0000000000000233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Porter P. C., Yang T., Luong A., Delclos G. L., Abramson S. L., Kheradmand F., and Corry D. B. (2011) Proteinases as molecular adjuvants in allergic airway disease. Biochim. Biophys. Acta 1810, 1059–1065 10.1016/j.bbagen.2011.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kheradmand F., Kiss A., Xu J., Lee S. H., Kolattukudy P. E., and Corry D. B. (2002) A protease-activated pathway underlying Th cell type 2 activation and allergic lung disease. J. Immunol. 169, 5904–5911 10.4049/jimmunol.169.10.5904 [DOI] [PubMed] [Google Scholar]
- 6. Porter P., Susarla S. C., Polikepahad S., Qian Y., Hampton J., Kiss A., Vaidya S., Sur S., Ongeri V., Yang T., Delclos G. L., Abramson S., Kheradmand F., and Corry D. B. (2009) Link between allergic asthma and airway mucosal infection suggested by proteinase-secreting household fungi. Mucosal Immunol. 2, 504–517 10.1038/mi.2009.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sharpe R. A., Bearman N., Thornton C. R., Husk K., and Osborne N. J. (2015) Indoor fungal diversity and asthma: a meta-analysis and systematic review of risk factors. J. Allergy Clin. Immunol. 135, 110–122 10.1016/j.jaci.2014.07.002 [DOI] [PubMed] [Google Scholar]
- 8. Burr M. L., Matthews I. P., Arthur R. A., Watson H. L., Gregory C. J., Dunstan F. D., and Palmer S. R. (2007) Effects on patients with asthma of eradicating visible indoor mould: a randomised controlled trial. Thorax 62, 767–772 10.1136/thx.2006.070847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pulimood T. B., Corden J. M., Bryden C., Sharples L., and Nasser S. M. (2007) Epidemic asthma and the role of the fungal mold Alternaria alternata. J. Allergy Clin. Immunol. 120, 610–617 10.1016/j.jaci.2007.04.045 [DOI] [PubMed] [Google Scholar]
- 10. Kumar P., Lopez M., Fan W., Cambre K., and Elston R. C. (1990) Mold contamination of automobile air conditioner systems. Ann. Allergy 64, 174–177 [PubMed] [Google Scholar]
- 11. Porter P. C., Lim D. J., Maskatia Z. K., Mak G., Tsai C. L., Citardi M. J., Fakhri S., Shaw J. L., Fothergil A., Kheradmand F., Corry D. B., and Luong A. (2014) Airway surface mycosis in chronic TH2-associated airway disease. J. Allergy Clin. Immunol. 134, 325–331 10.1016/j.jaci.2014.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Porter P., Roberts L., Fields A., Knight M., Qian Y., Delclos G. L., Han S., Kheradmand F., and Corry D. B. (2011) Necessary and sufficient role for T helper cells to prevent fungal dissemination during mucosal airway infection. Infect. Immun. 79, 4459–4471 10.1128/IAI.05209-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Denis O., van den Brûle S., Heymans J., Havaux X., Rochard C., Huaux F., and Huygen K. (2007) Chronic intranasal administration of mould spores or extracts to unsensitized mice leads to lung allergic inflammation, hyper-reactivity and remodelling. Immunology 122, 268–278 10.1111/j.1365-2567.2007.02636.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mak G., Porter P. C., Bandi V., Kheradmand F., and Corry D. B. (2013) Tracheobronchial mycosis in a retrospective case-series study of five status asthmaticus patients. Clin. Immunol. 146, 77–83 10.1016/j.clim.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sokol C. L., Barton G. M., Farr A. G., and Medzhitov R. (2008) A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat. Immunol. 9, 310–318 10.1038/ni1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tripathi P., Kukreja N., Singh B. P., and Arora N. (2009) Serine protease activity of Cur l 1 from Curvularia lunata augments Th2 response in mice. J. Clin. Immunol. 29, 292–302 10.1007/s10875-008-9261-9 [DOI] [PubMed] [Google Scholar]
- 17. Florsheim E., Yu S., Bragatto I., Faustino L., Gomes E., Ramos R. N., Barbuto J. A., Medzhitov R., and Russo M. (2015) Integrated innate mechanisms involved in airway allergic inflammation to the serine protease subtilisin. J. Immunol. 194, 4621–4630 10.4049/jimmunol.1402493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Balenga N. A., Klichinsky M., Xie Z., Chan E. C., Zhao M., Jude J., Laviolette M., Panettieri R. A. Jr., and Druey K. M. (2015) A fungal protease allergen provokes airway hyper-responsiveness in asthma. Nat. Commun. 6, 6763 10.1038/ncomms7763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wan H., Winton H. L., Soeller C., Tovey E. R., Gruenert D. C., Thompson P. J., Stewart G. A., Taylor G. W., Garrod D. R., Cannell M. B., and Robinson C. (1999) Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J. Clin. Invest. 104, 123–133 10.1172/JCI5844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nadeem A., Alharbi N. O., Vliagoftis H., Tyagi M., Ahmad S. F., and Sayed-Ahmed M. M. (2015) Proteinase activated receptor-2-mediated dual oxidase-2 up-regulation is involved in enhanced airway reactivity and inflammation in a mouse model of allergic asthma. Immunology 145, 391–403 10.1111/imm.12453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aubier M., Thabut G., Hamidi F., Guillou N., Brard J., Dombret M. C., Borensztajn K., Aitilalne B., Poirier I., Roland-Nicaise P., Taillé C., and Pretolani M. (2016) Airway smooth muscle enlargement is associated with protease-activated receptor 2/ligand overexpression in patients with difficult-to-control severe asthma. J. Allergy Clin. Immunol. 138, 729–739.e11 10.1016/j.jaci.2015.12.1332 [DOI] [PubMed] [Google Scholar]
- 22. Homma T., Kato A., Bhushan B., Norton J. E., Suh L. A., Carter R. G., Gupta D. S., and Schleimer R. P. (2016) Role of Aspergillus fumigatus in triggering protease-activated receptor-2 in airway epithelial cells and skewing the cells toward a T-helper 2 bias. Am. J. Respir. Cell Mol. Biol. 54, 60–70 10.1165/rcmb.2015-0062OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Millien V. O., Lu W., Shaw J., Yuan X., Mak G., Roberts L., Song L. Z., Knight J. M., Creighton C. J., Luong A., Kheradmand F., and Corry D. B. (2013) Cleavage of fibrinogen by proteinases elicits allergic responses through Toll-like receptor 4. Science 341, 792–796 10.1126/science.1240342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cho M., Lee J. E., Lim H., Shin H. W., Khalmuratova R., Choi G., Kim H. S., Choi W. S., Park Y. J., Shim I., Kim B. S., Kang C. Y., Kim J. O., Tanaka S., Kubo M., et al. (2018) Fibrinogen cleavage products and Toll-like receptor 4 promote the generation of programmed cell death 1 ligand 2-positive dendritic cells in allergic asthma. J. Allergy Clin. Immunol. 142, 530–541.e6 10.1016/j.jaci.2017.09.019 [DOI] [PubMed] [Google Scholar]
- 25. Hadebe S., Kirstein F., Fierens K., Chen K., Drummond R. A., Vautier S., Sajaniemi S., Murray G., Williams D. L., Redelinghuys P., Reinhart T. A., Fallert Junecko B. A., Kolls J. K., Lambrecht B. N., Brombacher F., and Brown G. D. (2015) Microbial ligand costimulation drives neutrophilic steroid-refractory asthma. PLoS ONE 10, e0134219 10.1371/journal.pone.0134219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hammad H., Chieppa M., Perros F., Willart M. A., Germain R. N., and Lambrecht B. N. (2009) House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med. 15, 410–416 10.1038/nm.1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trompette A., Divanovic S., Visintin A., Blanchard C., Hegde R. S., Madan R., Thorne P. S., Wills-Karp M., Gioannini T. L., Weiss J. P., and Karp C. L. (2009) Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature 457, 585–588 10.1038/nature07548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Flick M. J., Du X., Witte D. P., Jirousková M., Soloviev D. A., Busuttil S. J., Plow E. F., and Degen J. L. (2004) Leukocyte engagement of fibrin(ogen) via the integrin receptor αMβ2/Mac-1 is critical for host inflammatory response in vivo. J. Clin. Invest. 113, 1596–1606 10.1172/JCI20741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ling G. S., Bennett J., Woollard K. J., Szajna M., Fossati-Jimack L., Taylor P. R., Scott D., Franzoso G., Cook H. T., and Botto M. (2014) Integrin CD11b positively regulates TLR4-induced signalling pathways in dendritic cells but not in macrophages. Nat. Commun. 5, 3039 10.1038/ncomms4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhmurov A., Brown A. E., Litvinov R. I., Dima R. I., Weisel J. W., and Barsegov V. (2011) Mechanism of fibrin(ogen) forced unfolding. Structure 19, 1615–1624 10.1016/j.str.2011.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chiu L. L., Perng D. W., Yu C. H., Su S. N., and Chow L. P. (2007) Mold allergen, pen C 13, induces IL-8 expression in human airway epithelial cells by activating protease-activated receptor 1 and 2. J. Immunol. 178, 5237–5244 10.4049/jimmunol.178.8.5237 [DOI] [PubMed] [Google Scholar]
- 32. Lishko V. K., Podolnikova N. P., Yakubenko V. P., Yakovlev S., Medved L., Yadav S. P., and Ugarova T. P. (2004) Multiple binding sites in fibrinogen for integrin αMβ2 (Mac-1). J. Biol. Chem. 279, 44897–44906 10.1074/jbc.M408012200 [DOI] [PubMed] [Google Scholar]
- 33. Flick M. J., Du X., and Degen J. L. (2004) Fibrin(ogen)–αMβ2 interactions regulate leukocyte function and innate immunity in vivo. Exp. Biol. Med. 229, 1105–1110 10.1177/153537020422901104 [DOI] [PubMed] [Google Scholar]
- 34. Das A. M., Flower R. J., and Perretti M. (1997) Eotaxin-induced eosinophil migration in the peritoneal cavity of ovalbumin-sensitized mice: mechanism of action. J. Immunol. 159, 1466–1473 [PubMed] [Google Scholar]
- 35. Jia G. Q., Gonzalo J. A., Hidalgo A., Wagner D., Cybulsky M., and Gutierrez-Ramos J. C. (1999) Selective eosinophil transendothelial migration triggered by eotaxin via modulation of Mac-1/ICAM-1 and VLA-4/VCAM-1 interactions. Int. Immunol. 11, 1–10 10.1093/intimm/11.1.1 [DOI] [PubMed] [Google Scholar]
- 36. Larangeira A. P., Silva A. R., Gomes R. N., Penido C., Henriques M. G., Castro-Faria-Neto H. C., and Bozza P. T. (2001) Mechanisms of allergen- and LPS-induced bone marrow eosinophil mobilization and eosinophil accumulation into the pleural cavity: a role for CD11b/CD18 complex. Inflamm. Res. 50, 309–316 10.1007/PL00000249 [DOI] [PubMed] [Google Scholar]
- 37. Stroo I., Yang J., de Boer J. D., Roelofs J. J., van 't Veer C., Castellino F. J., Zeerleder S., and van der Poll T. (2017) Factor XI deficiency enhances the pulmonary allergic response to house dust mite in mice independent of factor XII. Am. J. Physiol. Lung Cell. Mol. Physiol. 312, L163–L171 10.1152/ajplung.00320.2016 [DOI] [PubMed] [Google Scholar]
- 38. Esnault S., Kelly E. A., Sorkness R. L., Evans M. D., Busse W. W., and Jarjour N. N. (2016) Airway factor XIII associates with type 2 inflammation and airway obstruction in asthmatic patients. J. Allergy Clin. Immunol. 137, 767–773.e6 10.1016/j.jaci.2015.05.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schuliga M., Royce S. G., Langenbach S., Berhan A., Harris T., Keenan C. R., and Stewart A. G. (2016) The coagulant factor Xa induces protease-activated receptor-1 and annexin A2-dependent airway smooth muscle cytokine production and cell proliferation. Am. J. Respir. Cell Mol. Biol. 54, 200–209 10.1165/rcmb.2014-0419OC [DOI] [PubMed] [Google Scholar]
- 40. Shinagawa K., Martin J. A., Ploplis V. A., and Castellino F. J. (2007) Coagulation factor Xa modulates airway remodeling in a murine model of asthma. Am. J. Respir. Crit. Care Med. 175, 136–143 10.1164/rccm.200608-1097OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Theopold U., Schmidt O., Söderhäll K., and Dushay M. S. (2004) Coagulation in arthropods: defence, wound closure and healing. Trends Immunol. 25, 289–294 10.1016/j.it.2004.03.004 [DOI] [PubMed] [Google Scholar]
- 42. Arnot C. J., Gay N. J., and Gangloff M. (2010) Molecular mechanism that induces activation of Spatzle, the ligand for the Drosophila Toll receptor. J. Biol. Chem. 285, 19502–19509 10.1074/jbc.M109.098186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rubel C., Gómez S., Fernández G. C., Isturiz M. A., Caamaño J., and Palermo M. S. (2003) Fibrinogen-CD11b/CD18 interaction activates the NF-κB pathway and delays apoptosis in human neutrophils. Eur. J. Immunol. 33, 1429–1438 10.1002/eji.200323512 [DOI] [PubMed] [Google Scholar]
- 44. Sitrin R. G., Pan P. M., Srikanth S., and Todd R. F. 3rd. (1998) Fibrinogen activates NF-κB transcription factors in mononuclear phagocytes. J. Immunol. 161, 1462–1470 [PubMed] [Google Scholar]
- 45. Smiley S. T., King J. A., and Hancock W. W. (2001) Fibrinogen stimulates macrophage chemokine secretion through Toll-like receptor 4. J. Immunol. 167, 2887–2894 10.4049/jimmunol.167.5.2887 [DOI] [PubMed] [Google Scholar]
- 46. Hodgkinson C. P., Patel K., and Ye S. (2008) Functional Toll-like receptor 4 mutations modulate the response to fibrinogen. Thromb. Haemost. 100, 301–307 [PubMed] [Google Scholar]
- 47. Köhler S., Schmid F., and Settanni G. (2015) The internal dynamics of fibrinogen and its implications for coagulation and adsorption. PLoS Comput. Biol. 11, e1004346 10.1371/journal.pcbi.1004346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reed C. E., and Kita H. (2004) The role of protease activation of inflammation in allergic respiratory diseases. J. Allergy Clin. Immunol. 114, 997–1008 10.1016/j.jaci.2004.07.060 [DOI] [PubMed] [Google Scholar]
- 49. Romani L. (2011) Immunity to fungal infections. Nat. Rev. Immunol. 11, 275–288 10.1038/nri2939 [DOI] [PubMed] [Google Scholar]
- 50. Afonina I. S., Müller C., Martin S. J., and Beyaert R. (2015) Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity 42, 991–1004 10.1016/j.immuni.2015.06.003 [DOI] [PubMed] [Google Scholar]
- 51. Goswami S., Angkasekwinai P., Shan M., Greenlee K. J., Barranco W. T., Polikepahad S., Seryshev A., Song L. Z., Redding D., Singh B., Sur S., Woodruff P., Dong C., Corry D. B., and Kheradmand F. (2009) Divergent functions for airway epithelial matrix metalloproteinase 7 and retinoic acid in experimental asthma. Nat. Immunol. 10, 496–503 10.1038/ni.1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cayrol C., Duval A., Schmitt P., Roga S., Camus M., Stella A., Burlet-Schiltz O., Gonzalez-de-Peredo A., and Girard J. P. (2018) Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat. Immunol. 19, 375–385 10.1038/s41590-018-0067-5 [DOI] [PubMed] [Google Scholar]
- 53. Boehm T. K., and DeNardin E. (2008) Fibrinogen binds IgG antibody and enhances IgG-mediated phagocytosis. Hum. Antibodies 17, 45–56 10.3233/HAB-2008-173-401 [DOI] [PubMed] [Google Scholar]
- 54. Yamanaka Y., Matsugano S., Yoshikawa Y., and Orino K. (2016) Binding analysis of human immunoglobulin G as a zinc-binding protein. Antibodies 5, 13 10.3390/antib5020013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Altieri D. C., Duperray A., Plescia J., Thornton G. B., and Languino L. R. (1995) Structural recognition of a novel fibrinogen chain sequence (117133) by intercellular adhesion molecule-1 mediates leukocyte-endothelium interaction. J. Biol. Chem. 270, 696–699 10.1074/jbc.270.2.696 [DOI] [PubMed] [Google Scholar]
- 56. Lee S. H., Prince J. E., Rais M., Kheradmand F., Shardonofsky F., Lu H., Beaudet A. L., Smith C. W., Soong L., and Corry D. B. (2003) Differential requirement for CD18 in T-helper effector homing. Nat. Med. 9, 1281–1286 10.1038/nm932 [DOI] [PubMed] [Google Scholar]
- 57. Lee S.-H., Prince J. E., Rais M., Kheradmand F., Ballantyne C. M., Weitz-Schmidt G., Smith C. W., and Corry D. B. (2008) Developmental control of integrin expression regulates Th2 effector homing. J. Immunol. 180, 4656–4667 10.4049/jimmunol.180.7.4656 [DOI] [PubMed] [Google Scholar]
- 58. Kanwar S., Smith C. W., Shardonofsky F. R., and Burns A. R. (2001) The role of Mac-1 (CD11b/CD18) in antigen-induced airway eosinophilia in mice. Am. J. Respir. Cell Mol. Biol. 25, 170–177 10.1165/ajrcmb.25.2.4295 [DOI] [PubMed] [Google Scholar]
- 59. Jeyaseelan S., Chu H. W., Young S. K., Freeman M. W., and Worthen G. S. (2005) Distinct roles of pattern recognition receptors CD14 and Toll-like receptor 4 in acute lung injury. Infect. Immun. 73, 1754–1763 10.1128/IAI.73.3.1754-1763.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barrera V., Skorokhod O. A., Baci D., Gremo G., Arese P., and Schwarzer E. (2011) Host fibrinogen stably bound to hemozoin rapidly activates monocytes via TLR-4 and CD11b/CD18-integrin: a new paradigm of hemozoin action. Blood 117, 5674–5682 10.1182/blood-2010-10-312413 [DOI] [PubMed] [Google Scholar]
- 61. Diamond M. S., Garcia-Aguilar J., Bickford J. K., Corbi A. L., and Springer T. A. (1993) The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J. Cell Biol. 120, 1031–1043 10.1083/jcb.120.4.1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Loftus P. A., and Wise S. K. (2015) Epidemiology and economic burden of asthma. Int. Forum Allergy Rhinol. 5, Suppl. 1, S7–S10 10.1002/alr.21547 [DOI] [PubMed] [Google Scholar]
- 63. Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., and Beutler B. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 10.1126/science.282.5396.2085 [DOI] [PubMed] [Google Scholar]
- 64. Vogel S. N., Hansen C. T., and Rosenstreich D. L. (1979) Characterization of a congenitally LPS-resistant, athymic mouse strain. J. Immunol. 122, 619–622 [PubMed] [Google Scholar]
- 65. Coxon A., Rieu P., Barkalow F. J., Askari S., Sharpe A. H., von Andrian U. H., Arnaout M. A., and Mayadas T. N. (1996) A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5, 653–666 10.1016/S1074-7613(00)80278-2 [DOI] [PubMed] [Google Scholar]
- 66. Weisel J. W., and Litvinov R. I. (2017) Fibrin formation, structure and properties. Subcell. Biochem. 82, 405–456 10.1007/978-3-319-49674-0_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available by the authors upon reasonable request.