

Abstract
Venomous marine cone snails produce peptide toxins (conotoxins) that bind ion channels and receptors with high specificity and therefore are important pharmacological tools. Conotoxins contain conserved cysteine residues that form disulfide bonds that stabilize their structures. To gain structural insight into the large, yet poorly characterized conotoxin H-superfamily, we used NMR and CD spectroscopy along with MS-based analyses to investigate H-Vc7.2 from Conus victoriae, a peptide with a VI/VII cysteine framework. This framework has CysI–CysIV/CysII–CysV/CysIII–CysVI connectivities, which have invariably been associated with the inhibitor cystine knot (ICK) fold. However, the solution structure of recombinantly expressed and purified H-Vc7.2 revealed that although it displays the expected cysteine connectivities, H-Vc7.2 adopts a different fold consisting of two stacked β-hairpins with opposing β-strands connected by two parallel disulfide bonds, a structure homologous to the N-terminal region of the human granulin protein. Using structural comparisons, we subsequently identified several toxins and nontoxin proteins with this “mini-granulin” fold. These findings raise fundamental questions concerning sequence–structure relationships within peptides and proteins and the key determinants that specify a given fold.
Keywords: toxin, protein structure, protein-disulfide isomerase, protein conformation, protein evolution, protein expression, disulfide, conotoxin, inhibitor cystine knot, antistasin, β-hairpin, CD spectroscopy, disulfide bond, granulin, NMR spectroscopy, toxin
Introduction
Venoms from a variety of animals such as snakes, spiders, scorpions, and marine snails contain diverse peptide toxins that bind with high specificity and affinity to their molecular targets. These characteristic features make venom peptide toxins potentially valuable pharmacological tools and drug leads (1). One toxin from a predatory marine cone snail, ω-MVIIA from Conus magus, is a Food and Drug Administration–approved drug (with the commercial name Prialt) used to treat severe and chronic pain (2), and several other conotoxins are in development for various pathologies, including pain, epilepsy, stroke, and diabetes (3–6). In addition, many venom peptides are used as affinity reagents and constitute valuable research tools.
Each of the ∼800 species of cone snails of the genus Conus produces a unique set of 100–400 venom peptides that are typically referred to as conotoxins or conopeptides. Conotoxins are ribosomally synthesized as preproproteins, containing a conserved signal sequence for endoplasmic reticulum (ER)4 localization, a propeptide region, and the C-terminal mature toxin that is released upon proteolytic removal of the propeptide. Conotoxins can be divided into gene superfamilies based on their conserved signal sequence (7, 8). To date, more than 53 gene superfamilies have been described in this genus (9). The peptide investigated in this study, H-Vc7.2, was first identified by transcriptome sequencing of the venom gland of Conus victoriae (10) and belongs to the H-superfamily of conotoxins.
Mature conotoxins display highly variable amino acid sequences at nearly all positions apart from the conserved cysteine residues that define their cysteine framework (11). Presently, the reported conotoxin sequences are divided into 26 cysteine frameworks (8). H-Vc7.2 adopts one of the most common cysteine frameworks, VI/VII, where the six cysteines are arranged in a C–C–CC–C–C pattern. Within this framework, disulfides are predicted to connect CysI with CysIV, CysII with CysV, and CysIII with CysVI. To date, this cysteine framework has only been observed to give rise to the inhibitor cystine knot (ICK) fold (also known as the “knottin” fold (12, 13)). This fold is a common structural motif found in many animal toxins, where a ring formed by two disulfide bonds (CysI–CysIV and CysII–CysV) and the intervening polypeptide backbone is threaded by the third disulfide (CysIII–CysVI) (12). The ICK fold is found in toxins of many different venomous species, such as cone snails, spiders, scorpions, centipedes and anemones, as well as in plants and viruses (14).
The application of next-generation sequencing technology to generate venom-gland transcriptomes has revolutionized venom peptide discovery (15) and led to a massive increase in the number of venom–peptide sequences reported, including many new classes of venom peptides. Accordingly, there are several newly described conotoxin gene families for which the structure and biological activity remain unknown (7). To gain a better understanding of the recently reported H-superfamily, we investigated H-Vc7.2 from C. victoriae and present the first 3D structure of a peptide from this superfamily. To generate sufficient material, we utilized a previously developed Escherichia coli expression system (CyDisCo) designed to produce disulfide-containing proteins (16, 17), modified here to include co-expression with a conotoxin foldase (conotoxin-specific protein-disulfide isomerase, csPDI) from Conus geographus (18). Using this system, fully oxidized H-Vc7.2 was recovered from the soluble fraction and purified for biophysical analysis. The solution structure of H-Vc7.2 determined by NMR spectroscopy unexpectedly showed that the peptide adopts a mini-granulin fold rather than an ICK fold. We identify the mini-granulin fold in a variety of proteins, indicating that it is more widespread than previously appreciated, and we discuss the implications of our work for sequence–structure relationships.
Results
Native H-Vc7.2 is devoid of post-translational modifications
Previous analyses of the venom gland transcriptome of C. victoriae revealed the uncharacterized conotoxin H-superfamily, from which three transcripts were identified (10). By searching high-resolution MS/MS spectra of reduced and alkylated C. victoriae venom against the translated venom gland transcriptome, the mature form of the H-Vc7.2 conotoxin was identified. H-Vc7.2 exists in the venom as a 25-amino acid–residue peptide with its six cysteine residues arranged in the type VI/VII framework (C–C–CC–C–C) (Fig. 1). No variants of H-Vc7.2, e.g. post-translationally-modified, or peptides of different chain length were detected.
Figure 1.

H-Vc7.2 is a 25-residue conotoxin devoid of post-translational modifications. Top, b/y ladder diagram summarizing observed b- and y-ions within the amino acid sequence of H-Vc7.2. Bottom, MS/MS spectrum of H-Vc7.2 in the reduced and alkylated venom of C. victoriae. *, internal fragment ions. Inset, H-Vc7.2 [M + 5H]5+ precursor ion selected for MS/MS had monoisotopic m/z of 627.46 (z = 5, predicted m/z 627.46).
H-Vc7.2 expressed in E. coli purifies as a single major species
Disulfide-containing proteins can be expressed in the oxidized state in the cytosol of E. coli using the CyDisCo system (16, 17), which can produce highly complex disulfide-bonded proteins (16, 19). In this system, the protein of interest is co-expressed with two redox enzymes, the mitochondrial oxidase Erv1p from Saccharomyces cerevisiae and human PDI. Erv1p provides the oxidizing equivalents to generate disulfide bonds de novo, whereas PDI isomerizes non-native disulfides. In contrast to the commonly used Shuffle and Origami cells, where the reducing pathways present in the cytoplasm are disrupted, the CyDisCo system thus uses an active enzyme system to make disulfide bonds. Published comparisons of CyDisCo versus Origami show the former system to be more efficient for the proteins investigated (16). Moreover, the CyDisCo system is highly versatile in the sense that it works in any strain and in any media tested to date.5
We modified the original CyDisCo system for targeted expression of conotoxins by co-expressing, in addition to human PDI and Erv1p, a conotoxin-specific PDI (Fig. 2A) (18). In previous work (18), we have shown that in vitro this csPDI enzyme greatly accelerates the generation of correctly folded conotoxins during oxidative folding. Therefore, we reasoned that co-expressing the csPDI may help improve expression levels of some conotoxins.
Figure 2.

NextH-Vc7. 2 expressed in E. coli was purified as a single major species. A, overview of the two plasmids used in this study. pLE577 encodes Erv1p, hPDI, and csPDI under control of a tac promoter and confers chloramphenicol resistance. pLE566 encodes Ub–His10–NextH-Vc7.2 under the control of a T7 promoter and confers kanamycin resistance. This vector contains the lacI gene, which encodes the lac repressor. B, overview of the Ub–His10–NextH-Vc7.2 fusion protein. C, nonreducing SDS-polyacrylamide gel of E. coli extracts from untreated cells (lane 1) and cells treated with 1 mm IPTG overnight at 30 °C (lanes 2–4). Total, soluble, and pellet fractions were loaded as indicated. The mobility of molecular mass marker bands in kDa is shown on the left-hand side of the gel. The expression of both PDI proteins was verified by MALDI MS (see “Experimental procedures”). D, preparative RP-HPLC indicating A215 (black line), A280 (gray line), and percent of solvent B (broken line). The material eluting over the volume indicated by the black line labeled C was reanalyzed by analytical RP-HPLC as shown in E. A single main peak eluted at 25 ml.
Recombinant H-Vc7.2 was expressed as a fusion protein with an N-terminal ubiquitin (Ub)-tag containing 10 consecutive histidines (Ub–His10) followed by a TEV protease recognition site (Fig. 2B) (20). Upon TEV cleavage, this recombinant H-Vc7.2 peptide contains four additional N-terminal amino acid residues, Gly–Ala–Met–Gly (GAMG), as compared with the native peptide because of the cloning procedure used. This N-terminally extended 29-residue recombinant peptide is hereafter referred to as NextH-Vc7.2, and was used for determination of the cysteine connectivity and structural analysis by NMR spectroscopy. For CD spectroscopy, comparison of reversed-phase HPLC (RP-HPLC) retention time with C. victoriae venom and bioactivity studies during this work, we also generated recombinant H-Vc7.2 (named rH-Vc7.2) devoid of the four N-terminal nonnative residues. All numbering throughout this study refers to the extended peptide, except for Fig. 1 where the numbering refers to the native sequence.
Overnight induction with IPTG resulted in clearly visible bands for hPDI and csPDI (that co-migrated by SDS-PAGE), Erv1p, and Ub–His10–NextH-Vc7.2 (Fig. 2C). Moreover, the majority of Ub–His10–NextH-Vc7.2 was found in the soluble fraction. NextH-Vc7.2 was purified as outlined in Fig. S1A. The His10-tagged fusion protein was purified from crude lysate by metal-affinity chromatography (Fig. S1B). Upon TEV protease cleavage, uncleaved fusion protein, His6–TEV protease, and the liberated Ub–His10-tag were removed by batch purification on cobalt resin, and the peptide was further purified by RP-HPLC. The obtained chromatogram (Fig. 2D) showed a major peak as well as smaller peaks with increased retention time. The fraction representing the major peak was collected as indicated in Fig. 2D and lyophilized. A small amount of the lyophilized product was re-analyzed, and the resulting chromatogram indicated a high purity of the peptide (Fig. 2E). MALDI-TOF MS was used to confirm full oxidation of the recombinant peptide (Fig. S2). The only assigned 13Cβ chemical shift, the one belonging to Cys-23, also strongly supported its involvement in disulfide-bond formation with a Cβ chemical shift value of 39.1 ppm (21).
Cysteines in NextH-Vc7.2 are connected in a CysI–CysIV, CysII–CysV, and CysIII–CysVI arrangement
To determine the cysteine connectivity in NextH-Vc7.2, we used sequential reduction and alkylation followed by mass spectrometric analyses as described recently (22). To suppress the risk of disulfide scrambling, reduction and alkylation steps were performed at low pH using a large molar excess of alkylating agent and with the peptide adsorbed to a solid phase, which further minimizes disulfide reconnection because of the restrained conformational freedom of the peptide. The sequential reduction and alkylation were performed with different maleimides resulting in samples where the cysteines of the individual disulfide bridges have distinct alkylation patterns. This approach allows unambiguous identification of the cysteine connectivity. In total, four different alkylation patterns with N-methylmaleimide (NMM), N-ethylmaleimide (NEM), and N-cyclohexylmaleimide (NCM) were obtained for NextH-Vc7.2. All alkylation patterns were compatible with a single cysteine connectivity in the peptide. The MS/MS spectra of NextH-Vc7.2 alkylated with two NMM, two NEM, and two NCM are shown in Fig. S3.
The NextH-Vc7.2 peptide produced mainly y-type ions with additional (y-NH3) and (y-H2O) fragments. Based on the MS/MS spectra in Fig. S3, the alkylation pattern was assigned as C8-NEM/C13-NCM/C17-NMM/C18-NEM/C23-NCM/C28-NMM. The resulting cysteine connectivity in the peptide consequently is C8–C18/C13–C23/C17–C28. Detailed information about the identified fragments and three additional alkylation patterns with identified fragments all supporting the same cysteine connectivity can be found in Fig. S4 and Tables S1–S4.
Recombinant H-Vc7.2 elutes with the same retention time as the native venom peptide, and disulfide bonds are critical for stability
To investigate whether rH-Vc7.2 (devoid of the four N-terminal nonnative residues) shared the same structural characteristics as the native peptide, we compared retention times of rH-Vc7.2 and the native peptide from C. victoriae venom by RP-HPLC. Moreover, the importance of the disulfide bonds for the stability of the rH-Vc7.2 peptide was investigated by CD spectroscopy.
The venom fraction that eluted at the same percent of solvent B as rH-Vc7.2 (24.3–24.7% solvent B) contained a peptide with the same mass as rH-Vc7.2 (observed [M + H]+ = 2785.15; calculated [M + H]+ = 2785.12) demonstrating that rH-Vc7.2 exhibits the same physicochemical properties as the native venom peptide (Fig. 3, A–C). Adjacent fractions were also subjected to MALDI-TOF MS. A corresponding mass was also observed in the fraction eluting immediately prior to rH-Vc7.2 (23.9–24.3% solvent B), but not in any other adjacent fractions. Future studies may determine whether the peptide with the same mass as H-Vc7.2 could represent the same peptide with an alternative fold (e.g. ICK or other) or a proline cis/trans isomer.
Figure 3.
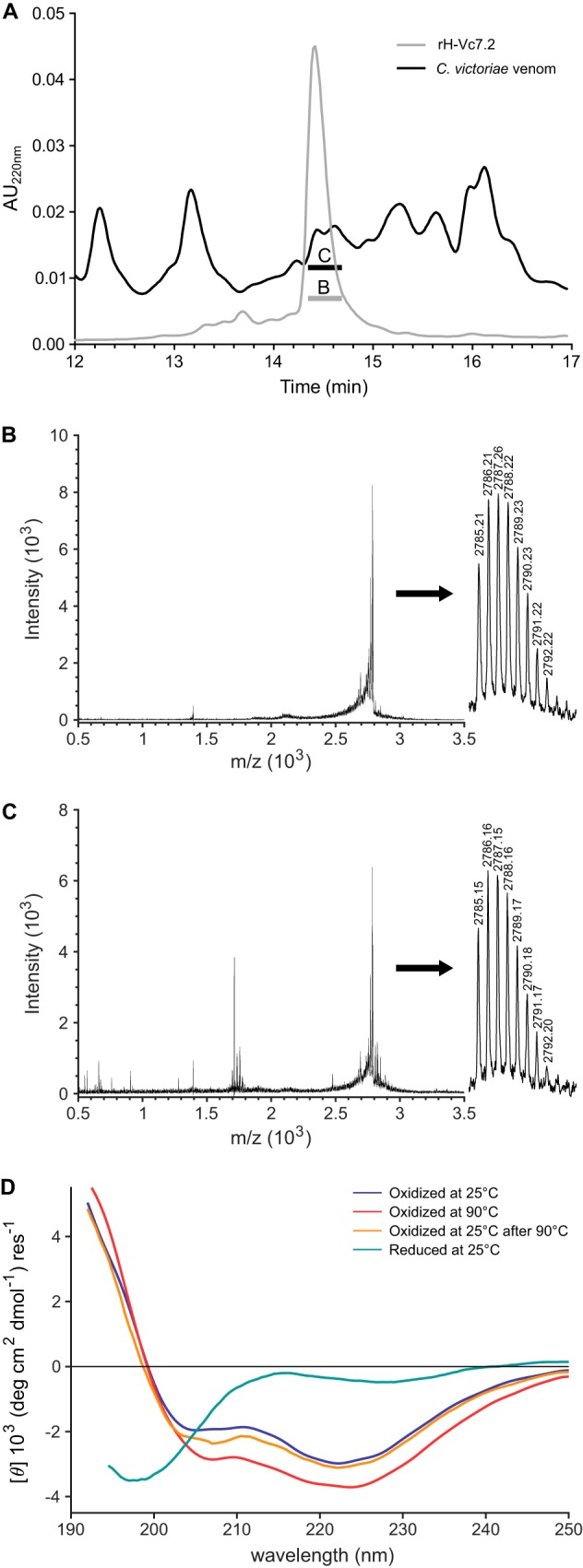
rH-Vc7.2 elutes with the same retention time as the native venom peptide and disulfide bonds are critical for stability. A, RP-HPLC profiles of rH-Vc7.2 peptide (gray line) and C. victoriae venom (black line). rH-Vc7.2 and venom fractions with the same retention time as rH-Vc7.2 were collected and analyzed by MALDI-TOF MS. B, MALDI-TOF spectra of rH-Vc7.2 showing a monoisotopic mass of [M + H]1+ = 2785.21 (inset, calculated [M + H]1+ = 2785.12). C, venom fraction eluting at the same retention time as rH-Vc7.2 contained a peptide of identical mass ([M + H]1+ = 2785.15) suggesting that the recombinant and native venom peptides have identical physicochemical properties. D, CD spectra of oxidized and reduced rH-Vc7.2 recorded at 25, 90, and again at 25 °C (after heating to 90 °C), and color-coded as indicated above the spectra.
We then used CD spectroscopy to characterize further structural properties of the peptide. CD spectra of rH-Vc7.2 were recorded at 25 and 90 °C and (after cooling) again at 25 °C, under both reducing and nonreducing conditions (Fig. 3D). The magnitude of the molar residual ellipticity in all spectra was low, indicating the presence of few regular secondary structure elements in the peptide. The spectrum of the oxidized peptide at 25 °C showed two minima, one at 205 nm and the other at 222 nm. A negative ellipticity minimum around 206 nm has been observed for small β-sheet and disulfide-rich proteins (23, 24). The combination of weak negative bands at around 208 and 222 nm is characteristic of both α-helical peptides and peptides with type I β-turns (25). Deconvolution of the CD spectrum predicted a protein devoid of α-helix but containing anti-parallel β-sheets (42.9%), turns (15.8%), and “others” (41.2%) (Fig. S5). Together, these CD data are consistent with the presence of β-turns and some β-sheet secondary structure in rH-Vc7.2 (see below). Upon reduction of the peptide, the CD spectrum changed considerably, displaying an ellipticity minimum at 198 nm and a shape of the curve suggesting an unstructured peptide.
To investigate the thermal stability of oxidized rH-Vc7.2, the change in ellipticity was followed at 224 nm from 10 to 90 °C. The experiment revealed a linear temperature dependence (data not shown), demonstrating that rH-Vc7.2 does not unfold cooperatively over this temperature range. The spectrum recorded at 90 °C displayed the same overall features as seen at 25 °C with a slightly lower ellipticity in the 205–250-nm range, indicating that the fold was preserved but that the conformational ensemble had changed. This change, however, was reversible upon cooling of the sample to 25 °C. We concluded that the folded peptide shows strong resistance to thermal denaturation and that the disulfide bonds are critical to maintain the structure of rH-Vc7.2.
NextH-Vc7.2 structure is characterized by two short, stacked β-hairpins
Having determined the cysteine connectivity and the importance of disulfide bonds for structural integrity, we used NMR spectroscopy to investigate the structure of NextH-Vc7.2. Using homonuclear 2D total correlation spectroscopy (TOCSY) and 2D NOE spectroscopy (NOESY) spectra in combination with triple resonance backbone spectra, 40% of backbone heavy atoms, and 60% of the nonexchangeable protons were assigned. Resonances from 20 out of 27 possible backbone amide groups were assigned. No peaks in the 15N heteronuclear single quantum coherence spectroscopy (HSQC) spectrum could be assigned to residues 6 and 7 and 14–18. The 15N HSQC (Fig. 4B) revealed a large variation in the intensity of observed peaks, with peaks from unassigned residues likely broadened beyond detection. Such peak broadening is the result of exchange between two or more conformations of the molecule on the millisecond time scale and indicates a dynamic nature of these regions of the structure. We note that residues 6 and 7 and residues 17 and 18, which are located in β-strands, are connected by a disulfide bond. If there are dynamics in the structure on one side of the disulfide bond, the other side likely also experiences these dynamics.
Figure 4.
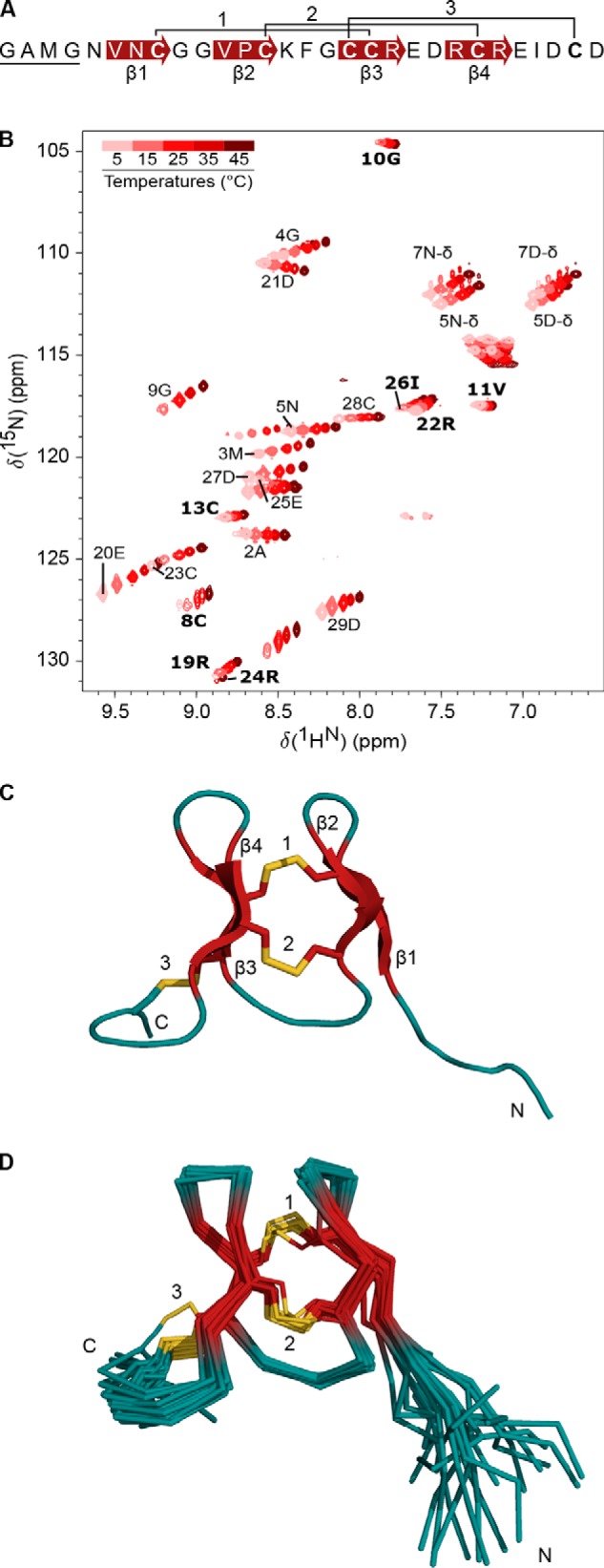
NextH-Vc7. 2 structure is characterized by two short, stacked β-hairpins. A, amino acid sequence of NextH-Vc7.2. The four N-terminal non-native residues are underlined; disulfide bonds are numbered, and β-strands are indicated with red arrows. B, 15N HSQC spectrum of NextH-Vc7.2 at different temperatures ranging from 5 to 45 °C and color-coded from pink to dark red as indicated by the color bar. The peaks from assigned backbone amides are labeled with one-letter code and residue number. Unassigned peaks are not labeled. Peaks with temperature coefficients higher than −4.6 ppb/K are labeled in bold. C, lowest energy NMR structure showing the three disulfide bonds connecting Cys-8 and Cys-18 (1), Cys-13 and Cys-23 (2), and Cys-17 and Cys-28 (3), and the two β-hairpins. Structural elements are colored with β-strands in red, sulfur atoms of the six cysteine residues in yellow, and loops in teal. Positions of the N and C termini are indicated. D, superimposition of the 20 lowest energy structures. The structures are shown in the same overall orientation as the conformer in C and with the same color-coding (see also Table 1).
One process that could cause the proposed peak broadening would be transient dimer formation (26). We therefore measured the diffusion coefficient for NextH-Vc7.2 by NMR to 1.96 ± 0.08 10−10 m2/s (data not shown). This is only slightly faster than the theoretical value of 1.54 10−10 m2/s calculated using HydroPro (27). This result therefore indicates that the peak broadening is not caused by the formation of dimers or higher-order oligomers. We also screened pH (in the range of pH 4.0–6.1), temperature (5–45 °C), and salt concentrations (up to 150 mm NaCl) to determine whether a single well-ordered conformation could be stabilized. However, we did not observe any large effect on the line shape of the different conditions tested, as exemplified by the temperature series shown in Fig. 4B.
Despite the missing assignments mentioned above, all Hα (except Gly-1 and Cys-17) and Cα shifts (except Phe-15 and Cys-17) were assigned, and there are TALOS restraints for residues 6 and 7 but not 14–18. So, NOEs could generally still be identified in the homonuclear NOESY spectrum, even for residues for which the HSQC peaks were not observed (except for Cys-17). The structure is thus well-defined even in the regions where the amide nitrogen is broadened beyond detection.
We calculated a structure of NextH-Vc7.2 using 152 distance restraints derived from a homonuclear 2D-NOESY spectrum (pH 6.1, 25 °C), 34 dihedral angle restraints, and the disulfide-bond restraints (Fig. 4C and Table 1). The structure shows that the core of the peptide is defined by the three disulfide bonds and two short, stacked β-hairpins (Fig. 4, C and D). The N-terminal β-hairpin comprises residues 6–13 and the C-terminal β-hairpin residues 17–24 (Fig. 4C). The overall fold of the calculated structure remained the same when the disulfide-bond restraints were omitted in the structure calculations. The five N-terminal residues (GAMGN) and the four residues at the C terminus (IDCD) were poorly defined. When excluding these residues, the backbone heavy atom pairwise root mean square deviation (RMSD) among the 20 lowest energy structures is 0.4 ± 0.2 Å (Fig. 4D and Table 1). The energy statistics from the final Xplor-NIH structure calculation are shown in Table S5. NextH-Vc7.2 and rH-Vc7.2 displayed almost identical 2D NOESY spectra with only minor chemical shift changes for some protons, strongly indicating that the structure of the two peptides is the same.
Table 1.
NMR structural restraints and structure statistics
None of the structures exhibited distance violations >0.3 Å or dihedral angle violations >5°.
| NextH-Vc7.2 | |
|---|---|
| Restraints | |
| NOE-based restraints | |
| Intraresidual (|i − j| = 0) | 60 |
| Sequential (|i − j| = 1) | 49 |
| Medium range (2 ≤ |i − j| ≤4) | 18 |
| Long (|i − j| >5) | 25 |
| Total | 152 |
| Dihedral angle restraints | 34 |
| Restraint statistics | |
| Mean RMSD from experimental restraints | |
| NOE-based distances, Å | 0.012 ± 0.002 |
| Dihedrals (°) | 0.18 ± 0.04 |
| Structure statisticsa | |
| Most favored regions | 84.1% |
| Allowed regions | 15.2% |
| Generously allowed regions | 0.5% |
| Disallowed regions | 0.2% |
| Coordinate precision, pairwise RMSD (Å)b | |
| H-Vc7.2 (6–25) | |
| Backbone heavy atoms (N, Cα, and C′) | 0.43 ± 0.23 |
| Heavy atoms | 1.37 ± 0.29 |
a PROCHECK was used.
b Molmol was used.
To gain further support for the presence of the two β-hairpins, we determined temperature coefficients for the amide proton chemical shifts (Fig. 5, A and B). Hydrogen-bonded protons typically have temperature coefficients closer to 0 than −4.6 ppb/K, whereas nonhydrogen-bonded protons typically have temperature coefficients lower than −4.6 ppb/K (28, 29). The temperature coefficients for rH-Vc7.2 were determined by linear regression using the amide hydrogen chemical shifts from a series of 15N HSQC spectra measured in the interval between 5 and 45 °C (Fig. 4B and 5A). Plotting the temperature coefficients onto the structure revealed that the amide protons predicted from the calculated structure to be involved in hydrogen bonding indeed had temperature coefficients of −4.6 ppb/K or higher (Fig. 5B). We note that although Gly-10 and Ile-26 are not involved in β-sheet formation, their temperature coefficients indicate hydrogen bonding. Based on analysis of the structure, the amide proton of Gly-10 could possibly form a hydrogen bond with the sulfur atom of Cys-8, whereas the most likely candidate for hydrogen bonding with the Ile-26 amide proton is the carbonyl oxygen of Arg-24.
Figure 5.
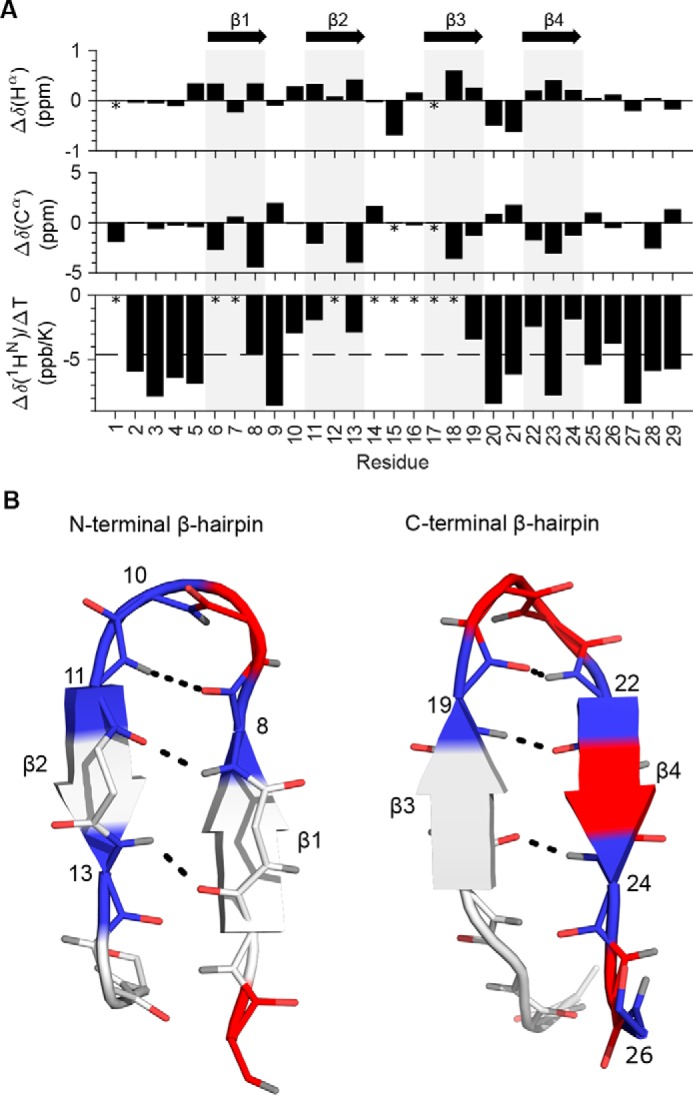
Secondary chemical shifts, amide hydrogen temperature coefficients, and β-sheet hydrogen bonds. A, NextH-Vc7.2 Hα and Cα secondary chemical shifts and temperature coefficients plotted per residue. Asterisks mark unassigned residues, and gray bars indicate the position of the four β-strands as indicated with arrows at the top. The dashed line at −4.6 ppb/K in the bottom plot indicates the cutoff for hydrogen bonding of amide hydrogens. B, residues of the β-hairpins formed by β-strands 1 and 2 (left) and β-strands 3 and 4 (right) are colored blue (with residue numbers labeled) if the temperature coefficient is higher than −4.6 ppb/K, red if lower than −4.6 ppb/K, and light gray if unassigned. Backbone atoms are shown with oxygens in red and hydrogens in gray. Black dashed lines indicate the hydrogen bonds between the β-strands.
Importantly, the raw NMR data were inconsistent with an ICK fold, which displays a two-stranded anti-parallel β-sheet that is often extended with an additional anti-parallel β-strand (12, 30). Instead, the data substantiated the presence of two short β-hairpins, the first comprising a type I and the second a type I′ β-turn as determined from the backbone dihedral angles.
Structural topology of NextH-Vc7.2 is unlike the ICK fold
On the basis of the disulfide framework VI/VII alone, it was predicted that H-Vc7.2 would have an ICK fold. However, in NextH-Vc7.2, the connections between β-strands β1, β2, and β3 and between β2, β3, and β4 are both left-handed in contrast to the ICK fold, where these connections are left-handed and right-handed, respectively. Furthermore, the crossing disulfides that are a hallmark of the ICK fold (Fig. 6A) are not present in NextH-Vc7.2 (Fig. 6B). The first disulfide in NextH-Vc7.2 connects β1 with the opposing β3, whereas the second disulfide connects the opposing β2 and β4. As a result, the first two disulfides in NextH-Vc7.2 are parallel. The third disulfide connects the beginning of β3 with the unstructured C-terminal region without threading the loop formed by the first two disulfide bonds, another hallmark of the ICK fold. The NextH-Vc7.2 structure therefore unexpectedly displays a fold that is clearly distinct from an ICK.
Figure 6.
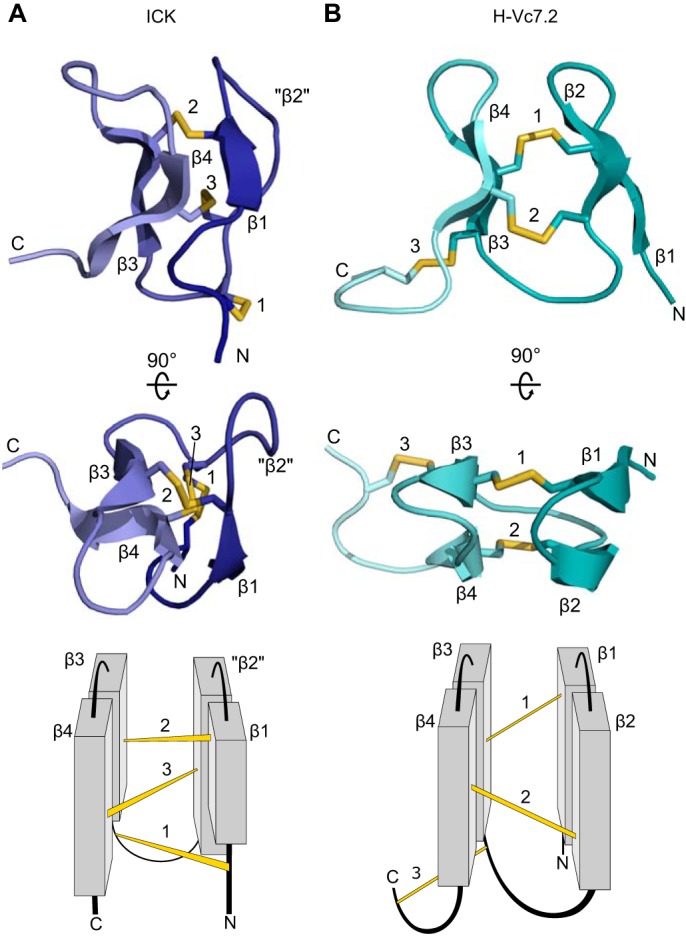
Structure topology of NextH-Vc7. 2 is unlike the ICK fold. Top and middle, 3D structures with β-strands, disulfides, and N and C termini labeled in two different orientations. Bottom, schematic topology representation of the structures shown above. A, a representative ICK (the SHL-I lectin (PDB code 1QK7) (64)), where “β2” indicates the position of the corresponding secondary structural element (β2) in NextH-Vc7.2, which is not present in the ICK. B, NextH-Vc7.2.
NextH-Vc7.2 adopts a fold similar to the N-terminal domain of granulin
Considering the results described above, we searched the Protein Data Bank (PDB) for structural homologs of NextH-Vc7.2. The results showed the NextH-Vc7.2 core structure to have the same fold as the N-terminal domain of the human progranulin A module (Fig. 7A). Although the second β-hairpin in the N-terminal domain of the human progranulin A module is longer than the corresponding β-hairpin in NextH-Vc7.2, the structures align well and display the same arrangement of disulfide bonds. Another conotoxin, Φ-MiXXVIIA, with structural homology to granulin was recently discovered (31). The two toxins have highly similar structures with the inter-β-hairpin disulfides placed similarly, whereas the remaining disulfides in the two toxins are at opposite ends of the peptides (Fig. 7B). Notably, H-Vc7.2, granulins, and Φ-MiXXVIIA are not the only peptides displaying the observed structure, which we identified in a number of other proteins. Thus, we found that domains from other toxins, such as the αD-GeXXa conotoxin and leech antistasins (serine protease inhibitors), as well as from the nontoxin proteins follistatin-related protein 3 (and other proteins with EGF-like domains, e.g. fibrillin) and the zinc-binding lobe of the E3 ubiquitin ligase, Pirh2, where cysteine residues coordinate Zn2+ ions rather than forming disulfide bonds, have similar folds (Fig. 7, C–F; see under “Discussion”). RMSD values for the structures aligned in Fig. 7, as well as an alignment of their sequences, are shown in Table S6.
Figure 7.

NextH-Vc7. 2 adopts a fold similar to the N-terminal domain of granulin. NextH-Vc7.2 (dark green) is aligned to different structural homologs (displayed in gray outside the region of structural homology). A, N-terminal domain of the human progranulin A module (dark blue, PDB code 2JYE) (33). B, Φ-MIXXVIIA conotoxin (violet) (31). C, C-terminal domain of the αD-GeXXa conotoxin (red, PDB code 4X9Z) (40). The second molecule of the αD-GeXXa dimer is depicted in light blue. D, N-terminal domain of the leech serine protease inhibitor bdellastasin (orange, PDB code 1C9T) (41). E, N-terminal domain of the follistatin-related protein 3 (FSTL-3) (yellow, PDB code 2KCX). F, zinc-binding β-hairpin repeat from the C-lobe of the N-terminal domain of Pirh2 (light green, PDB code 2K2C) (44). Top, alignment with the second and third β-hairpins; bottom, alignment with the first two β-hairpins. Zinc atoms are shown as purple spheres.
In conclusion, we find that the fold comprising two short, stacked β-hairpins stabilized by two parallel disulfide bonds across a small β-sandwich structure is present in a variety of toxins and other proteins. Given the wide distribution of this structure, which apparently constitutes an autonomous folding unit, and its homology with the N-terminal domain of granulins, we propose to name this structure the “mini-granulin” fold.
Investigation of bioactivity
rH-Vc7.2 was tested for bioactivity by intracranial injection in mice. In this assay, a dose of 3.5 nmol of peptide did not produce any strong reproducible behavioral changes compared with control animals injected with saline (data not shown). Similarly, rH-Vc7.2 (10 μm) did not produce any observable changes to normal or depolarization-induced intracellular Ca2+ levels in mouse dorsal root ganglion cells (data not shown).
Discussion
Whereas the same cysteine framework can give rise to different cysteine connectivities (32), the finding that the same cysteine connectivities can dictate different folds is unusual. This work therefore emphasizes that caution must be taken in assuming a specific structure based on a certain cysteine framework, or even a known disulfide pattern. Specifically, we demonstrate that, despite having the disulfide pattern expected of a peptide with a VI/VII cysteine framework, NextH-Vc7.2 did not adopt an ICK fold. Rather, the peptide displayed a mini-granulin fold, a structure composed of two short, stacked β-hairpins connected by two parallel disulfide bonds.
Progranulins are composed of separate disulfide-rich granulin modules of ∼55 residues each (33). Structurally, each module is constituted by stacks (four in most cases) of β-hairpins interconnected by stabilizing disulfide bonds (Fig. 7A) (34, 35). The N-terminal region comprising the first ∼30 residues of the human progranulin A module is structurally much better defined than the C-terminal region (33). Indeed, a fragment comprising residues 1–30 of carp granulin displays essentially the same structure as the corresponding region of the intact protein (36). Similarly, an N-terminal 35-residue peptide of a granulin-like module from a rice protease inhibitor constitutes a well-defined structure with the expected fold (37), and designed N-terminal peptides of human (38) and live fluke (39) granulins also adopt the same overall structure. Thus, it is well-established that the N-terminal region of several granulins constitutes an autonomous folding unit.
Here, we corroborate and extend the findings for granulin by identifying a variety of structural homologs of NextH-Vc7.2 where the mini-granulin fold constitutes a separate domain (Fig. 7). Our analyses show that the recent finding that the Φ-MiXXVIIA conotoxin adopts the mini-granulin fold (Fig. 7B) (31) is not a unique case. For instance, the C-terminal domain of the αD-GeXXa conotoxin, which has not previously been assigned to a particular fold, also constitutes a mini-granulin structure (Fig. 7C) (40). A domain with the mini-granulin fold is also found in the antistasins, i.e. leech serine protease inhibitors such as antistasin, hirustasin, and bdellastasin (Fig. 7D) (41). Another structural homolog of NextH-Vc7.2 is the EGF-like subdomain of follistatin-related protein 3 (42), a transforming growth factor β family antagonist (Fig. 7E). Structural homology between the N-terminal domain of granulin and the N-terminal subdomain of EGF-like molecules has been noted previously (36, 43). We also detected homology with the C-lobe of the N-terminal domain of the E3 ubiquitin ligase Pirh2, a zinc-binding region of the protein (Fig. 7F) (44), in which cysteines coordinate zinc atoms located between the β-hairpins rather than forming disulfide bonds. Overall, the mini-granulin structure is widely present as a separate domain in toxins as well as nontoxin proteins.
Although the presence of the mini-granulin fold in nature may well have been underappreciated to date, ICK peptides are still much more common (14). Their evolutionary success is likely a consequence of the high thermodynamic stability and resistance to proteolytic cleavage afforded by the ICK fold, and the fact that this fold can function as a scaffold for a vast array of functionalities (13, 45). In comparison, the core mini-granulin fold contains only two disulfides and is probably dependent on the acquisition of additional disulfides, as seen in e.g. H-Vc7.2 and Φ-MiXXVIIA, for added structural stability. Although Φ-MiXXVIIA has been demonstrated to display (weak) anti-apoptotic and cell-proliferative activities (31), as is the case for certain granulin modules, these are unlikely to constitute the native activities of this conotoxin, and the ability of granulin-like toxins to harbor different functionalities warrants further investigation.
Conotoxins and other disulfide-rich toxins have previously been produced successfully in E. coli using various expression systems (46, 47). NextH-Vc7.2 and rH-Vc7.2 were produced using the CyDisCo expression system that supports the formation of disulfide bonds in the reducing cytosol of E. coli. Based on our previous finding that the csPDI enzyme accelerates oxidative folding of selected conotoxins in vitro (18), we surmised that co-expression with csPDI could increase the folding efficiency of at least some conotoxins in this expression system. Further studies are required to address whether the csPDI–CyDisCo expression system is widely applicable to the expression of small disulfide-rich peptides and to investigate a potential synergism between hPDI and csPDI in disulfide-bond formation of conotoxins as we recently demonstrated in vitro for Conus PDI and csPDI (48).
Here, our results showed that when produced in the absence of the csPDI–CyDisCo system, a large fraction of NextH-Vc7.2 molecules ended up in the insoluble fraction (Fig. S6). A systematic investigation of the importance of this system for the production of oxidized NextH-Vc7.2, e.g. the specific effect of the csPDI as compared with human PDI or a comparison with other E. coli expression systems such as Shuffle and Origami cells, was beyond the scope of the current investigation. Ongoing work in our laboratory is aimed at understanding in more detail the effects of the csPDI–CyDisCo system on conotoxin production for a larger and diverse set of toxins.
The function of H-Vc7.2, and indeed all H-superfamily conotoxins, remains an open question. In the assays used here, no obvious bioactivity was detected. Potentially, H-Vc7.2 interacts with a receptor target in such a way as to not produce observable responses in these assays, or perhaps, the peptide is selective for one or more receptors not present in the assay systems used. Future in vivo activity assays using gastropods, the prey of C. victoriae, may shed light on the peptide's bioactivity.
From the many available structures of ICK peptides with large sequence variations in the loops between cysteine residues, it is clear that the disulfide bonds are crucial for directing the ICK fold (30). Apart from the side chain of Val-11 that packs against the first two disulfide bonds, the structure of NextH-Vc7.2 shows no evidence of a hydrophobic core that could help direct the folding of the peptide. Instead, as proposed for the Φ-MiXXVIIA conotoxin (31), the short loop lengths in H-Vc7.2 could well restrict the polypeptide conformation to preclude the formation of an ICK fold. An analysis of the more than 3300 sequences of presumed ICK proteins listed in the KNOTTIN database (http://www.dsimb.inserm.fr/KNOTTIN/6 (13)) supports this suggestion. Among these many sequences, only 10 contain four (or fewer) residues in the loop between CysI and CysII, three (or fewer) residues between CysII and CysIII, and no residues between CysIII and CysIV. No structures have been solved for any of these 10 proteins. Only when the loop length between CysIII and CysIV reaches three residues (while retaining ≤4 and ≤3 residues in the first two loops, respectively) have ICK structures been solved. However, a more thorough analysis of the consequences of varying loop lengths in cysteine framework VI/VII proteins is needed to determine the underlying features of the H-Vc7.2 sequence that govern its structure and lead to the formation of the mini-granulin fold rather than the ICK fold. Such work will be fundamental to allow the design of better structure prediction algorithms and help further our understanding of sequence–structure relationships in peptides and proteins.
Experimental procedures
Mass spectrometry (MS) on venom extract
C. victoriae snails were collected from Broome, Western Australia, under a commercial fishing license of the Western Australian Specimen Shell Managed Fishery (license number 2577). The venom was obtained by manual extrusion from freshly dissected venom glands. An aliquot (0.5 μg) of venom was reduced in 20 mm tris(2-carboxyethyl)phosphine (TCEP), pH 8, for 30 min at 60 °C, then alkylated by incubating in 40 mm iodoacetamide for 30 min. This reduced and alkylated venom was loaded onto a microfluidic trap column packed with ChromXP C18-CL 3-μm particles (300 Å nominal pore size). An analytical (15-cm × 75-μm ChromXP C18-CL 3-μm) microfluidic column was then switched in line, and venom was separated using a linear gradient of 0–80% acetonitrile, 0.1% formic acid over 90 min at a flow rate of 300 nl/min. Separated venom peptides were analyzed using an AB SCIEX 5600 TripleTOF mass spectrometer equipped with a Nanospray III ion source and accumulating 30 tandem MS (MS/MS) spectra/s. MS/MS data were analyzed with ProteinPilot software (version Beta 4.1.46) using the Paragon algorithm. The search databases comprised a six-frame translation of the C. victoriae venom gland transcriptome, as described previously (10), including the translated prepropeptide sequence of H-Vc7.2. MS/MS spectra identified by ProteinPilot as matching peptides were manually validated by comparison against a theoretical peak list (Protein Prospector MS-Product, University of California, San Francisco).
Plasmid generation
Based on the nucleotide sequence of H-Vc7.2 from C. victoriae (10), an E. coli codon-optimized H-Vc7.2 gene containing NcoI and BamHI restriction sites was synthesized (Eurofins Genomics GmbH) and cloned into the pET39_Ub19 plasmid (20) (a kind gift from V. Rogov and V. Dötsch, Goethe University, Frankfurt, Germany). The resulting plasmid expressing ubiquitin (Ub)–His10-tagged, N-terminally extended H-Vc7.2 (Ub–His10–NextH-Vc7.2) is referred to as pLE566. A plasmid (pLE879) differing only from pLE566 by encoding the native sequence devoid of the four non-native residues in NextH-Vc7.2 was purchased from TWIST Bioscience. The fusion protein produced from pLE879 was named Ub–His10–rH-Vc7.2.
A CyDisCo plasmid (pMJS205 (17)) encoding Erv1p and hPDI was a gift from L. W. Ruddock (Dept. of Biochemistry, University of Oulu, Finland). A codon-optimized sequence for E. coli expression of csPDIGH/GH from C. geographus (GenBankTM accession no. KT874567) (18) was synthesized with XbaI and XhoI restriction sites (Eurofins Genomics GmbH) and cloned into pMJS205, generating the pLE577 plasmid that encodes Erv1p, hPDI, and csPDIGH/GH (see Fig. 2).
Protein expression
Chemically competent E. coli BL21 Tuner(DE3) cells (Novagen) were co-transformed with pLE577 and pLE566 or pLE879 and plated on an LB agar plate containing kanamycin (50 μg/ml) and chloramphenicol (30 μg/ml). One colony was inoculated in LB medium containing the same antibiotics and incubated overnight at 37 °C at 200 rpm in an orbital shaker. Overnight cultures were diluted in LB or M9 media (3 g/liter KH2PO4, 15.1 g/liter Na2HPO4 12H2O, 5 g/liter NaCl, 1 mm MgSO4, 1 ml/liter M2 Trace element solution (203 g/liter MgCl2 6H2O, 2.1 g/liter CaCl2 2H2O, 2.7 g/liter FeSO4 7H2O, 20 mg/liter AlCl3 6H2O, 10 mg/liter CoSO4 7H2O, 2 mg/liter KCr(SO4)2 12H2O, 2 mg/liter CuCl2 2H2O, 1 mg/liter H3BO4, 20 mg/liter KI, 20 mg/liter MnSO4 H2O, 1 mg/liter NiSO4 6H2O, 4 mg/liter Na2MoO4 2H2O, 4 mg/liter ZnSO4 7H2O, 21 g/liter citric acid monohydrate), 1 g/liter [15N]NH4Cl, and 4 g/liter [13C]glucose) to an optical density at 600 nm (OD600) of 0.1 and incubated at 37 °C until OD600 reached ∼0.8. Protein expression was induced by 1 mm IPTG, and cultures were grown overnight at 30 °C. Expression of the 13C/15N-labeled peptide in 580 ml of M9 minimal medium was performed as described for the unlabeled peptide.
Cell harvest
Induced overnight cultures were centrifuged for 15 min at 6,000 × g; supernatants were discarded, and pellets were resuspended in 10 ml of lysis buffer (300 mm NaCl, 50 mm Na2HPO4/NaH2PO4, 10 mm imidazole, pH 8)/liter of culture medium. Sonication was performed on a Bandelin Sonopuls HD2200 equipped with a Boosterhorn SH 213 G and a 3 mm probe. Cells were sonicated 10× for 10 s at 30% power with 30 s on ice between pulses. After centrifugation for 50 min at 15,000 × g, the cleared lysates were transferred to fresh tubes, while pellets were dissolved in lysis buffer containing 8 m urea for SDS-PAGE analysis.
Protein purification
Ub–His10–NextH-Vc7.2 was purified from the cleared lysate on an ÄKTA purifier 900 chromatography system equipped with a GE Healthcare Tricorn 10/50 column packed with 5 ml of Qiagen Superflow Ni-NTA resin equilibrated in lysis buffer. Upon sample application, the column was washed with lysis buffer containing 20 mm imidazole until a stable baseline at A280 was achieved, and the protein was eluted with lysis buffer containing 400 mm imidazole. The pooled material (7 ml) was dialyzed twice against 2 liters of lysis buffer at 4 °C using Spectra/Por 1 dialysis tubing with a molecular mass cutoff of 6,000–8,000 Da (Spectrum Laboratories Inc.). The dialyzed sample was concentrated to ∼500 μl using Amicon Ultra 15-ml 3K centrifugal filters (Merck Millipore) according to the manufacturer's instructions.
Ni-NTA–purified Ub–His10–NextH-Vc7.2 was cleaved using His6–TEV protease (obtained as described below), which was activated by incubation with 10 mm dithiothreitol (DTT) for 30 min. To avoid reduction of disulfide bonds in the target fusion protein, DTT was diluted to 0.2 mm in the TEV solution by two rounds of dilution and concentration on an Amicon Ultra 15-ml 3K centrifugal filter. TEV cleavage was carried out in lysis buffer at room temperature overnight in a final concentration of 0.08 mm DTT. A Ub–His10–NextH-Vc7.2/His6–TEV molar ratio of 100:1 was used. To remove any uncleaved Ub–His10–NextH-Vc7.2, liberated Ub–His10 and His6–TEV, ∼500 μl of 80% slurry of Talon Superflow Metal Affinity Resin (Clontech) equilibrated with lysis buffer was added to the TEV-cleaved sample, and the mixture was incubated overnight at 4 °C on a rotating mixer. Talon beads were removed by centrifugation, and the supernatant was subjected to RP-HPLC. Trifluoroacetic acid (TFA) was added to 0.1%; pH was adjusted to ∼2 with 1 m HCl, and samples were spun at 16,100 × g for 10 min. RP-HPLC purification was performed on an ÄKTA purifier 900 chromatography system equipped with a Grace Vydac 218 TP C18 column (4.6 × 150 mm, 5 μm) using solvent A: 5% ethanol, 0.1% TFA, and solvent B: 90% ethanol, 0.085% TFA. The 0–60% solvent B gradient was developed over 60 min with a flow rate of 1 ml/min. Following purification, peptide-containing fractions from the major peak were pooled and lyophilized. RP-HPLCs (and plots of molar ellipticity; see below) were visualized in MATLAB (MathWorks®). Ub–His10–rH-Vc7.2 was purified using the same procedure.
His6–TEV protease expression and purification
An expression plasmid (pLE478) encoding maltose-binding protein (MBP) and His6-tagged TEV protease (MBP–His6–TEV) (20) was a gift from V. Rogov and V. Dötsch (Goethe University, Frankfurt, Germany). Upon expression, the fusion protein undergoes autocleavage to release TEV protease with an N-terminal His-tag (His6–TEV). Chemically competent BL21(DE3) cells (Invitrogen) were transformed with pLE478 and plated on an LB agar plate containing 100 μg/ml ampicillin. One colony was inoculated in 10 ml of LB medium containing 100 μg/ml ampicillin and grown overnight at 37 °C in an orbital shaker (200 rpm). The overnight culture was diluted to OD600 of 0.1 and incubated at 37 °C in an orbital shaker until the OD600 reached ∼1.0. Subsequently, IPTG was added to the culture to a concentration of 1 mm and incubated at 20 °C overnight. The His6–TEV protease was purified from overnight cultures essentially as the Ub–His10–NextH-Vc7.2 construct, but using different lysis buffer (50 mm Tris, 200 mm NaCl, 1% glycerol, 10 mm imidazole, pH 7.8), Ni-NTA–binding buffer (50 mm Tris, 200 mm NaCl, 20 mm imidazole, pH 7.8), elution buffer (50 mm Tris, 200 mm NaCl, 300 mm imidazole, pH 7.8), and dialysis buffer (50 mm Tris-HCl, 100 mm NaCl, pH 7.8). The concentration of dialyzed His6–TEV protease was calculated based on the A280 value and an extinction coefficient, ϵ280, of 32,290 m−1 cm−1. Purified His6–TEV protease was stored in dialysis buffer containing 5 mm DTT and 50% glycerol at −80 °C.
Concentration determination
As the H-Vc7.2 sequence contains no Trp or Tyr residues, the concentrations of NextH-Vc7.2 and rH-Vc7.2 were determined by measuring absorbance at 214 nm and using the extinction coefficient 34,621 m−1 cm−1 calculated as reported previously (49).
Confirmation of peptide oxidation state
The oxidation status of purified NextH-Vc7.2 was evaluated by MALDI-TOF MS using an Autoflex Smartbeam III instrument (Bruker) calibrated by external calibration (Peptide calibration standard I; Bruker Daltronics). Lyophilized rH-Vc7.2 was dissolved in 20 μl of 20 mm Tris, pH 8.0, containing 6 m guanidinium hydrochloride and incubated 2 h at 37 °C in the presence or absence of 50 mm TCEP. Subsequently, 100 mm iodoacetamide was added and samples were incubated for 2 h at room temperature in the dark to allow for S-carbamidomethylation of free thiols. For MS analysis, the samples were acidified by the addition of TFA and desalted by reversed-phase chromatography using Poros R1 microcolumns as described previously (50). The recovered samples were mixed with α-cyano-4-hydroxycinnamic acid prepared in 70% acetonitrile, 0.1% TFA, spotted on a stainless-steel target plate, and analyzed in positive reflectron mode. The generated data were evaluated using the GPMAW software and monoisotopic masses.
MS confirmation of csPDI and hPDI expression
Lysates from IPTG-induced E. coli cells were separated by reducing SDS-PAGE, and in-gel digestion was performed using porcine trypsin (Promega). Generated peptides were recovered by reversed-phase chromatography (StageTips, C18, Thermo Fisher Scientific) and eluted directly onto the target plate using α-cyano-4-hydroxycinnamic acid as described above. Peptides were analyzed in positive reflector mode, annotated, and interrogated by peptide mass fingerprinting using the MASCOT search engine (51). Cysteine propionamide was set as a fixed modification, due to the reaction of protein cysteine residues with acrylamide present in the gel support during electrophoresis. Oxidation of methionine residues was set as a variable modification. One missed cleavage was allowed, and the peptide mass tolerance was set to 50 ppm. Identifications were validated by LIFT analysis on selected peptides.
Determination of cysteine connectivity
The cysteine connectivity in NextH-Vc7.2 was determined following the method of Albert et al. (22). In brief, 5 nmol of the peptide were sequentially reduced with 500 mm TCEP and alkylated at pH 3.0 on an Empore C18 solid-phase extraction cartridge (3M, St. Paul, MN). For sequential alkylation, the maleimides NMM, NEM, and NCM were used (at 20 mm). Therefore, in each reduction/alkylation step one maleimide alkylated both cysteines of one disulfide bridge. The fully alkylated peptide was analyzed by LC tandem MS. In detail, the peptide was run on an ACQUITY BEH C18 UPLC column (1.0 × 150 mm, 1.7 μm) installed in an ACQUITY UPLC I-Class system (Waters) and coupled to a Xevo G2 Q-TOF mass spectrometer (Waters). Different alkylation patterns and subsequently the disulfide connectivity of the peptide were identified from characteristic combinations of y-type ions from obtained mass spectra.
RP-HPLC elution experiments with rH-Vc7.2 and C. victoriae venom
Approximately 1 nmol of rH-Vc7.2 was analyzed by RP-HPLC on an analytical C18 column (5-μm particle size, 4.6 × 250 mm, Vydac) using a gradient of 15–45% solvent B (90% acetonitrile, 0.1% TFA) over 30 min. The HPLC column was repeatedly washed with 5–100% solvent B to ensure that no more recombinant peptide was bound to the column prior to venom analysis.
Venom was extruded from a single frozen venom duct of C. victoriae and extracted in 400 μl of 40% acetonitrile, 0.1% TFA with a disposable plastic pestle. The mixture was centrifuged for 5 min at 10,000 × g to remove insoluble material. A 10-μl aliquot of the supernatant was diluted in 0.1% TFA, water and analyzed by RP-HPLC using the same conditions described above. Fractions eluting at similar % of solvent B as rH-Vc7.2 were collected in 0.25% B intervals for subsequent mass spectrometric analysis.
Fractions were lyophilized and submitted to the Proteomics Core Facility at the University of Utah. MALDI-TOF MS analysis was performed on rH-Vc7.2 and venom fractions eluting at a similar percent of solvent B. MALDI-MS measurements were performed on a Bruker Daltonics UltrafleXtreme ToF mass spectrometer in positive ion reflectron mode using pulsed ion extraction set at 100 ns. The instrument was calibrated using polyalanine, and samples were analyzed by averaging ∼500 single-shot spectra. Data were analyzed using flexAnalysis 1.4 (Bruker).
CD spectroscopy
CD spectroscopy of oxidized and reduced rH-Vc7.2 (40 μm in 10 mm KH2PO4/K2HPO4, pH 6.1, in the absence of TCEP or 25 mm NaH2PO4/Na2HPO4, pH 6.1, in the presence of 5 mm TCEP) was carried out using a JASCO J-810 spectropolarimeter. The fully reduced sample was prepared by incubating rH-Vc7.2 overnight with 10 mm DTT and 10 mm TCEP before purification by RP-HPLC. The lyophilized sample was dissolved in H2O for concentration determination before dilution into phosphate buffer. For thermostability assays, ellipticity at 224 nm was recorded from 10 to 90 °C at a rate of 1 °C/min followed by cooling to 5 °C. CD spectra were recorded at 25 °C in the wavelength range of 190–250 nm before and after heating. The final spectra were obtained after averaging over 15 spectra recorded at a scan rate of 10 nm/min, and baseline (buffer only, same conditions) was subtracted. High-frequency noise was removed using a Fourier transformation filter. The region from 190 to 200 nm was omitted due to a high-tension voltage value exceeding 600 Hz in the presence of TCEP.
NMR spectroscopy
Samples of ∼1 mm of both unlabeled and 13C,15N-labeled NextH-Vc7.2 were prepared in 25 mm sodium phosphate, 10% D2O, pH 6.1. Chemical shifts were assigned by standard sequential assignment based on the following recorded spectra: 2D 1H-1H-TOCSY and 2D 1H-1H-NOESY (on the unlabeled sample), and 15N HSQC, 13C HSQC, HNCA, HNCO, HNCOCA, HNCACO, and CBCACONH (on the double-labeled sample). All spectra were recorded at 25 °C on Bruker Avance III-HD NMR spectrometers operating at 750 and 600 MHz, equipped with TCl and QCl cryoprobes, respectively. The data were processed with nmrPipe (52) and analyzed in CCPNMR (53).
NMR structure calculations
CYANA was used for automated NOE assignment of the 2D 1H-1H NOESY spectrum and initial structure calculation, also including dihedral angles calculated with TALOS+ (54) and the disulfide pattern (C8–C18/C13–C23/C17–C28, determined as described above) as restraints (55). The final structure refinement was done using XPLOR-NIH with the NOE-derived distance restraints, dihedral angle restraints, and disulfide bond restraints (56). The 20 lowest energy structures out of 100 were selected and contained no NOE violations (>0.3Å) or dihedral angle violations >5°. Ramachandran-plot statistics were calculated using PROCHECK (57). Structure alignment was done using MOLMOL (58), and structure visualization was performed with PyMOL (DeLano Scientific). The structure has been deposited in the PDB (59) with PDB code 6Q5Z and chemical shifts and chemical shifts have been deposited in the Biological Magnetic Resonance Bank (http://www.bmrb.wisc.edu/(60)) with ID 34335.
Identification of structural homologs
To identify structural homologs of NextH-Vc7.2, we first downloaded all structures in the PDB, and the sequences from the structure files were extracted using an in-house automated Matlab script. The extracted sequences were then searched with a CX1–5CX1–5CX0–2C sequence motif (where X denotes any residue), representing a fragment of the H-Vc7.2 sequence covering the first four cysteine residues. All structures containing a sequence matching this motif were inspected in PyMOL. Structures containing a fold homologous to that of NextH-Vc7.2 were aligned to the NextH-Vc7.2 structure using the CLICK alignment server (61, 62).
Mouse bioassay and calcium imaging
All experiments involving the use of animals were approved by the Institutional Animal Care and Use Committee of the University of Utah. Swiss Webster mice (14–21 days old) were injected intracranially with 3.5 nmol of rH-Vc7.2 peptide (dissolved in 25 mm NaHPO4 buffer, pH 6.1, containing 10% D2O, 0.25 mm 4,4-dimethyl-4-silapentane-1-sulfonic acid, 0.02% NaN3). Following intracranial injection, mouse behavior was observed for 1 h to determine differences between treated and control animals. Calcium imaging on mouse lumbar dorsal root ganglion neurons, using 10 μm peptide, was carried out as described previously (63).
Author contributions
L. D. N., R. S. N., T. V., K. T., and L. E. supervision; L. D. N., M. M. F., A. A., A. B. B., C. L. S., S. D. R., S. V. P., H. S.-H., and K. T. investigation; L. D. N., M. M. F., S. V. P., T. V., H. S.-H., K. T., and L. E. methodology; L. D. N., M. M. F., T. V., H. S.-H., and L. E. writing-original draft; L. D. N., M. M. F., C. L. S., S. D. R., S. V. P., A. W. P., B. M. O., R. S. N., H. S.-H., K. T., and L. E. writing-review and editing; A. A., A. B. B., C. L. S., S. D. R., B. M. O., and R. S. N. data curation; A. W. P., B. M. O., R. S. N., T. V., H. S.-H., K. T., and L. E. funding acquisition; H. S.-H. and L. E. conceptualization.
Supplementary Material
Acknowledgments
We thank Nick Williamson, University of Melbourne, and the Mass Spectrometry and Proteomics Core Facility at the University of Utah (spectrometry equipment obtained through a Shared Instrumentation Grant 1 S10 OD018210 01A1) for technical assistance with MS. Norut Northern Research Institute was supported by Tromsø County Council Grant TFK2013-252.
This work was supported in part by Danish Council for Independent Research Technology and Production Sciences Grant 7017-00288, Ulla og Mogens Folmer Andersens Fond, Fondation Juchum and Hørslev-Fonden (L. E.), and the Velux Foundations (K. T.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6, Tables S1–S6, and supporting Ref. 1.
The atomic coordinates and structure factors (code 6Q5Z) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Chemical shifts have been deposited in the Biological Magnetic Resonance Bank with ID 34335.
L. Ruddock, University of Oulu, Finland, personal communication.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- ER
- endoplasmic reticulum
- ICK
- inhibitor cystine knot
- PDI
- protein-disulfide isomerase
- csPDI
- conotoxin-specific PDI
- hPDI
- human PDI
- Ub
- ubiquitin
- Ub–His10
- Ub containing 10 consecutive histidines
- TEV
- tobacco etch virus
- RP-HPLC
- reversed-phase high–performance liquid chromatography
- NMM
- N-methylmaleimide
- NEM
- N-ethylmaleimide
- NCM
- N-cyclohexylmaleimide
- RMSD
- root mean square deviation
- HSQC
- heteronuclear single quantum coherence spectroscopy
- EGF
- epidermal growth factor
- TCEP
- tris(2-carboxyethyl)phosphine
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- MBP
- maltose-binding protein
- Ni-NTA
- nickel-nitrilotriacetic acid
- PDB
- Protein Data Bank
- TOCSY
- total correlation spectroscopy
- NOESY
- NOE spectroscopy
- HSQC
- heteronuclear single quantum coherence.
References
- 1. Pennington M. W., Czerwinski A., and Norton R. S. (2018) Peptide therapeutics from venom: Current status and potential. Bioorg. Med. Chem. 26, 2738–2758 10.1016/j.bmc.2017.09.029 [DOI] [PubMed] [Google Scholar]
- 2. Miljanich G. P. (2004) Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 11, 3029–3040 10.2174/0929867043363884 [DOI] [PubMed] [Google Scholar]
- 3. Robinson S. D., and Safavi-Hemami H. (2017) Venom peptides as pharmacological tools and therapeutics for diabetes. Neuropharmacology 127, 79–86 10.1016/j.neuropharm.2017.07.001 [DOI] [PubMed] [Google Scholar]
- 4. Safavi-Hemami H., Brogan S. E., and Olivera B. M. (2019) Pain therapeutics from cone snail venoms: from Ziconotide to novel non-opioid pathways. J. Proteomics 190, 12–20 10.1016/j.jprot.2018.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balsara R., Dang A., Donahue D. L., Snow T., and Castellino F. J. (2015) Conantokin-G attenuates detrimental effects of NMDAR hyperactivity in an ischemic rat model of stroke. PLoS One 10, e0122840 10.1371/journal.pone.0122840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Menting J. G., Gajewiak J., MacRaild C. A., Chou D. H., Disotuar M. M., Smith N. A., Miller C., Erchegyi J., Rivier J. E., Olivera B. M., Forbes B. E., Smith B. J., Norton R. S., Safavi-Hemami H., and Lawrence M. C. (2016) A minimized human insulin-receptor-binding motif revealed in a Conus geographus venom insulin. Nat. Struct. Mol. Biol. 23, 916–920 10.1038/nsmb.3292 [DOI] [PubMed] [Google Scholar]
- 7. Robinson S. D., and Norton R. S. (2014) Conotoxin gene superfamilies. Mar. Drugs 12, 6058–6101 10.3390/md12126058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaas Q., Westermann J. C., and Craik D. J. (2010) Conopeptide characterization and classifications: an analysis using ConoServer. Toxicon 55, 1491–1509 10.1016/j.toxicon.2010.03.002 [DOI] [PubMed] [Google Scholar]
- 9. Olivera B. M., Safavi-Hemami H., Raghuraman S., and Teichert R. W. (2017) in Chemical Biology of Natural Products (Newman D. J., Cragg G. M., and Grothaus P. G., eds) 1st Ed., pp. 425–489, CRC Press, Taylor & Francis Group, Boca Raton, FL [Google Scholar]
- 10. Robinson S. D., Safavi-Hemami H., McIntosh L. D., Purcell A. W., Norton R. S., and Papenfuss A. T. (2014) Diversity of conotoxin gene superfamilies in the venomous snail, Conus victoriae. PLoS One 9, e87648 10.1371/journal.pone.0087648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Woodward S. R., Cruz L. J., Olivera B. M., and Hillyard D. R. (1990) Constant and hypervariable regions in conotoxin propeptides. EMBO J. 9, 1015–1020 10.1002/j.1460-2075.1990.tb08204.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pallaghy P. K., Nielsen K. J., Craik D. J., and Norton R. S. (1994) A common structural motif incorporating a cystine knot and a triple-stranded β-sheet in toxic and inhibitory polypeptides. Protein Sci. 3, 1833–1839 10.1002/pro.5560031022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Postic G., Gracy J., Périn C., Chiche L., and Gelly J. C. (2018) KNOTTIN: the database of inhibitor cystine knot scaffold after 10 years, toward a systematic structure modeling. Nucleic Acids Res. 46, D454–D458 10.1093/nar/gkx1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Undheim E. A., Mobli M., and King G. F. (2016) Toxin structures as evolutionary tools: using conserved 3D folds to study the evolution of rapidly evolving peptides. BioEssays 38, 539–548 10.1002/bies.201500165 [DOI] [PubMed] [Google Scholar]
- 15. Robinson S. D., Undheim E. A. B., Ueberheide B., and King G. F. (2017) Venom peptides as therapeutics: advances, challenges and the future of venom–peptide discovery. Expert Rev. Proteomics 14, 931–939 10.1080/14789450.2017.1377613 [DOI] [PubMed] [Google Scholar]
- 16. Nguyen V. D., Hatahet F., Salo K. E., Enlund E., Zhang C., and Ruddock L. W. (2011) Pre-expression of a sulfhydryl oxidase significantly increases the yields of eukaryotic disulfide bond containing proteins expressed in the cytoplasm of E. coli. Microb. Cell Fact. 10, 1 10.1186/1475-2859-10-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gaciarz A., Veijola J., Uchida Y., Saaranen M. J., Wang C., Hörkkö S., and Ruddock L. W. (2016) Systematic screening of soluble expression of antibody fragments in the cytoplasm of E. coli. Microb. Cell Fact. 15, 22 10.1186/s12934-016-0419-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Safavi-Hemami H., Li Q., Jackson R. L., Song A. S., Boomsma W., Bandyopadhyay P. K., Gruber C. W., Purcell A. W., Yandell M., Olivera B. M., and Ellgaard L. (2016) Rapid expansion of the protein-disulfide isomerase gene family facilitates the folding of venom peptides. Proc. Natl. Acad. Sci. U.S.A. 113, 3227–3232 10.1073/pnas.1525790113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moilanen A., Korhonen K., Saaranen M. J., and Ruddock L. W. (2018) Molecular analysis of human Ero1 reveals novel regulatory mechanisms for oxidative protein folding. Life Sci. Alliance 1, e201800090 10.26508/lsa.201800090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rogov V. V., Rozenknop A., Rogova N. Y., Löhr F., Tikole S., Jaravine V., Güntert P., Dikic I., and Dötsch V. (2012) A universal expression tag for structural and functional studies of proteins. Chembiochem 13, 959–963 10.1002/cbic.201200045 [DOI] [PubMed] [Google Scholar]
- 21. Sharma D., and Rajarathnam K. (2000) 13C NMR chemical shifts can predict disulfide bond formation. J. Biomol. NMR 18, 165–171 10.1023/A:1008398416292 [DOI] [PubMed] [Google Scholar]
- 22. Albert A., Eksteen J. J., Isaksson J., Sengee M., Hansen T., and Vasskog T. (2016) General approach to determine disulfide connectivity in cysteine-rich peptides by sequential alkylation on solid phase and mass spectrometry. Anal. Chem. 88, 9539–9546 10.1021/acs.analchem.6b02115 [DOI] [PubMed] [Google Scholar]
- 23. Gao B., Harvey P. J., Craik D. J., Ronjat M., De Waard M., and Zhu S. (2013) Functional evolution of scorpion venom peptides with an inhibitor cystine knot fold. Biosci. Rep. 33, e00047 10.1042/BSR20130052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ram N., Weiss N., Texier-Nogues I., Aroui S., Andreotti N., Pirollet F., Ronjat M., Sabatier J. M., Darbon H., Jacquemond V., and De Waard M. (2008) Design of a disulfide-less, pharmacologically inert, and chemically competent analog of maurocalcine for the efficient transport of impermeant compounds into cells. J. Biol. Chem. 283, 27048–27056 10.1074/jbc.M804727200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Woody R. W. (1995) Circular dichroism. Methods Enzymol. 246, 34–71 10.1016/0076-6879(95)46006-3 [DOI] [PubMed] [Google Scholar]
- 26. Yao S., Howlett G. J., and Norton R. S. (2000) Peptide self-association in aqueous trifluoroethanol monitored by pulsed field gradient NMR diffusion measurements. J. Biomol. NMR 16, 109–119 10.1023/A:1008382624724 [DOI] [PubMed] [Google Scholar]
- 27. Ortega A., Amorós D., and García de la Torre J. (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 10.1016/j.bpj.2011.06.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baxter N. J., and Williamson M. P. (1997) Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 9, 359–369 10.1023/A:1018334207887 [DOI] [PubMed] [Google Scholar]
- 29. Cierpicki T., and Otlewski J. (2001) Amide proton temperature coefficients as hydrogen bond indicators in proteins. J. Biomol. NMR 21, 249–261 10.1023/A:1012911329730 [DOI] [PubMed] [Google Scholar]
- 30. Lavergne V., Alewood P. F., Mobli M., and King G. F. (2015) The Structural Universe of Disulfide-rich Venom Peptides (King G. F., ed) pp. 37–79, Royal Society of Chemistry, London, UK [Google Scholar]
- 31. Jin A. H., Dekan Z., Smout M. J., Wilson D., Dutertre S., Vetter I., Lewis R. J., Loukas A., Daly N. L., and Alewood P. F. (2017) Conotoxin φ-MiXXVIIA from the superfamily G2 employs a novel cysteine framework that mimics granulin and displays anti-apoptotic activity. Angew. Chem. Int. Ed. Engl. 56, 14973–14976 10.1002/anie.201708927 [DOI] [PubMed] [Google Scholar]
- 32. Khoo K. K., and Norton R. S. (2012) in Amino Acids, Peptides and Proteins in Organic Chemistry, Analysis and Function of Amino Acids and Peptides (Hughes A. B., ed) pp. 395–410, Wiley-VCH Verlag GmbH & Co, Darmstadt, Germany [Google Scholar]
- 33. Tolkatchev D., Malik S., Vinogradova A., Wang P., Chen Z., Xu P., Bennett H. P., Bateman A., and Ni F. (2008) Structure dissection of human progranulin identifies well-folded granulin/epithelin modules with unique functional activities. Protein Sci. 17, 711–724 10.1110/ps.073295308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hrabal R., Chen Z., James S., Bennett H. P., and Ni F. (1996) The hairpin stack fold, a novel protein architecture for a new family of protein growth factors. Nat. Struct. Biol. 3, 747–752 10.1038/nsb0996-747 [DOI] [PubMed] [Google Scholar]
- 35. Palfree R. G., Bennett H. P., and Bateman A. (2015) The evolution of the secreted regulatory protein progranulin. PLoS One 10, e0133749 10.1371/journal.pone.0133749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vranken W. F., Chen Z. G., Xu P., James S., Bennett H. P., and Ni F. (1999) A 30-residue fragment of the carp granulin-1 protein folds into a stack of two β-hairpins similar to that found in the native protein. J. Pept. Res. 53, 590–597 10.1034/j.1399-3011.1999.00048.x [DOI] [PubMed] [Google Scholar]
- 37. Tolkatchev D., Xu P., and Ni F. (2001) A peptide derived from the C-terminal part of a plant cysteine protease folds into a stack of two β-hairpins, a scaffold present in the emerging family of granulin-like growth factors. J. Pept. Res. 57, 227–233 10.1111/j.1399-3011.2001.00828.x [DOI] [PubMed] [Google Scholar]
- 38. Tolkatchev D., Ng A., Vranken W., and Ni F. (2000) Design and solution structure of a well-folded stack of two β-hairpins based on the amino-terminal fragment of human granulin A. Biochemistry 39, 2878–2886 10.1021/bi992130u [DOI] [PubMed] [Google Scholar]
- 39. Bansal P. S., Smout M. J., Wilson D., Cobos Caceres C., Dastpeyman M., Sotillo J., Seifert J., Brindley P. J., Loukas A., and Daly N. L. (2017) Development of a potent wound healing agent based on the liver fluke granulin structural fold. J. Med. Chem. 60, 4258–4266 10.1021/acs.jmedchem.7b00047 [DOI] [PubMed] [Google Scholar]
- 40. Xu S., Zhang T., Kompella S. N., Yan M., Lu A., Wang Y., Shao X., Chi C., Adams D. J., Ding J., and Wang C. (2015) Conotoxin αD-GeXXA utilizes a novel strategy to antagonize nicotinic acetylcholine receptors. Sci. Rep. 5, 14261 10.1038/srep14261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rester U., Bode W., Moser M., Parry M. A., Huber R., and Auerswald E. (1999) Structure of the complex of the antistasin-type inhibitor bdellastasin with trypsin and modelling of the bdellastasin-microplasmin system. J. Mol. Biol. 293, 93–106 10.1006/jmbi.1999.3162 [DOI] [PubMed] [Google Scholar]
- 42. Cash J. N., Angerman E. B., Kattamuri C., Nolan K., Zhao H., Sidis Y., Keutmann H. T., and Thompson T. B. (2012) Structure of myostatin·follistatin-like 3: N-terminal domains of follistatin-type molecules exhibit alternate modes of binding. J. Biol. Chem. 287, 1043–1053 10.1074/jbc.M111.270801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cheek S., Krishna S. S., and Grishin N. V. (2006) Structural classification of small, disulfide-rich protein domains. J. Mol. Biol. 359, 215–237 10.1016/j.jmb.2006.03.017 [DOI] [PubMed] [Google Scholar]
- 44. Sheng Y., Laister R. C., Lemak A., Wu B., Tai E., Duan S., Lukin J., Sunnerhagen M., Srisailam S., Karra M., Benchimol S., and Arrowsmith C. H. (2008) Molecular basis of Pirh2-mediated p53 ubiquitylation. Nat. Struct. Mol. Biol. 15, 1334–1342 10.1038/nsmb.1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Norton R. S., and Pallaghy P. K. (1998) The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 36, 1573–1583 10.1016/S0041-0101(98)00149-4 [DOI] [PubMed] [Google Scholar]
- 46. Klint J. K., Senff S., Saez N. J., Seshadri R., Lau H. Y., Bende N. S., Undheim E. A., Rash L. D., Mobli M., and King G. F. (2013) Production of recombinant disulfide-rich venom peptides for structural and functional analysis via expression in the periplasm of E. coli. PLoS One 8:e63865 10.1371/journal.pone.0063865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Turchetto J., Sequeira A. F., Ramond L., Peysson F., Brás J. L., Saez N. J., Duhoo Y., Blémont M., Guerreiro C. I., Quinton L., De Pauw E., Gilles N., Darbon H., Fontes C. M., and Vincentelli R. (2017) High-throughput expression of animal venom toxins in Escherichia coli to generate a large library of oxidized disulphide-reticulated peptides for drug discovery. Microb. Cell Fact. 16, 6 10.1186/s12934-016-0617-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. O'Brien H., Kanemura S., Okumura M., Baskin R. P., Bandyopadhyay P. K., Olivera B. M., Ellgaard L., Inaba K., and Safavi-Hemami H. (2018) Ero1-mediated reoxidation of protein-disulfide isomerase accelerates the folding of cone snail toxins. Int. J. Mol. Sci. 19, 3418 10.3390/ijms19113418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kuipers B. J., and Gruppen H. (2007) Prediction of molar extinction coefficients of proteins and peptides using UV absorption of the constituent amino acids at 214 nm to enable quantitative reverse phase high-performance liquid chromatography-mass spectrometry analysis. J. Agric. Food Chem. 55, 5445–5451 10.1021/jf070337l [DOI] [PubMed] [Google Scholar]
- 50. Gobom J., Nordhoff E., Mirgorodskaya E., Ekman R., and Roepstorff P. (1999) Sample purification and preparation technique based on nano-scale reversed-phase columns for the sensitive analysis of complex peptide mixtures by matrix-assisted laser desorption/ionization mass spectrometry. J. Mass Spectrom. 34, 105–116 [DOI] [PubMed] [Google Scholar]
- 51. Perkins D. N., Pappin D. J., Creasy D. M., and Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 52. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 53. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 54. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 10.1007/s10858-009-9333-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Güntert P., and Buchner L. (2015) Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 62, 453–471 10.1007/s10858-015-9924-9 [DOI] [PubMed] [Google Scholar]
- 56. Schwieters C. D., Kuszewski J. J., Tjandra N., and Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 10.1016/S1090-7807(02)00014-9 [DOI] [PubMed] [Google Scholar]
- 57. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., and Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 58. Koradi R., Billeter M., and Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55, 29–32 10.1016/0263-7855(96)00009-4 [DOI] [PubMed] [Google Scholar]
- 59. Berman H., Henrick K., and Nakamura H. (2003) Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 10, 980 10.1038/nsb1203-980 [DOI] [PubMed] [Google Scholar]
- 60. Ulrich E. L., Akutsu H., Doreleijers J. F., Harano Y., Ioannidis Y. E., Lin J., Livny M., Mading S., Maziuk D., Miller Z., Nakatani E., Schulte C. F., Tolmie D. E., Kent Wenger R., Yao H., and Markley J. L. (2008) BioMagResBank. Nucleic Acids Res. 36, D402–D408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nguyen M. N., Tan K. P., and Madhusudhan M. S. (2011) CLICK–topology-independent comparison of biomolecular 3D structures. Nucleic Acids Res. 39, W24–W28 10.1093/nar/gkr393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nguyen M. N., and Madhusudhan M. S. (2011) Biological insights from topology independent comparison of protein 3D structures. Nucleic Acids Res. 39, e94 10.1093/nar/gkr348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Robinson S. D., Safavi-Hemami H., Raghuraman S., Imperial J. S., Papenfuss A. T., Teichert R. W., Purcell A. W., Olivera B. M., and Norton R. S. (2015) Discovery by proteogenomics and characterization of an RF-amide neuropeptide from cone snail venom. J. Proteomics 114, 38–47 10.1016/j.jprot.2014.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lü S., Liang S., and Gu X. (1999) Three-dimensional structure of Selenocosmia huwena lectin-I (SHL-I) from the venom of the spider Selenocosmia huwena by 2D-NMR. J. Protein Chem. 18, 609–617 10.1023/A:1020663619657 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.