

Abstract
The cohesin complex regulates sister chromatid cohesion, chromosome organization, gene expression, and DNA repair. Cohesin is a ring complex composed of four core subunits and seven regulatory subunits. In an effort to comprehensively identify additional cohesin-interacting proteins, we used gene editing to introduce a dual epitope tag into the endogenous allele of each of 11 known components of cohesin in cultured human cells, and we performed MS analyses on dual-affinity purifications. In addition to reciprocally identifying all known components of cohesin, we found that cohesin interacts with a panoply of splicing factors and RNA-binding proteins (RBPs). These included diverse components of the U4/U6.U5 tri-small nuclear ribonucleoprotein complex and several splicing factors that are commonly mutated in cancer. The interaction between cohesin and splicing factors/RBPs was RNA- and DNA-independent, occurred in chromatin, was enhanced during mitosis, and required RAD21. Furthermore, cohesin-interacting splicing factors and RBPs followed the cohesin cycle and prophase pathway of cell cycle–regulated interactions with chromatin. Depletion of cohesin-interacting splicing factors and RBPs resulted in aberrant mitotic progression. These results provide a comprehensive view of the endogenous human cohesin interactome and identify splicing factors and RBPs as functionally significant cohesin-interacting proteins.
Keywords: RNA-binding protein, cell cycle, mitosis, RNA splicing, protein–protein interaction, cell division, genome structure, CKAP5, cohesin, gene editing, MGA, STAG2
Introduction
Cohesin is a ubiquitously expressed multiprotein complex best known for its involvement in sister chromatid cohesin, but it also plays important roles in chromosome organization, gene expression, and DNA repair (1–5). In vertebrate cells, cohesin is a ring-like structure encircling chromatin composed of four core subunits: SMC1A, SMC3, RAD21, and either STAG1 or STAG2. Several additional subunits serve to regulate the core complex, including NIPBL, MAU2, WAPL, PDS5A, PDS5B, and sororin. The cohesin complex is highly conserved in both prokaryotic and eukaryotic unicellular organisms, as well as in metazoans. Somatic mutations of cohesin are present in a wide range of human cancers, and inherited mutations result in developmental disorders known as cohesinopathies (6–8).
Current models of cohesin function focus on the ability of the cohesin ring to encircle DNA in a cell cycle and developmentally-regulated fashion. This entrapment of DNA by cohesin is thought to result in several important mechanistic outcomes: (i) encirclement of homologous regions of sister chromatids (in trans) enforces sister chromatid cohesion until cohesin is degraded at the metaphase to anaphase transition, and (ii) encirclement of otherwise distant regions of contiguous DNA (in cis) enables the formation of loop structures important in regulating gene expression and chromosomal organization. The mechanistic implications of the cohesin ring model drive most current research into cohesin function and into the consequences of its inactivation in human disease.
In an effort to further our understanding of cohesin function using an unbiased approach, we set out to generate a complete cohesin interactome in human cells. To do this, gene editing was used to create a panel of 11 isogenic human cell lines in which each of the genes encoding the 11 known components of cohesin were modified with the addition of a dual epitope tag in either the C or N terminus of the encoded protein. Mass spectrometry was then performed on dual-affinity purifications to generate an endogenous human cohesin interactome. This effort, together with subsequent confirmation of the interactions and functional validation, revealed that cohesin interacts with a panoply of splicing factors and RNA-binding proteins (RBPs)2 required for mitotic progression, identifying splicing factors and RBPs as a novel class of functionally significant cohesin-interacting proteins.
Results
Generation of an endogenous cohesin interactome in human cells
To obtain a comprehensive picture of human cohesin protein–protein interactions, gene editing was used to add a dual FLAG–streptavidin-binding peptide (SBP) epitope tag to an endogenous allele of each of the genes encoding the 11 known components of cohesin in HCT116 cells, a human cell line with WT cohesin genes and intact sister chromatid cohesin (Fig. 1A and Fig. S1). Gene editing efficiencies are shown in Table S1. AAV-based gene editing is a reliably efficient technique for the introduction of precise sequence changes in cultured cells via homologous recombination (9–11). Although endogenous tagging has been used to generate interactomes in genetically tractable model organisms (12), this is, to our knowledge, the first application of endogenous tagging to each of the components of an entire protein complex or signaling pathway in human cells.
Figure 1.

Endogenous epitope tagging and generation of the cohesin interactome. A, SMC1A epitope-tagging vector was designed to modify the final coding exon (exon 25), adding a 1× FLAG-SBP (F-SBP) dual epitope tag immediately prior to the natural stop codon (TAG). Subsequent Cre-mediated recombination removed the FLOXed IRES-NeoR gene, leaving behind a single LoxP site in intron 24 and F-SBP in exon 25. See Fig. S1 for schematics and details of epitope-tagging vectors for the other 10 components of cohesin. B, nuclear extracts from parental HCT116 cells and cohesin epitope-tagged derivatives were immunoprecipitated (IP) with FLAG-M2 beads followed by peptide elution. Western blotting was performed with FLAG antibodies to measure the relative abundance of the tagged cohesin subunits and with antibodies to each of the other components of cohesin to measure their relative efficiency of co-purification. Asterisks denote the epitope-tagged protein, which is slightly larger than the untagged protein. Because PDS5B, sororin, NIPBL, and MAU2 are much less abundant than the other components of cohesin, more protein was used for immunoprecipitations in cells with epitope-tagged alleles of these genes as indicated. C, mass spectrometry data were analyzed using STRING software for the identification of functional protein interaction networks. Nodes represent proteins identified by MS from dual-affinity purifications of endogenous epitope-tagged cells. Blue nodes represent each of the 11 cohesin subunits used as baits. Red nodes represent interacting splicing factors and proteins with RNA-binding domains as identified by GO, KEGG, and Pfam analysis. Edges represent protein–protein interactions, with teal and pink representing known interactions from curated databases and experimentally determined, respectively; green, red, and blue represent predicted interactions via gene neighborhoods, gene fusions, and gene co-occurrences, respectively; and light green, black, and violet represent interactions predicted by text mining, co-expression, and protein homology analysis, respectively. D, heatmap of interactions between cohesin subunits (columns) and interacting splicing factors/RNA-binding proteins (rows). Proteins identified in two or more affinity purifications are shown. Colors as shown on the key represent the relative abundance of each protein (as measured by ion area) in each of the 11 affinity purifications.
We then used the epitope-tagged cells to determine the relative abundance of each of the individual components of cohesin in human cells. To do this, cohesin complexes were purified via FLAG immunoprecipitation from nuclear extracts derived from parental HCT116 cells and each of the 11 cohesin endogenous epitope-tagged derivatives. Western blotting was then performed with FLAG antibodies (Fig. 1B). This experiment demonstrated that core subunits SMC1A, SMC3, STAG2, as well as PDS5A are expressed in HCT116 cells at similarly high levels; RAD21, STAG1, and WAPL are expressed at similar intermediate levels, and PDS5B, sororin, NIPBL, and MAU2 are expressed at much lower levels. Next, each of the immunoprecipitates was interrogated by Western blotting with antibodies to each of the 11 known components of cohesin (Fig. 1B). This experiment demonstrated that SMC1A, SMC3, STAG2, and sororin are most efficient at co-purifying the other known components of cohesin and confirmed the known mutual exclusivity of STAG1 and STAG2 in cohesin complexes (13).
Dual-affinity purification was then performed on nuclear extracts from the 11 epitope-tagged cell lines and parental HCT116 cells. Initially, affinity purifications were separated using SDS-PAGE and stained with silver, demonstrating that this purification approach made it possible to purify cohesin to high levels of homogeneity using each of the 11 known subunits as baits (Fig. S2). The protein composition of individual-affinity purifications was then interrogated by GeLC-MS/MS following Coomassie staining as described in detail under “Experimental procedures.” This approach results in sensitive protein identification by concentrating the sample and removing nonprotein contaminants (14). Mass spectrometry raw data were analyzed using Proteome Discoverer 2.1.0.81 as described under “Experimental procedures.” Proteins passing the 1% FDR cutoff and that were represented by two or more unique peptides in affinity purifications from epitope-tagged cells but absent in isogenic parental cells are listed in the 11 tabs of Table S2. All known components of cohesin (but no other known structural components of chromatin) were reciprocally identified in this analysis (Table S3), as well as other known cohesin-interacting proteins such as components of the MCM replication licensing factor complex and components of the Mediator complex (15, 16).
Protein–protein interaction networks were then identified using STRING (Fig. 1C) (17). The interaction network was by far the most highly enriched for proteins involved in RNA splicing, RNA binding, and/or containing known RNA-recognition motifs than for any other pathway or protein domain (FDR 1.79 × 10−42) (Fig. 1D and Tables S4 and S5). Notable splicing factors/RBPs included the proteins encoded by the SF3B1 oncogene, the RBM10 tumor suppressor gene, and the EFTUD2, SNRNP200, and PRPF31 components of the U4/U6.U5 tri-snRNP complex, inherited mutations of which are a major cause of retinitis pigmentosa (18–20). These data were particularly intriguing in light of the recent observation that depletion of splicing factors in human cells can result in a loss of sister chromatid cohesin (21–24).
Two additional nonsplicing factor/RBP novel putative cohesin interactors were particularly robustly co-purified with cohesin, identified in affinity purifications from seven or more of the 11 epitope-tagged cell lines. These included the CKAP5 microtubule-binding protein and the MGA Myc-associated transcription factor. The details and functions of their interactions with cohesin will be described elsewhere. A complete list of noncohesin, nonsplicing factor proteins identified in affinity purifications from four or more epitope-tagged cell lines is presented in Table S6.
Validation of splicing factors and RNA-binding proteins as cohesin-interacting proteins
To confirm the interaction between cohesin and splicing factors/RBPs, Western blotting with antibodies to splicing factors and RBPs was performed on dual-affinity purifications from nuclear extracts of parental HCT116 cells and SMC3 epitope-tagged derivatives (Fig. 2A). This experiment confirmed the interaction between endogenous cohesin and endogenous SF3B1, SF3B3, ADAR1, PRPF31, SNRNP200, EFTUD2, HNRNPU, RBM10, RBM15, HNRNPH, HSPA8, PDCD11, THRAP3, DDX47, and PRPF6. To generalize these findings, endogenous cohesin was immunoprecipitated with SMC1A antibodies from HeLa cells and untransformed human epithelial cells (RPE–hTERT), and Western blotting was performed with a representative subset of these splicing factor/RBP antibodies (Fig. 2B). This experiment further confirmed the interaction between cohesin and splicing factors/RBPs and generalized it to genetically unmodified human cells.
Figure 2.

Interaction of cohesin with diverse splicing factors and RNA-binding proteins. A, dual-affinity purifications from nuclear extracts of HCT116 cells and SMC3 epitope-tagged derivatives were separated by SDS-PAGE, and Western blotting was performed with the antibodies indicated. B, immunoprecipitations (IP) with IgG and SMC1A antibodies from HeLa and RPE–hTERT nuclear extracts were separated by SDS-PAGE, and Western blotting was performed with the antibodies indicated. C, dual-affinity purifications from nuclear extracts of HCT116 cells and 11 cohesin epitope-tagged derivatives were separated by SDS-PAGE, and Western blotting was performed with the antibodies indicated.
To determine whether splicing factors/RBPs interact preferentially with cohesin complexes defined by the presence of specific cohesin subunits, Western blotting with splicing factor/RBP antibodies was performed on dual-affinity purifications from parental HCT116 cells and each of the 11 epitope-tagged cell lines (Fig. 2C). This experiment demonstrated that splicing factors/RBPs interact efficiently with SMC1A, SMC3, STAG2, WAPL, and PDS5A-containing cohesin complexes. Of these, splicing factors and RBPs appear to interact most efficiently with PDS5A-containing cohesin complexes.
Co-localization of cohesin and splicing factors in HeLa cells
To provide additional evidence for the interaction between cohesin and splicing factors/RBPs, we next tested whether cohesin and splicing factors/RBPs are co-localized in the nuclei of human cells. Double immunofluorescence was performed on HeLa cells using antibodies to cohesin (STAG2) together with antibodies to several interacting splicing factors (HNRNPH, EFTUD2, or SF3B1). The localization of cohesin and splicing factors in HeLa cell nuclei was identical (Fig. 3A). Next, we tested the cell cycle dependence of this co-localization. To do this, we performed cohesin/splicing factor double immunofluorescence on HeLa cells that had been treated with hydroxyurea, RO-3306 (25), and nocodazole to arrest cells in S phase, G2, and mitosis, respectively (Fig. 3B). This experiment demonstrated that the co-localization between cohesin and splicing factors is maintained in the S, G2, and M phases of the cell cycle.
Figure 3.
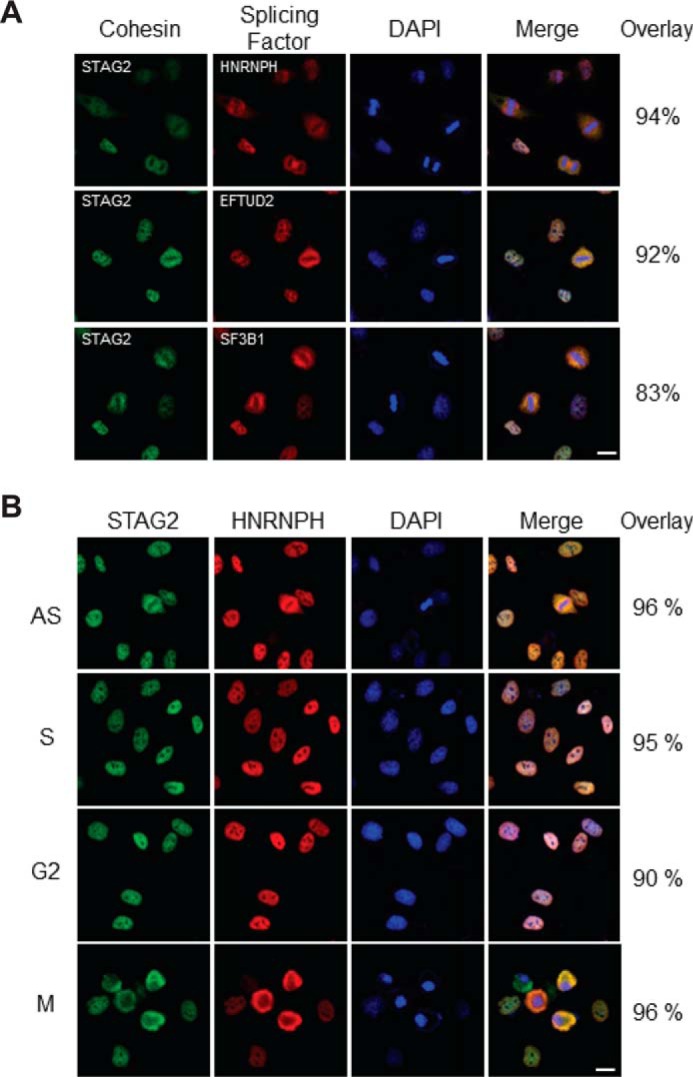
Co-localization of splicing factors with cohesin. A, proliferating HeLa cells were fixed, permeabilized, and double-stained with antibodies to cohesin (STAG2) and three different interacting splicing factors (HNRNPH, EFTUD2, and SF3B1). Imaging was performed on a Leica TCS SP8 confocal laser-scanning microscope, and co-localization was quantified using ImageJ software. For technical details of immunofluorescence and co-localization analysis, see “Experimental procedures.” B, HeLa cells were treated with DMSO vehicle alone (AS), hydroxyurea (S), RO-3306 (G2), and nocodazole (M) for 24 h to arrest cells at S, G2, and M phases of the cell cycle, respectively. Cells were then fixed, permeabilized, and double-stained with antibodies to cohesin (STAG2) and a representative interacting splicing factor (HNRNPH). Microscopy and co-localization analysis were performed as in A. DAPI, 4′,6-diamidino-2-phenylindole. Bar = 10 μm.
Interaction between cohesin and splicing factors occurs in chromatin but requires neither DNA nor RNA
We then tested whether spliceosomal small nuclear RNA and/or genomic DNA is required for maintenance of the cohesin-splicing factor/RBP complex. To test this, nuclear extracts were prepared from SMC3-tagged HCT116 cells and treated with RNase, DNase, or both. SMC3-containing cohesin complexes were purified by dual-affinity purification, and Western blotting with antibodies to representative splicing factors was performed (Fig. 4A). This experiment demonstrated that the interaction between cohesin and splicing factors/RBPs requires neither RNA nor DNA.
Figure 4.
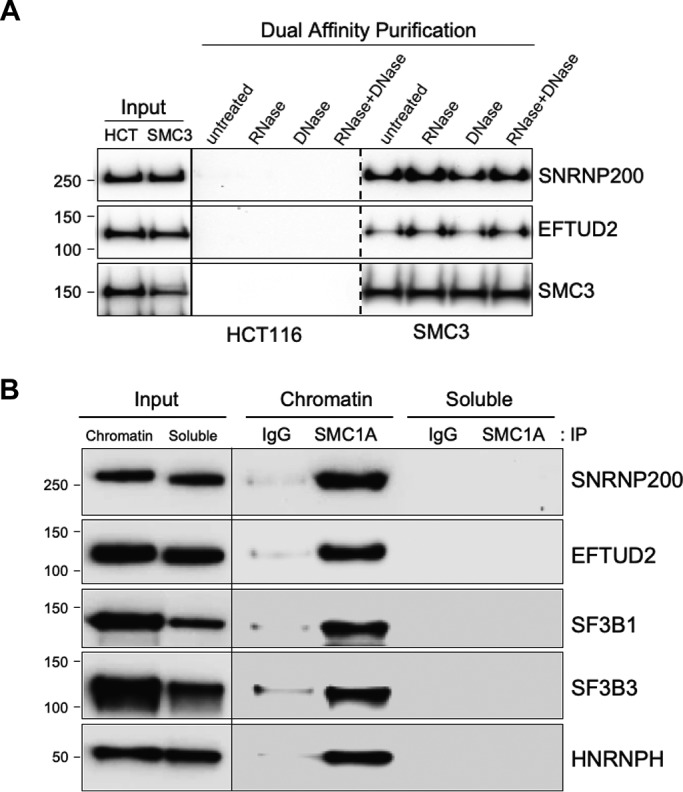
Interaction between cohesin and splicing factors occurs in chromatin and does not require DNA or RNA. A, dual-affinity purifications from RNase- and DNase-treated nuclear extracts of HCT116 cells and SMC3 epitope-tagged derivatives were separated by SDS-PAGE, and Western blotting was performed with the antibodies indicated. B, fractionated chromatin and soluble lysates were prepared from HeLa cells, immunoprecipitated (IP) with SMC1A antibodies or IgG control antibodies, and separated by SDS-PAGE, and Western blotting was performed with the antibodies indicated.
Next, we sought to determine whether the interaction between cohesin and splicing factors/RBPs occurs in chromatin or in the soluble fraction. To do this, fractionated HeLa cell lysates were prepared in which chromatin proteins and total soluble proteins were separated (see “Experimental procedures”), cohesin immunoprecipitated with SMC1A antibodies, and Western blotting performed with antibodies to representative interacting splicing factors/RBPs (Fig. 4B). This experiment demonstrated that the interaction between cohesin and splicing factors/RBPs occurs in the chromatin fraction, not in the soluble fraction. This experiment also further confirmed the DNA and RNA independence of the interactions, because they were maintained in chromatin fractions, which were treated with a universal nuclease prior to immunoprecipitation to reduce the viscosity of the lysates and release DNA- and RNA-bound proteins.
Interaction of cohesin with splicing factors requires RAD21
We next sought to determine which of the 11 known components of the cohesin complex were required for the interaction of cohesin with splicing factors/RBPs. Because our prior experiments had shown that splicing factors and RBPs interact most efficiently with PDS5A-containing cohesin complexes (Fig. 2C), we hypothesized that PDS5A might be particularly important, and therefore required, for the interaction between cohesin and splicing factors/RBPs. To test this, we used CRISPR to create PDS5A homozygous mutant derivatives of SMC1A epitope-tagged HCT116 cells (see “Experimental procedures”) and then performed dual-affinity purification and Western blotting with antibodies to representative splicing factors/RBPs (Fig. 5A). This experiment disproved our hypothesis, demonstrating that PDS5A is dispensable for the interaction of cohesin with splicing factors.
Figure 5.

Interaction of cohesin with splicing factors requires RAD21. A, nuclear extracts were prepared from the following: (i) HCT116 parental cells; (ii) HCT116 SMC1A-tagged cells; and (iii) HCT116 SMC1A-tagged, PDS5A KO cells. Dual-affinity purification was performed and separated by SDS-PAGE, and Western blotting was performed with the antibodies indicated. B, protein lysates were prepared from HCT116 cells and STAG2 mutant isogenic derivatives. Cohesin complexes were immunoprecipitated (IP) with SMC1A antibodies (or IgG control antibodies) and separated by SDS-PAGE, and Western blotting was performed with the antibodies indicated. C, HeLa cells were transfected with two independent STAG2 siRNAs (#1, s21089; #2, s21090) or scrambled siRNA and cultured for 72 h. Cells were then fixed, permeabilized, and double-stained with antibodies to SMC1A (cohesin) and HNRNPH (interacting splicing factor/RBP) for co-localization analysis. Cells were also stained with STAG2 antibodies to confirm depletion of STAG2. Microscopy and co-localization analysis were performed as in Fig. 3. DAPI, 4′,6-diamidino-2-phenylindole. Bar = 10 μm. D, HeLa cells were transfected with negative control siRNA (s4390843) or gene-specific siRNAs for SMC3 (s17426), STAG2 (s21090), RAD21 (s11726), STAG1 (s20074), sororin (s535461), NIPBL (s24588), WAPL (s22949), and PDS5B (s22912). Transfected cells were cultured for 3 days; protein lysates were prepared and separated by SDS-PAGE, and Western blotting was performed with the antibodies indicated to demonstrate depletion of the targeted proteins. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. E, HeLa cells were transfected with the siRNAs indicated and cultured for 3 days, and protein lysates were prepared. Cohesin complexes were then immunoprecipitated with SMC1A antibodies (or IgG control antibodies) and separated by SDS-PAGE, and Western blotting was performed with the antibodies indicated.
Next, we hypothesized that because STAG2 is the most commonly mutated cohesin subunit in human cancer, perhaps a key role for STAG2 is as a required bridge between cohesin and interacting splicing factors/RBPs. To test this hypothesis, we prepared protein lysates from a previously-described isogenic set of HCT116 parental cells and derivatives in which a bladder cancer–derived early truncating mutation (S97X) had been introduced by AAV-mediated gene editing (26). Cohesin was immunoprecipitated with SMC1A antibodies, and Western blotting was performed with antibodies to representative splicing factors and RBPs (Fig. 5B). This experiment demonstrated that STAG2 is not required for the interaction of cohesin with splicing factors/RBPs in HCT116 cells. To extend and generalize this result, we next tested whether STAG2 was required for the co-localization of cohesin and splicing factors/RBPs in HeLa cells. To test this, HeLa cells were transfected with STAG2 siRNAs and cultured for 72 h, and double immunofluorescence was performed with antibodies to cohesin (SMC1A) and a representative cohesin-interacting splicing factor/RBP (HNRNPH). Depletion of STAG2 did not alter the co-localization of SMC1A and HNRNPH (Fig. 5C). Together, these experiments demonstrated that STAG2 is dispensable for the interaction and co-localization of cohesin with splicing factors/RBPs in HCT116 and HeLa cells, respectively.
We next wanted to test additional components of cohesin to determine which were required for the interaction of cohesin with splicing factors/RBPs. Because stable inactivation of most of these subunits of cohesin is known to be lethal, we chose a transient approach. To do this, we validated siRNAs for SMC3, STAG2, RAD21, STAG1, sororin, NIPBL, WAPL, and PDS5B (Fig. 5D). Next, HeLa cells were transfected with the siRNAs, cultured for 3 days, and protein lysates prepared. Cohesin was then immunoprecipitated with SMC1A antibodies, and Western blotting was performed with antibodies to representative splicing factors/RBPs (Fig. 5E). This experiment demonstrated that RAD21 is the only individual component of cohesin tested that is specifically required for the interaction of cohesin with splicing factors/RBPs in HeLa cells.
Cohesin-interacting splicing factors and RNA-binding proteins follow the cohesin cycle of interaction with chromatin
Cohesin is loaded onto chromatin during DNA replication and released from chromatin during early mitosis in a process known as the “cohesin cycle” (27). Because the physical interaction between splicing factors and cohesin occurs on chromatin (Fig. 4B), we hypothesized that cohesin-interacting splicing factors and RBPs might be similarly loaded onto chromatin during DNA replication and released from chromatin during mitosis.
To test this hypothesis, HeLa cells were treated with hydroxyurea, RO-3306 (25), and nocodazole to arrest cells in S phase, G2, and mitosis, respectively, and chromatin, soluble, and total protein lysates were prepared. As expected, Western blotting with antibodies to SMC1A, STAG2, RAD21, SMC3, STAG1, PDS5A, PDS5B, and WAPL confirmed that cohesin was loaded onto chromatin during DNA replication and released from chromatin during mitosis (Fig. 6A). Remarkably, representative splicing factors and RBPs EFTUD2, ADAR1, SF3B1, SNRNP200, SF3B3, HNRNPH, and PRPF31 demonstrated a similar cell cycle–regulated interaction with chromatin. To generalize these findings, similar experiments were performed in RPE cells and HCT116 cells, with identical results (Fig. S3).
Figure 6.

Splicing factors and RNA-binding proteins follow the cohesin cycle and prophase pathway of cell cycle–regulated interactions with chromatin. A, HeLa cells were treated with DMSO (AS), hydroxyurea (S), RO-3306 (G2), and nocodazole (M) for 24 h to arrest cells at different stages of the cell cycle. Chromatin, soluble, and whole-cell lysates (WCL; RIPA) were prepared, and Western blotting was performed with the antibodies indicated. ND, not determined. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. B, HeLa cells were treated with 100 ng/ml nocodazole (NOC) for 24 h and then treated with the PLK1 inhibitor BI 2536 at 50 and 100 nm for 3 h. Cells were then harvested; chromatin lysates were prepared, and Western blotting was performed with the antibodies indicated.
To confirm that this was not just coincidence, we tested whether splicing factors and cohesin use the same biochemical mechanism for release from chromatin during mitosis. This mechanism, known as the “prophase pathway,” is defined by the requirement for polo-like kinase (PLK1) (28, 29). To determine whether the release of cohesin-interacting splicing factors and RBPs from chromatin in mitosis was similarly PLK1-dependent, nocodazole-treated HeLa cells were incubated in the presence or absence of PLK1 inhibitors, and chromatin extracts were prepared. Western blotting with antibodies to cohesin subunits and splicing factors demonstrated that the release of splicing factors/RBPs from chromatin in mitosis and the release of cohesin from chromatin in mitosis were equivalently PLK1-dependent (Fig. 6B). Together, these data show that the interaction of splicing factors/RBPs with chromatin mirrors that of cohesin itself, following the cohesin cycle and the prophase pathway.
Interaction between cohesin and splicing factors is enhanced in mitosis
We next tested whether the interaction between cohesin and splicing factors was cell cycle–regulated. To do this, HeLa cells were synchronized by double-thymidine block, released, and protein lysates prepared from cells at different stages of the cell cycle. Next, cohesin was immunoprecipitated with SMC1A antibodies and Western blotting performed with antibodies to representative splicing factors/RBPs (Fig. 7A; Fig. S4). This experiment demonstrated that the interaction between cohesin and splicing factors/RBPs is enhanced during mitosis. Identical results were obtained with cells that had been arrested by hydroxyurea, RO-3306, and nocodazole in the S, G2, and M phases of the cell cycle, respectively (Fig. 7B and Fig. S4).
Figure 7.

Interaction between cohesin and splicing factors is enhanced in mitosis. A, top, HeLa cells were synchronized by double thymidine block and whole-cell protein lysates (1% Nonidet P-40) prepared at different time points following release. Endogenous cohesin complexes were then immunoprecipitated with SMC1A antibodies or IgG control antibodies, and Western blotting with the antibodies indicated was performed. Bottom, direct Western blotting of the lysates studied at the top, demonstrating that the amount of cohesin and splicing factor proteins in the lysates does not change over the course of the cell cycle and demonstrating the efficiency of synchronization using cell cycle stage–specific antibodies (S phase, cyclin E1; G2, cyclin B1; M, phospho-H3). B, interaction between cohesin and splicing factors is enhanced in mitosis. Proliferating HeLa cells were treated with DMSO (AS), hydroxyurea (S), RO-3306 (G2), and nocodazole (M) for 24 h. Protein lysates were prepared (1% Nonidet P-40), subjected to immunoprecipitation (IP) with SMC1A antibodies, and studied by Western blotting with the antibodies indicated.
Depletion of cohesin-interacting splicing factors results in “dancing chromosomes”
Because the interaction between cohesin and splicing factors/RBPs is enhanced in mitosis, we hypothesized that inactivation of splicing factors might phenocopy the mitotic effects of cohesin inactivation. To test this hypothesis, we validated siRNAs for four representative cohesin-interacting splicing factors. EFTUD2, SNRNP200, and PRPF31 are components of the U4/U6.U5 tri-snRNP complex, which is the splicing complex most robustly co-purified by cohesin. SF3B3 is a component of the SF3b spliceosome complex, which is also particularly robustly co-purified by cohesin. HeLa cells were transfected with multiple siRNAs for these genes; cells were cultured for 3 days, and protein lysates were prepared and tested by Western blotting. As shown in Fig. 8A, transfection of HeLa cells with these siRNAs resulted in depletion of these splicing factors.
Figure 8.

Cohesin-interacting splicing factors are required for mitotic progression. A, validation of siRNAs targeting representative cohesin-interacting splicing factors in HeLa cells. HeLa cells were transfected with negative control siRNA (s4390843) or gene-specific siRNAs as indicated by the Ambion catalogue number, cultured for 72 h, and harvested in RIPA buffer, and Western blotting was performed with the antibodies indicated. B, HeLa-H2B-GFP cells were transfected with siRNAs for SNRNP200 (ss22846), EFTUD2 (s17862), PRPF31 (s25123), SF3B3 (s23849), and sororin (s535461) and studied by live-cell imaging as described under “Experimental procedures.” Representative aberrant mitoses in siRNA-transfected cells are shown. Bar = 10 μm. For time-lapse movies with images every 5 min from 36 to 72 h post-transfection, see Movies S1 and S2. C, quantification of aberrant mitoses in siRNA-transfected cells. Cells in four fields were counted over the 36-h period of observation for each siRNA. The data shown are from three independent biological replicates. Error bars indicate standard deviation.
Next, HeLa H2B-GFP cells were transfected with siRNAs and imaged every 5 min for 36 h using a Leica SP8 laser-scanning confocal microscope. Depletion of SNRNP200, EFTUD2, PRPF31, and SF3B3 resulted in a stereotyped prometaphase arrest we call “dancing chromosomes,” characterized by an inability of sister chromatids to maintain alignment at the metaphase plate and a failure to successfully initiate anaphase (Movies S1 and S2 and Fig. 8, B and C). This arrest is similar to the mitotic arrest caused by depletion of cohesin subunit sororin. Together, these data demonstrate that cohesin-interacting splicing factors are required for mitotic progression.
Discussion
In this study we used gene editing to introduce dual epitope tags into the endogenous allele of each of 11 known components of cohesin in human cells, and we performed dual-affinity purification and MS to define the endogenous cohesin interactome. We found the following: (i) cohesin interacts with a panoply of splicing factors and RBPs in chromatin; (ii) the interactions between splicing factors and cohesin and between splicing factors and chromatin are cell cycle-regulated, and (iii) cohesin and interacting splicing factors are similarly required for mitotic progression.
The data presented here build on prior studies that identified intriguing intersections between cohesin and splicing factors. In 2005, McCracken et al. (30) reported an evolutionarily conserved, robust physical interaction between cohesin and the SRRM1 splicing co-activator. Then, in 2014, four independent groups simultaneously reported that depletion of splicing factors, including many of those we reported here interact with cohesin, results in attenuation of sister chromatid cohesion that phenocopies depletion of cohesin itself (21–24). They further demonstrated that depletion of splicing factors results in aberrant splicing of the cohesin subunit sororin, resulting in its functional inactivation. However, the fact that these splicing factors interact physically with cohesin provides another possible mechanism for the observation that depletion of splicing factors results in loss of sister chromatid cohesion.
The molecular details of the physical interaction between cohesin and splicing factors/RBPs are of substantial interest to us. In this study, we show that the interactions occur in chromatin and require neither RNA nor DNA. We also demonstrate that RAD21, but no other individual component of cohesin, is required for the interaction with splicing factors and RBPs in HCT116 and HeLa cells. RAD21 is well-known to be an especially important component of cohesin, as it is required for the opening and closing of the cohesin ring around chromatin and is degraded by Separase to drive the metaphase to anaphase transition. Further studies are required to determine how these known functions for RAD21 interface mechanistically with the new function we uncover here, bridging the interaction between cohesin and interacting splicing factors/RBPs.
We are also interested in determining which, if any, individual splicing factors and RBPs are required for the interaction between cohesin and splicing factors/RBPs. However, because our MS data identified >30 such interacting splicing factors/RBPs, and none were overwhelmingly more abundant than others in the affinity purifications, we are at present unable to generate or test hypotheses regarding the identity of splicing factors required for maintenance of the complex.
We are intrigued by the possibility that splicing factors and RBPs are required for the loading and/or proper positioning of cohesin on chromatin. Our initial experiments testing this hypothesis demonstrated that depletion of splicing factors/RBPs does not alter the total amount of cohesin that interacts with chromatin in asynchronous proliferating HeLa cells (data not shown). These results are consistent with the findings of Sundaramoorthy et al. and van der Lelij et al. (21, 22), who demonstrated that splicing factors alter the interaction of cohesin with chromatin only in G2 cells. Further experiments using synchronized cells and more specific readouts such as cohesin ChIP-seq may prove informative.
Upon initially making the discovery reported here, we were surprised to find a large number of published studies that report discrete cell cycle phenotypes of splicing factor inactivation in a wide range of organisms, from Saccharomyces cerevisiae to metazoans (31–36). In most of these studies the authors propose that inactivation of splicing factors results in aberrant splicing of key cell cycle regulatory genes, resulting in cell cycle arrest and/or aberrant progression through the cell cycle. Such a splicing-dependent mechanism is consistent with current models of splicing factor function. However, inactivation of splicing factors is predicted to result in aberrant splicing of thousands of cellular mRNAs, a subset of which would be expected to be essential for viability. Therefore, why inactivation of splicing factors results in discrete and specific cell cycle abnormalities rather than rapid toxicity and cell death is a mystery.
We believe the findings reported here have the potential to provide insight into the mechanisms through which inactivation of splicing factors results in cell cycle arrest. Important next steps will include determining whether the requirement for splicing factors in mitotic progression depends on their interactions with cohesin and/or their splicing activities. However, such studies will require the development of techniques for rapid inactivation of cohesin-interacting splicing factors and the identification of splicing factor/RBP mutants that separate their splicing functions from their ability to interact with cohesin and to regulate mitotic progression.
This study also demonstrates the power of applying gene-editing approaches to the discovery of endogenous interactomes in human cells. To date, the addition of epitope tags to endogenous genes in human cells has been performed on an individual gene basis. This is, to our knowledge, the first application of endogenous tagging to an entire human protein complex or pathway.
In conclusion, here we demonstrate the existence of a robust physical interaction between endogenous cohesin and endogenous splicing factors/RBPs in the chromatin of human cells. Further studies are required to reveal the details of the biomolecular interactions within the complex and its importance in the various critical functions of cohesin and splicing factors in cell biology and human disease.
Experimental procedures
Cell lines
HCT116, HeLa, and RPE-hTERT cells were obtained from the ATCC. HeLa-H2B-GFP cells were obtained from Millipore. Cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C in 5% CO2.
Gene editing
AAV-based gene editing was used to modify an endogenous allele of each of the genes encoding the 11 known subunits of cohesin. The biological and technical principles underlying AAV-based gene editing are described in detail in Refs. 9–11.
Gene-editing vectors were designed as shown in Fig. 1A and Fig. S1. Homology arms were synthesized by Genscript and cloned sequentially into pAAV-SEPT, an AAV-based gene-editing acceptor vector we previously reported in which polylinkers for the cloning of left and right homology arms flank a promoterless splice–acceptor–IRES–neoR gene (10). Next, transient stocks of AAV-2 virions were created by co-transfection of 293T cells with gene-editing vectors together with pAAV-RC (Stratagene) and pHELPER (Stratagene) using X-tremeGENE 9 (Roche Applied Science). Two days after transfection, the medium was aspirated, and cell monolayers were scraped into 1 ml of PBS and subjected to four cycles of freeze/thaw. The lysate was then clarified by centrifugation at 12,000 rpm for 10 min in a benchtop microcentrifuge to remove cell debris, and the virus-containing supernatant was aliquoted and stored at −80 °C.
Virus was then used to infect HCT116 cells in T25 tissue culture flask, and 24 h later, cells were passaged at limiting dilution into 96-well plates in the presence of 1.0 mg/ml G418. After 14 days, individual G418-resistant clones were expanded, and genomic DNA was prepared in a 96-well format using the QIAamp 96 DNA blood kit (Qiagen). Clones were tested for homologous integration of the targeting vector using a primer pair specific for the targeted allele. Efficiencies of gene editing are shown in Table S1. Homologous integration of the dual epitope tag was then confirmed by DNA sequencing. Gene-edited clones were then infected with Cre-expressing adenovirus overnight and plated at limiting dilution into 96-well plates. Single colonies were expanded and tested for G418 sensitivity to identify clones in which the now gene-edited allele had been restored to its otherwise natural configuration. All homology arm and primer sequences are available from the authors upon request.
Protein preparations
Nuclear extracts for dual-affinity purification followed by GeLC-MS/MS were prepared using a modification of Dignam's nondetergent lysis method (37, 38). Whole-cell protein lysates for immunoprecipitation and Western blotting were prepared in either Nonidet P-40 lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Nonidet P-40) or RIPA buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS). Chromatin-associated proteins and total soluble proteins were prepared as described (39). Protein concentrations were determined using the bicinchoninic assay (Pierce).
Dual-affinity purification
Nuclear extracts were prepared from ∼109 cells (∼60 confluent 15-cm dishes) for parental HCT116 cells and each of the 11 cohesin epitope-tagged derivatives. Next, either 30 mg of nuclear extract (HCT116, SMC1A, SMC3, RAD21, STAG1, STAG2, WAPL, and PDS5A) or 100 mg of nuclear extract (HCT116, PDS5B, sororin, NIPBL, and MAU2) was incubated with FLAG M2 beads (Sigma) rotating at 4 °C for 1 h. Beads were then transferred to Poly-Prep® chromatography columns (Bio-Rad) and washed with 30× bed volume Tris-buffered saline (TBS) containing 150 mm NaCl. Proteins were then eluted with 5× bed volume 100 ng/μl 1× FLAG peptide (Sigma). FLAG eluents were then applied to Streptavidin Plus UltraLink resin (Pierce) rotating at 4 °C for 1 h. Beads were then transferred to Poly-Prep® chromatography columns and washed with 30× bed volume TBS containing 150 mm NaCl. Proteins were then eluted with 2× bed volume 1 mm d-biotin. Proteins were then concentrated by TCA precipitation, dissolved in LDS Sample Buffer (Thermo Fisher Scientific), and subjected to GeLC-MS/MS.
GeLC-MS/MS
Dual-affinity purified proteins derived from either 20 mg of nuclear extract (HCT116, SMC1A, SMC3, RAD21, STAG1, STAG2, WAPL, and PDS5A) or 75 mg of nuclear extract (HCT116, PDS5B, sororin, NIPBL, and MAU2) were separated by SDS-PAGE. Gels were fixed in 50% methanol, 7% acetic acid for 1 h at room temperature, washed with water, stained with Coomassie Brilliant Blue (Thermo Fisher Scientific) for 2 h, and destained with water overnight. Each lane was then excised in seven pieces. Gel slices were washed in 50% acetonitrile and rehydrated with a 50 mm ammonia bicarbonate/trypsin solution and digested at 37 °C overnight. Peptides were then extracted with a series of elutions, dried in a speed vac, and solubilized in 0.1% formic acid in water for analysis by tandem MS.
LC-MS/MS was performed on an LTQ Orbitrap Elite (Thermo Fisher Scientific) equipped with Waters (Milford, MA) NanoAcquity HPLC pump. Peptides were separated onto a 100-μm inner diameter microcapillary trapping column packed first with ∼5 cm of C18 Reprosil resin (5 μm, 100 Å, Dr. Maisch GmbH, Germany) followed by an analytical column with ∼20 cm of Reprosil resin (1.8 μm, 200 Å, Dr. Maisch GmbH, Germany). Separation was achieved by applying a gradient from 5 to 27% acetonitrile in 0.1% formic acid over 90 min at 200 nl/min. Electrospray ionization was enabled by applying a voltage of 1.8 kV using a homemade electrode junction at the end of the microcapillary column and sprayed from fused silica pico tips (New Objective, MA). The LTQ Orbitrap Elite was operated in the data-dependent mode for the MS methods. The MS survey scan was performed in the Orbitrap in the range of 395–1800 m/z at a resolution of 6 × 104, followed by the selection of the 20 most intense ions (TOP20) for CID-MS2 fragmentation in the ion trap using a precursor isolation width window of 2 m/z, AGC setting of 10,000, and a maximum ion accumulation of 200 ms. Singly charged ion species were not subjected to CID fragmentation. Normalized collision energy was set to 35 V and an activation time of 10 ms; AGC was set to 50,000, and the maximum ion time was 200 ms. Ions in a 10 ppm m/z window around ions selected for MS2 were excluded from further selection for fragmentation for 60 s.
Raw data were submitted for analysis in Proteome Discoverer 2.1.0.81 (Thermo Fisher Scientific) software. Assignment of MS/MS spectra was performed using the Sequest HT algorithm by searching the data against a protein sequence database, including all entries from the Human UniProt database (SwissProt 16,768, 2016) and other known contaminants such as human keratins and common lab contaminants. SEQUEST HT searches were performed using a 20-ppm precursor ion tolerance and requiring each peptide's N/C terminus to adhere to trypsin protease specificity while allowing up to two missed cleavages. Cysteine carbamidomethyl (+57.021) was set as static modifications, whereas methionine oxidation (+15.99492 Da) was set as variable modification. MS2 spectra assignment FDR of 1% on protein level was achieved by applying the target-decoy database search. Filtering was performed using Percolator (64-bit version). For quantification, a 0.02 m/z window centered on the theoretical m/z value of each of the six reporter ions and the intensity of the signal closest to the theoretical m/z value was recorded. Reporter ion intensities were exported in the result file of Proteome Discoverer 2.1 search engine as Excel tables.
STRING
Proteins identified by MS and listed in the 11 tabs of Table S2 were analyzed using the STRING database (http://string-db.org; version 10.0)3 (17) to construct a protein–protein interaction network. The network (Fig. 1C) was constructed using the highest confidence level setting, and unconnected nodes were removed.
Immunoprecipitation
For SMC1A immunoprecipitation, protein lysates were incubated with 3 μg of SMC1A antibody (clone H-6, Santa Cruz Biotechnology sc-393171) or IgG control antibody in TBS containing 0.1% Nonidet P-40 for 1 h rotating at 4 °C. Protein A/G-agarose beads (Santa Cruz Biotechnology) were then added, and the lysates were further incubated for 1 h rotating at 4 °C. Beads were then washed three times with TBS (either 150 or 350 mm NaCl) containing 0.1% Nonidet P-40. Proteins were then eluted into 2× LDS sample buffer at room temperature for 20 min. DTT was then added to a final concentration of 50 mm. Samples were boiled for 5 min and separated by SDS-PAGE.
For FLAG immunoprecipitation, FLAG M2 beads (Sigma) were washed once with TBS (150 mm NaCl) and then incubated with protein lysates for 1 h rotating at 4 °C. Beads were then washed three times in TBS, and bound proteins were eluted with 100 ng/μl 1× FLAG peptide (Sigma). Eluents were then concentrated by TCA precipitation, resuspended in sample buffer, and separated by SDS-PAGE.
Silver staining
Silver staining was performed with the SilverQuest staining kit (Thermo Fisher Scientific), following the manufacturer's instructions.
Western blotting
Protein lysates, affinity purifications, and immunoprecipitations were dissolved in LDS sample buffer, boiled for 5 min, and separated by SDS-PAGE. Proteins were transferred to polyvinylidene difluoride membranes, which were then probed with a 1:1000 dilution of primary antibodies overnight rotating at 4 °C. After incubation with horseradish peroxidase–conjugated secondary antibodies (Cell Signaling) rotating for 1 h at room temperature, membranes were developed with SuperSignal West Pico PLUS chemiluminescent substrate (Pierce) and imaged using a myECL imager (Pierce).
Immunofluorescence
HeLa cells were cultured on coverslips, fixed in 4% paraformaldehyde for 15 min, and then permeabilized with 0.5% Triton X-100 in PBS at room temperature for 15 min. Fixed and permeabilized cells were then blocked in 5% BSA and 0.25% Triton X-100 in PBS for 1 h at room temperature and then incubated with primary antibodies (1:200) in wash buffer (1% BSA and 0.25% Triton X-100 in PBS) overnight at 4 °C. Cells were then washed three times with wash buffer, then incubated with fluorescent secondary antibodies (Alexa Fluor 488 and Alexa Fluor 647, Thermo Fisher Scientific) in antibody dilution buffer for 1 h at room temperature, and then washed three times with wash buffer. The slides were mounted with antifade mountant with 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific) and imaged with a Leica TCS SP8 confocal laser-scanning microscope fitted with ×63/1.40 plan apochromat oil immersion objective. Images were processed and analyzed using ImageJ software (40).
Antibodies
Primary antibodies for immunoblotting were FLAG (M2) and tubulin (DM1α) from Sigma; sororin (ab192237) from Abcam; STAG2 (sc-81852), WAPL (sc-365189), HSPA8 (sc-7298), MAU2 (sc-243420), and NIPBL (sc-374625) from Santa Cruz Biotechnology; cyclin B1 (12231), cyclin E1 (20808), glyceraldehyde-3-phosphate dehydrogenase (2118), histone H3 (14269), and phospho-H3 (53348) from Cell Signaling Technologies; and ADAR1 (A303-884), DDX47 (A302-977), EFTUD2 (A300-957), HNRNPH (A300-511), PDCD11 (A303-804), PDS5A (A300-088), PDS5B (A300-538), PRPF31 (A303-919), RAD21 (A300-080), RBM10 (A301-006), RBM15 (A300-821), SF3B1 (A300-997), SF3B3 (A302-508), SMC1A (A300-055), SMC3 (A300-060), SNRNP200 (A303-453), and STAG1 (A302-579) from Bethyl Laboratories.
Primary antibodies for immunofluorescence were STAG2 (sc-81852) and SMC1A (sc-393171) from Santa Cruz Biotechnology, and HNRNPH (A300-511), EFTUD2 (A300-957), and SF3B1 (A300-997) from Bethyl Laboratories.
Double thymidine block for cell synchronization
HeLa cells (2 × 106) were plated in 10-cm2 dishes. After 24 h, cells were treated with 2 mm thymidine (Sigma) for 24 h to arrest cells in S phase. Cells were then released by washing in Hanks' buffered saline solution (HBSS) followed by an 8-h incubation in regular media. Cells were then treated again with 2 mm thymidine (Sigma) for 16 h to generate a synchronized population of cells arrested in S phase. Cells were then washed in HBSS, incubated in regular media to release them from the cell cycle arrest, and harvested at different time points for flow cytometry and protein preparation.
Drug treatments
Nocodazole (Sigma) was used at a final concentration of 100 ng/ml. RO-3306 (Sigma) was used at a final concentration of 10 μm. Hydroxyurea (Sigma) was used at a final concentration of 0.5 mm. BI 2536 (Selleckchem) was used at final concentrations of 50 and 100 nm.
Flow cytometry
Cells were fixed in 70% ethanol and stained in phosphate-buffered saline (PBS) containing 0.1% Triton X-100, 50 μg/ml RNase, and 50 μg/ml propidium iodide. DNA content was measured on a FACSort flow cytometer (BD Biosciences), and data were analyzed using ModFit software (Verity Software House). At least 20,000 cells were analyzed for each sample.
CRISPR
A guide RNA targeting the first coding exon of PDS5A (GTGGCGTCGTGAGTGCCGAC) was designed using the Feng Zhang lab's CRISPR guide RNA design tool (crispr.mit.edu) and cloned into lentiCRISPR version 2 (a gift from Feng Zhang, Addgene no. 52961). Lentivirus was packaged using pMD2.G (Addgene no. 12259) and psPAX2 (Addgene no. 12260) and was used to infect HCT116-SMC1A–tagged cells. Individual puromycinR colonies were obtained by limiting dilution and tested by Western blotting to identify clones in which expression of PDS5A had been lost. DNA sequencing was performed to confirm the presence of homozygous truncating mutations in the first exon of PDS5A, and the cells used for the experiments are shown in Fig. 5A.
siRNA
Cells were transfected with Silencer Select siRNAs (Ambion) using RNAiMax (Thermo Fisher Scientific) according to the manufacturer's instructions. Catalogue numbers for individual siRNAs and their depletion efficiencies are shown in Figs. 5D and 8A.
Live-cell imaging
Histone H2B-GFP–expressing HeLa cells (Millipore) were plated on μ-Slide 8-well glass bottom plate (Ibidi), transfected with siRNAs, and cultured for 36 h. The chamber slide was placed in a Leica TCS SP8 confocal laser-scanning microscope fitted with environmental chamber and stage-top CO2/humidity incubator held at 37 °C and 5% CO2 (Life Imaging Services). For each siRNA treatment, four positions were recorded every 5 min over a 36-h imaging session. Data acquisition was performed with Leica LAS X control software.
Author contributions
J.-S. K., X. H., J. L., Z. D., T. K., J. G., B. K., M. M. P., W. S. L., W. S. N., B. B., and T. W. investigation; T. W. and W. S. N. funding acquisition.
Supplementary Material
Acknowledgments
Mass spectrometry and bioinformatics analysis was conducted at the Mass Spectrometry and Proteomics Resource Laboratory, FAS Division of Science, Harvard University. The Lombardi Comprehensive Cancer Center was supported by National Institutes of Health Grant P30CA51008. We thank Peter Johnson and Shannon White for assistance with live-cell imaging
This work was supported by National Institutes of Health Grant R01CA169345 (to T. W.), Alex's Lemonade Stand (to T. W.), the Hyundai Hope on Wheels Foundation (to T. W.), and National Institutes of Health Common Fund 4D Nucleome Project Grant U54DK107979 (to W. S. N.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4, Tables S1–S6, and supporting Movies S1 and S2.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- RBP
- RNA-binding protein
- snRNP
- small nuclear ribonucleoprotein
- SBP
- streptavidin-binding peptide
- HBSS
- Hanks' buffered saline solution
- FDR
- false-discovery rate
- AAV
- adeno-associated virus.
References
- 1. Michaelis C., Ciosk R., and Nasmyth K. (1997) Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91, 35–45 10.1016/S0092-8674(01)80007-6 [DOI] [PubMed] [Google Scholar]
- 2. Guacci V., Koshland D., and Strunnikov A. (1997) A direct link between sister chromatid cohesin and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 91, 47–57 10.1016/S0092-8674(01)80008-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Losada A., Hirano M., and Hirano T. (1998) Identification of Xenopus SMC protein complexes required for sister chromatid cohesin. Genes Dev. 12, 1986–1997 10.1101/gad.12.13.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haarhuis J. H., Elbatsh A. M., and Rowland B. D. (2014) Cohesin and its regulation: on the logic of X-shaped chromosomes. Dev. Cell 31, 7–18 10.1016/j.devcel.2014.09.010 [DOI] [PubMed] [Google Scholar]
- 5. Uhlmann F. (2016) SMC complexes: from DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 17, 399–412 10.1038/nrm.2016.30 [DOI] [PubMed] [Google Scholar]
- 6. Solomon D. A., Kim T., Diaz-Martinez L. A., Fair J., Elkahloun A. G., Harris B. T., Toretsky J. A., Rosenberg S. A., Shukla N., Ladanyi M., Samuels Y., James C. D., Yu H., Kim J. S., and Waldman T. (2011) Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333, 1039–1043 10.1126/science.1203619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hill V. K., Kim J. S., and Waldman T. (2016) Cohesin mutations in human cancer. Biochim. Biophys. Acta 1866, 1–11 10.1016/j.bbcan.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zakari M., Yuen K., and Gerton J. L. (2015) Etiology and pathogenesis of the cohesinopathies. Wiley Interdiscip. Rev. Dev. Biol. 4, 489–504 10.1002/wdev.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khan I. F., Hirata R. K., and Russell D. W. (2011) AAV-mediated gene targeting methods for human cells. Nat. Protoc. 6, 482–501 10.1038/nprot.2011.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim J. S., Bonifant C., Bunz F., Lane W. S., and Waldman T. (2008) Epitope tagging of endogenous genes in diverse human cell lines. Nucleic Acids Res. 36, e127 10.1093/nar/gkn566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gaj T., Epstein B. E., and Schaffer D. V. (2016) Genome engineering using adeno-associated virus: basic and clinical research applications. Mol. Ther. 24, 458–464 10.1038/mt.2015.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krogan N. J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A. P., Punna T., Peregrín-Alvarez J. M., Shales M., Zhang X., Davey M., et al. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643 10.1038/nature04670 [DOI] [PubMed] [Google Scholar]
- 13. Losada A., Yokochi T., Kobayashi R., and Hirano T. (2000) Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J. Cell Biol. 150, 405–416 10.1083/jcb.150.3.405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paulo J. A. (2016) Sample preparation for proteomic analysis using a GeLC-MS/MS strategy. J. Biol. Methods 3, e45 10.14440/jbm.2016.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zheng G., Kanchwala M., Xing C., and Yu H. (2018) MCM2–7-dependent cohesin loading during S phase promotes sister-chromatid cohesion. Elife 7, e33920 10.7554/eLife.33920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kagey M. H., Newman J. J., Bilodeau S., Zhan Y., Orlando D. A., van Berkum N. L., Ebmeier C. C., Goossens J., Rahl P. B., Levine S. S., Taatjes D. J., Dekker J., and Young R. A. (2010) Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435 10.1038/nature09380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N. T., Roth A., Bork P., Jensen L. J., and von Mering C. (2017) The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 10.1093/nar/gkw937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anczuków O., and Krainer A. R. (2016) Splicing-factor alterations in cancers. RNA 22, 1285–1301 10.1261/rna.057919.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Agafonov D. E., Kastner B., Dybkov O., Hofele R. V., Liu W. T., Urlaub H., Lührmann R., and Stark H. (2016) Molecular architecture of the human U4/U6.U5 tri-snRNP. Science 351, 1416–1420 10.1126/science.aad2085 [DOI] [PubMed] [Google Scholar]
- 20. Daiger S. P., Sullivan L. S., and Bowne S. J. (2013) Genes and mutations causing retinitis pigmentosa. Clin. Genet. 84, 132–141 10.1111/cge.12203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sundaramoorthy S., Vázquez-Novelle M. D., Lekomtsev S., Howell M., and Petronczki M. (2014) Functional genomics identifies a requirement of pre-mRNA splicing factors for sister chromatid cohesin. EMBO J. 33, 2623–2642 10.15252/embj.201488244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van der Lelij P., Stocsits R. R., Ladurner R., Petzold G., Kreidl E., Koch B., Schmitz J., Neumann B., Ellenberg J., and Peters J. M. (2014) SNW1 enables sister chromatid cohesion by mediating the splicing of sororin and APC2 pre-mRNAs. EMBO J. 33, 2643–2658 10.15252/embj.201488202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Watrin E., Demidova M., Watrin T., Hu Z., and Prigent C. (2014) Sororin pre-mRNA splicing is required for proper sister chromatid cohesin in human cells. EMBO Rep. 15, 948–955 10.15252/embr.201438640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oka Y., Varmark H., Vitting-Seerup K., Beli P., Waage J., Hakobyan A., Mistrik M., Choudhary C., Rohde M., Bekker-Jensen S., and Mailand N. (2014) UBL5 is essential for pre-mRNA splicing and sister chromatid cohesion in human cells. EMBO Rep. 15, 956–964 10.15252/embr.201438679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vassilev L. T., Tovar C., Chen S., Knezevic D., Zhao X., Sun H., Heimbrook D. C., and Chen L. (2006) Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. U.S.A. 103, 10660–10665 10.1073/pnas.0600447103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim J. S., He X., Orr B., Wutz G., Hill V., Peters J. M., Compton D. A., and Waldman T. (2016) Intact cohesion, anaphase, and chromosome segregation in human cells harboring tumor-derived mutations in STAG2. PLoS Genet. 12, e1005865 10.1371/journal.pgen.1005865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peters J. M., and Nishiyama T. (2012) Sister chromatid cohesion. Cold Spring Harb. Perspect. Biol. 4, a011130 10.1101/cshperspect.a011130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sumara I., Vorlaufer E., Gieffers C., Peters B. H., and Peters J. M. (2000) Characterization of vertebrate cohesin complexes and their regulation in prophase. J. Cell Biol. 151, 749–762 10.1083/jcb.151.4.749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sumara I., Vorlaufer E., Stukenberg P. T., Kelm O., Redemann N., Nigg E. A., and Peters J. M. (2002) The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol. Cell 9, 515–525 10.1016/S1097-2765(02)00473-2 [DOI] [PubMed] [Google Scholar]
- 30. McCracken S., Longman D., Marcon E., Moens P., Downey M., Nickerson J. A., Jessberger R., Wilde A., Caceres J. F., Emili A., and Blencowe B. J. (2005) Proteomic analysis of SRm160-containing complexes reveals a conserved association with cohesin. J. Biol. Chem. 280, 42227–42236 10.1074/jbc.M507410200 [DOI] [PubMed] [Google Scholar]
- 31. Kittler R., Pelletier L., Heninger A. K., Slabicki M., Theis M., Miroslaw L., Poser I., Lawo S., Grabner H., Kozak K., Wagner J., Surendranath V., Richter C., Bowen W., Jackson A. L., et al. (2007) Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat. Cell Biol. 9, 1401–1412 10.1038/ncb1659 [DOI] [PubMed] [Google Scholar]
- 32. Neumann B., Walter T., Hériché J. K., Bulkescher J., Erfle H., Conrad C., Rogers P., Poser I., Held M., Liebel U., Cetin C., Sieckmann F., Pau G., Kabbe R., Wünsche A., et al. (2010) Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464, 721–727 10.1038/nature08869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huen M. S., Sy S. M., Leung K. M., Ching Y. P., Tipoe G. L., Man C., Dong S., and Chen J. (2010) SON is a spliceosome-associated factor required for mitotic progression. Cell Cycle 9, 2679–2685 10.4161/cc.9.13.12151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hofmann J. C., Tegha-Dunghu J., Dräger S., Will C. L., Lührmann R., and Gruss O. J. (2013) The Prp19 complex directly functions in mitotic spindle assembly. PLoS ONE 8, e74851 10.1371/journal.pone.0074851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hofmann J. C., Husedzinovic A., and Gruss O. J. (2010) The function of spliceosome components in open mitosis. Nucleus 1, 447–459 10.4161/nucl.1.6.13328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karamysheva Z., Díaz-Martínez L. A., Warrington R., and Yu H. (2015) Graded requirement for the spliceosome in cell cycle progression. Cell Cycle 14, 1873–1883 10.1080/15384101.2015.1039209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dignam J. D., Lebovitz R. M., and Roeder R. G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 10.1093/nar/11.5.1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jung S. Y., Malovannaya A., Wei J., O'Malley B. W., and Qin J. (2005) Proteomic analysis of steady-state nuclear hormone receptor co-activator complexes. Mol. Endocrinol. 19, 2451–2465 10.1210/me.2004-0476 [DOI] [PubMed] [Google Scholar]
- 39. Busslinger G. A., Stocsits R. R., van der Lelij P., Axelsson E., Tedeschi A., Galjart N., and Peters J. M. (2017) Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 544, 503–507 10.1038/nature22063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.