

Abstract
NOD1 and NOD2 are intracellular sensors of bacterial peptidoglycan that belong to the Nod-like receptor family of innate immune proteins. In addition to their role as direct bacterial sensors, it was proposed that the nucleotide-binding oligomerization domain (NOD) proteins could detect endoplasmic reticulum (ER) stress induced by thapsigargin, an inhibitor of the sarcoplasmic or endoplasmic reticulum calcium ATPase family that pumps Ca2+ into the ER, resulting in pro-inflammatory signaling. Here, we confirm that thapsigargin induces NOD-dependent pro-inflammatory signaling in epithelial cells. However, the effect was specific to thapsigargin, as tunicamycin and the subtilase cytotoxin SubAB from Shiga toxigenic Escherichia coli, which induce ER stress by other mechanisms, did not induce cytokine expression. The calcium ionophore A23187 also induced NOD-dependent signaling, and calcium chelators demonstrated a role for both intracellular and extracellular calcium in mediating thapsigargin-induced and NOD-dependent pro-inflammatory signaling, in part through the activation of plasma membrane-associated calcium release-activated channels. Moreover, our results demonstrate that both endocytosis and the addition of serum to the cell culture medium were required for thapsigargin-mediated NOD activation. Finally, we analyzed cell culture grade fetal calf serum as well as serum from laboratory mice using HPLC and MS identified the presence of various peptidoglycan fragments. We propose that cellular perturbations that affect intracellular Ca2+ can trigger internalization of peptidoglycan trace contaminants found in culture serum, thereby stimulating pro-inflammatory signaling. The presence of peptidoglycan in animal serum suggests that a homeostatic function of NOD signaling may have been previously overlooked.
Keywords: Nod-like receptor (NLR), innate immunity, cell signaling, cell biology, calcium intracellular release, endoplasmic reticulum stress (ER stress), NOD1, NOD2
Introduction
Detection of microbes by the innate immune system relies on several families of pattern recognition molecules (PRMs)3 that recognize conserved microbe-associated molecular patterns (MAMPs) that are highly conserved and are not produced by the noninfected host. Among those families of PRMs are the Toll-like receptors (TLRs) and Nod-like receptors (NLRs). In addition to the detection of MAMPs, certain NLR proteins, such as NLRP3, detect cellular perturbations or molecules, known as danger-associated molecular patterns that can arise as a result of an infection or following aseptic tissue damage (1).
NOD1 and NOD2 are two members of the NLR family of PRM that detect bacterial peptidoglycan (2). The specificity of NOD1 and NOD2 for peptidoglycan motifs is extremely high, as NOD1 detects meso-diaminopimelic acid-containing N-acetylmuramic acid (MurNAc)-tripeptide (Mur-TriDAP) found predominantly in Gram-negative bacteria (3–6), and NOD2 detects muramyl dipeptide (MDP) found in both Gram-negative and Gram-positive bacteria (3, 7, 8). Detailed studies on the minimal structural requirements of the peptidoglycan ligands needed for NOD1 or NOD2 activation revealed that the MurNAc moiety is not required for NOD1 activation as the d-Glu-meso-DAP dipeptide (iE-DAP) is sufficient for the detection and innate immune activation by this PRM (3, 9). In contrast, NOD2 can only be activated by muramyl dipeptides that have an intact MurNAc ring structure, and the sugar has to be attached to a dipeptide moiety (l-Ala-d-Glu or l-Ala-d-isoGln) (3, 10). Importantly, studies have shown that both NOD1 and NOD2 can directly bind to TriDAP and MDP, respectively, thus showing that NOD1 and NOD2 are bona fide cytoplasmic receptors (11–13), and that this interaction requires the leucine-rich repeat region of NOD1 and NOD2 proteins (14, 15).
Although functional binding studies demonstrate that NOD proteins have an extreme specificity for certain peptidoglycan fragments that is conserved in multiple vertebrates, recent studies have suggested that NOD signaling could also be triggered by viral infection (16), small Rho GTPases regulating cytoskeleton remodeling (17), and endoplasmic reticulum (ER) stress (18), implying that peptidoglycan-independent mechanisms of NOD stimulation may exist. This suggests that in addition to their high specificity toward peptidoglycan fragments, NOD1 and NOD2 may serve as promiscuous sensors of multiple and unrelated cellular stresses. Here, we aimed to better characterize how NOD proteins trigger pro-inflammatory signaling in response to ER stress. Although we confirm that thapsigargin, a specific inhibitor of the ER sarcoplasmic or endoplasmic reticulum calcium ATPase family (SERCA) calcium pump, triggers pro-inflammatory signaling in a NOD-dependent manner (18), our results suggest that this effect is actually mediated by the Ca2+-dependent internalization of peptidoglycan trace fragments found in the fetal calf serum added to cell culture media. Of note, low levels of peptidoglycan in human serum have already been reported and were shown to stimulate hyphal growth of Candida albicans (19). Together, our observations suggest that cellular perturbations that lead to increased intracellular Ca2+ levels may inadvertently trigger NOD-dependent signaling through the internalization of peptidoglycan contaminants, which offers an alternative explanation for the proposed promiscuous activation of NOD receptors by multiple unrelated stresses. These observations also suggest that chronic homeostatic peptidoglycan sensing by NOD proteins may impact multiple cellular processes in ways that have been overlooked previously and open up interesting questions in innate immunity, relating to understanding the physiological role of circulating peptidoglycan at homeostasis, both at the cellular and the tissue level.
Results and discussion
The human intestinal epithelial cell line HCT116, either wildtype (WT) or knockout through CRISPR-Cas9 for NOD1, NOD2, or both NOD1 and NOD2 (NOD1/2 double knockout or DKO) described previously (20), were stimulated with the ER stress inducer thapsigargin, which inhibits the SERCA pump that transports Ca2+ to the ER lumen. Although thapsigargin induced ER stress similarly in the four cell lines tested, as determined by the transcriptional up-regulation of the HSPA5 gene (that encodes heat-shock protein BiP/GRP78) (Fig. 1A), transcriptional up-regulation of the pro-inflammatory cytokines CXCL1 (Fig. 1A), IL8 (Fig. S1A), as well as the chemokine CCL20 (Fig. S1A) was significantly blunted in NOD2 KO and NOD1/2 DKO HCT116 cells, in line with previous results (18). In NOD1 KO cells, a nonsignificant trend for reduced expression of CXCL1 and IL8 was also noticed following thapsigargin stimulation, and significant reduction of CCL20 expression was observed (Fig. 1A and Fig. S1A). Together, these results suggest that, in HCT116 cells, NOD2 and to a lesser extent NOD1 contribute to the up-regulation of pro-inflammatory signaling induced by thapsigargin. On average, NOD1/2 DKO cells displayed an 8.35-fold reduction in CXCL1 induction following thapsigargin stimulation, a 6.33-fold reduction in IL8 expression, and a 2.85-fold reduction in CCL20 expression.
Figure 1.
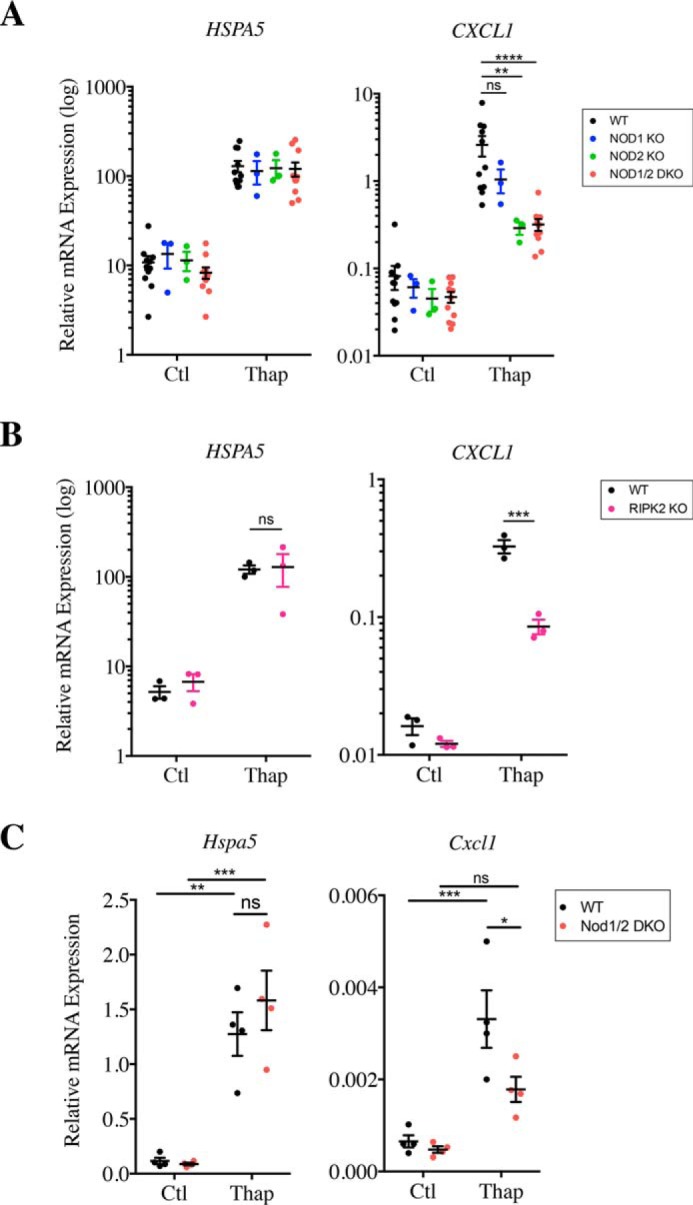
Thapsigargin induces pro-inflammatory cytokine expression in a NOD-dependent manner. A and B, expression of HSPA5 and CXCL1 in WT, NOD1 and NOD2 KO, and NOD1/2 DKO HCT116 cells (A) or WT and RIPK2 KO HCT116 cells (B) following stimulation with 0.1 μg/ml thapsigargin (Thap) for 4 h as measured by qRT-PCR C, expression of Hspa5 and Cxcl1 in primary intestinal organoids from WT and NOD1/2 DKO mice following stimulation with 1 μg/ml thapsigargin for 4 h. Gene expression analysis was carried out by qRT-PCR. In all data sets, each point is from an independent experiment and is the average of three technical replicates. *, **, ***, and ****, represents p < 0.05, p < 0.01, p < 0.001; and p < 0.0001, respectively. ns, not significant; Ctl, control.
To demonstrate that this effect requires the NOD adaptor protein RIP2, we engineered HCT116 RIPK2 KO cells by targeting the RIPK2 gene (encoding RIP2) using CRISPR-Cas9. As expected, RIPK2-targeted cells were insensitive to the synthetic NOD ligands MDP and iE-DAP in an NF-κB luciferase assay (Fig. S2A). Similar to NOD1/2 DKO cells, RIPK2 KO HCT116 cells displayed normal induction of ER stress-dependent HSPA5 (Fig. 1B) but significantly reduced up-regulation of CXCL1 (Fig. 1B), IL8, and CCL20 (Fig. S1B). On average, RIPK2 KO cells displayed a 3.82-fold reduction in CXCL1 induction following thapsigargin stimulation, a 13.48-fold reduction in IL8 expression, and a 3.04-fold reduction in CCL20 expression. To further validate our findings, we reproduced these results with other clones of NOD1/2 DKO and RIPK2 KO cells (Fig. S2, B and C). Finally, we isolated primary intestinal organoids from WT and Nod1/2 DKO mice and stimulated those with thapsigargin. In previous work, we identified using RNAseq that thapsigargin potently stimulated cytokine expression in murine organoids (21). Similar to our results with the human intestinal cell line HCT116, we observed that although thapsigargin triggered comparable expression of Hspa5 (encoding BiP) in WT and Nod1/2 DKO organoids, Cxcl1 expression was significantly blunted in Nod1/2 DKO organoids (Fig. 1C), although the effect was not as drastic as in HCT116 cells, suggesting that NOD-dependent signaling only represents a subset of thapsigargin-dependent pro-inflammatory cascades in murine organoids. Together, these results confirm the previous findings (18) that NOD1 and NOD2 are critical for inflammatory signaling induced by thapsigargin.
We next aimed to investigate whether other ER stress inducers trigger pro-inflammatory signaling in a NOD-dependent manner in HCT116 cells. We first used tunicamycin, a molecule that induces ER stress by preventing protein glycosylation in the ER lumen, thereby provoking the accumulation of misfolded proteins in the ER. This mechanism of ER stress induction is distinct from the one triggered by thapsigargin, which relies on the inhibition of Ca2+ accumulation in the ER lumen. Interestingly, although tunicamycin induced potent up-regulation of HSPA5 expression in WT, NOD1 KO, NOD2 KO, and NOD1/2 DKO HCT116 cells (Fig. 2A), it did not stimulate expression of CXCL1 or IL8 (Fig. 2A), suggesting that induction of pro-inflammatory signaling by thapsigargin is likely caused by the effect of the inhibitor on intracellular Ca2+ levels rather than caused by ER stress per se. To confirm these findings, we induced ER stress by a third mechanism, namely the targeting and degradation of the BiP protein by the SubAB toxin of Shiga toxigenic Escherichia coli, which causes ER stress as a consequence of reduced levels of luminal BiP chaperone (22, 23). SubAB strongly induced up-regulation of HSPA5 expression in WT, NOD1 KO, NOD2 KO, and NOD1/2 DKO HCT116 cells, whereas a mutant toxin unable to cleave BiP was unable to do so (Fig. 2B). However, SubAB did not trigger expression of CXCL1 or IL8, similar to tunicamycin (Fig. 2B). These results further demonstrated that at least in HCT116 cells, ER stress does not induce NF-κB–dependent pro-inflammatory cytokines such as CXCL1 and IL8 and that thapsigargin action on pro-inflammatory signaling is likely caused by its capacity to raise intracellular Ca2+ concentration.
Figure 2.

ER stress inducers that do not directly target Ca2+ stores do not trigger NOD-dependent pro-inflammatory cytokine expression. A and B, expression of HSPA5, CXCL1, and IL8 in WT, NOD1 and NOD2 KO, and NOD1/2 DKO HCT116 cells following stimulation with 1 μg/ml tunicamycin (Tunic) (A) or 50 ng/ml SubAB toxin from Shiga toxigenic E. coli, either WT (sAB) or mutated on position 272 (sA272B), which abolishes its activity, for 4 h (B) and measured by qRT-PCR. In all data sets, each point is from an independent experiment and is the average of three technical replicates. *, **, and ****, represents p < 0.05, p < 0.01, and p < 0.0001, respectively. ns, not significant; Ctl, control.
To validate the plausible role of intracellular Ca2+ levels in NOD-dependent pro-inflammatory signaling, WT and NOD1/2 DKO cells were stimulated with A23187, a Ca2+ ionophore that causes massive influx of Ca2+ into the cytosol. As expected, stimulation with A23187 induced up-regulation of HSPA5 in both WT and NOD1/2 DKO cells (Fig. 3A). Similarly to thapsigargin, A23187 also stimulated expression of CXCL1 and IL8, which was significantly blunted in NOD1/2 DKO cells (Fig. 3A), thus showing that the rise in intracellular Ca2+ levels caused NOD-dependent pro-inflammatory signaling. Additionally, using BAPTA-AM, a cell-permeant Ca2+ chelator, we directly tested the role of intracellular Ca2+ levels in thapsigargin-stimulated cells. Although BAPTA-AM did not significantly affect ER stress induced by thapsigargin (Fig. 3B), in line with the fact that BAPTA-AM does not prevent thapsigargin-dependent depletion of luminal ER stores of Ca2+ but neutralizes the effects of cytosolic Ca2+, it blunted the induction of CXCL1 and IL8 expression in WT cells but not in NOD1/2 DKO cells. As a result, thapsigargin-induced expression of pro-inflammatory cytokines was not significantly dependent on NOD1/2 in BAPTA-AM–treated cells, although a trend for decreased expression was still observed in NOD1/2 DKO cells (Fig. 3B), possibly because BAPTA-AM was unable to fully neutralize the effects of intracellular Ca2+. This suggests that the rise of intracellular Ca2+ levels caused NOD-dependent pro-inflammatory signaling in HCT116 cells.
Figure 3.

Increase in intracellular Ca2+ levels induces NOD-dependent pro-inflammatory signaling. A, expression of HSPA5, CXCL1, and IL8 in WT and NOD1/2 DKO HCT116 cells following stimulation with 1 μg/ml A23187. B and C, expression of HSPA5, CXCL1, and IL8 in WT and NOD1/2 DKO HCT116 cells following treatment with 0.1 μg/ml thapsigargin (Thap) either in the presence or absence of 15 μm BAPTA-AM, for 4 h (B) or in the presence or absence of 200 μm DMNP–EDTA for 1 h (C). In all panels, gene expressions were measured by qRT-PCR, and each point in all data sets is from an independent experiment and is the average of two or three technical replicates. *, **, ***, and ****, represents p < 0.05, p < 0.01, p < 0.001; and p < 0.0001, respectively. ns, not significant; Ctl, control.
A depletion in Ca2+ stores in the ER, associated with a rise in intracellular Ca2+ levels, acts as a signal for the activation of a family of plasma membrane calcium release-activated channels (CRACs), which triggers internalization of extracellular Ca2+ as a means to regenerate ER Ca2+ stores (24). Therefore, to determine whether extracellular Ca2+ was mobilized during thapsigargin-induced NOD-dependent cytokine expression, WT and NOD1/2 DKO HCT116 were stimulated with thapsigargin in the presence of DMNP–EDTA, a cell-impermeant Ca2+ chelator. Similar to the results obtained with BAPTA-AM, DMNP–EDTA did not impact HSPA5 expression induced by thapsigargin, but it significantly blunted CXCL1 (by 2.56-fold) and IL8 expression (by 2.16-fold) in WT cells without significantly affecting expression of these genes in NOD1/2 DKO cells (Fig. 3C). However, despite the effect of DMNP–EDTA, thapsigargin-induced expression of CXCL1 and IL8 remained significantly greater in WT as compared with NOD1/2 DKO cells (Fig. 3C). Finally, to explore the possible contribution of CRACs to the effects observed, the specific inhibitor GSK-7975A was used, which showed results similar to those obtained with DMNP–EDTA (Fig. S3). Taken together, we conclude that internalization of extracellular Ca2+ through CRAC channels contributes yet does not account for the whole NOD-dependent cytokine response to thapsigargin stimulation.
We next aimed to delineate the mechanism by which an increase in intracellular Ca2+ levels causes NOD-dependent pro-inflammatory signaling. We reasoned that because Ca2+ regulates vesicular trafficking, exocytosis, and recycling of lysosomes (25, 26), it could thus in turn impact the dynamic rate at which cells perform endocytosis. In addition, Ca2+ internalization through CRACs also contributes to endocytosis (27, 28). This led us to speculate that the Ca2+-dependent signal that triggers NOD-dependent activation in thapsigargin-stimulated cells could be coming from a factor within the extracellular milieu and be brought in by endocytosis. To test this hypothesis, WT and NOD1/2 DKO cells were grown in synthetic Hanks' balanced salt solution (HBSS) medium supplemented with Ca2+, in the presence or absence of 10% fetal calf serum (FCS), and were then stimulated with thapsigargin. Interestingly, NOD-dependent induction of IL8 following thapsigargin stimulation was only observed in the presence of serum, as thapsigargin was unable to trigger IL8 expression when cells were passaged in a medium containing only HBSS with Ca2+ prior to thapsigargin stimulation (Fig. 4A). This effect was Ca2+-dependent, because it was lost when serum was added to cells grown in HBSS without Ca2+ (Fig. S4A), again arguing for the role played by extracellular Ca2+ in these responses. Importantly, the absence of serum did not make the cells refractory to NOD-dependent cytokine expression nonspecifically, because addition of the NOD2 ligand L18-MDP to cells incubated in HBSS with Ca2+ could still induce expression of IL8 and CXCL1 (Fig. S4B). Finally, we boiled the FCS and filtered it to remove molecules with a molecular mass of >3 kDa. This boiled/filtered FCS was still able to potentiate NOD-dependent stimulation of IL8 following stimulation (Fig. S3C). This suggests that a heat-resistant small molecule present in FCS drives Ca2+-dependent stimulation of NOD1/2 proteins by thapsigargin.
Figure 4.

Endocytosis of small molecules from cell culture serum, which contains peptidoglycan fragments, triggers NOD-dependent activation of pro-inflammatory signaling induced by thapsigargin. A, expression of HSPA5 and IL8 in WT and NOD1/2 DKO HCT116 cells incubated overnight in HBSS medium supplemented with 140 μg/ml CaCl2 in the presence or absence of 10% FCS following stimulation with 0.1 μg/ml thapsigargin (Thap) for 4 h. B, expression of HSPA5 and IL8 in WT and NOD1/2 DKO HCT116 cells following stimulation with 0.1 μg/ml thapsigargin, in the presence or absence of 80 mm Dynasore, for 4 h. Gene expression analysis was carried out by qRT-PCR. C, LC-MS peak profile of three peptidoglycan fragments known to activate NOD1 and NOD2 (GM-TriDAP, GMDP, and TriDAP) identified in FCS. In all data sets, each point is from an independent experiment and is the average of three technical replicates. *, **, ***, and ****, represents p < 0.05, p < 0.01, p < 0.001; and p < 0.0001, respectively. ns, not significant; Ctl, control.
Because internalization of extracellular peptidoglycan into epithelial cells and subsequent activation of NOD1 and NOD2 occurs by clathrin- and dynamin-dependent endocytosis (29, 30), we next aimed to define whether the active molecule(s) in FCS needed to be internalized by endocytosis to drive thapsigargin-mediated stimulation of NOD proteins. To do so, WT and NOD1/2 DKO HCT116 cells were grown in a normal medium supplemented with 10% FCS and stimulated with thapsigargin in the presence or absence of dynasore, which specifically inhibits endocytosis. We observed that dynasore treatment potently decreased IL8 expression induced by thapsigargin and abolished the NOD dependence of this stimulation (Fig. 4B), implying that the effect required endocytosis. We concluded that a heat-resistant small molecule present in FCS is brought in by endocytosis to stimulate NOD1/2 proteins during thapsigargin stimulation.
All the experiments above point to the likely presence of peptidoglycan contaminants in the cell culture grade FCS in which our cells were grown and that these contaminants would be internalized by endocytosis at a higher rate following an increase in intracellular Ca2+, resulting in NOD-dependent stimulation of pro-inflammatory signaling. A previous study similarly reported that peptidoglycan traces found in human serum were responsible for hyphal growth of C. albicans (19). Moreover, while this manuscript was under evaluation, a new study demonstrated the presence of MDP and other peptidoglycan fragments in the blood of multiple warm-blooded animals (31). To directly determine whether our cell culture FCS contained peptidoglycan fragments, HPLC coupled to MS was conducted on two separate batches of FCS, which revealed the presence of multiple muramyl peptides and peptidoglycan-derived peptides (Fig. S5). In particular, several muramyl peptides identified, including GlcNAc-MurNAc-l-Ala-d-Gln-mesoDAP (GM-TriDAP), GlcNAc-MurNAc-l-Ala-d-Gln (GMDP), and l-Ala-d-Gln-mesoDAP (TriDAP) (Fig. 4C), are known activators of NOD1/2 (3). Similar results were obtained when sera from laboratory mice housed in our facility were tested (data not shown), suggesting that traces of circulating peptidoglycan in serum may be a common feature.
There are several direct implications of our observations. First, our results provide an alternative explanation for the previous implication of NOD proteins as mediators of inflammatory signaling in response to ER stress (18) by showing that an increase in intracellular Ca2+ levels, instead of ER stress itself, caused internalization of trace contaminants of peptidoglycan from the cell culture serum. Second, our results suggest that caution should be taken when analyzing whether a given stimulus induces NF-κB–dependent pro-inflammatory signaling in a NOD-dependent manner, because multiple pathways can cause transient increase in intracellular Ca2+ levels, which could in turn trigger internalization of peptidoglycan contaminants. In particular, it was shown that perturbation of the actin cytoskeleton through Rho GTPases could modulate NF-κB–dependent pro-inflammatory signaling in a NOD-dependent manner (17). To begin exploring the aforementioned hypothesis, WT and NOD1/2 DKO cells cultured in serum-supplemented HBSS were stimulated with cytochalasin D (CytD), which disrupts the actin cytoskeleton polymerization, in the presence or absence of extracellular Ca2+. Interestingly, we observed that stimulation with CytD in the presence of Ca2+ induced expression of IL8 and CXCL1 (albeit more modestly than thapsigargin did) in a NOD-dependent and calcium-dependent manner, whereas HSPA5 expression was not induced (Fig. S6). This suggests that multiple cellular perturbations could trigger NOD-dependent induction of pro-inflammatory pathways if they trigger mobilization of Ca2+ toward the intracellular compartment.
More generally, the presence of trace elements of peptidoglycan in animal serum may be of fundamental importance for physiology, although its implication has not yet been carefully evaluated. Interestingly, early studies have demonstrated that circulating peptidoglycan-derived muramyl peptides were able to regulate slow-wave sleep in rabbits (32–34). More recently, it was shown that translocation of peptidoglycan fragments from the intestinal microbiota into the circulation induced functional priming of neutrophils in a NOD1-dependent manner in mice (35), suggesting that these peptidoglycan fragments were sufficient to enhance systemic innate immunity. Because systemic administration of muramyl peptides has been shown to have numerous physiological consequences, from boosting innate and adaptive immune responses (36–38) to triggering protection against obesity-induced insulin tolerance (39) in the case of NOD2 agonists (and opposite effects in the case of NOD1 activators (39, 40)), it is tempting to speculate that tonic low-level activation of NOD1/2 systemically caused by circulating peptidoglycan fragments may have far-reaching implications for host physiology that need to be fully characterized.
Experimental procedures
Cell culture and reagents
The human epithelial HCT116 cell line (American Type Culture Collection) was cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (FCS), 2 mm l-glutamine, 50 IU penicillin, and 50 μg/ml streptomycin (Wisent Bio Products). Cells were maintained in 95% air, 5% CO2 at 37 °C. Endotoxin-free FCS and phosphate-buffered saline (PBS) were from Wisent (Saint-Bruno-de-Montarville, Quebec, Canada). In some experiments, cells were washed with PBS, and cell culture medium was replaced with Hanks' balanced salt solution (Thermo Fisher Scientific) supplemented or not with CaCl2 (Sigma; 140 μg/ml). In various experiments, cells were treated with 10 μg/ml MDP, 10 μg/ml iE-DAP, and 100 ng/ml L-18 MDP obtained from Invivogen; 0.1 μg/ml thapsigargin, 1 μg/ml tunicamycin, 1 μm A23187, 80 μm Dynasore monohydrate, 15 μm BAPTA-AM, 10 μm CRAC inhibitor (GSK7975A), and 5 μm cytochalasin D obtained from Sigma; and 200 μm DMNP–EDTA (cell impermeant) was purchased from Invitrogen and Thermo Fisher Scientific. SubAB and SubA272B, a kind gift from Drs James and Adrienne Paton (University of Adelaide, Australia), were used at a concentration of 50 ng/ml.
Primary murine organoids
To generate organoid cultures, crypts from the small intestines of mice were extracted as described previously (41). Briefly, the villi of the small intestine were removed by scraping, followed by washing with cold PBS. The remaining tissues were then homogenized and incubated in 2 mm EDTA in PBS for 30 min at 4 °C, followed by vigorous washing in PBS several times to obtain crypt-enriched supernatant fractions. The supernatant fractions were then passed through a cell strainer, pelleted at 300 × g for 5 min at 4 °C, and resuspended in 50 μl of Matrigel (Corning). The crypt-containing organoids were cultured by plating onto the center of a 24-well plate and grown in 500 μl of crypt culture medium supplemented with growth factors (R-spondin 1, Noggin, EGF). Organoids were allowed to grow 7 days, followed by passaging onto 6-well plates for stimulation (roughly 10–15 isolated organoids).
RNA isolation and quantitative RT-PCR
RNA samples were prepared using the GeneJETTM RNA purification kit (Thermo Fisher Scientific) according to the manufacturer's protocol. Eluted RNA was treated with DNase I (Fermentas) at 37 °C for 1 h to remove genomic DNA. cDNA was prepared from 1 μg of total RNA using oligo(dT), random hexamers, dNTPs, RNase OUT (Invitrogen), and Moloney murine leukemia virus reverse transcriptase (Sigma). cDNA was diluted accordingly and prepared in 12-μl reactions using PowerUp SYBR® Green Mastermix (Applied Biosystems). The CFX384 TouchTM real-time PCR detection system (Bio-Rad) was used to obtain the raw Ct values. Results were either analyzed using the 2−ΔCt formula normalizing target gene expression to the TATA sequence binding protein (TBP) housekeeping control, or fold change was calculated by 2−ΔΔCt formula.
NF-κB luciferase assay
To measure NF-κB luciferase activity, 250,000 HCT116 cells (WT or RIPK2 KO) were plated per well and transfected with β-gal, NF-κB luciferase reporter, and pcDNA3 plasmids for 24 h. 10 μg of MDP or iE-DAP was added directly to the cell culture medium at the time of transfection, as described previously (3). Cells were then gently washed with PBS and lysed in luciferase buffer, followed by incubation at room temperature for 10 min. 10 μl of each cell lysate was then added to a black 96-well plate along with 100 μl of luciferin buffer, and luminescence was read using the Victor3 plate reader. To measure expression of the β-gal construct (transfection control) for normalization, 10 μl of original cell lysates was added to 100 μl of the o-nitrophenyl β-d-galactopyranoside (ONPG) buffer and incubated at 37 °C for 30 min, and luminescence was read. The β-gal values were then used to normalize the absorbance values obtained for each sample.
LC/MS analysis of serum samples
Aliquots of FCS from different lots were tested as well as serum from laboratory mice kept in our facility in specific pathogen-free conditions. 20 μl of sera were loaded into the LC-MS platform. The LC-MS platform consisted of an Ultimate 3000 UHPLC coupled to a Q-Exactive mass spectrometer equipped with HESI II source (Thermo Fisher Scientific). Control of the system was performed using XCalibur 2.2 software (Thermo Fisher Scientific) and Chromeleon 7.2 software, with data processing conducted using Thermo Fisher Scientific Quan Browser. Separation by LC was conducted on a Hypersil Gold C18 column (50 × 2.1 mm, 1.9-μm particle size) (Thermo Fisher Scientific). The pump was run at a flow rate of 300 μl/min. Solvent A was water containing 0.1% formic acid; solvent B was acetonitrile containing 0.1% formic acid. The gradient uses was as follows: 0 min, 5% B; 1 min, 5% B; 2 min, 30% B; 3 min, 30% B; 4 min, 50% B; 7 min, 80% B; 9 min, 80% B; 10 min, 98% B; 11 min 98% B; 12 min, 5% B; 18 min, 5% B. Autosampler temperature was maintained at 10 °C, and injection volume was 20 μl. Data collection was done in positive ionization mode with MS1 scan range m/z 350–1200, resolution 70,000, AGC target of 3e6, and a maximum injection time of 100 ms, and MS2 data were collected using a TOP5 method, 0.4 m/z isolation window, 30 NCE, 17,500 resolution, AGC target 1e5, and a maximum injection time of 50 ms. Data collected were analyzed using Quan Browser.
Statistical analysis
Significant differences between mean values were evaluated using a two-way analysis of variance with multiple comparisons using Prism (GraphPad Inc). In all qRT-PCR experiments presented in this study, each point represents the average (from two or three technical replicates) from one experiment. Data from at least three independent experiments were pooled to generate each of the graphs presented (****, p < 0.0001; ***, p < 0.001; **, p < 0.01; *, p < 0.05).
qRT-PCR primers
The qRT-PCR primers used are as follows: mouse Rpl19 (forward), 5′-GCATCCTCATGGAGCACAAT-3′, and mouse Rpl19 (reverse), 5′-CTGGTCAGCCAGGAGCTT-3′; mouse Cxcl1 (forward), 5′-AGACCATGGCTGGGATTCAC-3′, and mouse Cxcl1 (reverse), 5′-AGTGTGGCTATGACTTCGGT-3′; mouse Hspa5 (forward), 5′-GCCTCATCGGACGACTT-3′, and mouse Hspa5 (reverse), 5′-GGGGCAAATGTCTTGGTT-3′; human TBP (forward), 5′-GGGCATTATTTGTGCACTGAGA-3′, and human TBP (reverse), 5′-TAGCAGCACGGTATGAGCAACT-3′; human IL8 (forward), 5′-CCACCGGAAGGAACCATCTC-3′, and human IL8 (reverse), 5′-TTCCTTGGGGTCCAGACAGA-3′; human CXCL1 (forward), 5′-CACACTCAAGAATGGGCGGA-3′, and human CXCL1 (reverse), 5′-ACTATGGGGGATGCAGGATTG-3′; human HSPA5 (forward), 5′-GAACGTCTGATTGGCGATGC-3′, and human HSPA5 (reverse), 5′-TCAACCACCTTGAACGGCAA-3′.
Author contributions
R. M., T. M., R. F., and S. E. G. data curation; R. M. and R. F. investigation; T. M., D. J. P., and S. E. G. conceptualization; T. M., D. J. P., and S. E. G. formal analysis; T. M., D. J. P., and S. E. G. writing-review and editing; D. J. P. and S. E. G. supervision; D. J. P. and S. E. G. funding acquisition; D. J. P. and S. E. G. validation; S. E. G. resources; S. E. G. writing-original draft; S. E. G. project administration.
Supplementary Material
Acknowledgments
We thank James and Adrienne Paton (University of Adelaide, Australia) for the kind gift of the SubAB toxins.
This work was supported by grants from the Canadian Institutes for Health Research (to the D. J. P. and S. E. G. laboratories) and by the Crohn's and Colitis Canada (to S. E. G.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6.
- PRM
- pattern recognition molecule
- TLR
- toll-like receptor
- NLR
- Nod-like receptor
- ER
- endoplasmic reticulum
- SERCA
- sarcoplasmic or endoplasmic reticulum calcium ATPase
- CRAC
- calcium release-activated channel
- MDP
- muramyl dipeptide
- FCS
- fetal calf serum
- MAMP
- microbe-associated molecular pattern
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester
- NOD
- nucleotide-binding oligomerization domain
- DMNP-EDTA
- 1-(4,5-dimethoxy-2-nitrophenyl)-1,2-diaminoethane-N,N,N′,N′-tetraacetic acid
- qRT
- quantitative real time
- CytD
- cytochalasin D
- DKO
- double knockout
- HBSS
- Hanks' balanced salt solution
- TBP
- TATA sequence binding protein
- Rpl19
- ribosomal protein L19.
References
- 1. Philpott D. J., Sorbara M. T., Robertson S. J., Croitoru K., and Girardin S. E. (2014) NOD proteins: regulators of inflammation in health and disease. Nat. Rev. Immunol. 14, 9–23 10.1038/nri3565 [DOI] [PubMed] [Google Scholar]
- 2. Mukherjee T., Hovingh E. S., Foerster E. G., Abdel-Nour M., Philpott D. J., and Girardin S. E. (2018) NOD1 and NOD2 in inflammation, immunity and disease. Arch. Biochem. Biophys. 2018, pii: S0003–9861(18)30937-8 10.1016/j.abb.2018.12.022 [DOI] [PubMed] [Google Scholar]
- 3. Girardin S. E., Travassos L. H., Hervé M., Blanot D., Boneca I. G., Philpott D. J., Sansonetti P. J., and Mengin-Lecreulx D. (2003) Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J. Biol. Chem. 278, 41702–41708 10.1074/jbc.M307198200 [DOI] [PubMed] [Google Scholar]
- 4. Girardin S. E., Boneca I. G., Carneiro L. A., Antignac A., Jéhanno M., Viala J., Tedin K., Taha M. K., Labigne A., Zähringer U., Coyle A. J., DiStefano P. S., Bertin J., Sansonetti P. J., and Philpott D. J. (2003) Nod1 detects a unique muropeptide from Gram-negative bacterial peptidoglycan. Science 300, 1584–1587 10.1126/science.1084677 [DOI] [PubMed] [Google Scholar]
- 5. Chamaillard M., Hashimoto M., Horie Y., Masumoto J., Qiu S., Saab L., Ogura Y., Kawasaki A., Fukase K., Kusumoto S., Valvano M. A., Foster S. J., Mak T. W., Nuñez G., and Inohara N. (2003) An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4, 702–707 10.1038/ni945 [DOI] [PubMed] [Google Scholar]
- 6. Girardin S. E., Tournebize R., Mavris M., Page A. L., Li X., Stark G. R., Bertin J., DiStefano P. S., Yaniv M., Sansonetti P. J., and Philpott D. J. (2001) CARD4/Nod1 mediates NF-κB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2, 736–742 10.1093/embo-reports/kve155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Inohara N., Ogura Y., Fontalba A., Gutierrez O., Pons F., Crespo J., Fukase K., Inamura S., Kusumoto S., Hashimoto M., Foster S. J., Moran A. P., Fernandez-Luna J. L., and Nuñez G. (2003) Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J. Biol. Chem. 278, 5509–5512 10.1074/jbc.C200673200 [DOI] [PubMed] [Google Scholar]
- 8. Girardin S. E., Boneca I. G., Viala J., Chamaillard M., Labigne A., Thomas G., Philpott D. J., and Sansonetti P. J. (2003) Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 278, 8869–8872 10.1074/jbc.C200651200 [DOI] [PubMed] [Google Scholar]
- 9. Magalhaes J. G., Philpott D. J., Nahori M. A., Jéhanno M., Fritz J., Le Bourhis L., Viala J., Hugot J. P., Giovannini M., Bertin J., Lepoivre M., Mengin-Lecreulx D., Sansonetti P. J., and Girardin S. E. (2005) Murine Nod1 but not its human orthologue mediates innate immune detection of tracheal cytotoxin. EMBO Rep. 6, 1201–1207 10.1038/sj.embor.7400552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rubino S. J., Magalhaes J. G., Philpott D., Bahr G. M., Blanot D., and Girardin S. E. (2013) Identification of a synthetic muramyl peptide derivative with enhanced Nod2 stimulatory capacity. Innate Immun. 19, 493–503 10.1177/1753425912471691 [DOI] [PubMed] [Google Scholar]
- 11. Grimes C. L., Ariyananda Lde Z., Melnyk J. E., and O'Shea E. K. (2012) The innate immune protein Nod2 binds directly to MDP, a bacterial cell wall fragment. J. Am. Chem. Soc. 134, 13535–13537 10.1021/ja303883c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Laroui H., Yan Y., Narui Y., Ingersoll S. A., Ayyadurai S., Charania M. A., Zhou F., Wang B., Salaita K., Sitaraman S. V., and Merlin D. (2011) l-Ala-γ-d-Glu-meso-diaminopimelic acid (DAP) interacts directly with leucine-rich region domain of nucleotide-binding oligomerization domain 1, increasing phosphorylation activity of receptor-interacting serine/threonine-protein kinase 2, and its interaction with nucleotide-binding oligomerization domain 1. J. Biol. Chem. 286, 31003–31013 10.1074/jbc.M111.257501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mo J., Boyle J. P., Howard C. B., Monie T. P., Davis B. K., and Duncan J. A. (2012) Pathogen sensing by nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is mediated by direct binding to muramyl dipeptide and ATP. J. Biol. Chem. 287, 23057–23067 10.1074/jbc.M112.344283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Girardin S. E., Jéhanno M., Mengin-Lecreulx D., Sansonetti P. J., Alzari P. M., and Philpott D. J. (2005) Identification of the critical residues involved in peptidoglycan detection by Nod1. J. Biol. Chem. 280, 38648–38656 10.1074/jbc.M509537200 [DOI] [PubMed] [Google Scholar]
- 15. Tanabe T., Chamaillard M., Ogura Y., Zhu L., Qiu S., Masumoto J., Ghosh P., Moran A., Predergast M. M., Tromp G., Williams C. J., Inohara N., and Núñez G. (2004) Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J. 23, 1587–1597 10.1038/sj.emboj.7600175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lupfer C., Thomas P. G., Anand P. K., Vogel P., Milasta S., Martinez J., Huang G., Green M., Kundu M., Chi H., Xavier R. J., Green D. R., Lamkanfi M., Dinarello C. A., Doherty P. C., and Kanneganti T. D. (2013) Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol. 14, 480–488 10.1038/ni.2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Keestra A. M., Winter M. G., Auburger J. J., Frässle S. P., Xavier M. N., Winter S. E., Kim A., Poon V., Ravesloot M. M., Waldenmaier J. F., Tsolis R. M., Eigenheer R. A., and Bäumler A. J. (2013) Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature 496, 233–237 10.1038/nature12025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Keestra-Gounder A. M., Byndloss M. X., Seyffert N., Young B. M., Chávez-Arroyo A., Tsai A. Y., Cevallos S. A., Winter M. G., Pham O. H., Tiffany C. R., de Jong M. F., Kerrinnes T., Ravindran R., Luciw P. A., McSorley S. J., et al. (2016) NOD1 and NOD2 signalling links ER stress with inflammation. Nature 532, 394–397 10.1038/nature17631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu X. L., Lee R. T., Fang H. M., Wang Y. M., Li R., Zou H., Zhu Y., and Wang Y. (2008) Bacterial peptidoglycan triggers Candida albicans hyphal growth by directly activating the adenylyl cyclase Cyr1p. Cell Host Microbe 4, 28–39 10.1016/j.chom.2008.05.014 [DOI] [PubMed] [Google Scholar]
- 20. Gaudet R. G., Guo C. X., Molinaro R., Kottwitz H., Rohde J. R., Dangeard A. S., Arrieumerlou C., Girardin S. E., and Gray-Owen S. D. (2017) Innate recognition of intracellular bacterial growth is driven by the TIFA-dependent cytosolic surveillance pathway. Cell Rep. 19, 1418–1430 10.1016/j.celrep.2017.04.063 [DOI] [PubMed] [Google Scholar]
- 21. Tsalikis J., Pan Q., Tattoli I., Maisonneuve C., Blencowe B. J., Philpott D. J., and Girardin S. E. (2016) The transcriptional and splicing landscape of intestinal organoids undergoing nutrient starvation or endoplasmic reticulum stress. BMC Genomics 17, 680 10.1186/s12864-016-2999-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chong D. C., Paton J. C., Thorpe C. M., and Paton A. W. (2008) Clathrin-dependent trafficking of subtilase cytotoxin, a novel AB5 toxin that targets the endoplasmic reticulum chaperone BiP. Cell. Microbiol. 10, 795–806 10.1111/j.1462-5822.2007.01085.x [DOI] [PubMed] [Google Scholar]
- 23. Wolfson J. J., May K. L., Thorpe C. M., Jandhyala D. M., Paton J. C., and Paton A. W. (2008) Subtilase cytotoxin activates PERK, IRE1 and ATF6 endoplasmic reticulum stress-signalling pathways. Cell. Microbiol. 10, 1775–1786 10.1111/j.1462-5822.2008.01164.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yeung P. S., Yamashita M., and Prakriya M. (2017) Pore opening mechanism of CRAC channels. Cell Calcium 63, 14–19 10.1016/j.ceca.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Di Paola S., Scotto-Rosato A., and Medina D. L. (2018) TRPML1: the Ca2+retaker of the lysosome. Cell Calcium 69, 112–121 10.1016/j.ceca.2017.06.006 [DOI] [PubMed] [Google Scholar]
- 26. Ganley I. G., Wong P. M., and Jiang X. (2011) Thapsigargin distinguishes membrane fusion in the late stages of endocytosis and autophagy. Autophagy 7, 1397–1399 10.4161/auto.7.11.17651 [DOI] [PubMed] [Google Scholar]
- 27. Voronina S., Collier D., Chvanov M., Middlehurst B., Beckett A. J., Prior I. A., Criddle D. N., Begg M., Mikoshiba K., Sutton R., and Tepikin A. V. (2015) The role of Ca2+ influx in endocytic vacuole formation in pancreatic acinar cells. Biochem. J. 465, 405–412 10.1042/BJ20140398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zeng B., Chen G. L., Garcia-Vaz E., Bhandari S., Daskoulidou N., Berglund L. M., Jiang H., Hallett T., Zhou L. P., Huang L., Xu Z. H., Nair V., Nelson R. G., Ju W., Kretzler M., et al. (2017) ORAI channels are critical for receptor-mediated endocytosis of albumin. Nat. Commun. 8, 1920 10.1038/s41467-017-02094-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee J., Tattoli I., Wojtal K. A., Vavricka S. R., Philpott D. J., and Girardin S. E. (2009) pH-dependent internalization of muramyl peptides from early endosomes enables Nod1 and Nod2 signaling. J. Biol. Chem. 284, 23818–23829 10.1074/jbc.M109.033670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marina-García N., Franchi L., Kim Y. G., Hu Y., Smith D. E., Boons G. J., and Núñez G. (2009) Clathrin- and dynamin-dependent endocytic pathway regulates muramyl dipeptide internalization and NOD2 activation. J. Immunol. 182, 4321–4327 10.4049/jimmunol.0802197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang Z., Wang J., Xu X., Wang H., Qiao Y., Chu W. C., Xu S., Chai L., Cottier F., Pavelka N., Oosting M., Joosten L. A., Netea M., Ng C. Y., Leong K. P., et al. (2019) Antibody neutralization of microbiota-derived circulating peptidoglycan dampens inflammation and ameliorates autoimmunity. Nat. Microbiol. 2019, 10.1038/s41564-019-0381-1 [DOI] [PubMed] [Google Scholar]
- 32. Krueger J. M., Karnovsky M. L., Martin S. A., Pappenheimer J. R., Walter J., and Biemann K. (1984) Peptidoglycans as promoters of slow-wave sleep. II. Somnogenic and pyrogenic activities of some naturally occurring muramyl peptides; correlations with mass spectrometric structure determination. J. Biol. Chem. 259, 12659–12662 [PubMed] [Google Scholar]
- 33. Krueger J. M., Pappenheimer J. R., and Karnovsky M. L. (1982) Sleep-promoting effects of muramyl peptides. Proc. Natl. Acad. Sci. U.S.A. 79, 6102–6106 10.1073/pnas.79.19.6102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martin S. A., Karnovsky M. L., Krueger J. M., Pappenheimer J. R., and Biemann K. (1984) Peptidoglycans as promoters of slow-wave sleep. I. Structure of the sleep-promoting factor isolated from human urine. J. Biol. Chem. 259, 12652–12658 [PubMed] [Google Scholar]
- 35. Clarke T. B., Davis K. M., Lysenko E. S., Zhou A. Y., Yu Y., and Weiser J. N. (2010) Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat. Med. 16, 228–231 10.1038/nm.2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fraser-Smith E. B., and Matthews T. R. (1981) Protective effect of muramyl dipeptide analogs against infections of Pseudomonas aeruginosa or Candida albicans in mice. Infect. Immun. 34, 676–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leclerc C., Juy D., Bourgeois E., and Chedid L. (1979) In vivo regulation of humoral and cellular immune responses of mice by a synthetic adjuvant, N-acetyl-muramyl-l-alanyl-d-isoglutamine, muramyl dipeptide for MDP. Cell. Immunol. 45, 199–206 10.1016/0008-8749(79)90377-0 [DOI] [PubMed] [Google Scholar]
- 38. Löwy I., Leclerc C., Bourgeois E., and Chedid L. (1980) Inhibition of mitogen-induced polyclonal activation by a synthetic adjuvant, muramyl dipeptide (MDP). J. Immunol. 124, 320–325 [PubMed] [Google Scholar]
- 39. Cavallari J. F., Fullerton M. D., Duggan B. M., Foley K. P., Denou E., Smith B. K., Desjardins E. M., Henriksbo B. D., Kim K. J., Tuinema B. R., Stearns J. C., Prescott D., Rosenstiel P., Coombes B. K., Steinberg G. R., and Schertzer J. D. (2017) Muramyl dipeptide-based postbiotics mitigate obesity-induced insulin resistance via IRF4. Cell Metab. 25, 1063–1074.e3 10.1016/j.cmet.2017.03.021 [DOI] [PubMed] [Google Scholar]
- 40. Chan K. L., Tam T. H., Boroumand P., Prescott D., Costford S. R., Escalante N. K., Fine N., Tu Y., Robertson S. J., Prabaharan D., Liu Z., Bilan P. J., Salter M. W., Glogauer M., Girardin S. E., et al. (2017) Circulating NOD1 activators and hematopoietic NOD1 contribute to metabolic inflammation and insulin resistance. Cell Rep. 18, 2415–2426 10.1016/j.celrep.2017.02.027 [DOI] [PubMed] [Google Scholar]
- 41. Tattoli I., Killackey S. A., Foerster E. G., Molinaro R., Maisonneuve C., Rahman M. A., Winer S., Winer D. A., Streutker C. J., Philpott D. J., and Girardin S. E. (2016) NLRX1 acts as an epithelial-intrinsic tumor suppressor through the modulation of TNF-mediated proliferation. Cell Rep. 14, 2576–2586 10.1016/j.celrep.2016.02.065 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.