

Abstract
Drug discovery and development pipelines are long, complex and depend on numerous factors. Machine learning (ML) approaches provide a set of tools that can improve discovery and decision making for well-specified questions with abundant, high-quality data. Opportunities to apply ML occur in all stages of drug discovery. Examples include target validation, identification of prognostic biomarkers and analysis of digital pathology data in clinical trials. Applications have ranged in context and methodology, with some approaches yielding accurate predictions and insights. The challenges of applying ML lie primarily with the lack of interpretability and repeatability of ML-generated results, which may limit their application. In all areas, systematic and comprehensive high-dimensional data still need to be generated. With ongoing efforts to tackle these issues, as well as increasing awareness of the factors needed to validate ML approaches, the application of ML can promote data-driven decision making and has the potential to speed up the process and reduce failure rates in drug discovery and development.
Biological systems are complex sources of information during development and disease. This information is now being systematically measured and mined at unprecedented levels using a plethora of ‘omics’ and smart technologies. The advent of these high-throughput approaches to biology and disease presents both challenges and opportunities to the pharmaceutical industry, for which the aim is to identify plausible therapeutic hypotheses from which to develop drugs. However, recent advances in a number of factors have led to increased interest in the use of machine learning (ML) approaches within the pharmaceutical industry. Coupled with infinitely scalable storage, the large increase in the types and sizes of data sets that may provide the basis for ML has enabled pharmaceutical companies to access and organize many more data. Data types can include images, textual information, biometrics and other information from wearables, assay information and high-dimensional omics data1.
Over the past few years, the field of artificial intelligence (AI) has moved from largely theoretical studies to real-world applications. Much of that explosive growth has to do with the wide availability of new computer hardware such as graphical processing units (GPUs) that make parallel processing faster, especially in numerically intensive computations. More recently, advances in new ML algorithms, such as deep learning (DL)2, that build powerful models from data and the demonstrable success of these techniques in numerous public contests3,4 have helped to enormously increase the applications of ML within pharmaceutical companies in the past 2 years.
Although many consumer service industries have been early adopters of newer methods from the field of ML, uptake from the pharmaceutical industry has lagged until recently. It is well known that the success rate for drug development (as defined from phase I clinical trials to drug approvals) is very low across all therapeutic areas and across the global pharmaceutical industry. A recent study on 21,143 compounds found that the overall success rate was as low as 6.2%5. Hence, much of the rationale for the use of ML technologies within the pharmaceutical industry is driven by business needs to lower overall attrition and costs.
All stages of drug discovery and development, including clinical trials, have embarked on developing and utilizing ML algorithms and software (FIG. 1) to identify novel targets6, provide stronger evidence for target-disease associations7, improve small-molecule compound design and optimization8, increase understanding of disease mechanisms, increase understanding of disease and non-disease phenotypes9, develop new biomarkers for prognosis, progression and drug efficacy1, improve analysis of biometric and other data from patient monitoring and wearable devices, enhance digital pathology imaging10 and extract high-content information from images at all levels of resolution.
Fig. 1 |. Machine learning applications in the drug discovery pipeline and their required data characteristics.
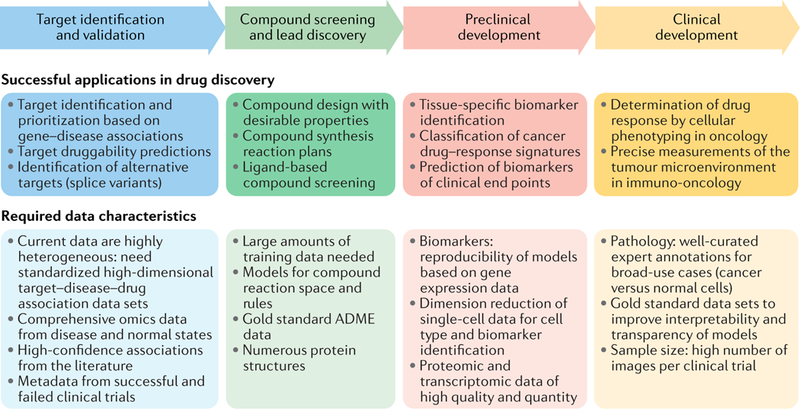
Several successful applications of machine learning in various stages of the drug development pipeline in pharmaceutical companies have been published. However, within each data domain, there are still challenges related to the standard of data quality and data quantity needed to capitalize on the full potential of these methods for discovery. ADME, absorption, distribution, metabolism and excretion.
Consequently, many pharmaceutical companies have begun to invest in resources, technologies and services to generate and curate data sets to support research in this area. Furthermore, technology giants such as IBM and Google, biotechnology start-ups and academic centres are not only providing cloud-based computation services but also working in the pharmaceutical and health-care space with industry partners. This Review provides an overview of current tools and techniques (the toolbox) used in ML, including deep neural nets, and an overview of progress so far in key pharmaceutical application areas.
The machine learning toolbox
Fundamentally, ML is the practice of using algorithms to parse data, learn from it and then make a determination or a prediction about the future state of any new data sets. So rather than hand-coding software routines with a specific set of instructions (pre-determined by the programmer) to accomplish a particular task, the machine is trained using large amounts of data and algorithms that give it the ability to learn how to perform the task. The programmer codes the algorithm used to train the network instead of coding expert rules.
The algorithms adaptively improve their performance as the quantity and quality of data available for learning increase. Hence, ML is best applied to solve problems for which a large amount of data and several variables are at hand but a model or formula relating these is not known.
There are two main types of technique that are used to apply ML: supervised and unsupervised learning. Supervised learning methods are used to develop training models to predict future values of data categories or continuous variables, whereas unsupervised methods are used for exploratory purposes to develop models that enable clustering of the data in a way that is not specified by the user. Supervised learning trains a model on known input and output data relationships so that it can predict future outputs for new inputs. Future outputs are typically models or results for data classification or an understanding of the most influential variables (regression). The unsupervised learning technique identifies hidden patterns or intrinsic structures in the input data and uses these to cluster data in meaningful ways.
Model selection concepts.
The aim of a good ML model is to generalize well from the training data to the test data at hand. Generalization refers to how well the concepts learned by the model apply to data not seen by the model during training. Within each technique, several methods exist (FIG. 2), which vary in their prediction accuracy, training speed and the number of variables they can handle. Algorithms must be chosen carefully to ensure that they are suitable for the problem at hand and the amount and type of data available. The amount of parameter tuning needed and how well the method separates signal from noise are also important considerations.
Fig. 2 |. Machine learning tools and their drug discovery applications.
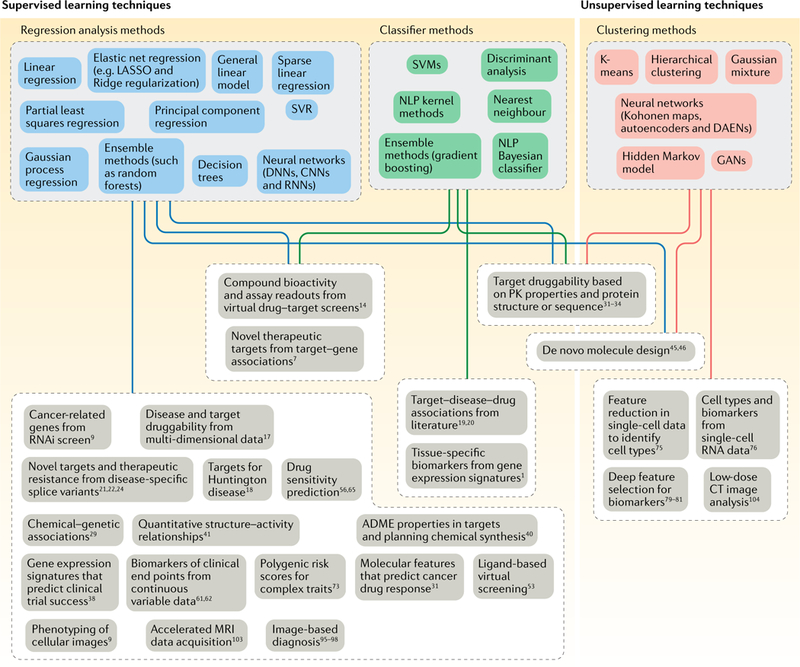
This figure gives an overview of the machine learning techniques that have been used to answer the drug discovery questions covered in this Review. A range of supervised learning techniques (regression and classifier methods) are used to answer questions that require prediction of data categories or continuous variables, whereas unsupervised techniques are used to develop models that enable clustering of the data. ADME, absorption, distribution, metabolism and excretion; CNN, convolutional neural network; CT, computed tomography; DAEN, deep autoencoder neural network; DNN, deep neural network; GAN, generative adversarial network; MRI, magnetic resonance imaging; NLP, natural language processing; PK, pharmacokinetic; RNAi, RNA interference; RNN, recurrent neural network; SVM, support vector machine; SVR, support vector regression.
Model overfitting happens when the model learns not only the signal but also some of the unusual features of the training data and incorporates these into the model, with a resulting negative impact on the performance of the model on new data. Underfitting refers to a model that can neither model the training data nor generalize to new data. Typical ways to limit overfitting are to apply resampling methods or to hold back part of the training data to use as a validation data set. Regularization regression methods (such as Ridge, LASSO or elastic nets) add penalties to parameters as model complexity increases so that the model is forced to generalize the data and not overfit. One of the most effective ways to avoid overfitting is the dropout method11, which randomly removes units in the hidden layer. Different ML techniques have different performance metrics. Basic evaluation metrics12 such as classification accuracy, kappa13, area under the curve (AUC), logarithmic loss, the F1 score and the confusion matrix can be used to compare performance across methods. The availability of gold standard data sets as well as independently generated data sets can be invaluable in generating well-performing models.
Several software libraries are now available for high-performance mathematical computation across a variety of hardware platforms (central processing units (CPUs), GPUs and tensor processing units (TPUs)), and from desktops to clusters of servers. Commonly used ML programmatic frameworks are the open-source framework TensorFlow, originally developed by researchers and engineers from the Google Brain team within Google’s AI organization (see Related links), as well as PyTorch, Keras and Scikit-learn.
Deep neural network architectures.
DL is a modern reincarnation of artificial neural networks from the 1980s and 1990s and uses sophisticated, multi-level deep neural networks (DNNs) to create systems that can perform feature detection from massive amounts of unlabelled or labelled training data2. The major difference between DL and traditional artificial neural networks is the scale and complexity of the networks used. In neural networks, input features are fed to an input layer, and after a number of nonlinear transformations using hidden layers, the predictions are generated by an output layer. This is typically done by using the back-propagation of errors to progressively reduce the difference between the obtained and the expected values of the output. Each output node corresponds to a task (or class) to be predicted. If there is only one node in the output layer, then the corresponding network is referred to as a single-task neural network. DL can have a large number of hidden layers because it uses more powerful CPU and GPU hardware, whereas traditional neural networks normally use one or two hidden layers because of hardware limitations. There are also many algorithmic improvements in DL.
The applications of DNNs in drug discovery have been numerous and include bioactivity prediction14, de novo molecular design, synthesis prediction and biological image analysis3. One advantage of DNNs is that they have several different flexible architectures described below and are thus used to answer a variety of questions. In the first architecture, deep convolutional neural networks (CNNs), some of the hidden layers are only locally (rather than globally) connected to the next hidden layer. CNNs achieve the best predictive performance in areas such as speech and image recognition by hierarchically composing simple local features into complex models. Graph convolutional networks are a special type of CNN that can be applied to structured data in the form of graphs or networks. The second architecture is the recurrent neural network (RNN), which takes the form of a chain of repeating modules of neural networks in which connections between nodes form a directed graph along a sequence. This allows for the analysis of dynamic changes over time where persistent information is needed. Long short-term memory neural networks are a special kind of RNN that are capable of learning long-term dependencies. The third example — fully connected feedforward networks — are networks in which every input neuron is connected to every neuron in the next layer. This is the opposite of an RNN in that, with fully connected feedforward networks, the gradient is clearly defined and computable through backpropagation. These models have been used in challenging predictive model building cases, such as with gene expression data, in which the number of samples is small relative to the number of features. The fourth network architecture is the deep autoencoder neural network (DAEN). This type of neural network is an unsupervised learning algorithm that applies backpropagation to project its input to its output with the purpose of dimension reduction15, thus trying to preserve the important random variables of the data while removing the non-essential parts. The fifth and final network architecture — generative adversarial networks (GANs) — consist of any two networks (although often a combination of feedforward neural networks and CNNs), where one is tasked to generate content and the other to classify that content.
Data characteristics.
The practice of ML is said to consist of at least 80% data processing and cleaning and 20% algorithm application. The predictive power of any ML approach is therefore dependent on the availability of high volumes of data of high quality. Data used for training need to be accurate, curated and as complete as possible in order to maximize predictability. Experimental design often involves discussions on the ideal sample size and the appropriate power calculations for correctly estimating this parameter. Whether the correct type of data is even available and what data should be experimentally generated are also key considerations for certain questions. ML applications are more powerful when used on data that have been generated in a systematic manner, with minimal noise and good annotation. As we discuss below, many applications are not particularly effective because data are combined from multiple sources with variable data quality. There are ongoing efforts to develop open annotated data in specific areas of drug discovery, such as target validation16. These aim to generate good quality positive and negative annotations in areas that are important to drug discovery and development to foster application of ML.
Applications in drug discovery
Target identification and validation.
The pre-eminent approach in drug discovery is to develop drugs (small molecules, peptides, antibodies or newer modalities including short RNAs or cell therapies) that will alter the disease state by modulating of the activity of a molecular target. Notwithstanding a recent resurgence in phenotypic screens, initiating a drug development programme requires identification of a target with a plausible therapeutic hypothesis: that modulation of the target will result in modulation of the disease state. Selecting this target on the basis of the available evidence is referred to as target identification and prioritization. Having made this preliminary choice, the next step is to validate the role of the chosen target in disease using physiologically relevant ex vivo and in vivo models (target validation). Although the ultimate validation of the target will only come later, through clinical trials, early target validation is crucial to focus efforts on potentially successful projects.
Modern biology is increasingly rich in data. This includes human genetic information in large populations, transcriptomic, proteomic and metabolomic profiling of healthy individuals and those with specific diseases and high-content imaging of clinical material. The ability to capture these large data sets and to re-use them via public databases presents new opportunities for early target identification and validation. However, these multi-dimensional data sets require appropriate analytical methods to yield statistically valid models that can make predictions for target identification, and this is where ML can be exploited. The range of experiments that can contribute to target identification and validation is wide, but if these experiments are data-driven, ML is increasingly being applied.
The first step in target identification is establishing a causal association between the target and the disease. Establishing causality requires demonstration that modulation of a target affects disease from either naturally occurring (genetic) variation or carefully designed experimental intervention. However, ML can be used to analyse large data sets with information on the function of a putative target to make predictions about potential causality, driven, for instance, by the properties of known true targets. ML methods have been applied in this way across several aspects of the target identification field. Costa et al.17 built a decision tree-based meta-classifier trained on network topology of protein-protein, metabolic and transcriptional interactions, as well as tissue expression and subcellular localization, to predict genes associated with morbidity that are also druggable. By inspecting the decision tree, they identified regulation by multiple transcription factors (TFs), centrality in metabolic pathways and extracellular location as key parameters. In other studies, ML models have focused on specific diseases or therapeutic areas. Jeon et al.6 built a support vector machine (SVM) classifier using various genomic data sets to classify proteins into drug targets and non-drug targets for breast, pancreatic and ovarian cancers. Key classification features were gene essentiality, mRNA expression, DNA copy number, mutation occurrence and protein-protein interaction network topology. In all, 122 global cancer targets were identified, 69 of which overlap with 116 known cancer targets. In addition, 266, 462 and 355 targets were identified as specific to breast, pancreatic and ovarian cancers, respectively. Two predicted targets were validated with peptide inhibitors that had strong anti-proliferative effects in cell culture models. Further, inhibitors for 137 predicted pancreatic cancer targets were almost twice as likely to show strong inhibition of cell viability as other compounds. Ament et al.18 built a model based on mouse TF binding sites and transcriptome profiling data to characterize transcriptional changes underlying Huntington disease. They reconstructed a genome-scale model of target genes for 718 TFs in the mouse striatum using a regression model and LASSO regularization. Overall, 13 of 48 identified TF modules were differentially expressed in striatal tissue in human disease and provided potential starting points for Huntington disease therapies. Molecular targets for tissue-specific antiageing therapies have been identified by Mamoshina et al.1. They compared gene expression signatures from young and old muscle. The comparison of several supervised ML methods revealed SVMs with linear kernel and deep feature selection to be best suited to the identification of ageing biomarkers. In each of these examples, ML generated a set of predictions of targets that have properties that suggest they are likely to bind drugs, or be involved in disease, but further validation is essential to generate a therapeutic hypothesis.
The literature is the primary source of knowledge on target association with disease. Automated processing of the literature unlocks information from unstructured text that would otherwise be inaccessible. Recent advances in natural language processing (NLP), an ML approach applied to text mining, have enabled more effective data mining to identify relevant papers. BeFree19 applies NLP Kernel methods to identify drug-disease, gene-disease and target-drug associations in Medline abstracts. This supervised learning approach relies on the manually annotated European Union adverse drug reactions (EU-ADR) database corpus of relationships and a semi-automatically annotated corpus based on the Genetic Association Database. DigSee20 identifies genes and diseases in Medline abstracts, uses NLP to extract biological events between these entities and ranks the evidence sentences with a Bayesian classifier.
One area with great scope for ML is in understanding basic aspects of biology to identify therapeutic opportunities through alternate modalities or novel targets. Understanding genetic variation in splicing signals is one example. DL splicing models are now able to accurately predict alternate splicing signals21. The latest integrative splicing models22 combine CLIP-seq assay data of splicing factor binding in vivo with RNA sequencing experiments in which these splicing factors have been knocked down or overexpressed. Combining splicing code models with predictions of de novo and complex splicing variations has allowed identification of splicing variants specific to Alzheimer disease23. Recent applications of similar approaches identified an escape mechanism from CART-19 immunotherapy24, rare genetic variants leading to deafness25 and splicing variants associated with autism26.
ML can also predict cancer-specific drug effects. Iorio et al.27 screened 990 cancer cell lines against 265 anticancer drugs and investigated how genome-wide gene expression, DNA methylation, gene copy number and somatic mutation data affect drug response. They used ANOVA, logic models and ML algorithms (elastic net regression and random forests) to identify molecular features that predict drug response. The most predictive data type across cancer types was gene expression, whereas the most predictive cancer-specific models included genomic features (driver mutations or copy number alterations) and were even better if they included DNA methylation data. Tsherniak et al.28 used data from RNA interference (RNAi) screens of 501 cancer cell lines to find molecular markers that predict cancer dependencies for 769 genes. They developed a nonlinear regression model based on conditional inference trees to generate predictive models based on gene expression, gene copy number and somatic gene mutations. McMillan et al.29 screened 222 chemicals against >100 heavily annotated cell models of diverse and characteristic somatic lung cancer lesions. They applied regularized ML (elastic net) and probability-based metrics (scanning Kolmogorov-Smirnov) to identify 171 chemical-genetic associations that revealed targetable mechanistic vulnerabilities in a range of oncotypes without effective therapies. These approaches suggest that there are opportunities for tumour-intrinsic precision medicine.
Another important question for drug developers is how likely it is that a drug can be made for any given target. For small-molecule drugs, this entails identifying targets that have features that suggest these proteins can bind small molecules30. Different target attributes can be used to generate these druggability models. Nayal and Honig31 trained a random forest classifier on physicochemical, structural and geometric attributes of 99 drug-binding and 1,187 non-drug-binding cavities from a set of 99 proteins. Size and shape of the surface cavities were the most important features. Several studies derived various physicochemical properties from protein sequences of known drug and non-drug targets and applied SVMs32,33 or biased SVMs with stacked autoencoders, a DL model34, to predict druggable targets. Druggable proteins have also been found to occupy specific regions of protein-protein interaction networks and tend to be highly connected6,17,35. Again, these examples of ML approaches generated sets of targets that are predicted as likely to bind drugs, hence reducing the potential search space, but these targets require further validation.
The holy grail for target identification or validation is the early prediction of future clinical trial success for a target-based drug discovery programme. Various non-ML analyses point to possible predictors of success5,36,37. Using ML, Rouillard et al.38 assessed omics data for a set of 332 targets that succeeded or failed phase III clinical trials by multivariate feature selection. They found gene expression data were particularly predictive of successful targets, characterized by low mean RNA expression and high variance across tissues. This study confirmed previous findings that ideal targets exhibit disease-specific expression in affected tissues39. Ferrero et al.7 trained a range of ML classifiers using target-disease associations from the open targets platform16 to predict de novo potential therapeutic targets. Assessment of feature importance identified the existence of an animal model, gene expression and genetic data as key data types for therapeutic target prediction independent of the indication. However, this approach is limited by the sparse nature of the data and the lack of information about reasons for failure of initiated programmes. More fundamentally, owing to the length of time between initiating a successful drug discovery programme and bringing the drug to market, successful programmes reflect earlier paradigms for drug development. The drivers of successful small-molecule programmes are unlikely to be the same today, as newer modalities, such as biologics (including antibodies), are available. The increasing focus on precision medicine introduces additional constraints. It is essential for future prediction approaches that extensive data on successful and failed drug discovery programmes are available with metadata in the public domain.
Small-molecule design and optimization.
The discovery of drug candidates that can block or activate the target protein of interest involves extensive virtual and experimental high-throughput screening of large compound libraries. Candidate structures are then further refined and modified to improve target specificity and selectivity, along with optimized pharmacodynamic, pharmacokinetic and toxicological properties. Importantly, though, the lack of sufficient high-quality data for new chemistry such as proteolysis-targeting chimeras (PROTACs) and macrocycles can limit the impact of ML on such chemistry.
Much work has been done to apply DL methods, such as multi-task neural networks, to ligand-based virtual screening. Given a lead compound, compounds that have a similar chemical structure can be identified computationally. This has typically been performed using classic statistical methods, but multi-task DNNs are proving to be more effective40. DNNs can significantly boost predictive power when inferring the properties and activities of small molecules41. The one-shot learning technique can be used to substantially reduce the amount of data required to make meaningful predictions about the readout of a molecule in a new experimental setup. Combining ML with Markov state models, this technique was used to identify the previously unknown mechanism of opiate binding to the μ-opioid receptor, revealing an allosteric site that is involved in its activation42. The benefits of multi-task models over single-task models are, however, highly data set-dependent. To help benchmark ML algorithms, Pande et al. compiled a large benchmarking data set, MoleculeNet43, which has been used for the comparison of different ML algorithms. MoleculeNet contains data on the properties of over 700,000 compounds. All data sets have been curated and integrated into the open-source DeepChem package (see Related links), which also includes other tools.
DNNs and modern tree search algorithms can also be used to plan efficient routes of chemical synthesis. To plan the synthesis of a target molecule, the molecule is formally decomposed using reversed reactions (retrosynthesis). This procedure results in a sequence of reactions that can then be executed in the laboratory in the forward direction to synthesize the target. A major challenge is to systematically apply synthetic chemistry knowledge to this process. The manual incorporation of transformation rules is prohibitive as the knowledge of chemistry grows exponentially, and the scope and limitations of many reactions are not completely understood. To automatically extract the rules, Segler et al.44 used the Reaxys database (~11 million reactions and ~300,000 rules) and performed a Monte Carlo tree search (MCTS) to score the tree nodes in conjunction with DNNs to steer the search in the most promising directions. In quantitative analyses, this method outperforms the gold standard, best first search, with two different implementations (heuristic method and neural). Furthermore, MCTS is 30 times faster than traditional computer-aided search methods for almost two-thirds of the molecules examined. Qualitative tests were also performed in a double-blind study. Organic chemists were asked to choose between literature-based and predicted synthesis routes without knowing how the route was obtained. Here, for the first time, chemists considered the quality of the predicted routes to be, on average, as good as routes taken from the literature.
Another valuable application of DL is molecular de novo design through reinforcement learning. Researchers at AstraZeneca45 made use of RNNs for expansion of the chemical space by tuning a sequence-based generative model to design compounds with almost optimal values for solubility, pharmacokinetic properties, bioactivity and other parameters. Kadurin et al.46 also developed similar models using deep GANs to perform molecular feature extraction on very large data sets. However, it must be noted that reinforcement learning might not help in identifying new and unprecedented synthetic routes47.
Community problem-solving competitions can be useful to advance method development in a particular area. Researchers at Merck Sharp & Dohme sponsored a Kaggle competition for the prediction of other relevant absorption, distribution, metabolism and excretion (ADME) parameters as well as some biochemical targets. The winning team used DNNs, which, in 13 out of 15 assay systems, performed slightly better than a standard random forest41. Some of their key learnings were that the optimization of the hyperparameters can improve DNNs, feature selection is not necessary, multi-task models perform better than single-task models and overfitting can be prevented by using dropout. Ramsundar et al.40 also observed that multi-task DNNs perform better than single-task DNNs. A comparison between single-task and multi-task DNNs and a comparison between different ML methods (random forest, SVM, naive Bayes and logic regression) were pursued by Lenselink et al.48 using one standardized data set obtained from ChEMBL49. Here, the DNN model performed best, and a multi-task DNN was also found to be better than a single-task DNN. Multi-task DNNs have also been shown to be better for predictions of lead optimization and lead identification, as they can synthesize information from many distinct biological sources50 owing to the presence of multiple nodes in the output layer.
Feature selection before model building can improve ML models, as shown in a study by Kramer and Gutlein51. They were also able to detect improvements in random forest models against other ML methods such as SVMs and naive Bayes, with faster performance and fewer features used while training models. In their view, one major benefit from filtering out chemical fingerprint bits is the improvement in model interpretability. If the fingerprint is not filtered, the interpretability is hindered owing to an effect called ‘bit collisions’. The crucial impact of filtering fingerprints was also independently shown by Landrum et al.8.
Hochreiter et al.52 also found that DNN-based models significantly outperformed all competing methods and that the predictive performance of DL, using a data set of all ChEMBL assays and target prediction based on a simplified molecular input line entry system (SMILES) input, is in many cases comparable to that of tests performed in wet laboratories. The Hochreiter group also showed that DNNs outperformed all other ML methods (k-nearest neighbour, naive Bayes, random forest and SVMs) and statistics-based methods (similarity ensemble approach53) for target prediction54. The same group won the majority of the challenges in the Tox21 Data Challenge 2014 (REF.55).
An unresolved challenge in the field of small-molecule design is how to best represent the chemical structure. A plethora of representations exist, from simple circular fingerprints such as the extended-connectivity fingerprint (ECFP) to sophisticated symmetry functions (FIG. 3). It is still not clear which structure representation works best for which small-molecule design problem. Therefore, it will be interesting to see if the rise in ML studies in the field of cheminformatics will give more guidance about the best choice for structure representation.
Fig. 3 |. The challenges of compound structure representation in machine learning models.
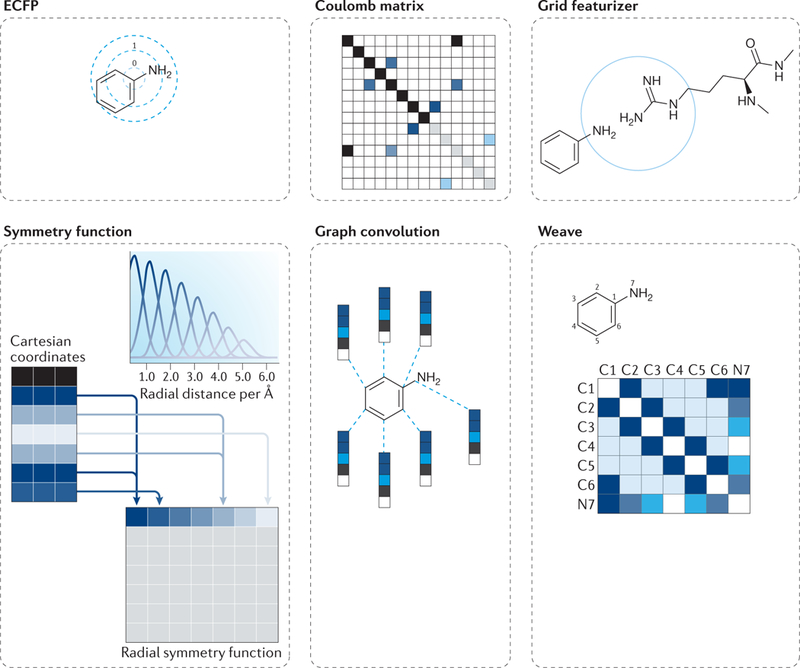
The appropriate representation of chemical structures and their features can take on many representations depending on the required application. Extended-connectivity fingerprints (ECFPs) contain information about topological characteristics of the molecule, which enables this information to be applied to tasks such as similarity searching and activity prediction. A Coulomb matrix encodes information about the nuclear charges of a molecule and their coordinates. The grid featurizer method incorporates structural features of both the ligand and the target protein as well as the intermolecular forces that contribute to binding affinity. Symmetry function is another common encoding of atomic coordinate information, which focuses on the distance between atom pairs and the on angles formed within triplets of atoms. The graph convolution method computes an initial feature vector and a neighbour list for each atom that summarizes the local chemical environment of an atom, including atom types, hybridization types and valence structures. Weave featurization calculates a feature vector for each pair of atoms in the molecule, including bond properties (if directly connected), graph distance and ring info, forming a feature matrix. Reproduced by permission of the Royal Society of Chemistry, Wu, Z. et al. MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 9, 513–530 (2018), REF.43.
Predictive biomarkers.
ML-based biomarker discovery and drug sensitivity predictive models are demonstrated approaches to help improve clinical success rates, to better understand the mechanism of action of a drug and to identify the right drug for the right patients56–58. Late-stage clinical trials take many years and millions of dollars to conduct, so it will be most beneficial to build, validate and apply predictive models earlier, using preclinical and/or early-stage clinical trial data. A translational biomarker can be predicted using ML approaches on preclinical data sets. After being validated using independent data sets (either preclinical or clinical), the model and its corresponding biomarker can be applied to stratify patients, identify potential indications and suggest the mechanisms of action of a drug (FIG. 4).
Fig. 4 |. Utilizing predictive biomarkers to support drug discovery and development.
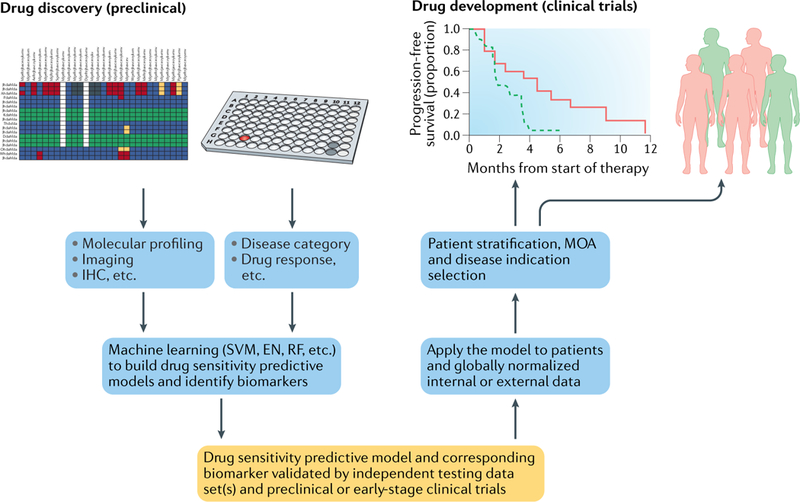
A drug sensitivity predictive model (yellow box) can be generated using machine learning approaches on preclinical data. The model could then be tested using data from early-stage clinical patient samples. Once validated, the model could be used for patient stratification and/or disease indication selection to support the clinical development of a drug, as well as to infer its mechanism of action. EN, elastic net; IHC, immunohistochemistry; MOA, mechanism of action; RF, random forest; SVM, support vector machine.
Although there are thousands of papers on biomarkers and predictive models in the literature, few of them have been used in clinical trials. Various factors contribute to this gap, including data quality, model selection, access to data and software, model reproducibility and the design of assays suitable for a clinical setting. To address some of the model-related issues, several community efforts have evaluated ML approaches to develop both classification and regression models. Several years ago, the US Food and Drug Administration (FDA) organized the MicroArray Quality Control II (MAQC II) initiative to evaluate various ML methods for predicting clinical end points from baseline gene expression data59. In the project, 36 independent teams analysed 6 microarray data sets to generate predictive models to classify a sample with 1 of13 clinical end points. General observations included the importance of the data quality control processes, the need for skilled scientists (some teams perform consistently better than other teams using the same ML methods) and the importance of selecting appropriate modelling approaches for clinical end points. For instance, a poor prediction of overall survival for patients with multiple myeloma could be partly due to applying an arbitrary survival cut-off of 24 months. Both gene expression and overall survival in multiple myeloma are continuous variables, and therefore, a regression-based prediction model is appropriate. Indeed, using a univariate Cox regression approach, a gene expression signature that significantly predicts a high-risk subgroup of patients was identified60. This signature was confirmed in several independent studies and from different regression-based approaches61–64, highlighting the advantage of a regression approach without predefined class membership.
The National Cancer Institute (NCI)-DREAM challenge was another community effort to evaluate regression methods for building drug sensitivity predictive models (defined as regression questions)65. Each participating team used their best modelling approaches and optimized their parameter sets on the same training data sets (35 breast cancer cell lines treated with 31 drugs) then tested the performance of their models on the same blinded testing data sets (18 breast cancer cell lines treated with the same 31 drugs). Six types of baseline profiling data were available for generating predictive models — RNA microarray, single nucleotide polymorphism (SNP) array, RNA sequencing, reverse phase protein array, exome sequencing and DNA methylation status — to which 44 participating teams applied various regression approaches such as kernel method, nonlinear regression (regression trees), sparse linear regression, partial least squares regression, principal component regression or ensemble methods. Consistent with the MAQC II results, some teams consistently outperformed other teams using the same approaches. The differential performance was likely reflective of the technical details used for quality control, data reduction, feature selection, splitting strategy and fine-tuning ML parameters, as well as potential incorporation of biological knowledge such as gene function information or clinical data into the construction of the predictive models. In addition, some drugs were easier to build predictive models for than others for all teams and methods. The NCI-DREAM challenge data sets and results continue to be used as validation data sets for method development and evaluation, for example, on new random forest ensemble frameworks66, group factor analyses67 and other approaches68,69.
Several successful case studies have now been published in which ML-generated predictive models and their corresponding biomarkers have played a critical role in drug discovery and development. Li et al.56 conducted a case study using standard-of-care drugs in which they first built models for drug sensitivity to erlotinib and sorafenib (one model for each drug) using cancer cell line screen data. They then applied the models to stratify patients from the BATTLE clinical trial70, who were treated with one of the two drugs, and demonstrated that the models were predictive and drug-specific. The model-derived biomarker genes were shown to be reflective of the mechanism of action of each drug, and when combined with globally normalized public domain data from various cancer types, the model predicted sensitivities of cancer types to each drug that were consistent with their FDA-approved indications. This study shows that using ML approaches to identify key features that contribute to drug sensitivity across various cancer types in a tissue-agnostic manner could be useful for drug development (in comparison with cancer type-based clinical trials followed by label expansions). In 2017, the FDA approved the programmed cell death 1 (PD1) inhibitor pembrolizumab for cancers with a specific genetic biomarker. This is the first FDA approval based on a cross-indication genetic biomarker rather than a cancer type71, highlighting the need for more mechanism-based biomarker discovery.
Recently, there has been much progress on ML-based predictive biomarkers in indications other than oncology using various types of input data. Tasaki et al.72 applied ML approaches to multi-omics data to better understand drug responses for patients with rheumatoid arthritis. Pare et al.73 developed a novel ML framework based on gradient boosted regression trees to build polygenic risk scores for predicting complex traits. Tested on the UK Biobank data set, their SNP-based models were able to explain 46.9% and 32.7% of overall polygenic variance for height and BMI, respectively. In addition, Khera et al.74 developed genome-wide polygenic scores to identify individuals at high risk of coronary artery disease, atrial fibrillation, type 2 diabetes, inflammatory bowel disease and breast cancer.
The rapid evolution of single-cell RNA sequencing technologies has been used for gene clustering and cell-specific biomarker discovery. Single-cell RNA sequencing techniques have been used to identify novel cell types, distinguish cell states, trace development lineages and integrate expression profiles with spatial resolution of cells. However, an unsolved challenge is the reduction in the gene expression measurements from tens of thousands of cells to low-dimension space, typically two or three variables. Ding et al.75 developed a probabilistic generative model, scvis, to reduce the high-dimensional space to the low-dimensional structures in single-cell gene expression data with uncertainty estimates. This tool was then used to analyse four single-cell RNA sequencing data sets and produced 2D representations of the multi-dimensional single-cell RNA sequencing data that could be interpreted to robustly identify cell types. In addition, Rashid et al.76 have used variational autoencoders (VAEs) to transform single-cell RNA sequencing data to a latent encoded feature space that more efficiently differentiates between the hidden tumour subpopulations. Analysis of the encoded feature space revealed subpopulations of cells and the evolutionary relationship between them. The method was completely unsupervised and required minimal pre-processing of the data. Additionally, the method is tolerant of gene expression dropout in single-cell RNA sequencing data sets. Wang and Gu77 proposed deep variational autoencoder for single-cell RNA sequencing data (VASC), a deep multi-layer generative model, for the unsupervised dimension reduction and visualization of this data. Tested on 20 data sets, VASC is superior and has broader data set compatibility than several state-of-the-art dimension-reduction methods such as ZIFA78 and SIMLR79.
One exciting recent development in ML is the rapid rise of feature selection for biomarker discovery. For example, researchers applied unsupervised DL models to extract meaningful representations of gene modules or sample clusters80. Way and Greene81 introduced a VAE model trained on The Cancer Genome Atlas (TCGA) pan-cancer RNA sequencing data and identified specific patterns in the VAE encoded features. Beck et al.82 conducted image analysis and data integration with gene expression and proteomics data to improve the identification of lung squamous cell carcinoma. Nirschl et al.83 showed that a CNN model could better predict the likelihood of cardiac failure from endomyocardial biopsy samples (AUC = 0.97) than two trained cardiac pathologists could (AUC = 0.73 and 0.75).
In all these examples, for ML-generated predictive biomarkers to be more successful, there are several key issues that still need to be addressed. At least some of these issues concern the interpretability of the classifier, considered by at least some end-users to be critical for clinical adoption. One of the other key issues is the need to validate these approaches in the context of multi-site, multi-institutional data sets to demonstrate the generalizability of the approach. The research community is actively addressing these issues and making rapid progresses, including the application of objective approaches and measures for model training and parameter optimization84, model interpretation and extraction of biological insights85, and model reproducibility86.
Computational pathology.
Pathology is a descriptive field, as a pathologist interprets what is seen on a glass slide by visual inspection. Analysis of these glass slides provides a vast amount of information, such as the type of cell present in the tissue and their spatial context. The interplay between tumour and immune cells within the tumour microenvironment is increasingly important in the study of immuno-oncology and is not captured by other technologies.
Pharmaceutical companies need to understand how drug treatments affect particular tissues and cells and need to test thousands of compounds before selecting a candidate for a clinical trial. Furthermore, as the number of clinical trials grows, discovering new biomarkers will be increasingly important to identify patients who will respond to a particular therapy. Increased use of computational pathology that may allow for the discovery of novel biomarkers and generate them in a more precise, reproducible and high-throughput manner will ultimately cut down drug development time and allow patients faster access to beneficial therapies.
Before DL, algorithms for tissue image analysis were often biologically inspired in collaboration with pathologists and required computer scientists to handcraft descriptive features for a computer to classify a certain type of tissue or cell. These studies were aimed at identifying morphological descriptors in widely used haemotoxylin and eosin (H&E)-stained images. Nuclear morphometry was among the earliest implementations of computational pathology, demonstrating the ability to determine associations between computer-generated features and prognosis87. Beck et al.88 looked at cells in the context of their spatial locations within the surrounding tumour stroma and showed associations between stromal features and survival in breast cancer. Lee et al.89 have also demonstrated that computational analysis of tumour-adjacent benign tissue in prostate cancer can reveal information that is typically ignored by pathologists but is associated with progression-free survival. More recently, Lu et al. showed that features that describe nuclear shape and nuclear orientation were strongly associated with survival in both oral cancers90 and early-stage oestrogen receptor-positive breast cancers91. In many cases, the availability of immunohisto-chemical stains, which use antibodies to target specific proteins in an image and mark specific cell and tissue types, circumvents the need for cell and tissue detection by morphology and thus enables the generation of sophisticated data without the use of DL tools. However, in the case of immuno-oncology, ML allows for high-throughput generation of features that describe spatial relationships for thousands of cells, an infeasible task for pathologists. Improvements in individual cell and tissue detection via DL methods allow for very precise measurements of the tumour microenvironment, so heterogeneous features that describe spatial relationships between cells and tissue structures can now be measured at scale (FIG. 5).
Fig. 5 |. Computational pathology tasks for machine learning applications.
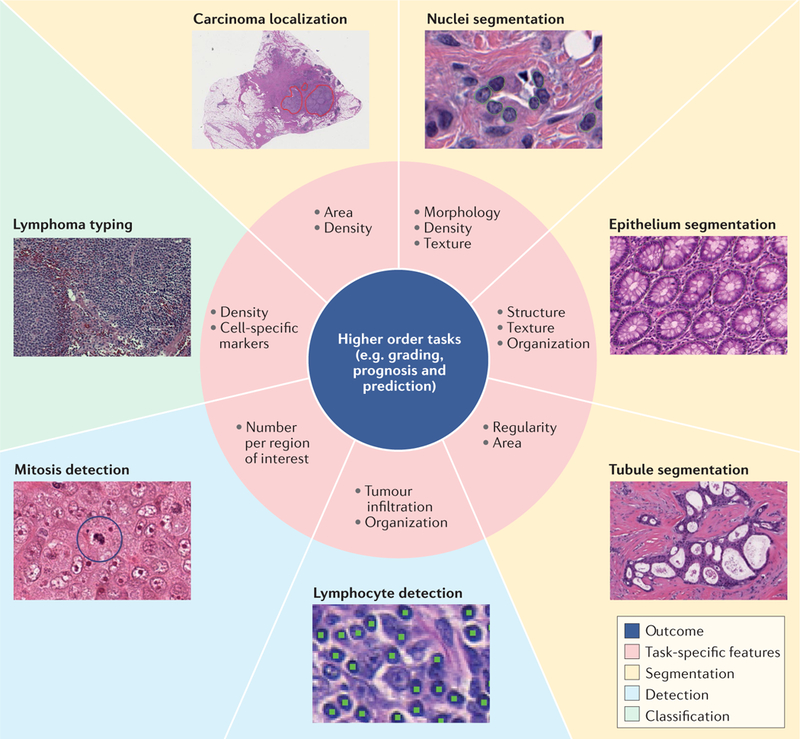
Deep learning frameworks can replace traditional handcrafted features in several basic pathology image-recognition tasks (such as segmentation of nuclei, epitheLia or tubules, lymphocyte detection, mitosis detection or classification of tumours) using image segmentation (yellow background), detection of specific features (blue background) or detection of a set of features used for classification (green background). Recognition is based on the task-specific features shown in the pink regions and can lead to more accurate prognosis or prediction of disease.
In a study by Mani et al.92, several markers for lymphocytes were utilized to understand the heterogeneity of these populations in breast cancer. Giraldo et al.93 examined cell-cell interactions and showed that, using cell densities and the relative location of PD1+ and CD8+ cells, they could identify patients with Merkel cell carcinoma who would respond to pembrolizumab. The trade-off for these types of experiment is that they use a lot of tissue, typically requiring additional slides for each stain; however, hundreds or thousands of features can be examined, and the number of possible cell-cell interactions increases with each stain used. In such a case, a combination of feature selection and ML methods is used to determine combinations that may be predictive of therapeutic response.
The application of CNNs to pathology images works well because there is a large number of viable pixels that can be used for training from a single biopsy or resection. Given enough well-curated exemplars, a DL algorithm can be designed to learn features automatically for a wide variety of classification tasks94. For example, a multi-scale convolutional neural network (M-CNN) was used in a supervised learning approach for phenotyping high-content cellular images9 in a single step as opposed to several, independent customized steps. Using solely pixel intensity values from the images to convert those images into phenotypes, the approach resulted in overall more accurate classification of the effects of a compound treatment at multiple concentrations. Many image analysis challenges have successfully used DL methods to identify areas within cancer tumours95–98, tubules99, mitotic activity100 and lymphocytes101,102 in breast and lung cancer.
Beyond pathology images, DL can also facilitate the integration of other modalities of information. DL can also be used to accelerate magnetic resonance imaging (MRI) data acquisition103 or reduce the radiation dose required for computed tomography (CT) imaging104. With improved imaging quality including temporal and spatial resolution and a high signal to noise ratio, the performance of image analysis may correspondingly improve in applications such as image quantification, abnormal tissue detection, patient stratification and disease diagnosis or prediction. Another recent study105 demonstrated the ability to use an inception DL framework to predict the presence of certain mutated genes from H&E-stained images of lung tumours.
However, although DL continues to excel in many specific image analysis tasks, in practice, a combination of DL and traditional image analysis algorithms is applied in most problem sets. This is done for several reasons. First, while DL has shown its ability to match or outperform humans in very specific problems (such as the detection of glomeruli), it is still not a great general-purpose image analysis tool. Development times remain long owing to this lack of flexibility. There is also an overall scarcity of expert labels available for a specific classification task, as these are expensive to generate. Approaches to mitigate this include using immunohistochemistry staining to provide additional information to pathologists for samples where annotations are challenging106 as well as efforts to increase the availability of well-curated expert annotations for broad-use cases (cancer cells versus normal cells), which is an ongoing community task.
Another challenge is the issue of transparency. DL methods are known for their black-box approach. The underlying rationale behind a decision for classification tasks is unclear. For drug development, it is important to understand mechanisms, and having an interpretable output can be useful for finding not only new potential drug targets but also new potential biomarkers to predict therapeutic response. The generation of many more handcrafted features is needed for increased trust in interpretability.
A further challenge is the large sample size needed in clinical trials to apply DL directly to infer therapeutic response. DL typically requires tens of thousands if not hundreds of thousands of examples to learn from, and clinical trials typically do not produce enough examples. In certain cases, it may be possible to combine data across clinical trials, but biases may exist that can make the results more difficult to interpret.
Examples of successful integration of DL and traditional image analysis workflows include work by Saltz et al.101 and Corredor et al.102, in which CNNs were used to detect lymphocytes in H&E-stained tissue and subsequent graph-based features were extracted to predict disease response. This will likely be a common role for DL in the near future, as its superior ability to detect cells and tissue can replace traditional segmentation and nuclear detection algorithms, and subsequent interpretable features can be applied to give spatial context to these features.
Outlook
ML approaches and recent developments in DL provide many opportunities to increase efficiency across the drug discovery and development pipeline. As such, we expect to see increasing numbers of applications for well-defined problems across the industry in the coming years. With available data becoming ‘bigger’, at least in the sense of more thoroughly covering the relevant variability of the whole data space, and as computers become increasingly more powerful, ML algorithms are going to systematically generate improved outputs, and new, interesting applications are expected to follow. This has been clearly exemplified in the previous sections, in which we have described some ML applications for target identification and validation, drug design and development, biomarker identification and pathology for disease diagnosis and therapy prognosis in the clinic.
These methods are also being applied within the health-care setting, which, when combined with drug discovery, could lead to significant advances in personalized medicine107. ML has also been applied to electronic health records108 and real-world evidence in order to improve clinical trial results and optimize the process of clinical trial eligibility assessment. For example, a recent study demonstrated that DNNs are a highly competitive approach for automatically extracting useful information from electronic medical records for disease diagnoses and classification109. Some studies have shown that ML models in electronic health records can outperform conventional models in predicting prognosis110. ML can also be applied to data now coming from sensors and wearables to understand disease and develop treatments, especially in the neurosciences111. Gkotsis et al.112 applied DL approaches to characterize mental health conditions on unstructured social media data, which is a difficult task for traditional ML approaches.
As shown in FIG. 1, ML approaches are beginning to be commonly used in the various steps of the discovery and development pipeline by pharmaceutical companies. This pervasive implementation of ML methods has a few but important known issues. A typical issue with deep-trained neural networks is the lack of inter-pretability, that is, the difficulties in obtaining a suitable explanation from the trained neural network on how it arrives at the result. If the system is used to diagnose a disease such as melanoma, for instance, on the basis of medical images, this lack of interpretability may hinder scientists, regulatory agencies, doctors and patients, even in situations in which neural networks perform better than human experts. Would a patient trust the ML diagnosis more than that of a human expert? Although much less dramatic, a similar situation may occur in drug design. Would a pharmaceutical company trust a neural network for choosing a small molecule for inclusion in their portfolio and investment to progress to the clinic, without a clear explanation for why the neural network has selected this molecule? In addition, there may be patent application issues with inventor-ship if compounds have been designed by computer algorithms. In any case, ML results have to be considered as only hypotheses or interesting starting points that are then further developed in studies by researchers. Complementary experiments that validate the ML result will help to build trust in approaches and outputs, but regulatory agencies have yet to clarify their view on the lack of interpretability for the clinical use of ML. However, even beyond the issue of trust, the lack of interpretability of the approaches makes it more difficult to troubleshoot these approaches when they unexpectedly fail on new unseen data sets.
Another important issue for neural networks is repeatability, which arises because ML outputs are highly dependent on the initial values or weights of the network parameters or even the order in which training examples are presented to the network, as all of them are typically chosen at random. Would the network always select the same disease target using the same expression data as the input? Would the structure of the drug proposed by the ML method always be the same? This lack of repeatability is particularly problematic for biomarker identification, as seen in situations where different tools generated different prognosis biomarkers for breast cancer on the basis of molecular expression signatures113. The fact that different ML methods can yield different results will add uncertainty to the adoption of these methods at scale. Some solutions to the problems of both interpretability and repeatability have been proposed. These usually centre on the use of a more complex or more time-consuming algorithm or averaging results from several network models, but this might be seen as adding only one more result to a range of existing results.
Another important point to consider is the availability of high-quality, accurate and curated data in large quantities to train and develop ML models. The requirements for the amounts and accuracy desired are dependent on the complexity of the data type and the question to be resolved. Thus, it can be expensive to generate these data sets. Pre-competitive consortia of pharmaceutical companies and academic institutions that use appropriate data standards and have the necessary operational and open data frameworks may be part of the solution to meet these data demands. Many of the data types that are used during drug discovery are far from comprehensive. For example, the knowledge of all folds and structures of proteins is not complete, and coverage of the data space is similarly incomplete. Thus, applications in which these structures are predicted, even if much progress has been made, are not yet as good as in other areas. The same applies for the prediction of reactions involved in the synthesis of small molecules for which the entire chemistry space is unknown.
Data curation is key to the provision of reusable and trustworthy data and can be expensive in terms of the time and skills required. Biological curation — the extraction of biological information from the scientific literature and its integration into a database — lies between an art and a science114, requiring a combination of computational skills with in-depth biological and domain expertise. Collaborative efforts to develop shared data resources and metadata (labels) may be ways by which high-quality data in the public domain can be made more available. This also includes metadata from both successful and failed drug discovery programmes that can enable prediction approaches and determination of factors that can reduce attrition in drug development. Much more pre-competitive collaboration is also needed to aggregate and generate large data resources of corporate bioactive data sets of investigational compounds as well as historic clinical trial data.
Another limitation in the application of ML models is in their use to predict alternative paradigms. Because the entire premise of ML relies on the use of training data to generate suitable models, ML models can only predict within the known framework of the training data. In medicinal chemistry, for example, the design of compounds with alternative mechanisms of action, such as macrocycles, protein-protein interaction inhibitors or PROTACs, can probably only be performed with traditional methods.
As well as data and models, the training of researchers that understand pharmaceutical science as well as computer science, computational statistics and statistical ML and are proficient in utilizing these methods needs to be accelerated. Competitions like the DREAM Challenges (see Related links), which have shown that team composition is a factor in performance, can also be useful to attract talent and advance methodology development. However, applications will need to be successful in the clinical setting in order to motivate further investment from large pharmaceutical and technology companies.
ML algorithms, including DL methods, have enabled the utilization of AI in the industry setting and in day to day life. The impact of ML methods in all areas of drug discovery and health care is already being felt, especially in the analysis of omics and imaging data. ML algorithms are also successful in speech recognition, NLP, computer vision and other applications. For example, Internet-enabled smart assistants are now commonplace and can transmit health-related information in the form of speech and images or videos. ML approaches applied to data collected from such an amalgamation of Internet-enabled technologies, coupled with biological data, have the potential to dramatically improve the predictive power of such algorithms and aid medical decision making about the therapeutic benefits, clinical biomarkers and side effects of therapies.
Graphical processing units
(GPUs). Processors designed to accelerate the rendering of graphics and that can handle tens of thousands of operations per cycle.
Central processing units
(CPUs). Processors designed to solve every computational problem in a general fashion and that can handle tens of operations per cycle. The cache and memory are designed to be optimal for any general programming problem.
Tensor processing units
(TPUs). Co-processors manufactured by Google that are designed to accelerate deep learning tasks developed using TensorFlow (a programming framework) and can handle up to 128,000 operations per cycle.
Support vector machine (SVM) classifier
A method that performs classification tasks by constructing separating lines to distinguish between objects with different class memberships in a multidimensional space.
CLIP–seq
Ultraviolet crosslinking immunoprecipitation (CLIP) followed by RNA sequencing to identify all RNA species bound by a protein of interest. This method can be used to map RNA protein binding sites or RNA modification sites on a genome-wide scale.
Heuristic method
A function that calculates the approximate cost of a problem (or ranks alternatives).
Chemical fingerprint
A concept used in chemical informatics to compare molecules with each other. The structure of a molecule is encoded in a series of binary digits (bits) that represent the presence or absence of particular substructures in the molecule.
Simplified molecular input line entry system (SMILES)
A line notation for entering and representing molecules and reactions; for example, carbon dioxide is represented as O=C= O.
Acknowledgements
The authors thank E. Birney and E. Papa for helpful comments, M. Segler for contributing to the small-molecule optimization subsection and A. Janowczyk for providing the pathology images in Figure 4.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
RELATED LiNKS
DeepChem: https://www.deepchem.io/
DREAM Challenges: http://dreamchallenges.org/
TensorFlow: https://www.tensorflow.org/
References
- 1.Mamoshina P et al. Machine learning on human muscle transcriptomic data for biomarker discovery and tissue-specific drug target identification. Front. Genet. 9, 242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LeCun Y, Bengio Y & Hinton G Deep learning. Nature 521, 436 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Engkvist O, Wang Y, Olivecrona M & Blaschke T The rise of deep learning in drug discovery. Drug Discov. Today 23, 1241–1250 (2018). This article is the first effort to highlight the recent applications of DL in drug discovery research and is an introduction to some popular DL architectures. [DOI] [PubMed] [Google Scholar]
- 4.Hinton G Deep learning — a technology with the potential to transform health care. JAMA 320, 1101–1102 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Wong CH, Siah KW & Lo AW Estimation of clinical trial success rates and related parameters. Biostatistics 10.1093/biostatistics/kxx069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeon J et al. A systematic approach to identify novel cancer drug targets using machine learning, inhibitor design and high-throughput screening. Genome Med. 6, 57 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrero E, Dunham I & Sanseau P In silico prediction of novel therapeutic targets using gene-disease association data. J. Transl Med. 15, 182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riniker S, Wang Y, Jenkins J & Landrum G Using information from historical high-throughput screens to predict active compounds. J. Chem. Inf. Model. 54, 1880–1891 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Godinez WJ, Hossain I, Lazic SE, Davies JW & Zhang X A multi-scale convolutional neural network for phenotyping high-content cellular images. Bioinformatics 33, 2010–2019 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Olsen T et al. Diagnostic performance of deep learning algorithms applied to three common diagnoses in dermatopathology. J. Pathol. Inform. 9, 32–32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava N, Hinton G, Krizhevsky A, Sutskever I & Salakhutdinov R Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 15, 1929–1958 (2014). [Google Scholar]
- 12.Jiao Y & Pufeng D Performance measures in evaluating machine learning based bioinformatics predictors for classifications. Quant. Biol. 4, 320 (2016). [Google Scholar]
- 13.Czodrowski P Count on kappa. J. Comput. Aided Mol. Des. 28, 1049–1055 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Rifaioglu AS et al. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief. Bioinform. 10.1093/bib/bby061 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinton GE & Salakhutdinov RR Reducing the dimensionality of data with neural networks. Science 313, 504 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Koscielny G et al. Open targets: a platform for therapeutic target identification and validation. Nucleic Acids Res. 45, D985–D994 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costa PR, Acencio ML & Lemke N A machine learning approach for genome-wide prediction of morbid and druggable human genes based on systems-level data. BMC Genomics 11, S9–S9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ament SA et al. Transcriptional regulatory networks underlying gene expression changes in Huntington’s disease. Mol. Systems Biol. 14, e7435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bravo A, Pinero J, Queralt-Rosinach N, Rautschka M & Furlong LI Extraction of relations between genes and diseases from text and large-scale data analysis: implications for translational research. BMC Bioinformatics 16, 55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J, Kim J. j. & Lee H An analysis of disease-gene relationship from Medline abstracts by DigSee. Sci. Rep. 7, 40154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leung MKK, Xiong HY, Lee LJ & Frey BJ Deep learning of the tissue-regulated splicing code. Bioinformatics 30, i121–i129 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jha A, Gazzara MR & Barash Y Integrative deep models for alternative splicing. Bioinformatics 33, i274–i282 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaquero-Garcia J et al. A new view of transcriptome complexity and regulation through the lens of local splicing variations. eLife 5, e11752 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sotillo E et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 5, 1282–1295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rohacek AM et al. ESRP1 mutations cause hearing loss due to defects in alternative splicing that disrupt cochlear development. Dev. Cell 43, 318–331 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiong HY et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347, 1254806 (2015). This article describes a computational model based on DL that predicts splicing regulation for any mRNA sequence and has been applied to more than half a million human mRNA splicing sequence variants. Thousands of known disease-causing mutations are identified as well as new disease-linked genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iorio F et al. A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754 (2016). This paper applies ML to data from somatic mutations, copy number alterations, DNA methylation and gene expression from 1,000 cancer cell lines to model drug response of the cell lines and demonstrates the importance of genomic features for prediction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsherniak A et al. Defining a cancer dependency map. Cell 170, 564–576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMillan EA et al. Chemistry-first approach for nomination of personalized treatment in lung cancer. Cell 173, 864–878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Lazikani B et al. in Bioinformatics — From Genomes to Therapies Ch. 36 (Wiley-VCH, 2008). [Google Scholar]
- 31.Nayal M & Honig B On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins 63, 892–906 (2006). This article describes a classifier to identify drug-binding cavities on the basis of physicochemical, structural and geometric attributes of proteins. [DOI] [PubMed] [Google Scholar]
- 32.Li Q & Lai L Prediction of potential drug targets based on simple sequence properties. BMC Bioinformatics 8, 353 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bakheet TM & Doig AJ Properties and identification of human protein drug targets. Bioinformatics 25, 451–457 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Wang Q, Feng Y, Huang J, Wang T & Cheng G A novel framework for the identification of drug target proteins: combining stacked auto-encoders with a biased support vector machine. PLOS ONE 12, e0176486 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kandoi G, Acencio ML & Lemke N Prediction of druggable proteins using machine learning and systems biology: a mini-review. Front. Physiol. 6, 366–366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson MR et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Morgan P et al. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat. Rev. Drug Discov. 17, 167–181 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Rouillard AD, Hurle MR & Agarwal P Systematic interrogation of diverse Omic data reveals interpretable, robust, and generalizable transcriptomic features of clinically successful therapeutic targets. PLOS Comput. Biol. 14, e1006142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar V, Sanseau P, Simola DF, Hurle MR & Agarwal P Systematic analysis of drug targets confirms expression in disease-relevant tissues. Sci. Rep. 6, 36205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramsundar B et al. Is multitask deep learning practical for pharma? J. Chem. Inf. Model. 57, 2068–2076 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Ma J, Sheridan RP, Liaw A, Dahl GE & Svetnik V Deep neural nets as a method for quantitative structure-activity relationships. J. Chem. Inf. Model. 55, 263–274 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Barati Farimani A, Feinberg E & Pande V Binding pathway of opiates to μ-opioid receptors revealed by machine learning. Biophys. J. 114, 62a–63a (2018). [Google Scholar]
- 43.Wu Z et al. MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 9, 513–530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Segler MHS, Preuss M & Waller MP Planning chemical syntheses with deep neural networks and symbolic AI. Nature 555, 604 (2018). This seminal paper describes a very thorough approach to retrosynthetic analysis. The authors show that their method can compete with retrosynthesis done by experienced chemists who are experts in this field. [DOI] [PubMed] [Google Scholar]
- 45.Olivecrona M, Blaschke T, Engkvist O & Chen H Molecular de-novo design through deep reinforcement learning. J. Cheminform. 9, 48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kadurin A, Nikolenko S, Khrabrov K, Aliper A & Zhavoronkov A druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol. Pharm. 14, 3098–3104 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Smith JS, Roitberg AE & Isayev O Transforming computational drug discovery with machine learning and AI. ACS Med. Chem. Lett. 9, 1065–1069 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lenselink EB et al. Beyond the hype: deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J. Cheminform. 9, 45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaulton A et al. The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramsundar B et al. Massively multitask networks for drug discovery. Preprint at arXiv https://arxiv.org/abs/1502.02072 (2015).
- 51.Gutlein M & Kramer S Filtered circular fingerprints improve either prediction or runtime performance while retaining interpretability. J. Cheminform. 8, 60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayr A et al. Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. Chem. Sci. 9, 5441–5451 (2018). This research paper describes the methodology being used by the winners of almost all categories of the Tox21 Challenge. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keiser MJ et al. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 25, 197 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Preuer K, Renz P, Unterthiner T, Hochreiter S & Klambauer G Frechet ChemNet Distance: a metric for generative models for molecules in drug discovery. J. Chem. Inf. Model. 58, 1736–1741 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Unterthiner T, Mayr A, Klambauer G & Hochreiter S Toxicity prediction using deep learning. Preprint at arXiv https://arxiv.org/abs/1503.01445 (2015). [Google Scholar]
- 56.Li B et al. Development of a drug-response modeling framework to identify cell line derived translational biomarkers that can predict treatment outcome to erlotinib or sorafenib. PLOS ONE 10, e0130700 (2015). In this paper, a translational predictive biomarker is used to demonstrate that predictive models can be generated from preclinical training data sets and then be applied to clinical patient samples to stratify patients, infer the mechanism of action of a drug and select appropriate disease indications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Gool AJ et al. Bridging the translational innovation gap through good biomarker practice. Nat. Rev. Drug Discov. 16, 587–588 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Kraus VB Biomarkers as drug development tools: discovery, validation, qualification and use. Nat. Rev. Rheumatol. 14, 354–362 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Shi L et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat. Biotechnol. 28, 827–838 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhan F et al. The molecular classification of multiple myeloma. Blood 108, 2020–2028 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shaughnessy JD Jr. et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 109, 2276–2284 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Zhan F, Barlogie B, Mulligan G, Shaughnessy JD Jr & Bryant, B. High-risk myeloma: a gene expression based risk-stratification model for newly diagnosed multiple myeloma treated with high-dose therapy is predictive of outcome in relapsed disease treated with single-agent bortezomib or high-dose dexamethasone. Blood 111, 968–969 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Decaux O et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the Intergroupe Francophone du Myelome. J. Clin. Oncol. 26, 4798–4805 (2008). [DOI] [PubMed] [Google Scholar]
- 64.Mulligan G et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 109, 3177–3188 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Costello JC et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 32, 1202–1212 (2014). This paper is an effort to collect and objectively evaluate various ML approaches by teams around the world on multi-omics data sets and various compounds. The data sets and results are continuously used as benchmarks for new method developments and validation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rahman R, Otridge J & Pal R IntegratedMRF: random forest-based framework for integrating prediction from different data types. Bioinformatics 33, 1407–1410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bunte K, Leppaaho E, Saarinen I & Kaski S Sparse group factor analysis for biclustering of multiple data sources. Bioinformatics 32, 2457–2463 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Huang C, Mezencev R, McDonald JF & Vannberg F Open source machine-learning algorithms for the prediction of optimal cancer drug therapies. PLOS ONE 12, e0186906 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hejase HA & Chan C Improving drug sensitivity prediction using different types of data. CPT Pharmacometrics Syst. Pharmacol. 4, e2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim ES et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 1, 44–53 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boyiadzis MM et al. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J. Immunother. Cancer 6, 35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tasaki S et al. Multi-omics monitoring of drug response in rheumatoid arthritis in pursuit of molecular remission. Nat. Commun. 9, 2755 (2018). This work identifies molecular signatures that are resistant to drug treatments and illustrates a multi-omics approach to understanding drug response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paré G, Mao S & Deng WQ A machine-learning heuristic to improve gene score prediction of polygenic traits. Sci. Rep. 7, 12665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Khera AV et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ding J, Condon A & Shah SP Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat. Commun. 9, 2002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rashid S, Shah S, Bar-Joseph Z & Pandya R Project Dhaka: variational autoencoder for unmasking tumor heterogeneity from single cell genomic data. Preprint at bioRxiv https://www.biorxiv.org/content/10.1101/183863v4 (2018). [DOI] [PMC free article] [PubMed]
- 77.Wang D & Gu J VASC: dimension reduction and visualization of single-cell RNA-seq data by deep variational autoencoder. Genomics Proteomics Bioinformatics 16, 320–331 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pierson E & Yau C ZIFA: dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol. 16, 241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang B, Zhu J, Pierson E, Ramazzotti D & Batzoglou S Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat. Methods 14, 414 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Tan J, Hammond JH, Hogan DA & Greene CA-O ADAGE-based integration of publicly available Pseudomonas aeruginosa gene expression data with denoising autoencoders illuminates microbe-host interactions. mSystems 1, e00025–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Way GP & Greene CS Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders. Pac. Symp. Biocomput. 23, 80–91 (2018). [PMC free article] [PubMed] [Google Scholar]
- 82.Casanova R et al. Morphoproteomic characterization of lung squamous cell carcinoma fragmentation, a histological marker of increased tumor invasiveness. Cancer Res. 77, 2585–2593 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Nirschl JJ et al. A deep-learning classifier identifies patients with clinical heart failure using whole-slide images of H&E tissue. PLOS ONE 13, e0192726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Angermueller C, Parnamaa T, Parts L & Stegle O Deep learning for computational biology. Mol. Syst. Biol. 12, 878 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Finnegan A & Song JS Maximum entropy methods for extracting the learned features of deep neural networks. PLOS Comput. Biol. 13, e1005836 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hutson M Artificial intelligence faces reproducibility crisis. Science 359, 725–726 (2018). [DOI] [PubMed] [Google Scholar]
- 87.Veltri RW, Partin AW & Miller MC Quantitative nuclear grade (QNG): a new image analysis-based biomarker of clinically relevant nuclear structure alterations. J. Cell. Biochem. Suppl. 35, S151–S157 (2000). [DOI] [PubMed] [Google Scholar]
- 88.Beck AH et al. Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Sci. Transl Med. 3, 108ra113 (2011). [DOI] [PubMed] [Google Scholar]
- 89.Lee G et al. Nuclear shape and architecture in benign fields predict biochemical recurrence in prostate cancer patients following radical prostatectomy: preliminary findings. Eur. Urol. Focus 3, 457–466 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu C et al. An oral cavity squamous cell carcinoma quantitative histomorphometric-based image classifier of nuclear morphology can risk stratify patients for disease-specific survival. Mod. Pathol. 30, 1655–1665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu C et al. Nuclear shape and orientation features from H&E images predict survival in early-stage estrogen receptor-positive breast cancers. Lab. Invest. 98, 1438–1448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mani NL et al. Quantitative assessment of the spatial heterogeneity of tumor-infiltrating lymphocytes in breast cancer. Breast Cancer Res. 18, 78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Giraldo NA et al. The differential association of PD-1, PD-L1, and CD8 + cells with response to pembrolizumab and presence of Merkel cell polyomavirus (MCPyV) in patients with Merkel cell carcinoma (MCC). Cancer Res. 77, 662 (2017). [Google Scholar]
- 94.Janowczyk A & Madabhushi A Deep learning for digital pathology image analysis: a comprehensive tutorial with selected use cases. J. Pathol. Informat. 7, 29 (2016). This article is the first comprehensive review of DL in the context of digital pathology images. The paper also systematically explains and presents approaches for training and validating DL classifiers for a number of image-based problems in digital pathology, including cell detection, segmentation and tissue classification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sharma H, Zerbe N, Klempert I, Hellwich O & Hufnagl P Deep convolutional neural networks for automatic classification of gastric carcinoma using whole slide images in digital histopathology. Comput. Med. Imaging Graph. 61, 2–13 (2017). [DOI] [PubMed] [Google Scholar]
- 96.Korbar B et al. Deep learning for classification of colorectal polyps on whole-slide images. J. Pathol. Informat. 8, 30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bychkov D et al. Deep learning based tissue analysis predicts outcome in colorectal cancer. Sci. Rep. 8, 3395 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cruz-Roa A et al. Accurate and reproducible invasive breast cancer detection in whole-slide images: A Deep Learning approach for quantifying tumor extent. Sci. Rep. 7, 46450 (2017). This is one of the first papers to apply DL to identify regions of breast cancer on digital pathology images and shows that the algorithmic approach outperforms breast cancer pathologists. It is one of the first studies to have a large data set of cases (>600) with independent training and validation sets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Romo-Bucheli D, Janowczyk A, Gilmore H, Romero E & Madabhushi A Automated tubule nuclei quantification and correlation with oncotype DX risk categories in ER + breast cancer whole slide images. Sci. Rep. 6, 32706 (2016). This article applies DL to identify the presence and location of tubules in breast pathology images and subsequently demonstrates that the number of detected tubules correlates with the risk assessments of breast cancer via a genomic test. It is one of the first papers to show how DL can be used to establish genotype–phenotype associations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Romo-Bucheli D, Janowczyk A, Gilmore H, Romero E & Madabhushi A A deep learning based strategy for identifying and associating mitotic activity with gene expression derived risk categories in estrogen receptor positive breast cancers. Cytometry A 91, 566–573 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saltz J et al. Spatial organization and molecular correlation of tumor-infiltrating lymphocytes using deep learning on pathology images. Cell Rep. 23, 181–193 (2018). This large-scale study utilizes DL to identify lymphocytes across all images and relate spatial characteristics of lymphocytes to molecular assessments. This article is key to the automatic quantification of immune cells from H&E slides and the identification of sub-categories of immune infiltrate as related to therapeutic outcome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Corredor G et al. Spatial architecture and arrangement of tumor-infiltrating lymphocytes for predicting likelihood of recurrence in early-stage non-small cell lung cancer. Clin. Cancer Res. 25, 1526–1534 (2018). In this paper, the spatial arrangement, and not just the density, of tumour-infiltrating lymphocytes in early-stage lung cancer pathology images is shown to be prognostic of recurrence. A comprehensive comparison is provided, showing that computer-extracted features of spatial arrangement of tumour-infiltrating lymphocytes are more prognostic than manual (pathologist) enumeration of tumour-infiltrating lymphocyte density. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cohen O, Zhu B & Rosen MS MR fingerprinting Deep Reconstruction NEtwork (DRONE). Magn. Reson. Med. 80, 885–894 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen H et al. Low-dose CT with a residual encoder-decoder convolutional neural network (RED-CNN). Preprint at arXiv https://arxiv.org/abs/1702.00288 (2017). [DOI] [PMC free article] [PubMed]
- 105.Coudray N et al. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat. Med. 24, 1559–1567 (2018). This paper uses DL frameworks to predict mutations from H&E images, which has implications for identifying key mechanistic insights from standard whole-slide imaging as well as for patient stratification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Turkki R, Linder N, Kovanen PE, Pellinen T & Lundin J Antibody-supervised deep learning for quantification of tumor-infiltrating immune cells in hematoxylin and eosin stained breast cancer samples. J. Pathol. Inform. 7, 38 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Norgeot B, Glicksberg BS & Butte AJ A call for deep-learning healthcare. Nat. Med. 25, 14–15 (2019). [DOI] [PubMed] [Google Scholar]
- 108.Esteva A et al. A guide to deep learning in healthcare. Nat. Med. 25, 24–29 (2019). [DOI] [PubMed] [Google Scholar]
- 109.Yang Z et al. Clinical assistant diagnosis for electronic medical record based on convolutional neural network. Sci. Rep. 8, 6329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Steele AJ, Denaxas SC, Shah AD, Hemingway H & Luscombe NM Machine learning models in electronic health records can outperform conventional survival models for predicting patient mortality in coronary artery disease. PLOS ONE 13, e0202344 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mohr DC, Zhang M & Schueller SM Personal sensing: understanding mental health using ubiquitous sensors and machine learning. Annu. Rev. Clin. Psychol. 13, 23–47 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gkotsis G et al. Characterisation of mental health conditions in social media using Informed Deep Learning. Sci. Rep. 7, 45141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Koscielny S Why most gene expression signatures of tumors have not been useful in the clinic. Sci. Transl Med. 2, 14ps12 (2010). [DOI] [PubMed] [Google Scholar]
- 114.Odell SG, Lazo GR, Woodhouse MR, Hane DL & Sen TZ The art of curation at a biological database: principles and application. Curr. Plant Biol. 11–12, 2–11 (2017). [Google Scholar]