

Key Clinical Message
Different mutations in glutamate receptor ionotropic delta 2 (GRID2) gene cause cerebellar ataxia in human. We report the largest homozygous deletion of the GRID2 gene reported to date, most probably causing complete loss of the gene product. Our patient presents mainly early onset cerebellar ataxia, cerebellar atrophy, nystagmus, and developmental delay with the least amount of intellectual disability.
Keywords: cerebellar ataxia, cerebellar atrophy, deletion of GRID2, developmental delay, oligo array CGH
1. INTRODUCTION
Glutamate receptor ionotropic delta 2 (GRID2) gene is known to be selectively expressed in Purkinje cells in the cerebellum where they play a key role in synaptogenesis, synaptic plasticity, and motor coordination. Different mutations in GRID2 including deletions cause cerebellar ataxia in human.
Here, we report the largest homozygous deletion of the GRID2 gene reported to date detected by oligo array Comparative Genomic Hybridization (oa‐CGH) in an Iranian family.
Our patient, who has this 1.3 Mb loss, encompassing exons 3‐16 of GRID2 gene presents mainly early onset cerebellar ataxia, cerebellar atrophy, nystagmus, and developmental delay with the least amount of intellectual disability in comparison to other reported cases; suggesting that the type of mutation is the source of phenotypic variability.
Glutamate receptor ionotropic delta 2 gene encodes glutamate receptor channel delta‐2 subunit, a member of glutamate receptor family. Ionotropic glutamate receptors are the predominant excitatory neurotransmitter receptors in the mammalian brain. The protein encoded by this gene is thought to be expressed selectively in cerebellar Purkinje cells and plays a role in synaptic organization between parallel fibers and Purkinje cells.1 Alternate splicing results in multiple transcript variants encoding distinct isoforms. The gene was mapped on chromosome 4q22 in 1998.2 A heterozygous point mutation in the mouse ortholog results in the phenotype named “lurcher”, leading to ataxia as a result of selective, cell‐autonomous apoptosis of cerebellar Purkinje cells during postnatal development.3 In mice, the homozygous state of this mutation leads to death shortly after birth from massive loss of mid‐ and hindbrain neurons during late embryogenesis. Deletions of the extracellular domain of the receptor cause hotfoot phenotype in mice that are characterized by cerebellar ataxia and mild abnormalities of the cerebellum.4, 5, 6
Mutations in GRID2 have been shown to cause cerebellar ataxia in human. Both heterozygous and homozygous deletions and point mutations of the gene have been reported.7, 8, 9 We report the largest homozygous deletion of GRID2 reported to date. We detected this deletion by oa‐CGH in an Iranian pedigree with two affected brothers (Figure 1).
Figure 1.
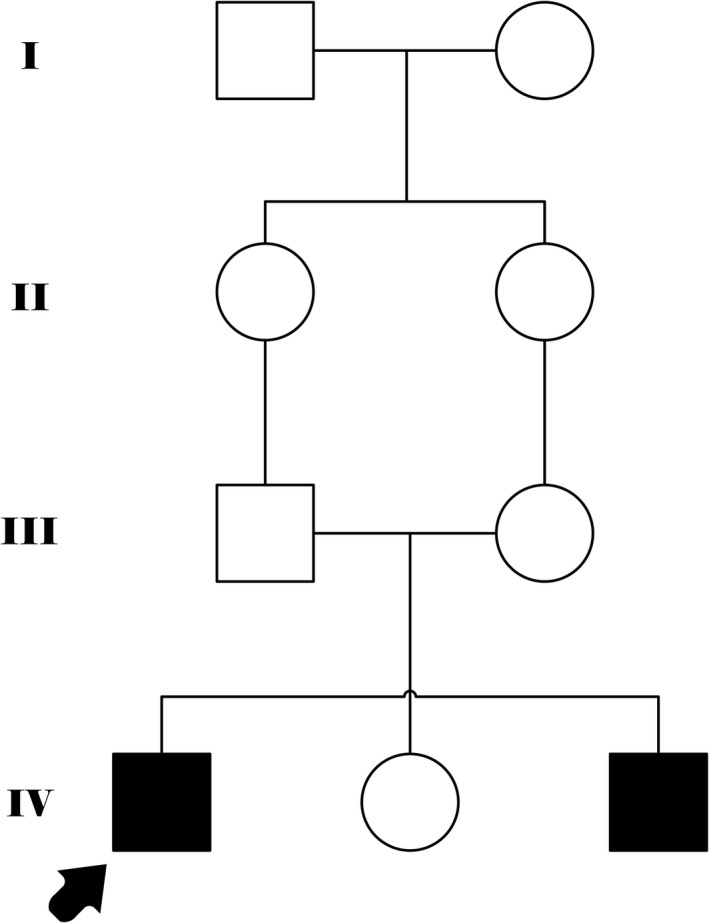
Family pedigree shows two affected brothers from the consanguineous marriage
2. METHODS
2.1. Patient reports
The proband is a 29‐year‐old male patient. He was referred for premarital genetic counseling with chief complaint of ataxia and distal muscle weakness. He is the firstborn son of a consanguineous first cousin couple. He has a normal sister and a similarly affected 11‐year‐old brother. According to his parents, pregnancy and delivery were uneventful, but early developmental milestones were delayed. He held neck at 9 months, sat at 4 years, and walked with support at six. Speech development was delayed. He was able to finish middle school (ninth grade).
At present, he walks with a walker due to severe ataxia and bilateral weakness of lower limbs. He can stand with support. His speech is nasal and delayed with a stutter. His present height is 168 cm (10th percentile), and he weighs 65 kg (between 25th and 50th percentile) with head circumference of 59 cm (over 95th percentile). He has horizontal nystagmus, and the other facial features are normal. The force of upper extremities and that of lower extremities is reduced. He has bilateral pes planus and hallux valgus. Deep tendon reflexes are slightly increased in lower extremities (3+). Babinski reflex is mute (signs of pyramidal involvement), and muscle tone is slightly spastic. The ataxia is truncal and appendicular. Finger to nose test is impaired. Brain MRI shows cerebellar atrophic changes (Figure 2).
Figure 2.
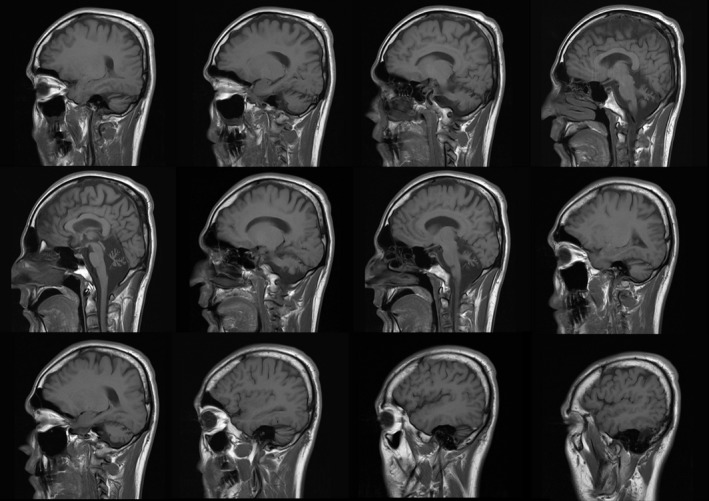
MRI images of the patient's brain; atrophic changes are noted in cerebellum diffusely
2.2. Genetic testing
Karyotype was performed and is normal. All molecular tests were performed on DNA extracted from peripheral blood. oa‐CGH of proband was performed on Cytochip 4X44 array. oa‐CGH of parents was performed on Cytochip 8X60 array. DNA was labeled and hybridized according to manufacturer's protocols. Slides were scanned on (Innopsys, Innoscan 910, Carbonne, France) laser scanner. All data were analyzed using Bluefuse Multi software version 2.0 (Blugnome Ltd., Cambridge, UK).
3. RESULTS
The results of oa‐CGH of proband showed a loss of 1.3 Mb on 4q22.1q22.2 from nucleotide 93 525 to 94 836 Kb. Due to the mean ratio of signal intensity, the loss was presumed to be homozygous. Identical loss of 1.3 Mb on 4q22.1q22.2 from nucleotide 93 184 to 94 518 Kb was detected in both parents. The loss in proband and parents includes exons 3‐16 of the GRID2 gene (Figure 3).
Figure 3.
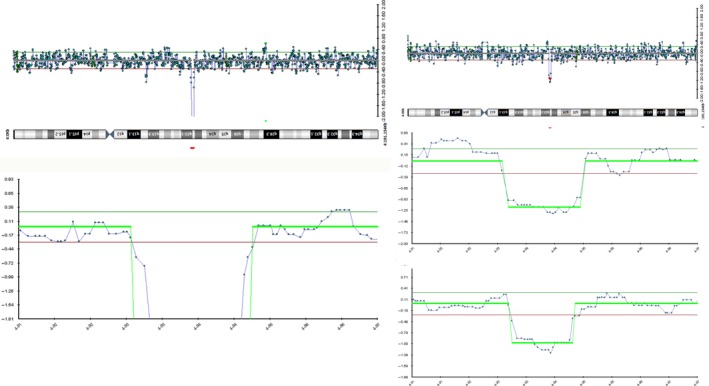
Oligo array comparative genomic hybridization reveals a 1.3 Mb loss on chromosome 4q22.1q22.2 in proband (right) and both parents (left middle: mother, left bottom father). The loss in the proband shows a ratio consistent with homozygosity
4. DISCUSSION
The homozygous loss detected in our patient is the largest homozygous deletion of the GRID2 gene reported to date. Homozygous deletion was reported for the first time by Utine et al in three members of a Turkish consanguineous family. The deletion in this family was detected by Affymetrix SNP array and reported to be approximately140 kb covering exons 3 and 4 of the GRID2 gene.7 In 2013, Hills et al reported homozygous deletions of the GRID2 gene in 2 unrelated families of Jordanian and Mexican descent. The deletion in the first family was of 37 kb including exon 4 and in the second family 50 kb including exon 2 of the GRID2 gene. In the second family, the affected individual also had additional maternal 335 kb loss of exon 2.10 In 2015, Van Schil et al11 reported a 331 kb homozygous deletion of exon 2 in a patient with cerebellar ataxia and retinal dystrophy born of a consanguineous family of Moroccan descent. The present case is a 1.3 Mb loss covering exons 3 to 16 of the gene and larger than any of the previously reported deletions, most probably causing complete loss of the gene product.
The phenotype reported by Utine et al7 includes nystagmus, hypotonia, truncal ataxia, developmental delay, oculomotor apraxia, and coordination defect with cerebellar and pyramidal tract involvement, accompanied by progressive cerebellar atrophy on serial neuroimaging studies. Hills et al10 reported developmental delay, hypotonia, tonic upgaze, truncal and appendicular ataxia, and progressive atrophy of cerebellar vermis and hemispheres. Their patients exhibit bursts of horizontal nystagmus. The presence of ataxia, cerebellar atrophy, nystagmus, and developmental delay appear to be consistent findings in all reported patients. The progression of cerebellar atrophy was documented in the cases reported by Hills et al10 and probably consistent with the diffuse atrophy detected in our patient. Considering that our patient is 29 years old and the oldest patient reported with congenital onset disease, the defects present in the lower distal extremities of our patient might be secondary to the ataxic gait and therefore not present in any of the other reported younger patients. The patient reported by Van Schil et al showed retinal dystrophy that was assumed to be related to the Grid2 deletion as other possible genomic causes were ruled out by next‐generation sequencing. As we became aware of the paper after the initial patient workup, we were unable to establish this point in our patient. Despite the early developmental delay in all patients reported and in our proband, the extent of intellectual disability is not consistent between previously reported cases and our patient. Our patient is the oldest one, yet he shows the least amount of intellectual disability. He was able to finish middle school in a normal school with average grades and is able to communicate with a slow and halted speech, suggesting that early developmental delay and progressive ataxia are more pronounced than intellectual disability. Considering the size of the deletion in our patient and the consequent possible lack of GRID2 gene product, it would be expected that our case would show the most severe phenotype. But as this is not the case, it could be postulated that the patients are educable and the severe developmental delay manifests as intellectual disability in the earlier years. It could also be postulated that lack of the protein caused by loss of the gene is less deleterious as in our patient than having a defective protein product caused by loss of fewer exons or point mutations. Coutelier et al report point mutations in an extensive family in which heterozygous carriers manifest a late onset progressive cerebellar ataxia and homozygous individuals show congenital onset phenotype.9 In the case of gene deletions, it is the homozygote carriers who show congenital onset disease. There have been no reports indicating that the heterozygote parents of these cases were also affected or showed signs of the cerebellar involvement. The only manifesting heterozygote deletion carrier was reported by Maier et al where there is loss of exon 1 in a patient with spastic paraplegia, ataxia, frontotemporal dementia, and lower motor neuron involvement. Further case studies and phenotype–genotype correlation are necessary to determine the mechanism and extent of intellectual disability in deletions of GRID2 gene and the age of onset of cerebellar ataxia. This case and the former cases also present evidence of the deleterious effect of copy number variations in autosomal recessive conditions by homozygosity resulting from consanguinity. It appears that the role of genomic imbalances must be taken into consideration as a possible mechanism for manifestation of autosomal recessive disorders.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
AUTHOR CONTRIBUTION
MT, MF, and AK: gathered all clinical materials and images, examined the patient, and correlated genotype to phenotype. GA and NS: performed oa‐CGH on patient and both parents and analyzed the results. AM: analyzed the results. KN: reviewed the literature and drafted the initial version of manuscript. RK: revised the manuscript and interpreted genetic results.
ACKNOWLEDGMENT
We would like to thank the family for their cooperation.
Taghdiri M, Kashef A, Abbassi G, et al. Further delineation of the phenotype caused by a novel large homozygous deletion of GRID2 gene in an adult patient. Clin Case Rep. 2019;7:1149–1153. 10.1002/ccr3.2020
REFERENCES
- 1. Takayama C, Nakagawa S, Watanabe M, Mishina M, Inoue Y. Light‐ and electron‐ microscopic localization of the glutamate receptor channel delta 2 subunit in the mouse Purkinje cell. Neurosci Lett. 1995;188:89‐92. [DOI] [PubMed] [Google Scholar]
- 2. Hu W, Zuo J, De Jager PL, Heintz N. The human glutamate receptor delta 2 gene (GRID2) maps to chromosome 4q22. Genomics. 1998;47:143‐145. [DOI] [PubMed] [Google Scholar]
- 3. Selimi F, Lohof AM, Heitz S, et al. Lurcher GRID2‐induced death and depolarization can be dissociated in cerebellar Purkinje cells. Neuron. 2003;37:813‐819. [DOI] [PubMed] [Google Scholar]
- 4. Lalouette A, Guénet JL, Vriz S. Hotfoot mouse mutations affect the delta 2 glutamate receptor gene and are allelic to lurcher. Genomics. 1998;50:9‐13. [DOI] [PubMed] [Google Scholar]
- 5. Wang Y, Matsuda S, Drews V, Torashima T, Meisler MH, Yuzaki M. A hot spot for hotfoot mutations in the gene encoding the delta2 glutamate receptor. Eur J Neurosci. 2003;17:1581‐1590. [DOI] [PubMed] [Google Scholar]
- 6. Lalouette A, Lohof A, Sotelo C, Guénet J, Mariani J. Neurobiological effects of a null mutation depend on genetic context: comparison between two hotfoot alleles of the delta‐2 ionotropic glutamate receptor. Neuroscience. 2001;105:443‐455. [DOI] [PubMed] [Google Scholar]
- 7. Utine GE, Haliloglu G, Salanci B, et al. A homozygous deletion in GRID2 causes a human phenotype with cerebellar ataxia and atrophy. J Child Neurol. 2013;28:926‐932. [DOI] [PubMed] [Google Scholar]
- 8. Maier A, Klopocki E, Horn D, et al. De novo partial deletion in GRID2 presenting with complicated spastic paraplegia. Muscle Nerve. 2014;49:289‐292. [DOI] [PubMed] [Google Scholar]
- 9. Coutelier M, Burglen L, Mundwiller E, et al. GRID2 mutations span from congenital to mild adult‐onset cerebellar ataxia. Neurology. 2015;84(17):1751‐1759. [DOI] [PubMed] [Google Scholar]
- 10. Hills LB, Masri A, Konno K, et al. Deletions in GRID2 lead to a recessive syndrome of cerebellar ataxia and tonic upgaze in humans. Neurology. 2013;81:1378‐1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Van Schil K, Meire F, Karlstetter M, et al. Early‐onset autosomal recessive cerebellar ataxia associated with retinal dystrophy: new human hotfoot phenotype caused by homozygous GRID2 deletion. Genet Med. 2014;17(4):291‐299. [DOI] [PubMed] [Google Scholar]