

Summary:
Autoimmune skin diseases are complex processes in which autoreactive cells must navigate through the skin tissue to find their targets. Regulatory T cells in the skin help to mitigate autoimmune inflammation and may in fact be responsible for the patchy nature of these conditions. In this review, we will discuss chemokines that are important for global recruitment of T cell populations to the skin during disease, as well as signals that fine-tune their localization and function. We will describe prototypical disease responses and chemokine families that mediate these responses. Lastly, we will include an overview of chemokine-targeting drugs that have been tested as new treatment strategies for autoimmune skin diseases.
Keywords: T Cells, Type 1/Type 2/Type 17 responses, Autoimmunity, Chemokines, Cell Trafficking, Skin
Introduction
The skin is a complex, multi-layered organ that serves as a first line of defense against infection. It provides immunity against pathogens through barrier function as well as anti-microbial peptide production, and discourages microbial growth through salinity and pH from perspiration. Resident immune cells in the skin monitor for infections, tumors, and tissue damage. It is becoming increasingly clear that the immune system in the skin and the non-immune cells that make up the skin itself are intrinsically connected during development and beyond, with immune cells like Tregs enhancing development of the epidermis (1). When skin homeostasis is perturbed, both hematopoietic and non-hematopoietic skin cells produce chemotactic signals to recruit additional immune cells from the blood to help fight infections, kill precancerous cells, or promote wound healing.
In autoimmune diseases, the immune system becomes inappropriately activated and targets self-tissues and cells. We have limited understanding about how the first immune signal breaks tolerance and initiate inflammation during autoimmunity; however, we have more data that address how immune cells then become recruited to the skin through chemotactic signals. These signals must guide immune cells as they navigate through the skin tissue to find their targets, as well as enhance their effector function. In this review, we will discuss how chemokines control T cell recruitment to the skin and fine-tune their migration within the skin at homeostasis, as well as during autoimmune inflammation. Specifically, we will discuss prototypical type 1 (vitiligo, alopecia areata), type 2 (atopic dermatitis, contact hypersensitivity), and type 17 (psoriasis) responses, as well as include brief descriptions of other types of pathologies (cutaneous lupus, morphea).
Overview of skin structure and chemokines at homeostasis
To understand how cells migrate to and through the skin during autoimmunity, it is important to understand the structure and landscape of the skin at homeostasis. In general, the skin is comprised of the epidermis, a tight stratified epithelium, the dermis, a loose, hypocellular and vascularized layer, and the subcutaneous fat. The outermost layer of the epidermis is called the stratum corneum, which is comprised of dead, enucleated keratinocytes. This layer is lipophilic and continually sloughed off to reduce the potential for infection. Below the stratum corneum, the intermediate layer contains keratinocytes in various stages of differentiation that are tightly bound to one another through integrins and other structural proteins that form a barrier to external elements. The deepest layer of the epidermis is comprised of basal keratinocytes (stratum basale), which continually divide and move superficially, forming a “conveyor belt” of differentiating keratinocytes to replace those that are lost. Keratinocytes are important chemokine producers in the skin, which we will discuss further in the context of specific diseases.
Interspersed throughout the epidermal layer are specialized antigen presenting cells called Langerhans cells. These are a subset of dendritic cells (DCs) that may be replenished from bone marrow DC precursors, but are originally derived from fetal liver (2). These cells process and present antigen to other immune cells, most notably T cells.
T cells are also a predominant cell in the epidermis and dermis, with some studies estimating there are more T cells in adult human skin than in peripheral blood (3). Many of these T cells have been labeled tissue resident memory T cells (Trm), as they do not recirculate through the blood and lymph but rather persist in skin for years to protect against potential recurrent infections. Of note, several studies have demonstrated that Trm function at least in part through chemokine production, which was coined their “sentinel alarm function” (4–8). Specific chemokine requirements for resident memory formation are under investigation, although CCR8 is implicated in developing epidermal resident memory (9); however, the upregulation of chemokine receptors in different disease states that allow the recruitment of various T cell subtypes is probably important for seeding the skin with antigen specific T cells in the first place and may explain why others argue for the requirement of other chemokine receptors, such as CXCR3 in the context of vitiligo (10).
Below the epidermis is the dermis. This layer is structurally comprised primarily of fibroblasts and extracellular matrix proteins such as collagen and elastin. Like their keratinocyte counterparts in the epidermis, fibroblasts can be key sources of chemokines in the dermis. Importantly, they also secrete extracellular matrix proteins that can bind to chemokines and trans-present them to immune cells. Formation of the matrix is integral to the function of some chemokines that need to oligomerize to be bioactive (11).
Interspersed throughout the dermis are endothelial cells, which form the blood vessels required for transport of nutrients and wastes to and from the tissue, respectively. Endothelial cells play key roles in skin immunity, including upregulation of adhesion molecules that facilitate rolling, tight adhesion, and extravasation of immune cells into the tissue from the blood. T cells in particular use cutaneous lymphocyte antigen to enter the skin along P-selectin expressing endothelium (12). Lymphatics play key roles in immune cell exit from tissues and migration to skin draining lymph nodes where antigens are filtered and processed for efficient immune activation and coordination of immune responses (13). Nerves innervate the skin and transmit signals that mediate sensation of touch, pain, heat, cold, etc. The interactions between nerves and immune cells have become better appreciated in recent studies, including those that promote itch (14–18) or inflammation characteristic of psoriasis (19).
As in the epidermis, there are dermal-resident immune cells, including antigen presenting cell (APCs) subsets that include CD103+ Langerin+ DCs, CD11b+ DCs, and macrophages (20–22). These cells may provide antigen depots to restimulate lymphocytes in situ for repriming once they enter the skin (23). APCs produce large quantities of chemokines on a per cell basis (despite their representing only a small fraction of total cells) and are therefore key sources of chemokines in tissues (24, 25).
T cells can be found throughout the dermis, patrolling (26) and maintaining disease in the context of autoimmunity (8). Like those in the epidermis, dermal T cells also produce chemokines themselves to promote recruitment of additional T cells (8) and/or to promote clustering (27), although dermal T cells are much more mobile in the looser tissue as compared to the tightly-packed epidermis (28). This activity is somewhat similar to how ants produce trails of pheromones to direct additional ants toward a newly discovered food source.
The epidermis and dermis are joined at what is termed the dermal-epidermal junction, or DEJ, which is comprised mostly of tight type VII collagen fibers. Melanocytes, which are the pigment-producing cells in the skin, and Merkel cells, which are specialized nerve fibers that mediate sensation of light touch, reside at the DEJ. Several diseases we will discuss are characterized by an “interface dermatitis,” which is essentially inflammation at the DEJ.
The subcutaneous fat layer is just below the dermis, and its role in skin immunity is becoming better appreciated especially in specific diseases such as scleroderma and morphea (skin-limited sclerosis). Fat cells directly produce chemokines and release of specific lipid mediators that help direct immune cell movement and activation status via changes in metabolism.
Hair follicles are interspersed throughout the skin, span the epidermis, dermis, and subcutaneous fat, and have recently been described as “entry portals” for bone-marrow derived Langerhans cell precursors (29). Further, regulatory T cells (Tregs) accumulate around hair follicles and may prevent immune responses to these constantly changing organs as they routinely grow, rest, fall out and regenerate depending on the stage of the hair cycle (anagen, telogen, catagen, etc; (1)). Tregs reside near hair follicles (30, 31) and help to tolerize against commensal skin microbiota (32). Nagao et al has described CCL8 being produced by the hair follicle, which may be important for recruiting Tregs into the hair follicle, as Tregs express the cognate receptor CCR8 (29). Hair follicles also provide a niche for stem cells, including melanocyte precursor cells that can repopulate and repigment the skin.
Recirculating immune cells follow homeostatic chemotactic signals to enter the skin, such as CXCL14, CXCL16, CCL17/CCL22, and CCL27 (reviewed in (33)). Cytokines, which can induce chemokinesis by inducing the release of chemokines, have recently been described to be essential for resident memory T cells and Langerhans cells: IL-15 is homeostatically produced by keratinocytes and presented on CD215 in trans to promote establishment of Trm ((34), reviewed in (35)). Together with IL-7, it is also important for directing migration of Langerhans cell precursors through the hair follicle and up into the epidermis (29). Future studies will need to be conducted to ascertain which chemokine families are acting downstream of IL-7 and IL-15 in the directed migration of Trm and LHC to the epidermis. CXCL14, whose receptor is unknown, facilitates recirculation of immune cells in the skin and provides antimicrobial function (reviewed in (36)). CXCL16 is constitutively produced on the surface of keratinocytes, but is proteolytically cleaved to recruit CXCR6+ T cells and NKT cells (37, 38). TSLP, which was extensively studied in the context of allergic disease, enhances barrier protection of the skin and increases Treg numbers during homeostasis (39–41)(39, 40). CCL17/CCL22 are ligands for CCR4 that promote recirculation of T cells to the epidermis at homeostasis (42) or towards antigen presenting cells (APCs) in the dermis during inflammatory states (43). The majority of blood CLA+ T cells express CCR4 to respond to these skin-homing ligands (44). CCL27 is specifically produced in the epidermis and helps position CCR10 expressing lymphocytes superficially in the skin (45, 46) and reviewed in (47). Skin DCs use vitamin D3 to program T cells to respond to CCL27 via upregulation of CCR10 (48). Of note, many human T cells express both CCR10 and CCR8, and are likely recruited to the skin in a stepwise fashion: at baseline, dermal cell types produce CCL1 that helps recruit CCR8-expressing T cells into dermis (9, 49) reviewed in (50), followed by CCL27 to position them in the epidermis via CCR10 signaling. Tregs first seed the skin utilizing CCR6 and presumably are maintained by the hair follicles utilizing the same signals (32). Together, all of these homeostatic chemokines facilitate T cell recruitment to and through the skin (Figure 1).
Figure 1. Homeostatic chemokines mediate T cell migration to and through the skin.
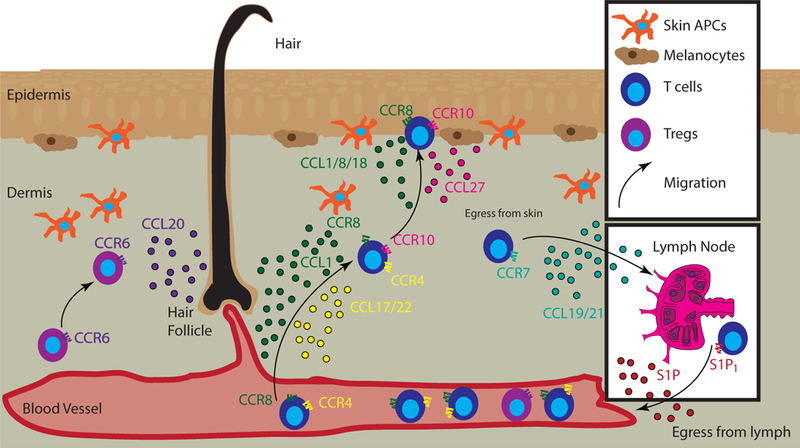
T cells (blue) that express CCR8 and/or CCR4 follow their respective ligand gradients, CCL1 (green dots) and/or CCL17/CCL22 (yellow dots) to enter the dermis, respectively. Epidermal positioning is mediated by CCL1/CCL8/CCL18 (green dots), which all bind to CCR8 on T cells. This second step of migration can also be mediated by CCL27 (pink) and its receptor CCR10. CCL20 (purple dots) is produced predominantly near the hair follicles, and serves to recruit and retain CCR6+ Tregs. T cells expressing CCR7 and CD62L follow CCL19/21 (turquoise) to exit the skin and enter the lymph node via afferent lymphatics. S1P1 mediates egress from the lymph node to the blood via an S1P gradient (red).
To egress from skin, T cells follow CCL19 and CCL21 signals (CCR7 ligands) presented on the lymphatic endothelium to enter skin-draining lymph nodes (reviewed in (51)). To recirculate back to the skin, T cells egress from lymph nodes by following sphingosine-1-phosphate (S1P) gradients to re-enter the circulation and then follow chemokine signals to re-enter the skin (52). Homeostatic signals may be modulated in disease states to facilitate enhanced trafficking of T cells to tissues as well as their retention there.
Homeostatic chemokine and cytokine signals in the skin also help to maintain and bolster the function of resident immune cell populations we have described above (reviewed in (53)). IL-15 is important for maintenance and function of Trm (6, 7). TGFβ is another important homeostatic skin mediator that helps to maintain multiple skin cell populations including Tregs (54), resident memory T cells (55), keratinocytes (56), fibroblasts (57) and langerhans cells (55, 58).
Another recently appreciated portion of the skin for chemokine function is the interstitial fluid (59). This fluid bathes the cells of the skin and transmits signals throughout the tissue, including chemokines and antigens. Of note, this tissue can be sampled in healthy controls or in lesional/nonlesional skin from patients with autoimmune skin conditions using a blister biopsy technique to measure chemokine levels, cellular infiltrates, and more (7, 60–65).
Inflammatory chemokines in the skin
Inflammation is initiated following activation of innate immune sensors/receptors in skin-resident cells (reviewed in (66)). Downstream signaling pathways activate key transcription factors that induce the production of proinflammatory cytokines and chemokines. Generally, some of these signals can be viewed as “recruit” signals whereas others are used more for fine-tuning of localization and/or tethering of immune cells in the skin.
Some chemokines that are used for global skin recruitment include CXCL9, which will be described in detail in the context of vitiligo, and TSLP. As mentioned above, TSLP can serve homeostatic functions, but it has also been shown to drive allergic responses and atopic dermatitis (67, 68). One study reported that in atopic dermatitis patients, this is likely due to promoter demethylation, with subsequent overexpression of TSLP (69). This hypothesis was supported experimentally by overexpressing TSLP in the skin of mice, which resulted in an atopic dermatitis-like disease characterized by massive dermal infiltrates of type 2 cells (70). In an OVA-induced model of AD, TSLPR−/− CD4+ T cells were able to properly home to skin, but were unable to transfer disease to recipient animals due to impaired type 2 cytokine production, indicating that TSLP signaling is required for optimal type 2 effector function (71). Similarly, intradermal injection of TSLP antibody blocked development of disease.
A classical example of a tethering chemokine is CXCL10, which can facilitate CXCR3+ T cell:DC interactions in lymph nodes (72–74) and promotes T cell localization to the epidermis in vitiligo, which we will discuss in detail below (75). CCR4 ligands also facilitate T cell:DC clustering in the lymph node and T cell:macrophage clustering in the skin (43, 76). We will discuss the roles of CCR4 and its ligands in the context of type 2 inflammation below.
CCL5 is important for recruitment and clustering of T cells in the dermis (11, 27, 77). CCL5 binds to CCR5, which can be expressed on CD4+ T cells, CD8+ T cells, and Tregs (reviewed in (78)). CCL5 has also been reported to interact with CCR3 and CCR1 (79). Like CCL5, CCR5 is promiscuous in that it can bind CCL3 and CCL4 as well (78). These ligands facilitate CD4+ T cell interactions with DCs in lymph nodes (80), though their roles in the skin have yet to be fully elucidated.
CCL18 mediates its effects through its cognate receptor CCR8 (81) and appears to have important roles both during inflammation and at homeostasis. Intriguingly, CCL18 expression is under control of both STAT1 and STAT6 promoters (82), which partially explains why it is enriched in both type 2-mediated skin disease (ie. atopic dermatitis) (83, 84) as well as type 1-mediated diseases (ie. alopecia areata) (85, 86). It is expressed predominantly in the lung, but can also be found in lymphoid compartments and in plasma. In addition to specific increases during skin inflammation, CCL18 expression is elevated in many autoinflammatory and fibrotic diseases in the skin and other barrier tissues such as bullous pemphigoid (87), scleroderma (88), Sjogren’s syndrome (89), and vitiligo (see GEO dataset from (75)). Owing to the absence of this gene in non-primate animals, the mechanistic role of CCL18 in these diseases processes has been difficult to dissect.
An important factor to consider when examining inflammatory chemokines in the skin is that different chemokine ligands can bind to the same receptor, but may mediate different signals depending upon where, and how strongly, they bind (90). One example of this is the dichotomy of CXCL9 as a recruit signal and CXCL10 as a localization signal, which is exemplified by previous work in vitiligo. Though both of these chemokines bind CXCR3, CXCL9 deficiency resulted in significantly fewer antigen-specific CD8+ T cells in the skin, whereas CXCL10 deficiency impacted epidermal localization and/or function (75). These data are discussed further below in the vitiligo section.
Overview of T cell-driven autoimmune skin diseases
Historically, T cell-mediated immune responses have been categorized based on which group of cytokines the T cells produce. In the case of CD4+ helper T cells, type 1 responses are mediated by Tbet transcription factor programming, production of IFN-γ/TNF/IL-2, and expression of the chemokine receptors CXCR3 and CCR5. Type 2 responses are mediated by GATA3, IL-4/5/13 production, and CCR4 and CCR8. Type 17 responses are mediated by RORgT, IL-17 production, and CCR6. CD8+ T cell responses are designated similarly, but are referred to as cytotoxic responses. However, in the case of autoimmunity, many mixed pathologies exist, with production of several proinflammatory cytokine mediators by CD4+ and/or CD8+ T cells. Here, we will discuss autoimmune skin diseases that exhibit prototypical T cell responses, as well as some with mixed pathologies.
Chemokine-mediated T cell recruitment in vitiligo, a prototypical cytotoxic type 1 disease
Vitiligo is an autoimmune skin disease in which melanocytes are targeted for destruction by autoreactive CD8+ T cells. Vitiligo affects approximately 1% of the population worldwide. Melanocytes normally reside at the dermal/epidermal junction (DEJ) within the basal layer of keratinocytes. Thus, autoreactive T cells must navigate through the dermis, up to the DEJ, and proximal to their melanocyte targets to kill them. Of note, T cell infiltration is most often observed just beyond the lesional border (perilesion) (91, 92): T cells kill melanocytes and migrate outward while pigment in keratinocytes takes weeks to turn over. Therefore, it is likely that the killing event precedes clinically observable depigmentation. Consistent with these observations, examination of chemokines and T cells are most clear just beyond the border of the lesions, whereas the lesion center represents stable or burned-out disease.
Vitiligo progression is driven by IFN-γ. Transcripts of IFN-γ dependent genes are enriched in vitiligo skin and include the chemokines CXCL9, CXCL10, CXCL11 and CCL5 (75, 93). Elevated protein levels of these chemokines have been confirmed in blood and skin samples from patients (60, 94–99). Autoreactive T cells express the cognate receptors for these chemokines, namely CCR5 and CXCR3. While CCR5 is just beginning to be studied in vitiligo (95), the functional role of the CXCR3 chemokine system was rigorously tested by genetic manipulation and antibody blockade in mouse models of disease. CXCR3-deficient autoreactive T cells fail to induce disease and targeting the receptor via antibody blockade or depletion both protects mice from vitiligo and reverses it (24, 75, 100–102). Similarly, targeting JAK signaling, which drives expression of CXCR3 ligands and other gene products downstream of IFN-γ, also halts and reverses disease in vitiligo patients (103–106).
Studies in mouse models of vitiligo have helped elucidate complementary roles for CXCL9 and CXCL10 in pathogenesis and have revealed insight into their relative functions. Mice deficient in CXCL9 or treated with CXCL9 blocking antibody have a T cell recruitment deficit to the skin, but the mice still develop full clinical disease. In contrast, mice deficient in CXCL10 or treated with a CXCL10 blocking antibody are protected from disease, although equal numbers of dermal T cells were observed (75). These observations, combined with studies examining the lymph node in the context of immunization and viral infection (73, 74), suggest that CXCL9 serves as a “recruit” signal primarily in the dermis, whereas CXCL10 provides either a tethering signal that enhances T cell function, and/or helps to localize the T cells to their targets residing at the DEJ (Figure 2). The role of CXCL11 in vitiligo has yet to be elucidated because C57BL/6J mice have a null mutation that prevents expression (107).
Figure 2. Chemokines promote T cell localization in the skin during vitiligo.
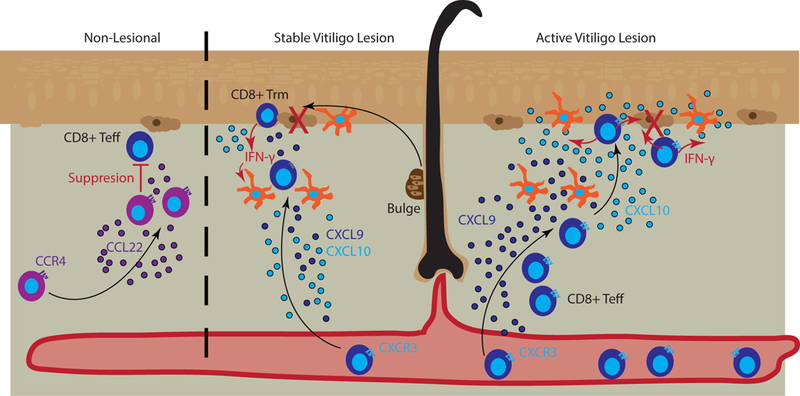
Left panel represents control of lesions by Tregs that naturally occupy the skin and suppress T effector cells. Tregs can be recruited via CCL22/CCR4 interactions (purple). Middle panel represents a stable vitiligo lesion in which resident memory T cells (Trm) are retained within the epidermis and maintain depigmentation. Melanocytes (brown) migrate out of the bulge of hair follicles and are recognized by Trm that secrete IFN-γ (red arrows), CXCL9 (dark blue), and CXCL10 (light blue) to recruit additional effector and central memory T cells to kill (large red X) new emigrants. Dermal antigen presenting cells (APCs) can respond to Trm IFN-γ and aid T cell recruitment through CXCL9 and CXCL10 release. Right panel represents an active vitiligo lesion in which high amounts of CXCL9, produced by dermal APCs, serve to recruit T cells to the skin, and CXCL10, produced by Langerhans cells (epidermal APCs) and keratinocytes (lighter brown cells in the epidermis), serves to facilitate localization up to the epidermis and destruction of melanocytes.
We used the reporter of expression of CXCR3 ligands mouse (REX3) (73) to determine the source of CXCL9 and CXCL10 in mice during vitiligo in both the transferred T cells (8) and in the host-derived skin cells (24). Consistent with studies describing chemokine production by Trm (4, 108), we found REX3+ Trm cells producing CXCL9 and CXCL10. However, Tcm were also REX3+, indicating that multiple phenotypes of T cells in the skin produce CXCR3 ligands (8). In studies using REX3+ hosts, many cell types in the skin produced these chemokines. In the epidermis, all cells produced CXCR3 ligands, but by cell number keratinocytes made up the majority of total chemokines. On a per-cell basis, the chemokine production was higher in antigen presenting cells found in the dermis as well as Langerhans cells in the epidermis. To test the contribution of these cellular sources of chemokine in vitiligo, we used genetically targeted mice to prevent IFN-γ signaling in specific cell types (STAT1 flox, with various cell-specific Cre expression) and found that IFN-γ signaling in keratinocytes primarily recruit autoreactive T cells during vitiligo and are required for disease pathogenesis (24). Endothelial cells provided an intermediate phenotype, whereas STAT1 was not required in melanocytes themselves for disease induction (unpublished observations).
In addition to the IFN-γ-CXCR3 signaling axis, the CXCR6 chemokine system has also been implicated in non-redundant recruitment of T cells during vitiligo. Investigators reported increased CXCL16 in the lesional skin of vitiligo patients, and intralesional T cells express its receptor CXCR6 (109). Intriguingly, CXCL16 is produced by keratinocytes in response to reactive oxygen species (ROS), suggesting that intrinsic stress or toxic intermediates produced during the inflammatory response can help recruit more T cells. Many investigators have focused on the role of cellular stress in vitiligo; however, it remains unknown whether ROS precipitates disease or is a product of inflammation.
CXCL8, also known as IL-8, is increased in the serum of patients with active vitiligo (94). CXCL8 binds to CXCR1 and CXCR2, which are highly expressed on neutrophils and eosinophils, but can also be expressed at low levels on T cells (110, 111). The functional role of CXCL8 in vitiligo has yet to be elucidated, as neutrophils and eosinophils are absent from vitiligo skin lesions.
Chemokine-mediated T cell recruitment in atopic or allergic dermatitis represents type 2 disease
Atopic dermatitis (AD) and allergic contact dermatitis (ACD) represent two inflammatory conditions primarily driven by T cells that respond inappropriately to environmental factors. AD is primarily driven by a defect in skin barrier function, which allows penetration and over sensitization to allergens and skin microflora, in particular Staphylococcus aureus. Repeated inflammatory responses are driven by CD4+ T cells that help to recruit other cellular mediators that together produce inflammation and clinical skin eruptions recognized as irritated, scaly skin with erythema. Resolution of the lesions can be observed following treatment with moisturizers (to repair the skin barrier), antibiotics (to eliminate bacterial stimuli), and/or anti-inflammatory drugs (to decrease inflammation). Pediatric patients typically “outgrow” AD, indicating age-related factors are also at play (112). While the barrier defect is common to most individuals with AD, the environmental factors and underlying mediators of disease may be heterogeneous (113). In contrast, ACD (discussed below) is characterized by T cell responses against specific haptens formed from chemical exposure, including metal ions (114) that drive inappropriate immune responses. Only avoidance of the skin allergen leads to full resolution.
AD is characterized by CD4 T cell infiltrates and at least initially exhibits a type 2 cytokine profile. Earlier translational studies suggested that AD skin gene expression signature shifted from a type 2 response to type 1 (115–118) and later studies corroborated this finding, but suggested that the type 2 response was always strongly present, even in advanced lesions as microarray screening techniques missed transcripts that were only detectable by RT-qPCR (119). When AD and ACD are directly compared, both have some type 2 signals but ACD has higher levels of type 1-associated chemokines, including CXCL10 (Kamsteeg et al.). A recent study found that AD patients with food allergies exhibit higher IgE titers and stronger type 2 cytokine responses than patients without food allergy (120), indicating subtypes or spectrums exist within the AD entity that could account for differences in cytokine and chemokine profiles. Nevertheless, the success of IL-4/IL-13 inhibition in AD by dupilumab indicates that type 2 cytokines drive disease pathogenesis (121, 122).
In support of these findings, translational studies of the recruitment of T cells in AD suggest a role for CCR4 (123, 124), which responds to IL-4 driven chemokine ligands. Moreover, additional translational profiling of AD skin highlights a prominent role for type 2, sometimes with type 1 signals (125, 126). The shift in the profiles may be explained because barrier defects allow for multiple antigens from a myriad of sources to penetrate the skin and elicit a mixed inflammatory response (Figure 3).
Figure 3. Chemokines promote T cell accumulation in atopic dermatitis.
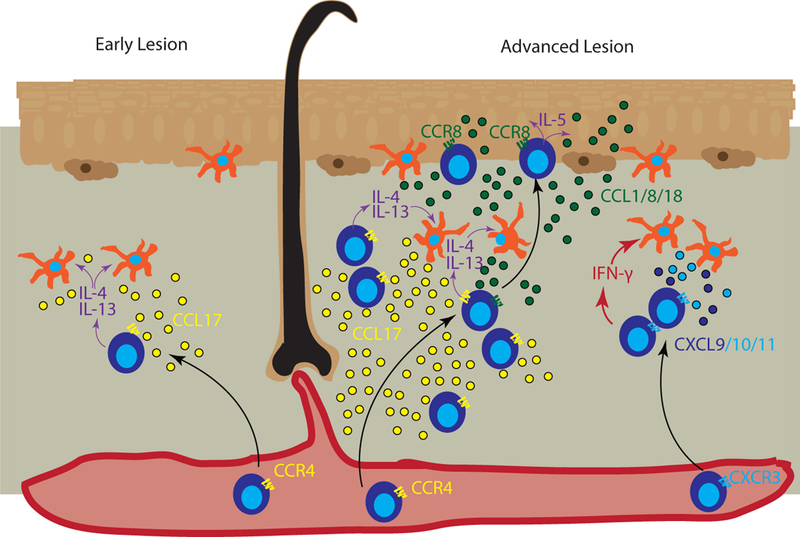
Left panel represents early or developing atopic dermatitis (AD) lesions, in which CCR4+ T cells migrate into the skin and secrete type 2 cytokines IL-4/IL-13 (purple arrows), which act on skin antigen presenting cells (APCs) and other cells that produce CCL17 (yellow) to recruit additional CCR4+ T cells. Right panel represents advanced AD, in which IL-4-activated APCs secrete CCR4 ligands, predominantly CCL17, to recruit additional T cells. Keratinocytes in lesional skin secrete CCL1/CCL8/CCL18 (green dots) to recruit CCR8-expressing T cells into the epidermis. T cells in the epidermal compartment continue secreting IL-5 (purple arrows), which induces keratinocytes to secrete more CCL1/CCL8/CCL18, creating a positive feedback loop. In advanced lesions, additional inflammatory signals are increased including Type 1-skewed T cells that secrete IFN-γ (red arrows), which induces CXCL9/CXCL10 (dark and light blue dots) production by APCs to recruit additional CXCR3+ T cells, resulting in a mixed type 1/type 2 chemokine profile seen in chronic exacerbated AD.
In light of the skin barrier defect in AD, it is not surprising that many homeostatic mediators are upregulated, including CCL1, CCL17, and CCL27 (46, 127). Once the epidermis becomes leaky, multiple antigens become available to the skin immune system, including bacteria, viruses, and allergens. The protease activity of allergens from dust mites, cockroaches, parasites, etc. are well-documented as contributing to type 2-type responses and likely account for why late AD lesions still predominantly express type 2-associated cytokines and chemokines (128–133) reviewed in (134). While studies are conflicted about which antigen presenting cell is most important for sensing allergen proteases, the end result is the same: increased recruitment and effector function of Type 2 cytokine-producing CD4+ T cells.
Type 2 cytokines provide a newly-defined link between the nervous and immune systems in AD: IL-4Ra on skin sensory neurons sense immune-derived IL-4 to promote itch in AD, and blocking this response with JAK1 inhibition reverses disease in patients recalcitrant to standard treatments (16). In contrast, different sensory neuron channel proteins contribute to itch and inflammation separately in a model of ACD (17). Furthermore, itching as a response to light touch, or alloknesis, is mediated by Piezo2 channels on Merkel cells (135). Thus, positive feedback loops between itch sensation and inflammation drive pathogenesis of AD and ACD (reviewed in (14)). The therapeutic potential of blocking Type 2 cytokine-induced migration to mitigate feedback loops that potentiate itch needs further investigation.
Many mouse models of AD are formed by inducing skin inflammation using common techniques: barrier disruption, sensitization and elicitation reactions, or model antigens. Regardless, Type 2 skewing appears to be important in all of these models, and a transgenic mouse that expresses IL-4 under a keratinocyte driven promoter that develops AD-like disease (136). In models using a delayed type hypersensitivity challenge, CCR4-bearing cells are the cells most efficiently recruited to the skin (44), and adoptively transferred antigen-specific T cells require CCR4 to migrate to the skin during antigen challenge (137).
However, CCR4 is not always the sole contributor to recruiting T cells in AD-like skin reactions. Blocking CCR4/CCL17 or CCR10/CCL27 abrogates recruitment to skin challenges (138, 139). Of note, injection or overexpression of either CCL17 or CCL27 alone into the dermis is enough to drive AD-like disease in mice, presumably through increased recruitment of immune cells that then react against normal microbiota (46, 140–142). Additional models of AD show a stronger reliance, although not always complete dependence, on CCR4 mediated recruitment of T cells (143–149). When chronic AD was modeled in mice, CCR8/CCL8 recruitment of T cells to the skin promoted inflammation, but not CCR4 (150). Since CCR8 may be required for resident memory T cell formation in the skin (9), it may be that acute inflammation amplifies signals present during homeostasis and that other chemokine receptors, like CCR8, are important for positioning in the skin for maintenance of disease in a chronic setting. Altogether, these animal studies corroborate the Type 2 and CCR4 signatures seen in many AD patient skin, but in addition to CCR4, CCR8 and CCR10 on T cells play important roles in maintaining the disease.
In contrast, ACD relies principally on IFN-γ induced chemokine ligands. After allergen elicitation, resident T cells produce IFN-y and lead to the upregulation of chemokines CXCL9, CXCL10, and CXCL11, driving a mixed recruitment of additional T cells and responders that contribute to inflammation. Mouse models of ACD utilizing dinitrofluorobenzene induce characteristic inflammation with a strong skin signature of CXCL9, CXCL10, and CXCL11, which is also present in Nickel allergic individuals (151) and replicated in other translational studies (152). Similarly, expanded T cells from allergic, dintorfluorbenze-exposed individuals produce IFN-γ rapidly after challenge (151).
Common to both AD and ACD, cytokines that induce migration/chemokinesis including TSLP and IL-16 are elevated in lesional skin. TSLP is induced in epithelial cells following trauma or following exposure to microbes or inflammation and thus provides a feed-forward loop in AD/ACD (153). IL-16 is elevated in patients with AD and ACD (154–156), and IL-16 is a pleomorphic cytokine that can induce CD4+ T cell migration when secreted, but in its pro-form can regulate cell cycle control (reviewed in (157)). In AD, higher IL-16 levels correlated to higher degrees of sensitization (154, 155). It is important to note that IL-16 preferentially induces migration of Tregs (158), so it is possible that its upregulation in inflamed skin represents an attempt at restoring homeostasis. In ACD but not AD, IL-16 promoter polymorphisms are associated with development of disease (159), and treatment with anti-IL-16 antibody reduces ear swelling in mouse models of ACD (160). Of note, a neuronal form of IL-16 exists (reviewed in (161)) that serves as a link between the nervous system and the immune system, and this was studied in the context of Multiple Sclerosis (reviewed in (162)). The role of IL-16 in neuroimmunology is especially relevant to skin disease in light of the recent studies examining inflammation and itch (14, 15), but will need to be further elucidated.
Chemokine-mediated T cell recruitment in psoriasis, a prototypical type 17 disease
Psoriasis is a type 17-mediated disease in the skin that appears to be autoimmune or autoinflammatory in nature. A number of putative antigens or danger-associated molecular patterns have been reported to be secreted by keratinocytes and, more recently, melanocytes (163–165), but no antigen has yet been confirmed. Nevertheless, IL-17 producing T cells drive pathogenesis and are found in high numbers in patient skin, adopting various phenotypes including Trm that persist in lesions even after treatment (166, 167). Psoriasis is dependent upon a feedback loop of TNFα, IL-6, IL-21, IL-22, IL-23, IL-17 and CCL20/CCR6 (reviewed in (168)). Blockade of TNFα (169), IL-17, and IL-23 reverse disease while blocking other cytokines appear less effective, highlighting the principal IL-17 signalling axis in driving disease pathogenesis (reviewed in (170)). CCL20/CCR6 interactions are required for dermal clustering of T cells and dendritic cells in psoriatic lesions (171). CCR6 was required for induction of disease in a mouse model of psoriasis-like inflammation, and facilitated localization of T cells to the epidermis (172). CCR6 expression marks IL-17-producing T cells in humans (173), and IL-17 induces production of CCL20 by keratinocytes (174), thus exacerbating disease in a feed-forward loop. Other cytokines produced during this response stimulate dendritic cells of the skin and damage the keratinocytes, which further promote the positive feedback loop and increase inflammation (Figure 4).
Figure 4. Chemokines in developing psoriatic lesions.
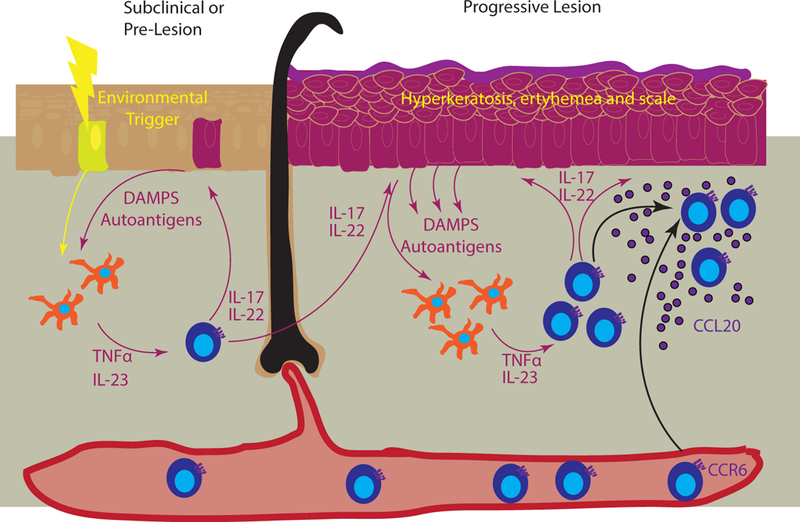
Left panel represents the precipitation of psoriasis in subclinical skin, where an environmental trigger (yellow bolt) induces release of danger associated molecular patterns (DAMPs) and autoantigens that are sensed by dermal APCs (yellow cells). These in turn secrete TNFα and IL-23, which activate skin T cells to produce IL-17 and IL-22, creating a feedforward loop (represented by magenta arrows). These cytokines in turn continue to damage keratinocytes, which results in the release of additional DAMPs and potential autoantigens. Right panel depicts a progressive lesion, with hyperkeratosis, erythema, and scale (magenta epidermis). T cell recruitment to the skin is primarily through keratinocyte-derived CCL20 (purple) that recruits CCR6-bearing cells.
IL-16 was reported to be elevated in psoriasis patients, though to a lesser extent than in AD patients. IL-16 was elevated in perilesional psoriatic skin at both the mRNA and protein level, whereas lesional skin only had increased protein levels (175). Thus, the edges of psoriatic lesions may represent areas of higher T cell activity and recruitment as is observed in vitiligo. CXCL16/CXCR6 are also elevated in psoriasis (176) and correlate with increased CD8+ T cells in lesional skin (177). Although IL-17/IL-22 producing CD4+ T cells primarily mediate psoriasis (178), CD8+ T cells may also help drive pathology (179).
Interface dermatitis: T cell recruitment in cutaneous lupus
Interface dermatitis describes the appearance of immune infiltration at the DEJ. This type of infiltration is found in many diseases of the skin, including vitiligo, lichen planus, and others, but is a hallmark of cutaneous lupus erythematosus (CLE) (180). CLE can be a component of systemic lupus, or it can exist on its own (181, 182). Understanding how CLE progresses to systemic lupus is important for considering aggressive treatment options, and successfully treating skin manifestations can ameliorate systemic symptoms (183, 184). CLE can be triggered by certain drugs (185, 186), UV light (187, 188), hormonal imbalances (189–191), and likely additional environmental causes that have yet to be explored.
There are 3 main clinical subtypes of CLE, including acute (ACLE), subacute (SCLE), and chronic (CCLE) (192, 193). All 3 subtypes look virtually the same histologically, with an interface dermatitis characterized by a lymphocytic infiltrate. These subtypes also share chemokine expression profiles, with CXCR3 ligands being highly upregulated in an IFN-dependent manner (194–196). There is a paucity of mouse models of CLE, as the most frequently used lupus models do not exhibit skin manifestations of disease. However, a recently published mouse model of cutaneous lupus appears to recapitulate CCLE, and nanostring analysis revealed both human CCLE and mouse CCLE skin samples exhibited increased expression of CCL3, CCL4, CCL7, CCL8, and, most notably, very high expression of CXCR3 ligands (197). Future studies will focus on the functional roles of these chemokines in the murine model and in human disease.
A recent study uncovered a cellular mechanism for sensing skin damage following UVB irradiation in two mouse models of systemic lupus (MRL/lpr and B6.SLE1yaa, (198)): ADAM17 is upregulated in Langerhans cells following UVB exposure, which in turn increases the release EGFR ligands in an attempt to protect keratinocytes from further UVB damage. While this study did not directly focus on the impact of this cellular sensing loop on chemokine expression and immune cell recruitment, we hypothesize that ADAM17 upregulation will increase the release of chemokines such as CX3CL1 from the surface of resident skin cells, as was previously reported in HUVECs and NIH3T3 cells (199). Functionally distinct subsets of CD8+ T cells that are important for immune surveillance express the cognate receptor of CX3CL1, namely CX3CR1 (200), and could therefore be recruited to sites of UVB induced injury. Of note, CX3CL1/CX3CR1 have been implicated in mediating immune complex deposition (201), and an antagonist of CX3CL1 can delay and ameliorate kidney damage in a mouse model of systemic lupus (MRL/lpr, (202)). Additional investigation will need to be conducted to determine whether the CX3CR1 system plays a role in CLE.
Fibrotic disease: T cell recruitment in morphea
Another type of autoinflammatory process that occurs in the skin is fibrosis, which in settings other than wounding represents a distinct inflammatory process that causes significant morbidity (reviewed in (203)). This occurs in the diseases scleroderma (also known as systemic sclerosis or SSc) and morphea, in which inflammation of the dermis precedes fibroblast activation and collagen deposition (204, 205). Historically, morphea was referred to as a manifestation of skin-limited scleroderma (termed localized scleroderma or LSc); however, it is becoming apparent that the pathophysiology of these diseases may be distinct with overlapping elements in their skin fibrotic processes (206). On the cellular level, skin-resident macrophages appear to become improperly activated, and secrete chemokines to recruit T cells. These T cells in turn produce proinflammatory cytokines and chemokines, which further activates macrophages and fibroblasts themselves. This results in a positive feedback loop of inflammation as the fibroblasts then secrete more chemokines, as well as collagen and other gene products to promote fibrotic responses. Thus, this trio of cells, macrophages, T cells, and fibroblasts, appears to drive skin fibrosis during morphea and scleroderma (see also (207)).
A common element in autoinflammatory fibrosis is the CXCR3 chemokine family, which is highly upregulated in lesional skin of both morphea and scleroderma patients (206, 208, 209). CXCR3 expression on fibroblasts plays a role in wound healing and re-epithelialization (210–214). Interestingly, CXCL10 attenuates, rather than promotes, fibrosis in the lung in mouse models of systemic sclerosis due to its ability to limit fibroblast migration (215, 216) and in models of urethral fibrosis due to its ability to cross-talk to pro-fibrotic TGFβ pathways (217). CXCL9, but not CXCL10, was also deemed critical for driving kidney disease in a mouse model of immune mediated kidney damage due to its ability to activate T cells and macrophages in renal tissue (218). Thus, understanding the specific downstream effects of chemokine ligands with shared receptors is important when designing drugs appropriate for targeted effects: in the case of morphea, reducing the pro-fibrotic effects of CXCL9 while leaving the anti-fibrotic effects of CXCL10 is paramount to halting disease (Figure 5).
Figure 5. Chemokines promote T cell recruitment and fibrotic inflammation in morphea.
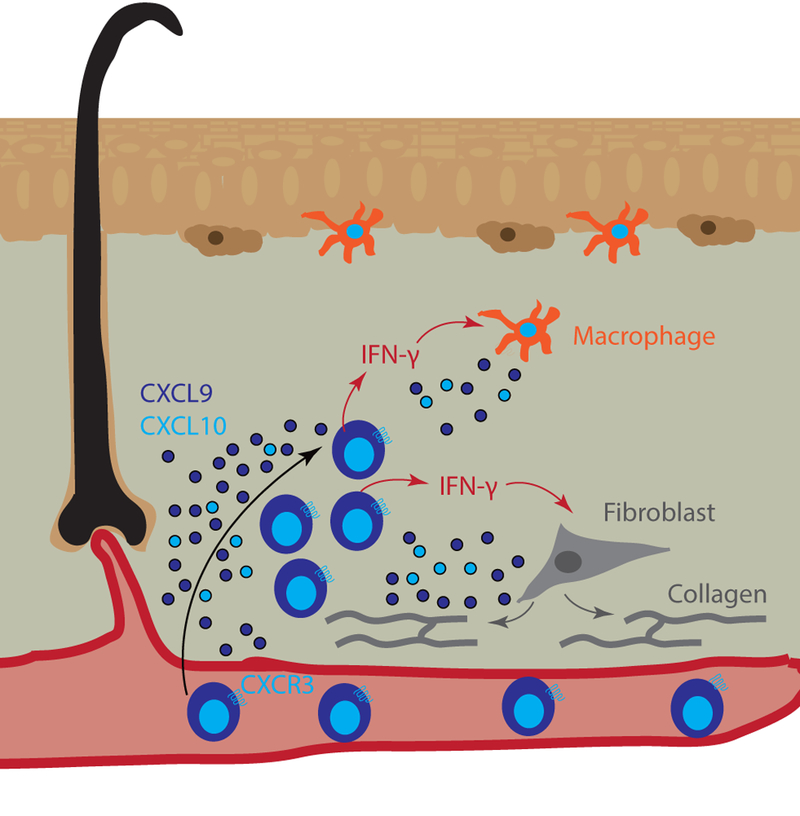
Morphea is characterized by deep dermal infiltrates and fibrosis, which are comprised primarily of T cells and macrophages (orange dermal APC) that may act directly upon fibroblasts (grey cells). Reports indicate that CXCR3 ligands are highly induced within lesional skin and serve as biomarkers of disease. These ligands recruit T cells (blue), which secrete IFN-γ (red arrows) to activate dermal macrophages (orange cells). Macrophages secrete additional CXCR3 ligands (CXCL9 dark blue, CXCL10 light blue dots) to further recruit T cells. Fibroblasts become activated by signals from profibrotic T cells and produce collagen (grey lines/arrows) to form fibrotic plaques.
CXCL16 is upregulated in the serum and skin of patients with systemic sclerosis (219, 220). As mentioned above, CXCL16 is homeostatically produced by keratinocytes in a transmembrane form that can be cleaved from the surface following metalloproteinase activation. Other chemokines that have been implicated in fibrotic skin disease include CCL2 (binds to CCR2), CCL3 (binds CCR1, CCR4 and CCR5) and CCL4 (binds CCR5) (reviewed in (203)). These chemokines have been studied in models of lung, liver or urethral fibrosis, in which their expression and subsequent fibrosis was dependent upon the production of IL-4 and IL-13 production (221–223). Of note, IL-21 was reported to induce IL-4 and IL-13 receptors on macrophages, thus making them more sensitive to these cytokines and poised to amplify signals via chemokine production (224). IL-21 can also induce profibrotic CD8+ Type 2 responses, thus amplifying the T cell-macrophage-fibroblast loop (225). These studies, along with those that have identified the IFN-γ-mediated CXCR3 ligand production discussed above, indicate that autoinflammatory fibrosis is a mixed pathology. Direct links between T cell production of these type 1 and 2 cytokines and subsequent production of chemokines have yet to be definitively studied in dermal fibrosis.
Another layer of complexity in fibrotic disease lies in the recruitment and activation of fibroblasts themselves, in addition to activation or differentiation into myofibroblasts, which are fibroblasts that express smooth muscle actin and other transcripts usually characteristic of muscle cells. Lysophosphatidic acid and its receptor, LPA1, are important for dermal fibrosis in mouse models of scleroderma (226), though this appears to be due to enhanced fibroblast migration and function independent of T cell recruitment (227). TGFβ family members are important for activation of fibroblasts, though the relationship to T cell-derived TGFβ production and T cell infiltration is unclear (reviewed in (203)).
Autoimmunity of the hair follicle: T cell recruitment in alopecia areata
Alopecia areata (AA) is an autoimmune disease characterized by hair loss as a result of immune activation in the hair follicle (reviewed in (228, 229)). Like psoriasis, there is no clear consensus on the autoantigens involved in this disease (one hypothesis is presented here: (230)). The inflammatory infiltrate in the hair bulb primarily features CD4 and CD8 T cells (231, 232), but natural killer cells (233), macrophages (232, 234), mast cells (232, 235) and other cell types (232) are sometimes found on histological examination and may be involved in disease pathogenesis (reviewed in (236)). Animal models of AA replicate human immune infiltration and indicate a pivotal role for T cells in mediating hair loss (237, 238).
Translational studies of the blood and skin of patients reveals that the pathogenesis of AA exhibit a strong type 1 signature (239), including the production of IFN-γ by T cells in disturbed hair follicles (231) and the expression of CXCR3 and its ligands, particularly CXCL10 (240–242). Studies in mice revealed that IFN-y deficient mice were protected from hair loss (243) while exogenous injection of IFN-γ could induce disease (244). Parallelling human translational studies, CXCR3 and its ligands increase in mice prone to developing AA (245).
These studies drove investigators to test if blocking IFN-y, CXCR3 or its ligands could reverse disease. Indeed, animal models depend on CXCR3-mediated recruitment for development of AA (246). Moreover, targeting IFN-y signaling via JAK inhibition can reverse disease in animals (247) and people (247–250). These findings support the idea that CXCR3 mediated recruitment of pathogenic T cells is central to AA pathogenesis.
At homeostasis, the hair follicle expresses CCL2 in the isthmus; CCL20 in the the infundibulum; and CCL8 in the bulge (29). Thus, T cell recruitment to and through the hair follicle is likely coordinated by multiple ligands. A few studies have reported upregulation of additional chemokine families in AA including CCL5, CCL17, CCL18, and CCL26, as well as cytokines that induce migration/chemokinesis such as TSLP, periostin, and IL-16 (85, 86, 251–253). CCL17 serum levels correlated with disease severity and responsiveness to treatment, and immunostaining of skin showed colocalization of CCL17 with CD68+ cells (234). As in ACD, IL-16 promoter polymorphisms are associated with increased risk of AA (254). The functional roles of the other chemokine families have yet to be elucidated.
Like AD, there is a neuroimmune component of the disease, as substance P is highly upregulated in lesional skin (232). Substance P is a neuropeptide that is secreted by neurons and immune cells, and has been associated with pain reception and inflammation (reviewed in (255)). Substance P can induce the migration and/or activation of many different immune cell subsets. In vitro studies in human skin culture models demonstrated that substance P injection was sufficient to induce MHC-I upregulation while simultaneously downregulating pro-survival signals in the hair follicle including NGF-R, which the authors postulated represents loss of immune privilege ((256), reviewed in (257)). In the C3H/HeJ mouse model of AA, substance P is upregulated in early lesions, but not chronic lesions, and infiltrating T cells and macrophages express its receptor, NK-1R (258). Stress has been implicated as a driving factor in this feedback loop (259), as opposed to itch in AD (260–262). Further studies will need to be conducted to better map this pathway during initiation and progression of AA.
Treg recruitment during autoimmune skin disease
Tregs serve to resolve inflammation in multiple autoimmune diseases and promote tissue healing during homeostasis (reviewed in (263)). They also follow chemokines in order to mediate suppression of effector T cells in peripheral tissues, which were reported in numerous models of inflammation (264–273). In the context of inflammation, Tregs downregulate lymphoid homing markers and upregulate CCR2, CCR4, CCR5, CCR6, CCR8, CCR9, CXCR3, CXCR5 and CXCR6 as well as P- and E-selectins, depending upon the signals they receive (274). For example, in a contact hypersensitivity model, CXCR3 is required for Treg trafficking to the skin, and CXCR3−/− mice exhibit delayed recovery (275, 276). Similar effects were observed for CCR5−/− Tregs in models of graft-versus-host disease (271).
In vitiligo, CCL22 expression was reported to be significantly reduced in lesional skin (277). This was hypothesized to lead to reduced recruitment of Tregs, which has since been reported in other studies as well (278, 279). To this end, Eby et al overexpressed CCL22 in the skin in two different mouse models of vitiligo and observed increased recruitment of Tregs, which correlated with a concomitant improvement of disease (280) (see also Figure 2). A similar defect in CCL22 expression with reduced Tregs in skin was observed in pemphigus vulgaris (281).
Targeting chemokines in autoimmune skin diseases as therapeutics
In light of the importance of immune cell migration through the skin tissue to find target cells during autoimmunity, several investigators and companies have targeted chemokines and/or their receptors as potential therapeutics in hopes of providing new treatment options for patients. Here we will discuss several therapeutic strategies that have been tested in autoimmune skin diseases, as well as other chemokine-based therapeutics currently on the market for other indications. Refer to table 1 for a summary of specific FDA-approved inhibitors.
Table 1.
Current FDA-approved chemokine-related therapeutics
| Drug class | Drug name | Target | Indication | Potential other uses |
References |
|---|---|---|---|---|---|
| Small molecule | Tofacitinib | JAK3>>JAK1>JAK2 | RA, psoriatic arthritis, UC | Vitiligo, AA | (103, 104, 286, 289, 290, 294) |
| Ruxolitinib | JAK½ | Myelofibrosis, polycythemia vera | CLE, morphea, RA, vitiligo, AA | (105, 106, 247, 285, 287, 292, 307) | |
| Baracitinib | JAK½ | RA | CLE, morphea, AA, psoriasis | (248, 292, 308–310) | |
| Oclacitinib | Pan JAK inihbitor | AD | many | (311) | |
| Maraviroc | CCR5 | HIV | Type 1 diseases | (305) | |
| Antibody | Mogamulizumab | CCR4 | CTCL | AD | (296–299) |
In vitiligo, targeting the CXCR3 chemokine axis with CXCL10 or CXCR3 antibodies reverses disease in a preclinical model of vitiligo (75, 100). Inhibition of JAKs, which are upstream of CXCR3 ligand production, have reversed disease in several case studies of vitiligo, and one case in particular demonstrated that initiation of treatment directly correlated with a drop of CXCL10 protein in the serum (103–106). Interestingly, epigallocatechin-3-gallate (EGCG), a compound in green tea, was recently reported to inhibit JAK2 and subsequent release of CXCL10 and CCL2 from melanocytes in vitro (282). While EGCG has not been tested for the treatment of vitiligo, it has shown some efficacy in trials for acne (283) and telangectasias (284). Further trials would need to be conducted to determine the efficacy of EGCG for the treatment of autoimmune skin diseases.
JAK inhibitors have also exhibited efficacy in alopecia areata (247, 285–288), morphea (289), AD (16, 288, 290), hypereosinophilic syndrome (291), and CLE (292). In the TSK1 model of scleroderma/morphea, JAK2 inhibition had the added benefit of inhibiting fibroblast responses in addition to reducing T cell recruitment (293). A benefit of small molecule JAK inhibitors in autoimmune skin conditions is that they can be administered topically (285, 290, 294). A downside is that rebound phenomena have been observed (295), and they do not appear to have long-lasting, durable results (106). JAK inhibitors appear to confirm hypotheses in mice that targeting cytokine signaling upstream of chemokine function in vitiligo would be a viable treatment option for patients. However it is important to note that JAK inhibitors have pleiotropic effects, and therefore it is unclear how much of this mechanism of action can be directly attributed to effects on chemokines. Future human trials that target chemokines or their receptors directly will need to be conducted to ascertain this.
A CCR4 antibody, mogamulizumab, is approved to treat cutaneous T cell lymphoma (CTCL), and likely acts through a depleting mechanism ((296, 297), reviewed in (298, 299)). While there are not yet reports using this approach to treat type 2 mediated skin diseases, it may be useful for AD, ACD, and similar type 2-mediated pathologies. In diseases with other chemokine profiles, such as vitiligo, introducing CCL22 to recruit new populations of Tregs may prove to be another viable treatment option (280). Indeed, we hypothesize that part of what goes awry in autoimmune conditions is an inability to resolve inflammation because the immune system promotes a feed-forward loop mediated by specific cytokine and chemokine families. Providing mediators from a different type of response could help distract and/or reset the immune system to resolve inflammation (i.e. using a type 2-associated chemokine in a type 1 response as exemplified by CCL22 in vitiligo). In fact, this is one possible explanation for the efficacy of contact sensitization as a treatment for alopecia areata (300–302).
Treatments that disrupt the type 17 axis have had good success in treating psoriasis (reviewed in (170)). While there are no currently approved CCR6 inhibitors, CCR6 is an attractive target for psoriasis (303). There are numerous other compounds in pipelines and early phase trials for various indications, including inhibitors of CCR6, CXCR3 (304), and other GPCRs. There is an FDA-approved CCR5 inhibitor that is marketed to interfere with the ability of HIV to infect T cells following binding to CCR5 (305), though it has not yet been tested to block CCR5 signaling as a treatment for autoimmunity. In light of the possible proinflammatory role of CCR5 in the pathogenesis of several skin diseases (75, 94, 95), blocking CCL5 or CCR5 may be a future therapeutic avenue.
Summary and future outlook
Classifying skin diseases with shared pathologies could help translate treatment options faster, such that blocking common chemotactic mediators to resolve one disease might work in a disease with shared recruitment mechanisms (reviewed in (306) and exemplified here by JAK inhibitors). A challenge that remains is that several chemokine families may serve redundant roles in recruitment of T cells to the skin, and thus targeting a single chemokine may not be sufficient. Combined approaches may prove to be the best avenue of pharmacologic interventions. The future looks promising for chemokine therapies in autoimmune skin diseases.
Acknowledgments:
JMR is supported by a Career Development Award from the Dermatology Foundation and a Target Identification in Lupus Award from the Lupus Research Alliance. NIH training grants supported KIE (AI095213) and JPS (AI132152, AI095213). JEH is supported by NIH awards AR09114 and AR07302. We thank Marina Tuzova for insightful comments.
Footnotes
Conflict of interest: JMR, JPS and JEH are inventors on patent application #62489191, “Diagnosis and Treatment of Vitiligo” which covers targeting IL-15 and Trm for the treatment of vitiligo. JEH is scientific founder of Villaris Therapeutics, Inc, which is focused on developing treatments for vitiligo.JMR and JEH are inventors on patent application #15/851,651, “Anti-human CXCR3 antibodies for the Treatment of Vitiligo” which covers targeting CXCR3 for the treatment of vitiligo.
References:
- 1.Ali N, Zirak B, Rodriguez RS, et al. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell 2017; 169: 1119–1129.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoeffel G, Wang Y, Greter M, See P, Teo P Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac–derived macrophages. Journal of 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark RA, Chong B, Mirchandani N, et al. The vast majority of CLA+ T cells are resident in normal skin. J. Immunol. 2006; 176: 4431–4439. [DOI] [PubMed] [Google Scholar]
- 4.Schenkel JM, Fraser KA, Vezys V, Masopust D Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol. 2013; 14: 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackay LK, Rahimpour A, Ma JZ, et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013; 14: 1294. [DOI] [PubMed] [Google Scholar]
- 6.Cheuk S, Schlums H, Gallais Sérézal I, et al. CD49a Expression Defines Tissue-Resident CD8+ T Cells Poised for Cytotoxic Function in Human Skin. Immunity 2017; 46: 287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richmond JM, Strassner JP, Zapata L Jr., et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci. Transl. Med. 2018; 10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richmond JM, Strassner JP, Rashighi M, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J. Invest. Dermatol. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCully ML, Ladell K, Andrews R, et al. CCR8 Expression Defines Tissue-Resident Memory T Cells in Human Skin. J. Immunol. 2018; 200: 1639–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boniface K, Jacquemin C, Darrigade A-S, et al. Vitiligo Skin Is Imprinted with Resident Memory CD8 T Cells Expressing CXCR3. J. Invest. Dermatol. 2018; 138: 355–364. [DOI] [PubMed] [Google Scholar]
- 11.Proudfoot AEI, Handel TM, Johnson Z, et al. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U. S. A. 2003; 100: 1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuhlbrigge RC, Kieffer JD, Armerding D, Kupper TS. Cutaneous lymphocyte antigen is a specialized form of PSGL-1 expressed on skin-homing T cells. Nature 1997; 389: 978–981. [DOI] [PubMed] [Google Scholar]
- 13.Woodruff MC, Heesters BA, Herndon CN, et al. Trans-nodal migration of resident dendritic cells into medullary interfollicular regions initiates immunity to influenza vaccine. J. Exp. Med. 2014; 211: 1611–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trier AM, Kim BS. Cytokine modulation of atopic itch. Curr. Opin. Immunol. 2018; 54: 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L, Kaplan DH. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 2015; 43: 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oetjen LK, Mack MR, Feng J, et al. Sensory Neurons Co-opt Classical Immune Signaling Pathways to Mediate Chronic Itch. Cell 2017; 171: 217–228.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng J, Yang P, Mack MR, et al. Sensory TRP channels contribute differentially to skin inflammation and persistent itch. Nat. Commun. 2017; 8: 980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oetjen LK, Mack MR, Feng J, et al. Neuronal IL-4Rα and JAK1 signaling critically mediate atopic dermatitis-associated. J. Allergy Clin. Immunol. 2018; 141: AB92. [Google Scholar]
- 19.Riol-Blanco L, Ordovas-Montanes J, Perro M, et al. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 2014; 510: 157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helft J, Ginhoux F, Bogunovic M, Merad M Origin and functional heterogeneity of non-lymphoid tissue dendritic cells in mice. Immunol. Rev. 2010; 234: 55–75. [DOI] [PubMed] [Google Scholar]
- 21.Chu C-C, Di Meglio P, Nestle FO. Harnessing dendritic cells in inflammatory skin diseases. Semin. Immunol. 2011; 23: 28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malissen B, Tamoutounour S, Henri S The origins and functions of dendritic cells and macrophages in the skin. Nat. Rev. Immunol. 2014; 14: 417–428. [DOI] [PubMed] [Google Scholar]
- 23.Igyártó BZ, Haley K, Ortner D, et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 2011; 35: 260–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richmond JM, Bangari DS, Essien KI, et al. Keratinocyte-Derived Chemokines Orchestrate T-Cell Positioning in the Epidermis during Vitiligo and May Serve as Biomarkers of Disease. J. Invest. Dermatol. 2017; 137: 350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin H, Iwasaki A A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 2012; 491: 463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe R, Gehad A, Yang C, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci. Transl. Med. 2015; 7:279ra39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iijima N, Iwasaki A T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 2014; 346: 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ariotti S, Beltman JB, Chodaczek G, et al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc. Natl. Acad. Sci. U. S. A. 2012; 109: 19739–19744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagao K, Kobayashi T, Moro K, et al. Stress-induced production of chemokines by hair follicles regulates the trafficking of dendritic cells in skin. Nat. Immunol. 2012; 13: 744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez Rodriguez R, Pauli ML, Neuhaus IM, et al. Memory regulatory T cells reside in human skin. J. Clin. Invest. 2014; 124: 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gratz IK, Truong H-A, Yang SH-Y, et al. Cutting Edge: memory regulatory t cells require IL-7 and not IL-2 for their maintenance in peripheral tissues. J. Immunol. 2013; 190: 4483–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scharschmidt TC, Vasquez KS, Pauli ML, et al. Commensal Microbes and Hair Follicle Morphogenesis Coordinately Drive Treg Migration into Neonatal Skin. Cell Host Microbe 2017; 21: 467–477.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCully ML, Kouzeli A, Moser B Peripheral Tissue Chemokines: Homeostatic Control of Immune Surveillance T Cells. Trends Immunol. 2018; 39: 734–747. [DOI] [PubMed] [Google Scholar]
- 34.Sowell RT, Goldufsky JW, Rogozinska M, et al. IL-15 Complexes Induce Migration of Resting Memory CD8 T Cells into Mucosal Tissues. J. Immunol. 2017; 199: 2536–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006; 6: 595–601. [DOI] [PubMed] [Google Scholar]
- 36.Lu J, Chatterjee M, Schmid H, Beck S, Gawaz M CXCL14 as an emerging immune and inflammatory modulator. J. Inflamm. 2016; 13: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scholz F, Schulte A, Adamski F, et al. Constitutive expression and regulated release of the transmembrane chemokine CXCL16 in human and murine skin. J. Invest. Dermatol. 2007; 127: 1444–1455. [DOI] [PubMed] [Google Scholar]
- 38.Tohyama M, Sayama K, Komatsuzawa H, et al. CXCL16 is a novel mediator of the innate immunity of epidermal keratinocytes. Int. Immunol. 2007; 19: 1095–1102. [DOI] [PubMed] [Google Scholar]
- 39.Ziegler SF, Artis D Sensing the outside world: TSLP regulates barrier immunity. Nat. Immunol. 2010; 11: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y -J. TSLP in epithelial cell and dendritic cell cross talk. Adv. Immunol. 2009; 101: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leichner TM, Satake A, Harrison VS, et al. Skin-derived TSLP systemically expands regulatory T cells. J. Autoimmun. 2017; 79: 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kusumoto M, Xu B, Shi M, Matsuyama T, Aoyama K, Takeuchi T Expression of chemokine receptor CCR4 and its ligands (CCL17 and CCL22) in murine contact hypersensitivity. J. Interferon Cytokine Res. 2007; 27: 901–910. [DOI] [PubMed] [Google Scholar]
- 43.Katou F, Ohtani H, Nakayama T, et al. Macrophage-derived chemokine (MDC/CCL22) and CCR4 are involved in the formation of T lymphocyte-dendritic cell clusters in human inflamed skin and secondary lymphoid tissue. Am. J. Pathol. 2001; 158: 1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soler D, Humphreys TL, Spinola SM, Campbell JJ. CCR4 versus CCR10 in human cutaneous TH lymphocyte trafficking. Blood 2003; 101: 1677–1682. [DOI] [PubMed] [Google Scholar]
- 45.Morales J, Homey B, Vicari AP, et al. CTACK, a skin-associated chemokine that preferentially attracts skin-homing memory T cells. Proc. Natl. Acad. Sci. U. S. A. 1999; 96: 14470–14475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Homey B, Alenius H, Müller A, et al. CCL27-CCR10 interactions regulate T cell-mediated skin inflammation. Nat. Med. 2002; 8: 157–165. [DOI] [PubMed] [Google Scholar]
- 47.Kunkel EJ, Butcher EC. Chemokines and the tissue-specific migration of lymphocytes. Immunity 2002; 16: 1–4. [DOI] [PubMed] [Google Scholar]
- 48.Sigmundsdottir H, Pan J, Debes GF, et al. DCs metabolize sunlight-induced vitamin D3 to “program” T cell attraction to the epidermal chemokine CCL27. Nat. Immunol. 2007; 8: 285–293. [DOI] [PubMed] [Google Scholar]
- 49.Schaerli P, Ebert L, Willimann K, et al. A skin-selective homing mechanism for human immune surveillance T cells. J. Exp. Med. 2004; 199: 1265–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCully ML, Moser B The human cutaneous chemokine system. Front. Immunol. 2011; 2: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andrian UH von, Mempel, TR. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol. 2003; 3: 867–878. [DOI] [PubMed] [Google Scholar]
- 52.Pham THM, Okada T, Matloubian M, Lo CG, Cyster JG. S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity 2008; 28: 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andrian UH von, Mackay CR. T-cell function and migration—two sides of the same coin. N. Engl. J. Med. 2000; 343: 1020–1034. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Peters T, Sindrilaru A, et al. TGF-β–dependent suppressive function of Tregs requires wild-type levels of CD18 in a mouse model of psoriasis. J. Clin. Invest. 2008; 118: 2629–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohammed J, Beura LK, Bobr A, et al. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-β. Nat. Immunol. 2016; 17: 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsumoto K, Hashimoto K, Hashiro M, Yoshimasa H, Yoshikawa K Modulation of growth and differentiation in normal human keratinocytes by transforming growth factor-beta. J. Cell. Physiol. 1990; 145: 95–101. [DOI] [PubMed] [Google Scholar]
- 57.Denton CP, Khan K, Hoyles RK, et al. Inducible Lineage-Specific Deletion of TβRII in Fibroblasts Defines a Pivotal Regulatory Role during Adult Skin Wound Healing. J. Invest. Dermatol. 2009; 129: 194–204. [DOI] [PubMed] [Google Scholar]
- 58.Kaplan DH, Li MO, Jenison MC, Shlomchik WD, Flavell RA, Shlomchik MJ. Autocrine/paracrine TGFbeta1 is required for the development of epidermal Langerhans cells. J. Exp. Med. 2007; 204: 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benias PC, Wells RG, Sackey-Aboagye B, et al. Structure and Distribution of an Unrecognized Interstitium in Human Tissues. Sci. Rep. 2018; 8: 4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Strassner JP, Rashighi M, Ahmed Refat M, Richmond JM, Harris JE. Suction blistering the lesional skin of vitiligo patients reveals useful biomarkers of disease activity. J. Am. Acad. Dermatol. 2017; 76: 847–855.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caixia T, Hongwen F, Xiran L Levels of soluble interleukin-2 receptor in the sera and skin tissue fluids of patients with vitiligo. J. Dermatol. Sci. 1999; 21: 59–62. [DOI] [PubMed] [Google Scholar]
- 62.Anbar T, Zuel-Fakkar NM, Matta MF, Arbab MMI. Elevated homocysteine levels in suction-induced blister fluid of active vitiligo lesions. Eur. J. Dermatol. 2016; 26: 64–67. [DOI] [PubMed] [Google Scholar]
- 63.Ozdemir M, Yillar G, Wolf R, et al. Increased basic fibroblast growth factor levels in serum and blister fluid from patients with vitiligo. Acta Derm. Venereol. 2000; 80: 438–439. [DOI] [PubMed] [Google Scholar]
- 64.Gupta S, Shroff S, Gupta S Modified technique of suction blistering for epidermal grafting in vitiligo. Int. J. Dermatol. 1999; 38: 306–309. [DOI] [PubMed] [Google Scholar]
- 65.Babu A, Thappa DM, Jaisankar TJ. Punch grafting versus suction blister epidermal grafting in the treatment of stable lip vitiligo. Dermatol. Surg. 2008; 34: 166–78; discussion 178. [DOI] [PubMed] [Google Scholar]
- 66.Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat. Rev. Immunol. 2014; 14: 289–301. [DOI] [PubMed] [Google Scholar]
- 67.Leyva-Castillo JM, Hener P, Jiang H, Li M TSLP produced by keratinocytes promotes allergen sensitization through skin and thereby triggers atopic march in mice. J. Invest. Dermatol. 2013; 133: 154–163. [DOI] [PubMed] [Google Scholar]
- 68.Leyva-Castillo JM, Hener P, Michea P, et al. Skin thymic stromal lymphopoietin initiates Th2 responses through an orchestrated immune cascade. Nat. Commun. 2013; 4: 2847. [DOI] [PubMed] [Google Scholar]
- 69.Luo Y, Zhou B, Zhao M, Tang J, Lu Q Promoter demethylation contributes to TSLP overexpression in skin lesions of patients with atopic dermatitis. Clin. Exp. Dermatol. 2014; 39: 48–53. [DOI] [PubMed] [Google Scholar]
- 70.Yoo J, Omori M, Gyarmati D, et al. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J. Exp. Med. 2005; 202: 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He R, Oyoshi MK, Garibyan L, Kumar L, Ziegler SF, Geha RS. TSLP acts on infiltrating effector T cells to drive allergic skin inflammation. Proc. Natl. Acad. Sci. U. S. A. 2008; 105: 11875–11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferguson AR, Engelhard VH. CD8 T cells activated in distinct lymphoid organs differentially express adhesion proteins and coexpress multiple chemokine receptors. J. Immunol. 2010; 184: 4079–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Groom JR, Richmond J, Murooka TT, et al. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 2012; 37: 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sung JH, Zhang H, Moseman EA, et al. Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell 2012; 150: 1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rashighi M, Agarwal P, Richmond JM, et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci. Transl. Med. 2014; 6: 223ra–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Semmling V, Lukacs-Kornek V, Thaiss CA, et al. Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell-licensed DCs. Nat. Immunol. 2010; 11: 313–320. [DOI] [PubMed] [Google Scholar]
- 77.Collins N, Jiang X, Zaid A, et al. Skin CD4+ memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat. Commun. 2016; 7: 11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014; 32: 659–702. [DOI] [PubMed] [Google Scholar]
- 79.Struyf S, Menten P, Lenaerts JP, et al. Diverging binding capacities of natural LD78beta isoforms of macrophage inflammatory protein-1alpha to the CC chemokine receptors 1, 3 and 5 affect their anti-HIV-1 activity and chemotactic potencies for neutrophils and eosinophils. Eur. J. Immunol. 2001; 31: 2170–2178. [DOI] [PubMed] [Google Scholar]
- 80.Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell–dendritic cell interaction. Nature 2006; 440: 890. [DOI] [PubMed] [Google Scholar]
- 81.Islam SA, Ling MF, Leung J, Shreffler WG, Luster AD. Identification of human CCR8 as a CCL18 receptor. J. Exp. Med. 2013; 210: 1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Politz O, Kodelja V, Guillot P, Orfanos CE, Goerdt S Pseudoexons and regulatory elements in the genomic sequence of the beta-chemokine, alternative macrophage activation-associated CC-chemokine (AMAC)-1. Cytokine 2000; 12: 120–126. [DOI] [PubMed] [Google Scholar]
- 83.Günther C, Bello-Fernandez C, Kopp T, et al. CCL18 is expressed in atopic dermatitis and mediates skin homing of human memory T cells. J. Immunol. 2005; 174: 1723–1728. [DOI] [PubMed] [Google Scholar]
- 84.Pivarcsi A, Gombert M, Dieu-Nosjean M-C, et al. CC chemokine ligand 18, an atopic dermatitis-associated and dendritic cell-derived chemokine, is regulated by staphylococcal products and allergen exposure. J. Immunol. 2004; 173: 5810–5817. [DOI] [PubMed] [Google Scholar]
- 85.Fuentes-Duculan J, Gulati N, Bonifacio KM, et al. Biomarkers of alopecia areata disease activity and response to corticosteroid treatment. Exp. Dermatol. 2016; 25: 282–286. [DOI] [PubMed] [Google Scholar]
- 86.Suárez-Fariñas M, Ungar B, Noda S, et al. Alopecia areata profiling shows TH1, TH2, and IL-23 cytokine activation without parallel TH17/TH22 skewing. J. Allergy Clin. Immunol. 2015; 136: 1277–1287. [DOI] [PubMed] [Google Scholar]
- 87.Günther C, Carballido-Perrig N, Kopp T, Carballido JM, Pfeiffer C CCL18 is expressed in patients with bullous pemphigoid and parallels disease course. Br. J. Dermatol. 2009; 160: 747–755. [DOI] [PubMed] [Google Scholar]
- 88.Luzina IG, Atamas SP, Wise R, Wigley FM, Xiao HQ, White B Gene expression in bronchoalveolar lavage cells from scleroderma patients. Am. J. Respir. Cell Mol. Biol. 2002; 26: 549–557. [DOI] [PubMed] [Google Scholar]
- 89.Xanthou G, Polihronis M, Tzioufas AG, Paikos S, Sideras P, Moutsopoulos HM. “Lymphoid” chemokine messenger RNA expression by epithelial cells in the chronic inflammatory lesion of the salivary glands of Sjögren’s syndrome patients: possible participation in lymphoid structure formation. Arthritis Rheum. 2001; 44: 408–418. [DOI] [PubMed] [Google Scholar]
- 90.Colvin RA, Campanella GSV, Sun J, Luster AD. Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J. Biol. Chem. 2004; 279: 30219–30227. [DOI] [PubMed] [Google Scholar]
- 91.Le Poole IC, Wijngaard RM van den, Westerhof W, Das PK. Presence of T cells and macrophages in inflammatory vitiligo skin parallels melanocyte disappearance. Am. J. Pathol. 1996; 148: 1219–1228. [PMC free article] [PubMed] [Google Scholar]
- 92.Wijngaard R van den, Wankowicz-Kalinska A, C Le Poole, B Tigges, Westerhof W, Das P. Local immune response in skin of generalized vitiligo patients. Destruction of melanocytes is associated with the prominent presence of CLA+ T cells at the perilesional site. Lab. Invest. 2000; 80: 1299–1309. [DOI] [PubMed] [Google Scholar]
- 93.Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, Turka LA. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-γ for autoreactive CD8+ T-cell accumulation in the skin. J. Invest. Dermatol. 2012; 132: 1869–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang L, Yang S, Lei J, et al. Role of chemokines and the corresponding receptors in vitiligo: A pilot study. J. Dermatol. 2018; 45: 31–38. [DOI] [PubMed] [Google Scholar]
- 95.Rezk AF, Kemp DM, El-Domyati M, et al. Misbalanced CXCL12 and CCL5 Chemotactic Signals in Vitiligo Onset and Progression. J. Invest. Dermatol. 2017; 137: 1126–1134. [DOI] [PubMed] [Google Scholar]
- 96.Wang XX, Wang QQ, Wu JQ, et al. Increased expression of CXCR3 and its ligands in patients with vitiligo and CXCL10 as a potential clinical marker for vitiligo. Br. J. Dermatol. 2016; 174: 1318–1326. [DOI] [PubMed] [Google Scholar]
- 97.Ferrari SM, Fallahi P, Santaguida G, et al. Circulating CXCL10 is increased in non-segmental vitiligo, in presence or absence of autoimmune thyroiditis. Autoimmun. Rev. 2017; 16: 946–950. [DOI] [PubMed] [Google Scholar]
- 98.Ibrahim ZA, Gheida SF, Elbndary AS. Study of the Serum Level of the Monokine Induced by Interferon Gamma (MIG) in Vitiligo Patients. gulfdermajournal.net [Google Scholar]
- 99.Bertolotti A, Boniface K, Vergier B, et al. Type I interferon signature in the initiation of the immune response in vitiligo. Pigment Cell Melanoma Res. 2014; 27: 398–407. [DOI] [PubMed] [Google Scholar]
- 100.Richmond JM, Masterjohn E, Chu R, Tedstone J, Youd ME, Harris JE. CXCR3 Depleting Antibodies Prevent and Reverse Vitiligo in Mice. J. Invest. Dermatol. 2017; 137: 982–985. [DOI] [PubMed] [Google Scholar]
- 101.Riding RL, Richmond JM, Harris JE. Mouse Model for Human Vitiligo. Curr. Protoc. Immunol. 2018; e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gregg RK, Nichols L, Chen Y, Lu B, Engelhard VH. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice. J. Immunol. 2010; 184: 1909–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Craiglow BG, King BA. Tofacitinib Citrate for the Treatment of Vitiligo: A Pathogenesis-Directed Therapy. JAMA Dermatol. 2015; 151: 1110–1112. [DOI] [PubMed] [Google Scholar]
- 104.Liu LY, Strassner JP, Refat MA, Harris JE, King BA. Repigmentation in vitiligo using the Janus kinase inhibitor tofacitinib may require concomitant light exposure. J. Am. Acad. Dermatol. 2017; 77: 675–682.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rothstein B, Joshipura D, Saraiya A, et al. Treatment of vitiligo with the topical Janus kinase inhibitor ruxolitinib. J. Am. Acad. Dermatol. 2017; 76: 1054–1060.e1. [DOI] [PubMed] [Google Scholar]
- 106.Harris JE, Rashighi M, Nguyen N, et al. Rapid skin repigmentation on oral ruxolitinib in a patient with coexistent vitiligo and alopecia areata (AA). J. Am. Acad. Dermatol. 2016; 74: 370–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 2011; 89: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ariotti S, Hogenbirk MA, Dijkgraaf FE, et al. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 2014; 1254803. [DOI] [PubMed] [Google Scholar]
- 109.Li S, Zhu G, Yang Y, et al. Oxidative stress drives CD8+ T-cell skin trafficking in patients with vitiligo through CXCL16 upregulation by activating the unfolded protein response in keratinocytes. J. Allergy Clin. Immunol. 2017; 140: 177–189.e9. [DOI] [PubMed] [Google Scholar]
- 110.Takata H, Tomiyama H, Fujiwara M, Kobayashi N, Takiguchi M Cutting edge: expression of chemokine receptor CXCR1 on human effector CD8+ T cells. J. Immunol. 2004; 173: 2231–2235. [DOI] [PubMed] [Google Scholar]
- 111.Lippert U, Zachmann K, Henz BM, Neumann C Human T lymphocytes and mast cells differentially express and regulate extra- and intracellular CXCR1 and CXCR2. Exp. Dermatol. 2004; 13: 520–525. [DOI] [PubMed] [Google Scholar]
- 112.Weidinger S, Novak N Atopic dermatitis. Lancet 2016; 387: 1109–1122. [DOI] [PubMed] [Google Scholar]
- 113.Bieber T, D’Erme AM, Akdis CA, et al. Clinical phenotypes and endophenotypes of atopic dermatitis: Where are we, and where should we go? J. Allergy Clin. Immunol. 2017; 139: S58–S64. [DOI] [PubMed] [Google Scholar]
- 114.Schmidt M, Raghavan B, Müller V, et al. Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat. Immunol. 2010; 11: 814–819. [DOI] [PubMed] [Google Scholar]
- 115.Thepen T, Langeveld-Wildschut EG, Bihari IC, et al. Biphasic response against aeroallergen in atopic dermatitis showing a switch from an initial TH2 response to a TH1 response in situ: an immunocytochemical study. J. Allergy Clin. Immunol. 1996; 97: 828–837. [DOI] [PubMed] [Google Scholar]
- 116.Grewe M, Walther S, Gyufko K, Czech W, Schöpf E, Krutmann J Analysis of the cytokine pattern expressed in situ in inhalant allergen patch test reactions of atopic dermatitis patients. J. Invest. Dermatol. 1995; 105: 407–410. [DOI] [PubMed] [Google Scholar]
- 117.Grewe M, Gyufko K, Schöpf E, Krutmann J Lesional expression of interferon-gamma in atopic eczema. Lancet 1994; 343: 25–26. [DOI] [PubMed] [Google Scholar]
- 118.Werfel T, Morita A, Grewe M, et al. Allergen specificity of skin-infiltrating T cells is not restricted to a type-2 cytokine pattern in chronic skin lesions of atopic dermatitis. J. Invest. Dermatol. 1996; 107: 871–876. [DOI] [PubMed] [Google Scholar]
- 119.Gittler JK, Shemer A, Suárez-Fariñas M, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012; 130: 1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Leung DYM, Calatroni A, Zaramela LS, et al. A multi-omics evaluation of the non-lesional skin surface identifies atopic dermatitis with food allergy (AD FA+) as a unique endotype. J. Allergy Clin. Immunol. 2019; 143: AB125. [Google Scholar]
- 121.Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N. Engl. J. Med. 2014; 371: 130–139. [DOI] [PubMed] [Google Scholar]
- 122.Simpson EL, Bieber T, Guttman-Yassky E, et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N. Engl. J. Med. 2016; 375: 2335–2348. [DOI] [PubMed] [Google Scholar]
- 123.Nouri-Aria KT, Wilson D, Francis JN, et al. CCR4 in human allergen-induced late responses in the skin and lung. Eur. J. Immunol. 2002; 32: 1933–1938. [DOI] [PubMed] [Google Scholar]
- 124.Mizukawa Y, Takahashi R, Yamazaki Y, Kimishima M, Shiohara T Fucosyltransferase VII-positive, skin-homing T cells in the blood and skin lesions of atopic dermatitis patients. Exp. Dermatol. 2008; 17: 170–176. [DOI] [PubMed] [Google Scholar]
- 125.Dyjack N, Goleva E, Rios C, et al. Minimally invasive skin tape strip RNA sequencing identifies novel characteristics of the type 2-high atopic dermatitis disease endotype. J. Allergy Clin. Immunol. 2018; 141: 1298–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Suárez-Fariñas M, Tintle SJ, Shemer A, et al. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 2011; 127: 954–64.e1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stutte S, Gerbitzki N, Novak N, & Förster I (2012). Expression and function of CCL17 in atopic dermatitis. Atopic Dermatitis-Disease Etiology and Clinical Management. [Google Scholar]
- 128.Sokol CL, Chu N-Q, Yu S, Nish SA, Laufer TM, Medzhitov R. Basophils function as antigen-presenting cells for an allergen-induced T helper type 2 response. Nat. Immunol. 2009; 10: 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sokol CL, Barton GM, Farr AG, Medzhitov R A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat. Immunol. 2008; 9: 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tang H, Cao W, Kasturi SP, et al. The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling. Nat. Immunol. 2010; 11: 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hammad H, Plantinga M, Deswarte K, et al. Inflammatory dendritic cells—not basophils—are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J. Exp. Med. 2010; 207: 2097–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Asokananthan N, Graham PT, Stewart DJ, et al. House dust mite allergens induce proinflammatory cytokines from respiratory epithelial cells: the cysteine protease allergen, Der p 1, activates protease-activated receptor (PAR)-2 and inactivates PAR-1. The Journal of Immunology 2002; 169: 4572–4578. [DOI] [PubMed] [Google Scholar]
- 133.Wada K, Matsuwaki Y, Moriyama H, Kita H Cockroach induces inflammatory responses through protease-dependent pathways. Int. Arch. Allergy Immunol. 2011; 155 Suppl 1: 135–141. [DOI] [PubMed] [Google Scholar]
- 134.Paul WE, Zhu J How are T(H)2-type immune responses initiated and amplified? Nat. Rev. Immunol. 2010; 10: 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Feng J, Luo J, Yang P, Du J, Kim BS, Hu H Piezo2 channel-Merkel cell signaling modulates the conversion of touch to itch. Science 2018; 360: 530–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chen L, Martinez O, Overbergh L, Mathieu C, Prabhakar BS, Chan LS. Early up-regulation of Th2 cytokines and late surge of Th1 cytokines in an atopic dermatitis model. Clin. Exp. Immunol. 2004; 138: 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Campbell JJ, O’Connell DJ, Wurbel M-A. Cutting Edge: Chemokine receptor CCR4 is necessary for antigen-driven cutaneous accumulation of CD4 T cells under physiological conditions. J. Immunol. 2007; 178: 3358–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Reiss Y, Proudfoot AE, Power CA, Campbell JJ, Butcher EC. CC chemokine receptor (CCR)4 and the CCR10 ligand cutaneous T cell-attracting chemokine (CTACK) in lymphocyte trafficking to inflamed skin. J. Exp. Med. 2001; 194: 1541–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mirshahpanah P, Li Y-YY, Burkhardt N, Asadullah K, Zollner TM. CCR4 and CCR10 ligands play additive roles in mouse contact hypersensitivity. Exp. Dermatol. 2008; 17: 30–34. [DOI] [PubMed] [Google Scholar]
- 140.Vestergaard C, Deleuran M, Gesser B, Larsen CG. Thymus-and activation-regulated chemokine (TARC/CCL17) induces a Th2-dominated inflammatory reaction on intradermal injection in mice. Exp. Dermatol. 2004; 13: 265–271. [DOI] [PubMed] [Google Scholar]
- 141.Tsunemi Y, Saeki H, Nakamura K, et al. CCL17 transgenic mice show an enhanced Th2-type response to both allergic and non-allergic stimuli. Eur. J. Immunol. 2006; 36: 2116–2127. [DOI] [PubMed] [Google Scholar]
- 142.Kagami S, Saeki H, Tsunemi Y, et al. CCL27-transgenic mice show enhanced contact hypersensitivity to Th2, but not Th1 stimuli. Eur. J. Immunol. 2008; 38: 647–657. [DOI] [PubMed] [Google Scholar]
- 143.Matsuo K, Nagakubo D, Komori Y, et al. CCR4 Is Critically Involved in Skin Allergic Inflammation of BALB/c Mice. J. Invest. Dermatol. 2018; 138: 1764–1773. [DOI] [PubMed] [Google Scholar]
- 144.Fülle L, Steiner N, Funke M, et al. RNA Aptamers Recognizing Murine CCL17 Inhibit T Cell Chemotaxis and Reduce Contact Hypersensitivity In Vivo. Mol. Ther. 2018; 26: 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Murray C, Ahrens K, Devalaraja M, et al. Use of a Canine Model of Atopic Dermatitis to Investigate the Efficacy of a CCR4 Antagonist in Allergen-Induced Skin Inflammation in a Randomized Study. J. Invest. Dermatol. 2016; 136: 665–671. [DOI] [PubMed] [Google Scholar]
- 146.Gehad A, Al-Banna NA, Vaci M, et al. Differing requirements for CCR4, E-selectin, and α4β1 for the migration of memory CD4 and activated T cells to dermal inflammation. J. Immunol. 2012; 189: 337–346. [DOI] [PubMed] [Google Scholar]
- 147.Matsui K, Nishikawa A Percutaneous application of peptidoglycan from Staphylococcus aureus induces infiltration of CCR4+ cells into mouse skin. J. Investig. Allergol. Clin. Immunol. 2011; 21: 354–362. [PubMed] [Google Scholar]
- 148.Wang X, Fujita M, Prado R, et al. Visualizing CD4 T-cell migration into inflamed skin and its inhibition by CCR4/CCR10 blockades using in vivo imaging model. Br. J. Dermatol. 2010; 162: 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Onoue A, Kabashima K, Kobayashi M, Mori T, Tokura Y Induction of eosinophil- and Th2-attracting epidermal chemokines and cutaneous late-phase reaction in tape-stripped skin. Exp. Dermatol. 2009; 18: 1036–1043. [DOI] [PubMed] [Google Scholar]
- 150.Islam SA, Chang DS, Colvin RA, et al. Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat. Immunol. 2011; 12: 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Meller S, Lauerma AI, Kopp FM, et al. Chemokine responses distinguish chemical-induced allergic from irritant skin inflammation: memory T cells make the difference. J. Allergy Clin. Immunol. 2007; 119: 1470–1480. [DOI] [PubMed] [Google Scholar]
- 152.Kamsteeg M, Jansen PAM, Vlijmen-Willems IMJJ van, et al. Molecular diagnostics of psoriasis, atopic dermatitis, allergic contact dermatitis and irritant contact dermatitis. Br. J. Dermatol. 2010; 162: 568–578. [DOI] [PubMed] [Google Scholar]
- 153.Allakhverdi Z, Comeau MR, Jessup HK, et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 2007; 204: 253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nagy G, Gáspár K, Irinyi B, et al. Association between serum IL-16 levels and the degree of sensitization in patients with atopic dermatitis. Int. Arch. Allergy Immunol. 2011; 156: 69–74. [DOI] [PubMed] [Google Scholar]
- 155.Zheng HY, Zhao L, Li CX, Li SH. Correlation between serum IL-16 and atopic dermatitis. Genet. Mol. Res. 2016; 15: [DOI] [PubMed] [Google Scholar]
- 156.Koppes SA, Ljubojevic Hadzavdic S, Jakasa I, et al. Stratum corneum profiles of inflammatory mediators in patch test reactions to common contact allergens and sodium lauryl sulfate. Br. J. Dermatol. 2017; 176: 1533–1540. [DOI] [PubMed] [Google Scholar]
- 157.Richmond J, Tuzova M, Cruikshank W, Center D Regulation of cellular processes by interleukin-16 in homeostasis and cancer. J. Cell. Physiol. 2014; 229: 139–147. [DOI] [PubMed] [Google Scholar]
- 158.McFadden C, Morgan R, Rahangdale S, et al. Preferential migration of T regulatory cells induced by IL-16. J. Immunol. 2007; 179: 6439–6445. [DOI] [PubMed] [Google Scholar]
- 159.Reich K, Westphal G, König IR, et al. Association of allergic contact dermatitis with a promoter polymorphism in the IL16 gene. J. Allergy Clin. Immunol. 2003; 112: 1191–1194. [DOI] [PubMed] [Google Scholar]
- 160.Masuda K, Katoh N, Soga F, Kishimoto S The role of interleukin-16 in murine contact hypersensitivity. Clin. Exp. Immunol. 2005; 140: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Glass WG, Sarisky RT, Vecchio AMD. Not-so-sweet sixteen: the role of IL-16 in infectious and immune-mediated inflammatory diseases. J. Interferon Cytokine Res. 2006; 26: 511–520. [DOI] [PubMed] [Google Scholar]
- 162.Skundric DS. Chemotactic signaling and beyond: link between interleukin-16 and axonal degeneration in multiple sclerosis. Neural Regeneration Res. 2015; 10: 1761–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lande R, Botti E, Jandus C, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 2014; 5: 5621. [DOI] [PubMed] [Google Scholar]
- 164.Arakawa A, Siewert K, Stöhr J, et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J. Exp. Med. 2015; 212: 2203–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Strassner JP, Rashighi M, Harris JE. Melanocytes in psoriasis: convicted culprit or bullied bystander? Pigment Cell Melanoma Res. 2016; 29: 261–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cheuk S, Wikén M, Blomqvist L, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J. Immunol. 2014; 192: 3111–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Matos TR, O’Malley JT, Lowry EL, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing αβ T cell clones. J. Clin. Invest. 2017; 127: 4031–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Singh TP, Lee CH, Farber JM. Chemokine receptors in psoriasis. Expert Opin. Ther. Targets 2013; 17: 1405–1422. [DOI] [PubMed] [Google Scholar]
- 169.Yost J, Gudjonsson JE. The role of TNF inhibitors in psoriasis therapy: new implications for associated comorbidities. F1000 Med. Rep. 2009; 1: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Frieder J, Kivelevitch D, Menter A Secukinumab: a review of the anti-IL-17A biologic for the treatment of psoriasis. Ther. Adv. Chronic Dis. 2018; 9: 5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kim T-G, Jee H, Fuentes-Duculan J, et al. Dermal clusters of mature dendritic cells and T cells are associated with the CCL20/CCR6 chemokine system in chronic psoriasis. J. Invest. Dermatol. 2014; 134: 1462–1465. [DOI] [PubMed] [Google Scholar]
- 172.Hedrick MN, Lonsdorf AS, Shirakawa A-K, et al. CCR6 is required for IL-23–induced psoriasis-like inflammation in mice. J. Clin. Invest. 2009; 119: 2317–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Singh SP, Zhang HH, Foley JF, Hedrick MN, Farber JM. Human T cells that are able to produce IL-17 express the chemokine receptor CCR6. The Journal of Immunology 2008; 180: 214–221. [DOI] [PubMed] [Google Scholar]
- 174.Harper EG, Guo C, Rizzo H, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J. Invest. Dermatol. 2009; 129: 2175–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Purzycka-Bohdan D, Szczerkowska-Dobosz A, Zablotna M, et al. Assessment of Interleukin 16 Serum Levels and Skin Expression in Psoriasis Patients in Correlation with Clinical Severity of the Disease. PLoS One 2016; 11: e0165577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Oh S-T, Schramme A, Tilgen W, Gutwein P, Reichrath J. Overexpression of CXCL16 in lesional psoriatic skin. Dermatoendocrinol. 2009; 1: 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Günther C, Carballido-Perrig N, Kaesler S, Carballido JM, Biedermann T CXCL16 and CXCR6 are upregulated in psoriasis and mediate cutaneous recruitment of human CD8+ T cells. J. Invest. Dermatol. 2012; 132: 626–634. [DOI] [PubMed] [Google Scholar]
- 178.Eyerich S, Eyerich K, Pennino D, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Invest. 2009; 119: 3573–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Res PCM, Piskin G, Boer OJ de, et al. Overrepresentation of IL-17A and IL-22 producing CD8 T cells in lesional skin suggests their involvement in the pathogenesis of psoriasis. PLoS One 2010; 5: e14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kirchhof MG, Dutz JP. The immunopathology of cutaneous lupus erythematosus. Rheum. Dis. Clin. North Am. 2014; 40: 455–74, viii. [DOI] [PubMed] [Google Scholar]
- 181.Stannard JN, Kahlenberg JM. Cutaneous lupus erythematosus: updates on pathogenesis and associations with systemic lupus. Curr. Opin. Rheumatol. 2016; 28: 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hejazi EZ, Werth VP. Cutaneous Lupus Erythematosus: An Update on Pathogenesis, Diagnosis and Treatment. Am. J. Clin. Dermatol. 2016; 17: 135–146. [DOI] [PubMed] [Google Scholar]
- 183.Wieczorek IT, Propert KJ, Okawa J, Werth VP. Systemic symptoms in the progression of cutaneous to systemic lupus erythematosus. JAMA Dermatol. 2014; 150: 291–296. [DOI] [PubMed] [Google Scholar]
- 184.Chong BF. Understanding how cutaneous lupus erythematosus progresses to systemic lupus erythematosus. JAMA Dermatol. 2014; 150: 296. [DOI] [PubMed] [Google Scholar]
- 185.Laurinaviciene R, Sandholdt LH, Bygum A Drug-induced cutaneous lupus erythematosus: 88 new cases. Eur. J. Dermatol. 2017; 27: 28–33. [DOI] [PubMed] [Google Scholar]
- 186.Sandholdt LH, Laurinaviciene R, Bygum A Proton pump inhibitor-induced subacute cutaneous lupus erythematosus. Br. J. Dermatol. 2014; 170: 342–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kuhn A, Wenzel J, Weyd H Photosensitivity, apoptosis, and cytokines in the pathogenesis of lupus erythematosus: a critical review. Clin. Rev. Allergy Immunol. 2014; 47: 148–162. [DOI] [PubMed] [Google Scholar]
- 188.Meller S, Winterberg F, Gilliet M, et al. Ultraviolet radiation--induced injury, chemokines, and leukocyte recruitment: an amplification cycle triggering cutaneous lupus erythematosus. Arthritis & Rheumatism 2005; 52: 1504–1516. [DOI] [PubMed] [Google Scholar]
- 189.Cohen-Solal JFG, Jeganathan V, Hill L, et al. Hormonal regulation of B-cell function and systemic lupus erythematosus. Lupus 2008; 17: 528–532. [DOI] [PubMed] [Google Scholar]
- 190.Yell JA, Burge SM. The effect of hormonal changes on cutaneous disease in lupus erythematosus. Br. J. Dermatol. 1993; 129: 18–22. [DOI] [PubMed] [Google Scholar]
- 191.Grimaldi CM, Michael DJ, Diamond B Cutting edge: expansion and activation of a population of autoreactive marginal zone B cells in a model of estrogen-induced lupus. J. Immunol. 2001; 167: 1886–1890. [DOI] [PubMed] [Google Scholar]
- 192.Bennion SD, Middleton MH, David-Bajar KM, Brice S, Norris DA. In three types of interface dermatitis, different patterns of expression of intercellular adhesion molecule-1 (ICAM-1) indicate different triggers of disease. J. Invest. Dermatol. 1995; 105: 71S–79S. [DOI] [PubMed] [Google Scholar]
- 193.Obermoser G, Sontheimer RD, Zelger B Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus 2010; 19: 1050–1070. [DOI] [PubMed] [Google Scholar]
- 194.Wenzel J, Zahn S, Mikus S, Wiechert A, Bieber T, Tüting T The expression pattern of interferon-inducible proteins reflects the characteristic histological distribution of infiltrating immune cells in different cutaneous lupus erythematosus subsets. Br. J. Dermatol. 2007; 157: 752–757. [DOI] [PubMed] [Google Scholar]
- 195.Flier J, Boorsma DM, Beek PJ van, et al. Differential expression of CXCR3 targeting chemokines CXCL10, CXCL9, and CXCL11 in different types of skin inflammation. J. Pathol. 2001; 194: 398–405. [DOI] [PubMed] [Google Scholar]
- 196.Wenzel J, Wörenkämper E, Freutel S, et al. Enhanced type I interferon signalling promotes Th1-biased inflammation in cutaneous lupus erythematosus. J. Pathol. 2005; 205: 435–442. [DOI] [PubMed] [Google Scholar]
- 197.Mande P, Zirak B, Ko W-C, et al. Fas ligand promotes an inducible TLR-dependent model of cutaneous lupus–like inflammation. J. Clin. Invest. 2018; 128: 2966–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Shipman WD, Chyou S, Ramanathan A, et al. A protective Langerhans cell–keratinocyte axis that is dysfunctional in photosensitivity. Sci. Transl. Med. 2018; 10: eaap9527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Garton KJ, Gough PJ, Blobel CP, et al. Tumor Necrosis Factor-α-converting Enzyme (ADAM17) Mediates the Cleavage and Shedding of Fractalkine (CX3CL1). J. Biol. Chem. 2001; 276: 37993–38001. [DOI] [PubMed] [Google Scholar]
- 200.Gerlach C, Moseman EA, Loughhead SM, et al. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity 2016; 45: 1270–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Morimura S, Sugaya M, Sato S Interaction between CX3CL1 and CX3CR1 regulates vasculitis induced by immune complex deposition. Am. J. Pathol. 2013; 182: 1640–1647. [DOI] [PubMed] [Google Scholar]
- 202.Inoue A, Hasegawa H, Kohno M, et al. Antagonist of fractalkine (CX3CL1) delays the initiation and ameliorates the progression of lupus nephritis in MRL/lpr mice. Arthritis Rheum. 2005; 52: 1522–1533. [DOI] [PubMed] [Google Scholar]
- 203.Wynn TA. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008; 214: 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Malviya N, Cyrus N, Turner J, Jacobe H 288 Immune dysregulation in morphea. J. Invest. Dermatol. 2017; 137: S49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tabib T, Morse C, Wang T, Chen W, Lafyatis R SFRP2/DPP4 and FMO1/LSP1 Define Major Fibroblast Populations in Human Skin. J. Invest. Dermatol. 2018; 138: 802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Torok KS, Kurzinski K, Kelsey C, et al. Peripheral blood cytokine and chemokine profiles in juvenile localized scleroderma: T-helper cell-associated cytokine profiles. Semin. Arthritis Rheum. 2015; 45: 284–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hams E, Bermingham R, Fallon PG. Macrophage and Innate Lymphoid Cell Interplay in the Genesis of Fibrosis. Front. Immunol. 2015; 6: 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.O’Brien JC, Rainwater YB, Malviya N, et al. Transcriptional and Cytokine Profiles Identify CXCL9 as a Biomarker of Disease Activity in Morphea. J. Invest. Dermatol. 2017; 137: 1663–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mirizio E, Mandal R, Yan Q, Horne W, Schollaert-Fitch K, & Torok KS (2018). Genetic Signatures from RNA Sequencing of Pediatric Localized Scleroderma (LS) Skin. ARTHRITIS & RHEUMATOLOGY, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kroeze KL, Boink MA, Sampat-Sardjoepersad SC, Waaijman T, Scheper RJ, Gibbs S Autocrine regulation of re-epithelialization after wounding by chemokine receptors CCR1, CCR10, CXCR1, CXCR2, and CXCR3. J. Invest. Dermatol. 2012; 132: 216–225. [DOI] [PubMed] [Google Scholar]
- 211.Yates CC, Whaley D, Kulasekeran P, et al. Delayed and deficient dermal maturation in mice lacking the CXCR3 ELR-negative CXC chemokine receptor. Am. J. Pathol. 2007; 171: 484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yates CC, Krishna P, Whaley D, Bodnar R, Turner T, Wells A Lack of CXC chemokine receptor 3 signaling leads to hypertrophic and hypercellular scarring. Am. J. Pathol. 2010; 176: 1743–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Yates CC, Whaley D, Hooda S, Hebda PA, Bodnar RJ, Wells A Delayed reepithelialization and basement membrane regeneration after wounding in mice lacking CXCR3. Wound Repair Regen. 2009; 17: 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Huen AC, Wells A The Beginning of the End: CXCR3 Signaling in Late-Stage Wound Healing. Adv. Wound Care 2012; 1: 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Jiang D, Liang J, Campanella GS, et al. Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan-4. J. Clin. Invest. 2010; 120: 2049–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Tager AM, Kradin RL, LaCamera P, et al. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am. J. Respir. Cell Mol. Biol. 2004; 31: 395–404. [DOI] [PubMed] [Google Scholar]
- 217.Xie H, Feng C, Fu Q, Sa Y -L, Xu, Y-M. Crosstalk between TGF-β1 and CXCR3 signaling during urethral fibrosis. Mol. Cell. Biochem. 2014; 394: 283–290. [DOI] [PubMed] [Google Scholar]
- 218.Menke J, Zeller GC, Kikawada E, et al. CXCL9, but not CXCL10, promotes CXCR3-dependent immune-mediated kidney disease. J. Am. Soc. Nephrol. 2008; 19: 1177–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Rabquer BJ, Tsou P-S, Hou Y, et al. Dysregulated expression of MIG/CXCL9, IP-10/CXCL10 and CXCL16 and their receptors in systemic sclerosis. Arthritis Res. Ther. 2011; 13: R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Yanaba K, Muroi E, Yoshizaki A, et al. Serum CXCL16 concentrations correlate with the extent of skin sclerosis in patients with systemic sclerosis. J. Rheumatol. 2009; 36: 1917–1923. [DOI] [PubMed] [Google Scholar]
- 221.Gao J-L, Wynn TA, Chang Y, et al. Impaired Host Defense, Hematopoiesis, Granulomatous Inflammation and Type 1–Type 2 Cytokine Balance in Mice Lacking CC Chemokine Receptor 1. J. Exp. Med. 1997; 185: 1959–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Blease K, Mehrad B, Standiford TJ, et al. Airway Remodeling Is Absent in CCR1−/− Mice During Chronic Fungal Allergic Airway Disease. The Journal of Immunology 2000; 165: 1564–1572. [DOI] [PubMed] [Google Scholar]
- 223.Anders H-J, Vielhauer V, Frink M, et al. A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J. Clin. Invest. 2002; 109: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Pesce J, Kaviratne M, Ramalingam TR, et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J. Clin. Invest. 2006; 116: 2044–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Brodeur TY, Robidoux TE, Weinstein JS, Craft J, Swain SL, Marshak-Rothstein A IL-21 Promotes Pulmonary Fibrosis through the Induction of Profibrotic CD8+ T Cells. J. Immunol. 2015; 195: 5251–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Castelino FV, Seiders J, Bain G, et al. Amelioration of dermal fibrosis by genetic deletion or pharmacologic antagonism of lysophosphatidic acid receptor 1 in a mouse model of scleroderma. Arthritis Rheum. 2011; 63: 1405–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Tager AM, LaCamera P, Shea BS, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008; 14: 45–54. [DOI] [PubMed] [Google Scholar]
- 228.Rork JF, Rashighi M, Harris JE. Understanding autoimmunity of vitiligo and alopecia areata. Curr. Opin. Pediatr. 2016; 28: 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gilhar A, Etzioni A, Paus R Alopecia areata. N. Engl. J. Med. 2012; 366: 1515–1525. [DOI] [PubMed] [Google Scholar]
- 230.Paus R, Slominski A, Czarnetzki BM. Is alopecia areata an autoimmune-response against melanogenesis-related proteins, exposed by abnormal MHC class I expression in the anagen hair bulb? Yale J. Biol. Med. 1993; 66: 541–554. [PMC free article] [PubMed] [Google Scholar]
- 231.Bodemer C, Peuchmaur M, Fraitaig S, Chatenoud L, Brousse N, De Prost Y Role of cytotoxic T cells in chronic alopecia areata. J. Invest. Dermatol. 2000; 114: 112–116. [DOI] [PubMed] [Google Scholar]
- 232.Cetin ED, Savk E, Uslu M, Eskin M, Karul A Investigation of the inflammatory mechanisms in alopecia areata. Am. J. Dermatopathol. 2009; 31: 53–60. [DOI] [PubMed] [Google Scholar]
- 233.Ito T, Ito N, Saatoff M, et al. Maintenance of hair follicle immune privilege is linked to prevention of NK cell attack. J. Invest. Dermatol. 2008; 128: 1196–1206. [DOI] [PubMed] [Google Scholar]
- 234.Inui S, Noguchi F, Nakajima T, Itami S Serum thymus and activation-regulated chemokine as disease activity and response biomarker in alopecia areata. J. Dermatol. 2013; 40: 881–885. [DOI] [PubMed] [Google Scholar]
- 235.Christoph T, Müller-Röver S, Audring H, et al. The human hair follicle immune system: cellular composition and immune privilege. Br. J. Dermatol. 2000; 142: 862–873. [DOI] [PubMed] [Google Scholar]
- 236.Alkhalifah A, Alsantali A, Wang E, McElwee KJ, Shapiro J Alopecia areata update: part I. Clinical picture, histopathology, and pathogenesis. J. Am. Acad. Dermatol. 2010; 62: 177–88, quiz 189–90. [DOI] [PubMed] [Google Scholar]
- 237.Gilhar A, Ullmann Y, Berkutzki T, Assy B, Kalish RS. Autoimmune hair loss (alopecia areata) transferred by T lymphocytes to human scalp explants on SCID mice. J. Clin. Invest. 1998; 101: 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Sundberg JP, Cordy WR, King LE Jr., Alopecia areata in aging C3H/HeJ mice. J. Invest. Dermatol. 1994; 102: 847–856. [DOI] [PubMed] [Google Scholar]
- 239.Jabbari A, Cerise JE, Chen JC, et al. Molecular signatures define alopecia areata subtypes and transcriptional biomarkers. EBioMedicine 2016; 7: 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ghoreishi M, Martinka M, Dutz JP. Type 1 interferon signature in the scalp lesions of alopecia areata. Br. J. Dermatol. 2010; 163: 57–62. [DOI] [PubMed] [Google Scholar]
- 241.Ito T, Hashizume H, Shimauchi T, et al. CXCL10 produced from hair follicles induces Th1 and Tc1 cell infiltration in the acute phase of alopecia areata followed by sustained Tc1 accumulation in the chronic phase. J. Dermatol. Sci. 2013; 69: 140–147. [DOI] [PubMed] [Google Scholar]
- 242.Ito T, Fujiyama T, Tokura Y Th1 chemokine CXCL10 and alopecia areata: the possible target for the treatment of alopecia areata. J. Clin. Cell. Immunol. 2016; 7: 2. [Google Scholar]
- 243.Freyschmidt-Paul P, McElwee KJ, Hoffmann R, et al. Interferon-γ-deficient mice are resistant to the development of alopecia areata. Br. J. Dermatol. 2006; 155: 515–521. [DOI] [PubMed] [Google Scholar]
- 244.Gilhar A, Kam Y, Assy B, Kalish RS. Alopecia areata induced in C3H/HeJ mice by interferon-gamma: evidence for loss of immune privilege. J. Invest. Dermatol. 2005; 124: 288–289. [DOI] [PubMed] [Google Scholar]
- 245.McPhee CG, Duncan FJ, Silva KA, et al. Increased expression of Cxcr3 and its ligands, Cxcl9 and Cxcl10, during the development of alopecia areata in the mouse. J. Invest. Dermatol. 2012; 132: 1736–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Dai Z, Xing L, Cerise J, et al. CXCR3 Blockade Inhibits T Cell Migration into the Skin and Prevents Development of Alopecia Areata. J. Immunol. 2016; 197: 1089–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Xing L, Dai Z, Jabbari A, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat. Med. 2014; 20: 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Jabbari A, Dai Z, Xing L, et al. Reversal of Alopecia Areata Following Treatment With the JAK½ Inhibitor Baricitinib. EBioMedicine 2015; 2: 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Liu LY, Craiglow BG, Dai F, King BA. Tofacitinib for the treatment of severe alopecia areata and variants: A study of 90 patients. J. Am. Acad. Dermatol. 2017; 76: 22–28. [DOI] [PubMed] [Google Scholar]
- 250.Jabbari A, Nguyen N, Cerise JE, et al. Treatment of an alopecia areata patient with tofacitinib results in regrowth of hair and changes in serum and skin biomarkers. Exp. Dermatol. 2016; 25: 642–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Tanyasiri K, Hira K, Mitsuishi K, Ueki R, Sekigawa I, Ogawa H Interleukin-16 in patients with alopecia areata. J. Dermatol. Sci. 2005; 37: 55–57. [DOI] [PubMed] [Google Scholar]
- 252.Song T, Pavel AB, Wen H-C, et al. An integrated model of alopecia areata biomarkers highlights both TH1 and TH2 upregulation. J. Allergy Clin. Immunol. 2018; 142: 1631–1634.e13. [DOI] [PubMed] [Google Scholar]
- 253.Subramanya RD, Coda AB, Sinha AA. Transcriptional profiling in alopecia areata defines immune and cell cycle control related genes within disease-specific signatures. Genomics 2010; 96: 146–153. [DOI] [PubMed] [Google Scholar]
- 254.Lew B-L, Chung J-H, Sim W-Y. Association between IL16 gene polymorphisms and susceptibility to alopecia areata in the Korean population. Int. J. Dermatol. 2014; 53: 319–322. [DOI] [PubMed] [Google Scholar]
- 255.O’Connor TM, O’Connell J, O’Brien DI, Goode T, Bredin CP, Shanahan F The role of substance P in inflammatory disease. J. Cell. Physiol. 2004; 201: 167–180. [DOI] [PubMed] [Google Scholar]
- 256.Peters EMJ, Liotiri S, Bodó E, et al. Probing the effects of stress mediators on the human hair follicle: substance P holds central position. Am. J. Pathol. 2007; 171: 1872–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Gilhar A, Paus R, Kalish RS. Lymphocytes, neuropeptides, and genes involved in alopecia areata. J. Clin. Invest. 2007; 117: 2019–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Siebenhaar F, Sharov AA, Peters EMJ, et al. Substance P as an Immunomodulatory Neuropeptide in a Mouse Model for Autoimmune Hair Loss (Alopecia Areata). J. Invest. Dermatol. 2007; 127: 1489–1497. [DOI] [PubMed] [Google Scholar]
- 259.García-Hernández MJ, Ruiz-Doblado S, Rodriguez-Pichardo A, Camacho F Alopecia areata, stress and psychiatric disorders: a review. J. Dermatol. 1999; 26: 625–632. [DOI] [PubMed] [Google Scholar]
- 260.Arck PC, Handjiski B, Peters EMJ, et al. Stress inhibits hair growth in mice by induction of premature catagen development and deleterious perifollicular inflammatory events via neuropeptide substance P-dependent pathways. Am. J. Pathol. 2003; 162: 803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Arck P, Paus R From the brain-skin connection: the neuroendocrine-immune misalliance of stress and itch. Neuroimmunomodulation 2006; 13: 347–356. [DOI] [PubMed] [Google Scholar]
- 262.Paus R, Theoharides TC, Arck PC. Neuroimmunoendocrine circuitry of the “brain-skin connection.” Trends Immunol. 2006; 27: 32–39. [DOI] [PubMed] [Google Scholar]
- 263.Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat. Immunol. 2018; 19: 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Coghill JM, Fowler KA, West ML, et al. CC chemokine receptor 8 potentiates donor Treg survival and is critical for the prevention of murine graft-versus-host disease. Blood 2013; 122: 825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Zhang N, Schröppel B, Lal G, et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity 2009; 30: 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Sather BD, Treuting P, Perdue N, et al. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J. Exp. Med. 2007; 204: 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Müller M, Carter SL, Hofer MJ, et al. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. The Journal of Immunology 2007; 179: 2774–2786. [DOI] [PubMed] [Google Scholar]
- 268.Yamazaki T, Yang XO, Chung Y, et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J. Immunol. 2008; 181: 8391–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Chaudhry A, Rudra D, Treuting P, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 2009; 326: 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Barth T, Schmidt D, Botteron C, et al. An early reduction in Treg cells correlates with enhanced local inflammation in cutaneous leishmaniasis in CCR6-deficient mice. PLoS One 2012; 7: e44499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, et al. Critical role for CCR5 in the function of donor CD4+ CD25+ regulatory T cells during acute graft-versus-host disease. Blood 2005; 106: 3300–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Cavassani KA, Campanelli AP, Moreira AP, et al. Systemic and local characterization of regulatory T cells in a chronic fungal infection in humans. J. Immunol. 2006; 177: 5811–5818. [DOI] [PubMed] [Google Scholar]
- 273.Moreira AP, Cavassani KA, Massafera Tristão FS, et al. CCR5-dependent regulatory T cell migration mediates fungal survival and severe immunosuppression. J. Immunol. 2008; 180: 3049–3056. [DOI] [PubMed] [Google Scholar]
- 274.Lee JH, Kang SG, Kim CH. FoxP3+ T Cells Undergo Conventional First Switch to Lymphoid Tissue Homing Receptors in Thymus but Accelerated Second Switch to Nonlymphoid Tissue Homing Receptors in Secondary Lymphoid Tissues. The Journal of Immunology 2007; 178: 301–311. [DOI] [PubMed] [Google Scholar]
- 275.Sugaya M, Suga H, Miyagaki T, Ohmatsu H, Okochi H, & Sato S (2013). CXCR3 deficiency prolongs Th1-type contact hypersensitivity. JOURNAL OF INVESTIGATIVE DERMATOLOGY, 133, S6–S6. [DOI] [PubMed] [Google Scholar]
- 276.Suga H, Sugaya M, Miyagaki T, Ohmatsu H, Okochi H, Sato S CXCR3 deficiency prolongs Th1-type contact hypersensitivity. J. Immunol. 2013; 190: 6059–6070. [DOI] [PubMed] [Google Scholar]
- 277.Klarquist J, Denman CJ, Hernandez C, et al. Reduced skin homing by functional Treg in vitiligo. Pigment Cell Melanoma Res. 2010; 23: 276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Dwivedi M, Laddha NC, Arora P, Marfatia YS, Begum R Decreased regulatory T-cells and CD 4+/CD 8+ ratio correlate with disease onset and progression in patients with generalized vitiligo. Pigment Cell Melanoma Res. 2013; 26: 586–591. [DOI] [PubMed] [Google Scholar]
- 279.Lili Y, Yi W, Ji Y, Yue S, Weimin S, Ming L Global activation of CD8+ cytotoxic T lymphocytes correlates with an impairment in regulatory T cells in patients with generalized vitiligo. PLoS One 2012; 7: e37513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Eby JM, Kang H-K, Tully ST, et al. CCL22 to Activate Treg Migration and Suppress Depigmentation in Vitiligo. J. Invest. Dermatol. 2015; 135: 1574–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Asothai R, Anand V, Das D, et al. Distinctive Treg associated CCR4-CCL22 expression profile with altered frequency of Th17/Treg cell in the immunopathogenesis of Pemphigus Vulgaris. Immunobiology 2015; 220: 1129–1135. [DOI] [PubMed] [Google Scholar]
- 282.Ning W, Wang S, Dong X, et al. Epigallocatechin-3-gallate (EGCG) Suppresses the Trafficking of Lymphocytes to Epidermal Melanocytes via Inhibition of JAK2: Its Implication for Vitiligo Treatment. Biol. Pharm. Bull. 2015; 38: 1700–1706. [DOI] [PubMed] [Google Scholar]
- 283.Yoon JY, Kwon HH, Min SU, Thiboutot DM, Suh DH. Epigallocatechin-3-gallate improves acne in humans by modulating intracellular molecular targets and inhibiting P. acnes. J. Invest. Dermatol. 2013; 133: 429–440. [DOI] [PubMed] [Google Scholar]
- 284.Domingo DS, Camouse MM, Hsia AH, et al. Anti-angiogenic effects of epigallocatechin-3-gallate in human skin. Int. J. Clin. Exp. Pathol. 2010; 3: 705–709. [PMC free article] [PubMed] [Google Scholar]
- 285.Craiglow BG, Tavares D, King BA. Topical Ruxolitinib for the Treatment of Alopecia Universalis. JAMA Dermatol. 2016; 152: 490–491. [DOI] [PubMed] [Google Scholar]
- 286.Kennedy Crispin M, Ko JM, Craiglow BG, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight 2016; 1: e89776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Mackay-Wiggan J, Jabbari A, Nguyen N, et al. Oral ruxolitinib induces hair regrowth in patients with moderate-to-severe alopecia areata. JCI Insight 2016; 1: e89790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Morris GM, Nahmias ZP, Kim BS. Simultaneous improvement of alopecia universalis and atopic dermatitis in a patient treated with a JAK inhibitor. JAAD Case Rep 2018; 4: 515–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Kim SR, Charos A, Damsky W, Heald P, Girardi M, King BA. Treatment of generalized deep morphea and eosinophilic fasciitis with the Janus kinase inhibitor tofacitinib. JAAD Case Rep 2018; 4: 443–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Bissonnette R, Papp KA, Poulin Y, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br. J. Dermatol. 2016; 175: 902–911. [DOI] [PubMed] [Google Scholar]
- 291.King B, Lee AI, Choi J Treatment of Hypereosinophilic Syndrome with Cutaneous Involvement with the JAK Inhibitors Tofacitinib and Ruxolitinib. J. Invest. Dermatol. 2017; 137: 951–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Kahn JS, Deverapalli SC. JAK‐STAT signaling pathway inhibition: a role for treatment of discoid lupus erythematosus and dermatomyositis. International journal of 2018; [DOI] [PubMed] [Google Scholar]
- 293.Dees C, Tomcik M, Palumbo-Zerr K, et al. JAK-2 as a novel mediator of the profibrotic effects of transforming growth factor β in systemic sclerosis. Arthritis Rheum. 2012; 64: 3006–3015. [DOI] [PubMed] [Google Scholar]
- 294.Alves de Medeiros AK, Speeckaert R, Desmet E, Van Gele M, De Schepper S, Lambert J JAK3 as an Emerging Target for Topical Treatment of Inflammatory Skin Diseases. PLoS One 2016; 11: e0164080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Fukuyama T, Ganchingco JR, Bäumer W Demonstration of rebound phenomenon following abrupt withdrawal of the JAK1 inhibitor oclacitinib. Eur. J. Pharmacol. 2017; 794: 20–26. [DOI] [PubMed] [Google Scholar]
- 296.Tobinai K, Takahashi T, Akinaga S Targeting chemokine receptor CCR4 in adult T-cell leukemia-lymphoma and other T-cell lymphomas. Curr. Hematol. Malig. Rep. 2012; 7: 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Ishida T, Utsunomiya A, Jo T, et al. Mogamulizumab for relapsed adult T-cell leukemia-lymphoma: Updated follow-up analysis of phase I and II studies. Cancer Sci. 2017; 108: 2022–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Yoshie O, Matsushima K CCR4 and its ligands: from bench to bedside. Int. Immunol. 2015; 27: 11–20. [DOI] [PubMed] [Google Scholar]
- 299.Duvic M, Evans M, Wang C Mogamulizumab for the treatment of cutaneous T-cell lymphoma: recent advances and clinical potential. Ther. Adv. Hematol. 2016; 7: 171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Freyschmidt-Paul P, Happle R, McElwee KJ, Hoffmann R Alopecia areata: treatment of today and tomorrow. J. Investig. Dermatol. Symp. Proc. 2003; 8: 12–17. [DOI] [PubMed] [Google Scholar]
- 301.Hoffmann R, Wenzel E, Huth A, et al. Cytokine mRNA levels in Alopecia areata before and after treatment with the contact allergen diphenylcyclopropenone. J. Invest. Dermatol. 1994; 103: 530–533. [DOI] [PubMed] [Google Scholar]
- 302.Harris JE, Seykora JT, Lee RA. Renbök Phenomenon and Contact Sensitization in a Patient With Alopecia Universalis. Arch. Dermatol. 2010; 146: 422–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Hedrick MN, Lonsdorf AS, Hwang ST, Farber JM. CCR6 as a possible therapeutic target in psoriasis. Expert Opin. Ther. Targets 2010; 14: 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Andrews SP, Cox RJ. Small Molecule CXCR3 Antagonists. J. Med. Chem. 2016; 59: 2894–2917. [DOI] [PubMed] [Google Scholar]
- 305.Tan Q, Zhu Y, Li J, et al. Structure of the CCR5 Chemokine Receptor–HIV Entry Inhibitor Maraviroc Complex. Science 2013; 341: 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Richmond JM & Harris JE (2014). The Pathobiologic Basis of Autoimmunity. Pathobiology of Human Disease, 39–48. [Google Scholar]
- 307.Patel D, Richmond JM, Garg M, et al. 102 CXCL9 drives morphea pathogenesis in mice. J. Invest. Dermatol. 2018; 138: S17. [Google Scholar]
- 308.Genovese MC, Kremer J, Zamani O, et al. Baricitinib in Patients with Refractory Rheumatoid Arthritis. N. Engl. J. Med. 2016; 374: 1243–1252. [DOI] [PubMed] [Google Scholar]
- 309.Taylor PC, Keystone EC, Heijde D van der, et al. Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis. N. Engl. J. Med. 2017; 376: 652–662. [DOI] [PubMed] [Google Scholar]
- 310.Papp KA, Menter MA, Raman M, et al. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br. J. Dermatol. 2016; 174: 1266–1276. [DOI] [PubMed] [Google Scholar]
- 311.Damsky W, King BA. JAK inhibitors in dermatology: The promise of a new drug class. J. Am. Acad. Dermatol. 2017; 76: 736–744. [DOI] [PMC free article] [PubMed] [Google Scholar]