

Abstract
Scope:
DNA repair inhibitors have broad clinical applications in tumor types with DNA repair defects, including colorectal cancer (CRC). Structural analogs of the anticancer agent sulforaphane (SFN) were investigated as modifiers of histone deacetylase (HDAC) and histone acetyltransferase (HAT) activity, and for effects on DNA damage/repair pertinent to human CRC.
Methods and results:
In the polyposis in rat colon (Pirc) model, single oral administration of SFN and structurally related long-chain isothiocyanates (ITCs) decreased histone deacetylase 3 (HDAC3) expression and increased pH2AX levels markedly in adenomatous colon polyps, extending prior observations on HDAC3 inhibition/turnover in cell-based assays. Colon cancer cells at a high initial plating density had diminished cytotoxicity from SFN, whereas novel tetrazole-containing heterocyclic analogs of SFN retained their efficacy. The potent SFN analogs triggered DNA damage, cell cycle arrest, apoptosis, and loss of a key DNA repair regulator, C-terminal binding protein (CtBP) interacting protein (CtIP). These SFN analogs also altered HAT/HDAC activities and histone acetylation status, lowered the expression of HDAC3, P300/CBP-associated factor (PCAF) and lysine acetyltransferase 2A (KAT2A/GCN5), and attenuated homologous recombination (HR)/non-homologous end joining (NHEJ) repair activities in colon cancer cells.
Conclusion:
Novel tetrazole-containing heterocyclic analogs of SFN provide a new avenue for chemosensitization in colon cancer cells via modulation of HAT/HDAC activities and associated DNA damage/repair signaling pathways.
Keywords: colon cancer, C-terminal binding protein (CtBP) interacting protein, DNA damage, DNA repair, histone acetyltransferase, histone deacetylase, sulforaphane analogs
1. Introduction
According to the American Cancer Society, about 135,430 people were diagnosed with colorectal cancer (CRC) and about 50,260 died of the disease in 2017, making it the second leading cause of cancer death that affects both men and women.[1] Therapeutic approaches to CRC are currently inadequate, and include surgery with radiation and/or chemotherapy.[2] A key mechanism of action of such therapy is through DNA damage,[3] but cancer cells counteract these events by activating DNA repair,[4] typically resulting in drug resistance and poor clinical responses. In the absence of alternative approaches, there is an urgent need to define adjunct therapies that interfere with the DNA repair machinery and increase the efficacy of current CRC treatments. Several poly(ADP-ribose) polymerase (PARP) inhibitors are under clinical investigation in cancers with DNA repair defects, and progress in PARP inhibitor development has led to the accelerated approval of Olaparib (Lynparza) for ovarian cancer.[4] Subsequently, DNA repair inhibitors are anticipated to have a much broader clinical application in malignancies characterized by DNA repair defects, such as CRC.[5] This utility may be enhanced via combination with radiation, chemotherapy, or targeted anticancer agents, such as those acting on the epigenome.
DNA double-strand breaks (DSBs) are among the most damaging lesions in DNA.[3–6] If left unrepaired, DSBs lead to cell death, whereas faulty DNA repair leads to genomic instability.[5] DSBs are repaired by two key pathways, homologous recombination (HR) and non-homologous end joining (NHEJ). DSB repair is initiated by DNA end resection, giving rise to single-strand DNA (ssDNA) tails that are immediately coated by the replication protein A (RPA) complex for protection.[3,6] C-terminal binding protein (CtBP) interacting protein (CtIP) is a major player in the choice between NHEJ and HR that initiates HR by activating DNA end resection together with the MRN complex.[6] Multiple signals converge on CtIP to initiate DNA end resection, the nature of which depends on the complexity of the break within the chromatin.[6,7] Subsequently, BRCA2 mediates the replacement of RPA by RAD51, leading to DNA repair using the intact sequence as a template.[3] Alternatively, DNA repair by NHEJ is initiated by binding of the Ku70/Ku80 heterodimer, which forms a complex with DNA ligase IV and DNA-PKcs.[3] A principle component of chromatin that plays a role in these processes is the modification of histones, and therefore there is growing interest in histone/protein post-translational modifications, such as acetylation, that impact DNA damage response pathways.[6,7] Protein acetylation is modulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), which influence chromatin dynamics and DNA damage response via changes in histone and non-histone proteins.[6–8]
We previously reported that dietary phytochemicals that affect the epigenome can trigger a DNA damage response in cancer cells, redefining their role as chemo- or radiosensitizers.[7] Among the cancer preventive agents found abundantly in cruciferous vegetables, sulforaphane (SFN) and related isothiocyanates (ITCs) inhibit HDAC activity[8] and increase HDAC protein turnover,[9] triggering DNA damage in colon cancer cells.[10] To extend these observations in vivo, we tested SFN and its structural analogs in the polyposis in rat colon (Pirc) model,[11] demonstrating loss of histone deacetylase 3 (HDAC3) protein expression in adenomatous colon polyps and increased pH2AX levels, a marker of DNA damage. We also examined RPA32, a protein hyperphosphorylated in response to DNA damage that binds ss-DNA, promoting DNA repair.[12] In order to assess the effect on key DNA repair pathways, we took advantage of the SeeSaw Reporter (SSR), a system designed to assess the balance between NHEJ and HR.[13] In cell-based assays, we further tested novel tetrazole-containing SFN analogs with increased potency toward cell cycle arrest, apoptosis induction, turnover of the DNA repair protein CtIP, and altered HAT/HDAC activities linked to deregulated homologous recombination (HR) and NHEJ.
2. Experimental Section
2.1. Animals
Preclinical studies were approved by the Institutional Animal Care and Use Committee (IACUC 2017–0236-IBT). Rat experiments used the Pirc model of familial adenomatous polyposis (FAP).[11] Animals were generated as published[11] by breeding F344/NTac ApcPirc/+ males and females (Taconic, NY) and housed in standard caging with free access to water and AIN93 diet, minus t-butylhydro-quinone (Research Diets, NJ). ITCs were formulated in corn oil at a concentration of 8 mg mL–1 before dosing. At 8 months of age, Pirc males (n = 3 per group) were assigned randomly to five different arms of the study. Rats were administered a single oral gavage of 60 mg kg–1 of ITC, or vehicle. The study was terminated 6 h after dosing, rats were euthanized, and colon polyps were collected and stored prior to immunoblotting and immunohistochemistry (IHC), as reported.[17]
2.2. Cells and Chemicals
Cell lines were obtained from ATCC and grown at 37 °C in 5% CO2 with 1% penicillin/streptomycin. HCT116 and SW480 colon cancer cells were cultured in McCoy’s 5A media (Invitrogen) supplemented with 10% heat inactivated fetal bovine serum, whereas CCD112 non-cancer colonic cells, HEK293 human embryonic kidney cells, and SH-SY5Y human bone marrow cells were maintained in Eagle’s minimum essential medium (EMEM) supplemented with 10% fetal bovine serum. Treatments were performed when cells were about 80% confluent, unless indicated otherwise.
SFN, 6-methylsulfinylhexyl isothiocyanate (6-SFN), and 9-methylsulfinylnonyl isothiocyanate (9-SFN) were procured from LKT laboratories, and allyl isothiocyanate (AITC) was from Sigma. Novel tetrazole-containing heterocyclic analogs of SFN analogs (chemical structures shown in Table 1) were synthesized at Tianjin Key Laboratory of Molecular Drug Research, as reported.[14] Aliquots of stock solutions in dimethylsulfoxide (DMSO; 56 mM) were stored at –20 °C and thawed for single use before each experiment. Stock solutions were diluted in growth medium and added to cells at a final concentration of 15 μM, unless indicated otherwise. Additional experiments included deletion of CtIP from colon cancer cells using CRISPR/Cas9, as reported previously.[15]
Table 1.
Structure and molecular weight of SFN and its analogs.
| Compound | Structure | Chemical formula and molecular weight |
|---|---|---|
| SFN | C6H11NOS2
MW: 177.2880 |
|
| SFN2 | C6H11NO2S2
MW: 193.2870 |
|
| 9A | C10H13NO3S2
MW: 259.3451 |
|
| B | 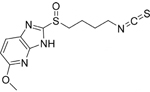 |
C12H14N4O2S2
MW: 310.3952 |
| 9B | 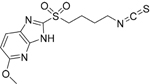 |
C12H14N4O3S2
MW: 326.3946 |
| 9C | 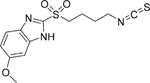 |
C13H15N3O3S2
MW: 325.4065 |
| 3D | 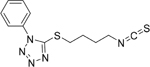 |
C12H13N5S2
MW: 291.3951 |
| 8D | 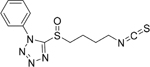 |
C12H13N5OS2
MW: 307.3945 |
| 9D | 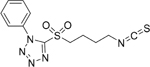 |
C12H13N5O2S2
MW: 323.3939 |
| 8E | 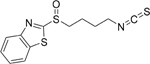 |
C12H12N2OS3
MW: 296.4315 |
2.3. Immunoblotting
Whole cell extracts were prepared and immunoblotted as described previously.[10] Frozen samples of colon tumors and/or adjacent tissue were thawed and subjected to immunoblotting using the reported methodology.[16] Briefly, proteins (20 μg per lane) were separated by SDS-PAGE on 4–12% Bis–Tris gel, and transferred to nitrocellulose membranes (Invitrogen). Membranes were saturated with 2% BSA for 1 h, followed by overnight incubation at 4 °C with primary antibodies for HDAC1 (#7872), HDAC2 (#7899), HDAC3 (#11417), HDAC6 (#11420), and histone acetyltransferase 1 (HAT1) (#8751) from Santa Cruz; pH2AX Ser139 (#2577), H2AX (#2595), CtIP (#9201), lysine acetyltransferase 2A (KAT2A/GCN5) (#3305), and cleaved caspase-3 (#9661) from Cell Signaling; pRPA32 S4/S8 (#A300– 245A), P300/CBP-associated factor (PCAF; #A301–666A), E1A binding protein P300 (P300; #A300–358A), and lysine acetyltransferase 8 (MOF) (#A300–992A) from Bethyl Labs; SIRT6 (#39911) from Active Motif; acetyl histone H4 (#06–866) from Millipore; histone H4 (#AB10158) from Abcam; and β-actin (#A5441) from Sigma. After washing, membranes were incubated with horseradish peroxidase-conjugated secondary anti-bodies (Bio-Rad) for 1 h. Bands were visualized using Western Lightning Plus-ECL Enhanced Chemiluminescence Substrate (Perkin Elmer, Inc.) and detected using a ChemiDoc imaging system (BioRad).
2.4. Immunohistochemistry
Formalin-fixed paraffin-embedded rat colon tumor was processed for IHC, as reported.[17] Slides containing 5 μm sections were rehydrated and placed in an Autostainer (Dako). After adding primary antibody to pH2AX (Abcam #ab23345) and HDAC3 (Santa Cruz #11417) for 30 min, and One Step HRP polymer anti-IgG (ImmunoBioscience) for 7 min, Nova Red (Vector Labs) was applied for 5 min followed by hematoxylin (Dako). Images were acquired on a Nikon E400 microscope equipped with a CCD camera.
2.5. Immunofluorescence
Cells were grown on glass coverslips (#1.5, VWR), pre-coated with poly-L-Lysine (Sigma, #P1399), and were treated with DMSO, SFN or its analogs in 6-well plates. Following treatment, cells were fixed with 2% phosphate buffered formalin for 10 min, and permeabilized with 2.1% citric acid and 0.5% Tween 20 in distilled water (pH 2.0) for 10 min at room temperature. Samples were blocked in 1% BSA and incubated overnight with pH2AX Ser139 (#2577) from Cell Signaling and pRPA32 S4/S8 (#A300–245A) from Bethyl Labs, followed by incubation with secondary antibody coupled to AlexaFluor 488 (1:250, Molecular Probes) for 1 h. DAPI (Prolong Gold antifade reagent, Molecular Probes) was used to counterstain the nuclei. Fluorescent images were captured on a fluorescence microscope (DeltaVision Elite) for image acquisition and analysis.
2.6. Cell Viability
Cell viability was determined using the CCK-8 assay (Dojindo) as reported previously.[10] The assay measures cell viability based on the ability of living cells to reduce soluble WST-8 to water-soluble formazan dye. Cells in the exponential growth phase were plated at a cell density of either 5000 cells per well (sub-confluent) or 20 000 cells per well (confluent) in 96-well tissue culture plates. After attachment overnight, cells were treated with compounds for 24 h in the concentration range of 0–60 μM (0, 0.9, 1.8, 3.7, 7.5,15, 30, and 60 μM). CCK-8 solution (10 μL) was added to each well of the 96-well plate and incubated for 4 h at room temperature. The absorbance was measured at 450 nm using a Cytation3 microplate reader (BioTek).
2.7. Flow Cytometry
Cell cycle analysis was done as reported before.[10] Briefly, cells were treated with DMSO, SFN, or its structural analogs at 15 μM. Adherent and non-adherent cells were collected at 24 h in cold phosphate buffered saline (PBS), fixed in 70% ethanol, and stored at 4 °C for at least 48 h. Fixed cells were washed with PBS and resuspended in propidium iodide (PI)/Triton X-100 staining solution containing RNaseA. Samples were incubated in the dark for 30 min before cell cycle analysis. DNA content was detected using the LSR II Flow cytometer (BD Biosciences).
2.8. HAT and HDAC Activity
Whole cell lysates were tested for HAT and HDAC activity using commercially available kits. HDAC activity was measured using the Fluor-de-Lys assay, as reported earlier.[10] The HAT activity assay was performed using a colorimetric assay kit according to manufacturer’s instructions (BioVision, Milpitas, California). Incubations were performed with whole cell extract (10 μg protein) of HCT116 cells following treatment with DMSO, SFN, or its structural analogs at 15 μM. Fluorescence- and absorbance-based read-outs of HDAC and HAT activity, respectively, were quantified using a microplate reader (Cytation3, BioTek).
2.9. Immunoprecipitation
The IP methodology was performed as reported earlier.[10] Whole cell extracts from adherent and non-adherent cells were prepared as previously described. Cell extract (500 μg) was pre-cleared with 100 μL Protein A Sepharose CL-4B beads (GE Healthcare Life sciences) on a rotator at 4 °C for 2 h. Pre-cleared supernatant was subjected to overnight IP with anti-acetyl lysine antibody (10 μg mg–1 protein, #AB3879, Millipore). Samples were incubated with 100 μL of beads on a rotator at 4 °C for 2 h and acetylated proteins bound to the beads were washed three times with PBST, denatured in standard loading buffer, and examined by immunoblotting with primary antibodies for CtIP (#9201) from Cell Signaling, as described above.
2.10. SeeSaw Reporter Assay
The impairment of DNA DSB repair (HR and NHEJ) due to treatment with SFN or its analogs was measured via the SSR assay, using a previously published methodology.[13] Briefly, ≈1 × 105 cells were plated in a 60 mm petri dish overnight followed by treatment with 5 μM of SFN, its structural analogs, or DMSO (vehicle control) for 6 h. Following treatment, cells were cotransfected with a combination of 2 μg each of SSR 2.0 (a gift from Pablo Huertas Sanchez, Addgene plasmid # 0393) and I-Scel expression plasmid (pCBAScel was a gift from Maria Jasin, Addgene plasmid #26477) using Lipofectamine 2000 reagent. After 24 h post-transfection, cells were fixed with 4% paraformaldehyde, stained, and washed with PBS prior to visualization using a fluorescent microscope. For flow cytometry, cells were harvested, washed with 1 × DPBS, incubated with 1 mL of TrypLE Express (Gibco) dissociation reagent at 37 °C for 2 min, followed by centrifugation at 900 RPM for 3 min, and re-suspended in 500 μL of 1X DPBS with 0.1% FBS solution. FACS analysis for green fluorescent protein (GFP) and red fluorescent protein (RFP) positive cells representing the NHEJ and HR repair frequencies, respectively, was performed using BD FACS Fortessa and BD FACS-Diva Software v5.0.3. The percent positivity for GFP and RFP positive cells from 10,000 events was measured for each sample. The assay was carried out in HCT116, HEK293, and SH-SY5Y cells following the same protocol.
2.11. Proximity Ligation Assays
HCT116 cells were grown overnight in 8-well chamber slides. After treatment for 6 h with test agents, cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and incubated with primary antibodies for CtIP (#61142, mouse monoclonal; Active Motif), PCAF (#A301–666A, rabbit polyclonal; Bethyl Labs), or GCN5 (#3305, rabbit polyclonal, cell signaling), as per reported methodologies.[18] Proximity ligation assays (PLA) were carried out using the Duolink PLA kit (Sigma-Aldrich, St. Louis, MO), according to the manufacturer’s recommendations. The nuclei were counterstained with DAPI, and the PLA signals were visualized and quantified with the aid of a fluorescence microscope (DeltaVision Elite) at 40× magnification.
2.12. Statistics
Results are representative of at least three independent assays unless otherwise indicated, and expressed as mean ± SE. Student’s t-test was used for paired comparisons, while differences between groups were determined by analysis of variance (ANOVA) followed by Bonferroni’s test using GraphPad Prism software version 5.04, and indicated as such in the respective figure legends. Statistical significance is indicated in the figures as follows: p < 0.05 (*), p < 0.01 (**), p < 0.001 (***).
3. Results
3.1. ITCs Decrease HDAC3 Expression and Increase DNA Damage in Pirc colon Polyps
We previously demonstrated that SFN, 6-SFN, and 9-SFN targeted HDAC3 for inhibition and turnover, and caused DNA damage in human colon cancer cells.[10] In the current study, we extended these observations in vivo, using the Pirc model of colon cancer. Immunoblotting (Figure 1A) and IHC analyses (Figure 1B) revealed that adenomatous colon polyps in the Pirc model had HDAC3 overexpression and increased pH2AX levels, a DNA damage biomarker, compared to adjacent normal-looking colon. When Pirc rats were administered ITCs by single oral gavage, the acute dosing regimen caused increased pH2AX levels in colon polyps, coincident with decreased HDAC3 (Figure 1C). Densitometry measurements from the immunoblotting analyses revealed a significant increase in pH2AX/H2AX by ITC treatment, compared to vehicle controls (p < 0.01, Figure 1D). In parallel, HDAC3/β-actin was significantly decreased by ITC treatment (p < 0.01, Figure 1E), whereas SIRT6/β-actin was unchanged by the test agents (Figure 1F). In Pirc colon polyps, IHC analyses further corroborated the increase in pH2AX levels after ITC treatment (Figure 1G).
Figure 1.
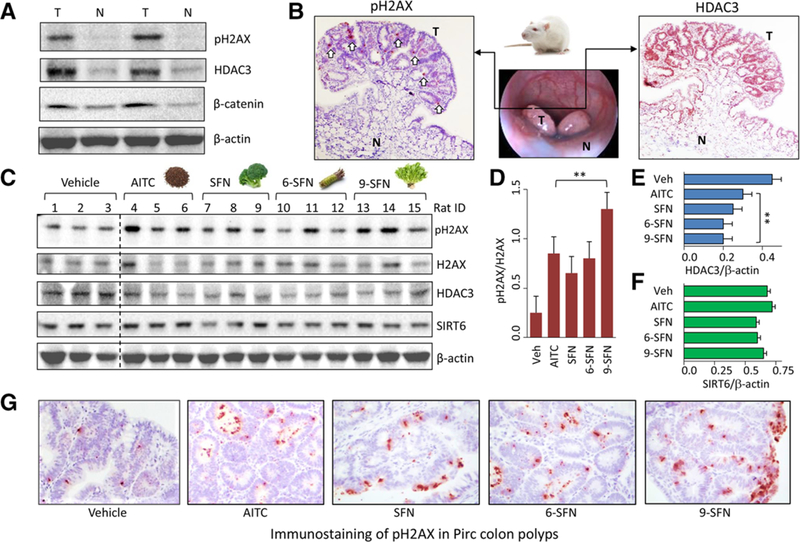
Decreased HDAC3 expression and increased pH2AX in the Pirc model. Tumor and adjacent normal colon from Pirc males (≈8 months old) were probed for HDAC3 and pH2AX by A) immunoblotting with β-actin as loading control and B) immunohistochemistry (IHC); T, tumor; N, normal; white arrows, pH2AX immunostaining. C) Pirc males received corn oil vehicle or ITC (60 mg kg–1) by single oral gavage and were sacrificed 6 h later. Tumor tissue lysates (n = 3) were immunoblotted for pH2AX, H2AX, HDAC3, and SIRT6, with β-actin as loading control. D–F) Immunoblots were quantified by densitometry and the data plotted as mean ± SEM (n = 3) by one-way ANOVA (**p < 0.01, compared to vehicle). G) Immunostaining of pH2AX in Pirc colon polyps after IHC analyses.
3.2. Plating Density Dictates the Differential Response to SFN and Its Structural Analogs in Colon Cancer Cells
In human colon cancer cells, SFN-induced cytotoxicity was highly dependent on initial cell seeding density (Figure 2). Compared to sub-confluent HCT116 cells seeded at 5000 cells per well, confluent HCT116 cells seeded at 20,000 cells per well were significantly less sensitive to SFN treatment (p < 0.05 for 3.75–7.5 μM SFN, and p < 0.01 for 15–60 μM SFN). The reduced sensitivity of confluent cells to SFN treatment provided a platform to screen for more potent analogs under the experimental conditions employed. Shi et al.[14] synthesized several heterocyclic ring-containing SFN analogs—for structures and molecular weights refer to Table 1. Structural analogs were evaluated for cytotoxicity against confluent HCT116 and SW480 colon cancer cells (Tables 2 and 3). Under these experimental conditions, the tetrazole side-chain analogs 3D, 8D, and 9D were significantly more effective than SFN (p < 0.05), with 8D that retained the sulfoxide group, being the most potent. No such difference in efficacy between compounds was observed when cells were plated at sub-confluent cell density (Table S1, Supporting Information). Notably, 3D, 8D, and 9D were less toxic to colonic non-transformed/normal CCD112 cells (IC50 > 60 μM, Table 3) as compared to SW480 colon cancer cells. 9A, 9C (Table 1) and 3D, 9D (Table 2) have lower IC50 compared to SFN, but the high interexperimental variability suggests the need for future lead optimization to improve issues, such as, poor solubility and stability.
Figure 2.
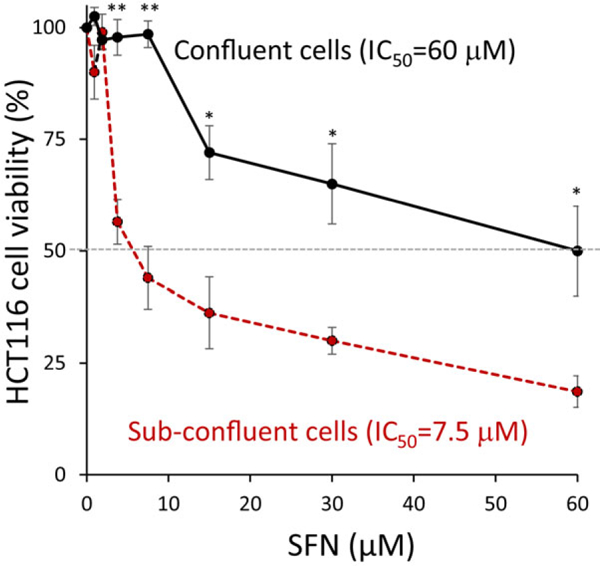
Cytotoxicity of SFN is dependent on plating density. HCT116 colon cancer cells were seeded at two different cell densities (30%, sub-confluent; 80%, confluent) and treated for 24 h with SFN in the dose range 0–60 μm. Cell viability was assessed using the CCK-8 assay (n = 4 experiments); *p < 0.05, < 0.01, confluent versus the corresponding sub-confluent cell density data point using Student’s t-test.
Table 2.
Cytotoxicity of SFN analogs in HCT116 colon cancer cell line (confluent cell density).
|
|
Cell viability of HCT116 cells IC50 [μm] (mean ± SE) |
||
|---|---|---|---|
| Compound ID | R | n | Confluent cell density |
| 8D | 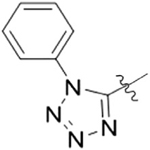 |
1 | 10.6 ± 2.2a) |
| 3D | 0 | 12.3 ± 3.3a) | |
| 9D | 2 | 11.5 ± 4.3a) | |
| 9C | 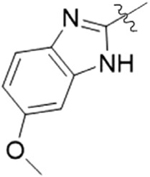 |
2 | 14.9 ± 7.9 |
| 9A | 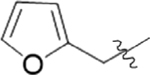 |
2 | 18.2 ± 5.2 |
| 8E | 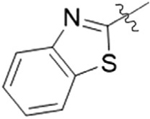 |
1 | 30.1 ± 4.1 |
| 8B |  |
1 | 36.6 ± 7.5 |
| 9B | 2 | 56.5 ± 3.5 | |
| SFN2 | methyl | 2 | 54.5 ± 5.5 |
| SFN | methyl | 1 | 56.4 ± 1.8 |
p < 0.05 compared to SFN.
Table 3.
Cytotoxicity of SFN analogs in cancer (SW480) versus normal (CCD112) colon cells.
| Cell viability IC50
[μm] (mean ± SE) |
||||
|---|---|---|---|---|
| Compound ID | R | n | Colon cancer SW480 |
Colon normal CCD112 |
| 8D | 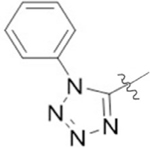 |
1 | 5.0 ± 0.9a) | >60 |
| 3D | 0 | 16.2 ± 2.6 | >60 | |
| 9D | 2 | 23.1 ± 5.8 | >60 | |
| SFN | Methyl | 1 | 45.1 ± 10.9 | >60 |
p < 0.01 compared to SFN.
Compared to vehicle controls, fluorescence-activated cell sorting (FACS) revealed no significant effect of SFN on cell cycle kinetics in the confluent plating protocol (Figure 3A). However, under the same experimental conditions, HCT116 cells treated for 24 h with the structural analogs underwent cell cycle arrest with potency in the order 3D > 8D > 9D > SFN (Figure 3A). The analogs 3D, 8D, and 9D increased 25%–35% of cells in G2M phase (Figure 3B), which were statistically higher than vehicle treatment (p < 0.05).
Figure 3.
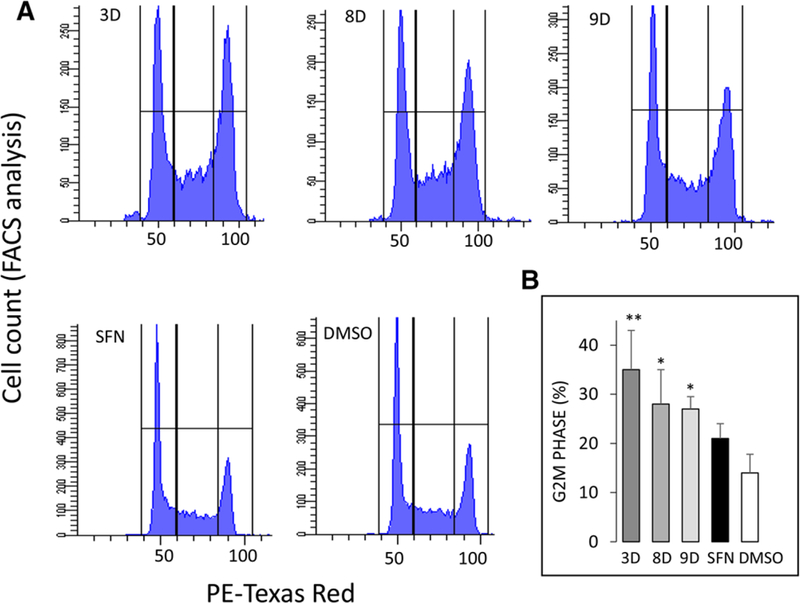
Cell cycle arrest by SFN and structural analogs. HCT116 cells were seeded overnight at confluent cell density and treated for 24 h with 15 μm SFN or selected structural analogs from Table 1. A) DNA content via flow cytometry and B) FACS data quantified (mean ± SEM), *p < 0.05, **p < 0.01 vs. DMSO vehicle control, by one-way ANOVA (n = 3 experiments).
3.3. Tetrazole SFN Analogs Enhance DNA Damage, Reduce DNA Repair, Trigger CtIP Loss, and Cause Programed Cell Death
SFN-treated HCT116 cells (15 μm, 24 h) had increased nuclear pH2AX and pRPA32 levels, indicating enhanced DNA damage and repair, respectively (Figure 4A, arrows). Immunoblotting revealed that, at the early time point of 6 h, SFN and its analogs 3D, 8D, and 9D increased pH2AX, with 8D being most effective (Figure 4B), which was confirmed by immunofluorescence (Figure S1, Supporting Information). In addition, 8D-treated cells had increased cleaved caspase-3, indicative of apoptotic cell death (Figure 4B). However, unlike SFN, pRPA32 was not increased by 3D, 8D, and 9D (Figure 4B), revealing that DNA repair signaling was impeded by the SFN analogs. These results coincided with the loss of a key DNA repair protein, CtIP, after cells were treated with SFN analogs, with 8D causing nearly complete loss of CtIP protein (Figure 4B).
Figure 4.
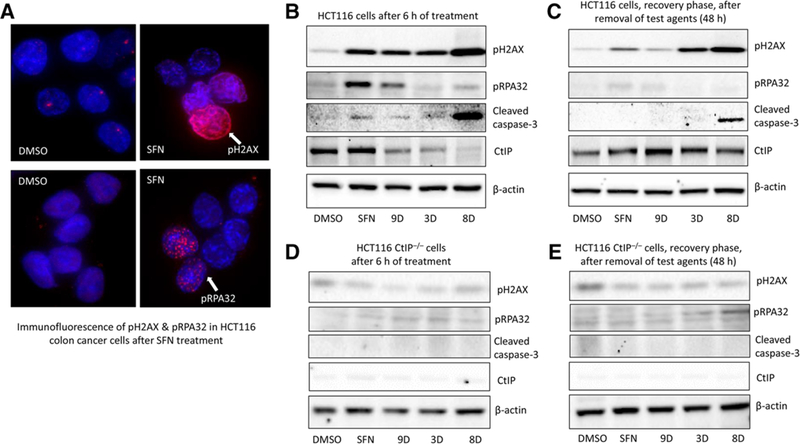
DNA damage response by SFN and structural analogs. A) HCT116 cells were treated with DMSO or 15 μm SFN for 24 h and examined for pH2AX and pRPA32 immuno-fluorescence (arrows); DAPI stained nuclei (blue). Confluent HCT116 cells were treated with DMSO (vehicle), 15 μm SFN, or its analogs (3D, 8D, and 9D), and whole cell lysates were assessed by immunoblotting B) 6 h after treatment with compounds or C) 48 h after treatment, which includes 6 h of treatment followed by compound-free media for 42 h (recovery). D,E) Repeat of experiments in (B) and (C), using HCT116 CtIP knockout cells (HCT116 CtIP–/–), generated via CRISPR/Cas9 genome editing. Antibodies detected DNA damage signaling (pH2AX and pRPA32), apoptosis (cleaved caspase-3), and DNA repair (CtIP). β-actin, loading control. Data are representative of at least three independent experiments.
To assess the recovery from DNA damage, HCT116 cells were treated for 6 h with SFN or its analogs, as above, and the cell culture medium was replaced with ITC-free medium for an additional 42 h. In contrast to the findings at 6 h (Figure 4B), there was minimal pH2AX induction detected at 48 h following SFN and 9D removal (Figure 4C), indicating efficient recovery from the DNA damaging effects of these compounds. However, pH2AX expression continued to be induced markedly in 3D- and 8D-treated HCT116 cells at 48 h (Figure 4C), representing a more sustained DNA damaging effect of these two compounds. Further, cleaved caspase-3 continued to be detected in 8D-treated cells at 48 h (Figure 4C). Notably, CtIP turnover was no longer evident in the recovery samples (Figure 4C).
To further dissect the role of CtIP, we generated HCT116 cells lacking CtIP using CRISPR/Cas9. Remarkably, SFN and its analogs (3D, 8D, and 9D) had reduced cytotoxicity in HCT116 CtIP–/– cells, compared to parental HCT116 cells (Table S2, Supporting Information), and pH2AX was not increased by any of the treatments (Figure 4D,E). CtIP–/– cells were resistant to SFN and its analogs, and did not undergo DNA damage, repair, or cell death by apoptosis (Figure 4D,E).
3.4. Tetrazole SFN Analogs Interfere with HR and NHEJ Pathways in Colon Cancer Cells
Since CtIP is a key player in the HR pathway,[19] we employed the SSR assay, which is highly sensitive to DNA end resection.[13] With the SSR 2.0 construct, DSBs induced by the meganuclease I-SceI can be monitored as homology-driven and homology-independent repair events based on distinct fluorescent signals of red for HR and green for NHEJ (Figure 5A,B). Under these experimental conditions, cells were treated with 5 μm SFN analogs for 6 h, and fluorescence was measured by flow cytometry (Figure 5C). Quantification of the percentage DNA repair from three independent experiments confirmed that both HR and NHEJ pathways were decreased by SFN analogs 3D, 8D, and 9D (Figure 5D); with 8D and 9D significantly reducing both HR and NHEJ pathways (p < 0.05).
Figure 5.
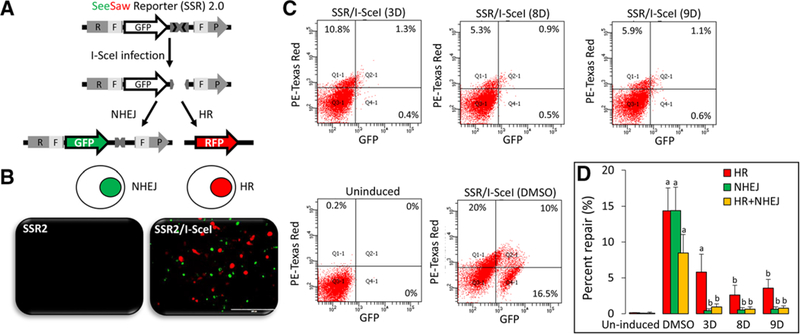
Inhibition of DNA repair by SFN analogs. A) A schematic representation of the See Saw reporter SSR 2.0, as published.[18] B) Images of HCT116 cells transfected with SSR 2.0 for 24 h (left panel), followed by transfection with I-SceI for 6 h (right panel) to induce DNA damage response, as indicated by HR-mediated (red) or NHEJ-mediated repair (green) fluorescence. C) SH-SY5Y cells were co-transfected with SSR 2.0/I-SceI and treated with 5 μm of SFN analogs for 6 h, as described in Experimental Section. Percent NHEJ (GFP) or HR (RFP) was measured via flow cytometry in negative controls without I-SceI (Uninduced), positive controls (SSR 2.0/I-SceI, DMSO), and test samples (SSR 2.0/I-SceI, treated with SFN analogs). D) Percentage NHEJ or HR quantified from three independent experiments, mean ± variance. a) p < 0.05 (DMSO vs. uninduced), b) p < 0.05 (DMSO vs. treatment), analyzed by one-way ANOVA.
3.5. Tetrazole SFN Analogs Target Specific HDACs and HATs in Colon Cancer Cells
Colon cancer cells plated at high seeding density were treated with SFN or its analogs for 6 h, and whole cell lysates were examined for HAT/HDAC activity and protein expression. HDAC activity was unchanged by SFN, unlike in the sub-confluent assays,[9,10] but was reduced significantly after treatment of HCT116 cells with 3D, 8D, and 9D (p < 0.05, Figure 6A, upper panel). Immunoblotting of whole cell lysates revealed a marked loss of HDAC3 (Figure 6A, lower panel), with little or no changes in other class I (HDAC½), class II (HDAC6), and class III (SIRT6) HDACs.
Figure 6.
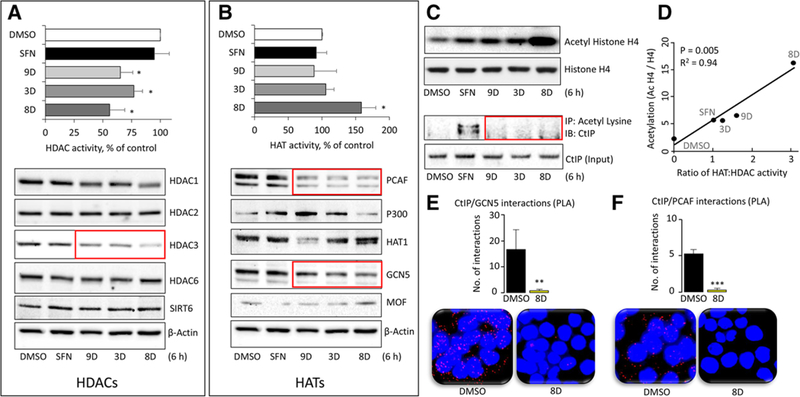
HAT and HDAC changes produced by SFN analogs. HCT116 cells were treated with DMSO (vehicle) or 15 μm SFN or its analogs for 6 h and whole cell lysates were examined for A) HDAC activity (top panel) or HDAC protein expression, with β-actin as loading control in the immunoblots (bottom panel). B) Repeat of the experiments in (A), but for HATs. C) Histone H4 acetylation, normalized to histone H4 (top panel) assessed by immunoblotting, and CtIP acetylation changes normalized to CtIP input controls (bottom panel) by immunoprecipitation after Ac-Lys pull-down. D) The ratio of HAT:HDAC activity (from A,B) and the level of histone acetylation (densitometry from C) were plotted for each compound. Data are representative ofthree experiments (*p < 0.05 vs. DMSO). E,F) In HCT116 cells, proximity ligation assays (PLA) identified endogenous interactions ofCtIP:PCAF and CTIP:GCN5 proteins in situ (red dots). DAPI-stained nuclei (blue). Cells were imaged 6 h after treatment with DMSO (vehicle) or analog 8D, and the interactions per nuclei were quantified and averaged across five different microscopic fields. Data = mean ± SE, **p < 0.01, ***p < 0.001 for 8D versus vehicle controls using Student’s t-test. Data are representative of at least three independent experiments.
Under similar experimental conditions, we observed consistently increased HAT activity by 8D, but not by SFN or the other analogs (p < 0.05, Figure 6B, upper panel). Immunoblotting of whole cell lysates revealed a loss of PCAF and GCN5 protein expression by 9D, 3D, and 8D (Figure 6B, red boxes), which was not observed for SFN. Induction of p300 was detected for SFN, 3D, and 9D, whereas 9D also produced a marked loss of HAT1 protein expression. Loss of MOF expression was detected in SFN-treated cells, whereas the structural analogs 3D, 8D, and 9D had minimal effects on MOF, compared to the vehicle controls (Figure 6B, lower panel).
As a read-out of HAT versus HDAC changes, we evaluated histone and non-histone protein acetylation. We observed an increase in histone H4 acetylation after treatment with SFN, 9D, and 3D, and a striking induction by the structural analog 8D (Figure 6C, top panel). Combining data from Figure 6A–C, histone H4 acetylation was highly correlated with the ratio of HAT:HDAC activities (Figure 6D). Acetylation of the non-histone protein CtIP was detected for SFN, as reported,[10] but not for the structural analogs 3D, 8D, and 9D (Figure 6C, red box). Subsequently, interactions of endogenous proteins in situ were examined via PLA, revealing for the first time abundant CtIP/GCN5 (Figure 6E) and CtIP/PCAF (Figure 6F) associations in HCT116 colon cancer cells that were inhibited significantly by 8D treatment.
4. Discussion
Beneficial effects of cruciferous vegetables, such as broccoli, mustard, watercress, and wasabi (Figure 1), have been attributed to their ITC content, and to compounds characterized by an –N = C = S functional group.[20,21] We recently discovered that through the inhibition of deacetylase activity, ITCs such as SFN, 6-SFN, and 9-SFN target HDAC3 for inhibition and turnover, and cause DNA damage in colon cancer cells, but not in normal cells.[10] In the current study, we first sought to extend these observations in vivo, using the Pirc model of colon cancer. Based on an initial dose-finding study that showed SFN was safe after single oral gavage of 35–140 mg kg–1 body weight,[22,23] Pirc males were administered 60 mg ITC kg–1 body weight by single oral gavage. This acute dose was well tolerated by the rats, and 6 h after dosing, ITCs decreased HDAC3 expression in colon polyps coincident with increased pH2AX levels (Figure 1). These in vivo results confirm that SFN and related ITCs cause DNA damage in colon cancer. The class III deacetylase SIRT6 was immunoblotted and compared with the class I deacetylase HDAC3, since both enzymes reportedly act as CtIP deacetylases.[10] However, unlike results obtained in colon cancer cells, SIRT6 levels were unchanged after treatment with ITCs in vivo (Figure 1C,F). The mechanistic reasons are worthy of further investigation.
SFN has been evaluated in several human trials (www.clinicaltrials.gov), including the testing of SFN-enriched broccoli sprout extract (BSE) supplements,[16] but opportunities exist to improve efficacy.[16] We turned to medicinal chemistry approaches that modified SFN as a lead compound. For this, we tested SFN analogs that were chemically modified by the replacement of the methyl group in SFN with heterocyclic moieties such as furan, methoxypyrimidine, tetrazole, and thiazole, as well as replacement of the sulfoxide group with sulfide or sulfone (Table 1). Confluence-dependent resistance has been shown during drug treatment,[24] differentiation of human embryonic cells,[25] and stem cells.[26] Based on our results, we speculate that confluent cells have reduced proliferative activity, enhanced cell–cell communication, and altered HDAC/HAT activities that collectively dictate the responsiveness to ITC treatment. In fact, after reaching confluency by 48–72 h, HCT116 cells underwent cell cycle arrest in G0/G1, and HDAC activity reached a plateau.[9] Given the differential effects of test agents on cytotoxicity versus cell proliferation,[24–26] we plated HCT116 cells at different initial cell seeding densities and demonstrated that high seeding density (80% confluence) lessened the cytotoxicity of SFN (IC50 > 50 μm) compared to low seeding density (30% confluence, IC50 ≈ 10 μm; Figure 2). In marked contrast, however, 3D, 8D, and 9D retained their potency (IC50 ≈ 10 μm) even when HCT116 cells were plated at high seeding density (Table 2). These analogs were at least fivefold more potent than SFN, with 8D exhibiting an IC50 of 10.6 ± 2.2 μm in HCT116 cells (Table 2), and 5.0 ± 0.9 μm in SW480 cells (Table 3). Importantly, SFN and its analogs were less cytotoxic to CCD112 normal colon cells (Table 3, IC50 > 60 μm) compared to SW480 colon cancer cells. The high proliferative activity in cancer cells, along with dysregulated HAT, HDAC, and DNA repair proteins, might explain the differential response compared with normal cells. HDACs have been implicated in DNA damage and/or repair, and HDAC3 knockdown recapitulated some of the changes associated with DNA damage.[10] Notably, pH2AX induction occurred within 6 h, the same timeframe as HDAC3 turnover in SFN-treated colon cancer cells.[10] HDAC3 is overexpressed in colon tumor compared to adjacent normal tissues,[16] which might explain why we observe a marked inhibitory effect by ITCs that preferentially target HDAC3 for inhibition and turnover.[9,10,16] In HCT116 cells, treatment with SFN analogs 3D, 9D, and 8D led to G2M cell cycle arrest (Figure 3). Moreover, SFN analogs, in particular 8D, i) increased cleaved caspase-3 indicative of apoptosis induction (Figure 4B), ii) enhanced pH2AX levels indicative of DNA damage (Figure 4B), iii) reduced pRPA32 and CtIP indicative of reduced DNA repair (Figure 4B), and iv) had poor recovery of pH2AX (Figure 4C).
We previously discovered that through the inhibition of deacetylase activity, SFN targets CtIP for early acetylation in colon cancer cells, interfering with CtIP as a key regulator of DNA repair.[10] This was supported by the observation that ITC-induced CtIP acetylation and turnover coincided with the activation of an autophagic response.[10] We determined that HDAC3 interacts directly with CtIP, and that the turnover of HDAC3 by SFN causes N-terminal acetylation and turnover of CtIP protein (Rajendran et al., manuscript in preparation). Interestingly, the novel compounds 3D, 8D, and 9D also targeted CtIP for turnover (Figure 4B), but in contrast to SFN, no CtIP acetylation was detected (Figure 6C, red box, bottom panel). This indicates that other protein turnover pathways might be involved, such as via ubiquitination.[27] Nonetheless, regardless of the specific pathway(s) involved in CtIP turnover, it appears that SFN and its analogs require CtIP for triggering DNA damage (Figure 4D,E). Since CtIP is necessary for H2AX phosphorylation,[28] it is possible that SFN and its analogs engage CtIP as a chemosensitizer for activating pH2AX. Experiments using CtIP–/– cells (Figure 4D,E) established that pH2AX is not triggered by SFN and its analogs in the absence of CtIP.
Using a unique reporter assay that is sensitive to DNA end resection,[13] we examined the early steps shared by the various HR pathways, and observed that the potent SFN analogs inhibited both HR and NHEJ pathways (Figure 5). Effects of SFN analogs on CtIP turnover point to roles in HR,[29] whereas inhibition of NHEJ might implicate other proteins, such as cell cycle and apoptosis regulator 2 (CCAR2), which is evolving as a “master regulator” of DNA repair.[13] In order to further characterize the role of HR/NHEJ, other important players in the HR/NHEJ pathway, such as Ku70/80, BRCA1, RAD51 need to be tested in future experiments.
The DNA damage response is modulated by the acetylation status of histone and non-histone proteins, and by the opposing activities of HAT and HDAC enzymes.[30] Competing HAT versus HDAC regulatory mechanisms of ITCs have received relatively scarce attention, especially in the context of DNA damage/repair defects in cancer cells. We found that HCT116 cells treated with SFN analogs 3D, 8D, and 9D had reduced HDAC activity (Figure 6A) and increased HAT activity, particularly for 8D (Figure 6B). HDAC inhibition coincided with the loss of HDAC3 protein expression (Figure 6A, lower panel, red box), whereas HAT activation involved a complex interplay of increased and decreased HAT proteins, depending on the SFN analog tested (Figure 6B, lower panel). An exhaustive screen of all potential HATs was not undertaken, and it remains to be clarified as to why histone H4 acetylation linked to an open chromatin state[31] was observed for all of the test agents (Figure 6C, top panel), whereas only SFN triggered CtIP acetylation (Figure 6C, bottom panel). Rather than protein expression per se, it might be protein–protein interactions that are of greater importance.
In this context, ITCs have been shown to exploit faulty DNA repair events,[10] via loss of HDAC3 maintenance of chromatin structure and genome stability in colon cancer cells.[32] Moreover, bromodomain (BRD)-containing HATs (p300, GCN5, and PCAF) are recruited to DNA damage sites and regulate DSB repair,[33] with perturbation of acetylation signaling negatively impacting HR.[27] Currently, it is unknown if these HATs facilitate HR exclusively through their catalytic activities, or through their BRD acetyl “reader” functionality, affecting the transcription of DNA repair genes.[33] For example, GCN5 was previously implicated in CtIP acetylation by ITCs,[10] and loss of GCN5 and PCAF interactions with CtIP (Figure 6E,F) provides one explanation for the lack of CtIP acetylation after treatment with 3D, 8D, and 9D (Figure 6C, bottom panel). Also, turnover of HAT1 by analog 9D might promote HR by facilitating histone turnover.[34]
Collectively, our studies indicate that potent tetrazole-containing SFN analogs might destabilize a critical repair complex containing CtIP, HDAC3, PCAF, and GCN5 (Figure 7), providing an avenue for synthetic lethality.[19] Future investigations are warranted in order to delineate their individual roles, and to fully understand the relevance of these targets for combination chemotherapy using drugs + ITCs to enhance chemosensitization in the clinical setting.
Figure 7.
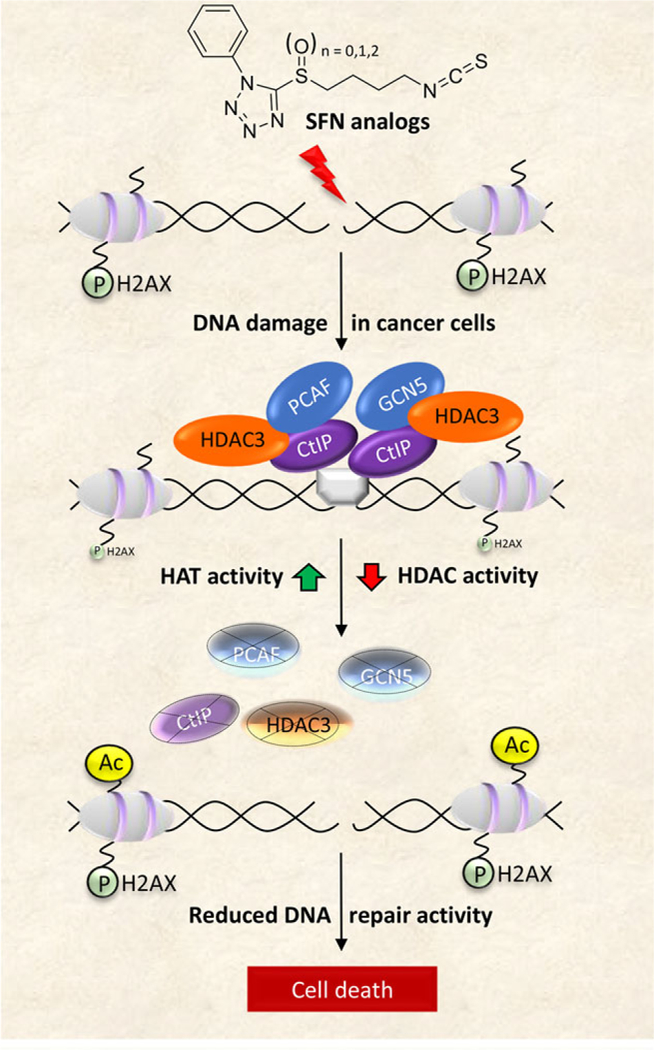
Working model of HDAC/HAT modulation by SFN analogs impacting DNA repair pathways, leading to cancer cell death. Ac, acetylation; P, phosphorylation. Through various mechanisms, not well characterized, SFN analogs trigger DSB in DNA that become flanked by increased pH2AX levels. A functional DNA damage response requires the formation of an efficient repair complex, typically comprising an MRN complex (gray box bridging the DNA break point) bound by the DNA resection and repair factor, CtIP. HATs and HDACs, such as PCAF, GCN5, and HDAC3, regulate the acetylation status of CtIP and maintain its activity/stability, leading to efficient DNA repair and reduced pH2AX levels. Treatment with SFN analogs causes an imbalance in HAT/HDAC activity and protein expression, leading to increased histone acetylation. This also adversely affects the stability of CtIP and its binding partners, including PCAF, GCN5, and HDAC3. As a crucial factor for DNA end resection, reduced levels of CtIP negatively influence homologous recombination (HR), concluding in impairment of DSB repair and eventual cell death. As described in the text, normal cells escape these outcomes, and are refractory to the mechanisms that trigger DNA damage and apoptosis in cancer cells.
Supplementary Material
Acknowledgements
P.R., A.O., and R.H.D. were responsible for the concept and design of the studies. J.M. and M.H. performed DNA repair experiments. C.Y. synthesized and provided the SFN analogs. L.L. and G.S.J. performed PLA, immunofluorescence, and microscopy. A.O. performed cell-based mechanistic experiments, immunoblotting, and immunoprecipitation experiments. W.M.D. assisted with the animal experiments. P.R. and R.H.D. drafted, edited, and finalized the manuscript, with input from all co-authors. The authors thank Margie Moczygemba and Sevinj Iskandarova of the Flow Cytometry Core (Texas A&M Health Science Center, Houston, TX) for technical assistance. Christiane V. Lohr and Kay A. Fischer performed IHC on Pirc tissues (Oregon Veterinary Diagnostic Laboratory, Oregon State University, Corvallis, OR). This research was supported in part by NIH grants CA090890 and CA122959, as well as the John S. Dunn Foundation, Chancellor’s Research Initiative, for R.H.D., and R01 NS088645 for M.H.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Adaobi Okonkwo, Center for Epigenetics and Disease Prevention, Institute of Biosciences and Technology, Texas A&M Health Science Center, Texas A&M College of Medicine, Houston, TX, USA, 77030.
Dr. Joy Mitra, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX, USA, 77030
Gavin S. Johnson, Center for Epigenetics and Disease Prevention, Institute of Biosciences and Technology, Texas A&M Health Science Center, Texas A&M College of Medicine, Houston, TX, USA, 77030
Li Li, Center for Epigenetics and Disease Prevention, Institute of Biosciences and Technology, Texas A&M Health Science Center, Texas A&M College of Medicine, Houston, TX, USA, 77030.
Wan Mohaiza Dashwood, Center for Epigenetics and Disease Prevention, Institute of Biosciences and Technology, Texas A&M Health Science Center, Texas A&M College of Medicine, Houston, TX, USA, 77030.
Dr. Muralidhar L. Hegde, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX, USA, 77030 Weill Cornell Medical College of Cornell University NY, USA, 10065.
Dr. Chen Yue, The State Key Laboratory of Elemento-Organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), College of Pharmacy, and Tianjin Key Laboratory of Molecular Drug Research Nankai University Tianjin, China, 300071
Dr. Roderick H. Dashwood, Department of Clinical Cancer Prevention University of Texas MD Anderson Cancer Center Houston, TX, USA, 77030 Department of Nutrition and Food Science, Texas A&M University, College Station, TX, USA, 77843; Department of Molecular and Cellular Medicine Texas A&M College of Medicine College Station, TX, USA, 77843; Center for Epigenetics and Disease Prevention, Institute of Biosciences and Technology, Texas A&M Health Science Center, Texas A&M College of Medicine, Houston, TX, USA, 77030.
Dr. Praveen Rajendran, Center for Epigenetics and Disease Prevention, Institute of Biosciences and Technology, Texas A&M Health Science Center, Texas A&M College of Medicine, Houston, TX, USA, 77030
References
- [1].American Cancer Society. Cancer Facts & Figures 2018. Atlanta, Ga: American Cancer Society, 2018. Retrieved from https://www.cancer.org/cancer/colon-rectal-cancer/about/key-statistics.html [Google Scholar]
- [2].Wolpin BM, Mayer RJ, Gastroenterology, 2008, 134, 1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Huertas P, Nat. Struct. Mol. Biol. 2010, 17, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lord CJ, Ashworth A, Curr. Opin. Pharmacol. 2008, 8, 363. [DOI] [PubMed] [Google Scholar]
- [5].Sinicrope FA, Nat. Rev. Clin. Oncol. 2010, 7, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Roth-stein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, Foiani M, Nature 2011, 471, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rajendran P, Ho E, Williams DE, Dashwood RH, Clin. Epigenetics 2011, 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Myzak MC, Karplus PA, Chung FL, Dashwood RH, Cancer Res. 2004, 64, 5767. [DOI] [PubMed] [Google Scholar]
- [9].Rajendran P, Delage B, Dashwood WM, Yu TW, Wuth B, Williams DE, Ho E, Dashwood RH, Mol. Cancer. 2011, 10, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rajendran P, Kidane A, Yu TW, Dashwood WM, Bisson WH, Lohr CV, Ho E, Williams DE, Dashwood RH, Epigenetics. 2013, 8, 612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Amos-Landgraf JM, Kwong LN, Kendziorski CM, Reichelderfer M, Torrealba J, Weichert J, Haag JD, Chen KS, Waller JL, Gould MN, Dove WF, Proc. Natl. Acad. Sci. 2007, 104, 4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Binz SK, Sheehan AM, Wold MS, DNA Repair (Amst). 2004, 3, 1015. [DOI] [PubMed] [Google Scholar]
- [13].López-Saavedra A, Gómez-Cabello D, Domínguez-Sánchez MS, Mejías-Navarro F, Fernández-Ávila MJ, Dinant C, Martínez-Macías MI, Bartek J, Huertas P, Nat. Commun. 2016, 7, 12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shi YH, Dai DF, Li J, Dong YW, Jiang Y, Li HG, Gao Y, Chong CK, Li HY, Chu XQ, Yang C, Zhang Q, Tong ZS, Bai CG, Chen Y, Molecules, 2016, 21, 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Johnson GS, Li J, Beaver LM, Dashwood WM, Sun D, Rajendran P, Williams DE, Ho E, Dashwood RH, Mol. Nutr. Food Res. 2017, 61, 1600769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rajendran P, Dashwood WM, Li L, Kang Y, Kim E, Johnson G, Fischer KA, Lohr CV, Williams DE, Ho E, Yamamoto M, Lieberman DA, Dashwood RH, Clin. Epigenetics, 2015, 7, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ertem FU, Zhang W, Chang K, Mohaiza Dashwood W., Rajendran P, Sun D, Abudayyeh A, Vilar E, Abdelrahim M, Dashwood RH, Int.J. cancer, 2016, 140, 460. [DOI] [PubMed] [Google Scholar]
- [18].Vasquez V, Mitra J, Hegde PM, Pandey A, Sengupta S, Mitra S, Rao KS, Hegde ML,Alzheimers Dis J. 2017, 60, S133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chernikova SB, Game JC, Brown JM, Cancer Biol. Ther. 2012, 13, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Herr I, Buchler MW, Cancer Treat. Rev. 2010, 36, 377. [DOI] [PubMed] [Google Scholar]
- [21].Higdon JV, Delage B, Williams DE, Dashwood RH, Pharmacol. Res. 2007, 55, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hu R, Hebbar V, Kim BR, Chen C, Winnik B, Buckley B, Soteropoulos P, Tolias P, Hart RP, Kong AN,J. Pharmacol. Exp. Ther. 2004, 310, 263. [DOI] [PubMed] [Google Scholar]
- [23].Cornblatt BS, Ye L, Dinkova-Kostova AT, Erb M, Fahey JW, Singh NK, Chen MS, Stierer T, Garrett-Mayer E, Argani P, Davidson NE, Talalay P, Kensler TW, Visvanathan K, Carcinogenesis 2007, 28, 1485. [DOI] [PubMed] [Google Scholar]
- [24].Caviglia C, Zór K, Canepa S, Carminati M, Larsen LB, Raiteri R, Andresen TL, Heiskanen A, Emneus J, Analyst 2015, 140, 3623. [DOI] [PubMed] [Google Scholar]
- [25].Gage BK, Webber TD, Kieffer TJ, PLoS One 2013, 8, e82076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wilson HK, Canfield SG, Hjortness MK, Palecek SP, Shusta EV, Fluids Barriers CNS 2015, 12, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ferretti LP, Himmels SF, Trenner A, Walker C, von Aesch C, Eggenschwiler A, Murina O, Enchev R, Peter M, Freire R, Porro A, Sartori AA, Nat. Commun. 2016, 7, 12628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Duquette ML, Zhu Q, Taylor ER, Tsay AJ, Shi LZ, Berns MW, McGowan CH, PLoS Genet. 2012, 8, e1003050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yuan J, Chen J,J. Biol. Chem. 2009, 284, 31746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gong F, Miller KM, Mutat. Res. 2013, 750, 23. [DOI] [PubMed] [Google Scholar]
- [31].Eot-Houllier G, Fulcrand G, Magnaghi-Jaulin L, Jaulin C, Cancer Lett. 2009, 274, 169. [DOI] [PubMed] [Google Scholar]
- [32].Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, Yenamandra A, Locke K, Yuan JL, Bonine-Summers AR, Wells CE, Kaiser JF, Washington MK, Zhao Z, Wagner FF, Sun ZW, Xia F, Holson EB, Khabele D, Hiebert SW, Cancer Cell 2010, 18, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chiu LY, Gong F, Miller KM, Philos. Trans. R Soc. Lond. B Biol. Sci. 2017, 372, 1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yang X, Li L, Liang J, Shi L, Yang J, Yi X, Zhang D, Han X, Yu N, Shang Y,J. Biol. Chem. 2013, 288, 18271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.