

Abstract
There has been a rise in the prevalence of nonalcoholic fatty liver disease (NAFLD), paralleling a worldwide increase in diabetes and metabolic syndrome. NAFLD, a continuum of liver abnormalities from nonalcoholic fatty liver (NAFL) to nonalcoholic steatohepatitis (NASH), has a variable course but can lead to cirrhosis and liver cancer. Here we review the pathogenic and clinical features of NAFLD, its major comorbidities, clinical progression and risk of complications and in vitro and animal models of NAFLD enabling refinement of therapeutic targets that can accelerate drug development. We also discuss evolving principles of clinical trial design to evaluate drug efficacy and the emerging targets for drug development that involve either single agents or combination therapies intended to arrest or reverse disease progression.
There has been an explosive interest in NAFLD and its more advanced stage, NASH, because of their growing impact on world health. In the United States, the number of NAFLD cases is projected to expand from 83.1 million in 2015 (~25% of the population) to 100.9 million in 2030. An increased proportion of these cases will be NASH, rising from 20% to 27% of adults with NAFLD during this interval1. This rising disease prevalence will exact a growing economic burden2 and will be accompanied by both an increasing number of patients with cirrhosis and end-stage liver disease necessitating liver transplantation3,4 and an alarming increase in hepatocellular carcinoma5. Compared to incidence in other liver diseases, a larger percentage (~35–50%) of HCCs that arise in NASH occur before patients are cirrhotic and routine screening for cancer is conducted5,6. As a result, these tumors tend to be larger and less amenable to curative therapies than those with other etiologies7.
Globally, the prevalence of NAFLD is estimated at ~25% and is highest in the Middle East and South America and lowest in Africa8. Whereas NAFLD typically is accompanied by central obesity in North America and Europe (~83% of patients), in Asia there is a sizable percentage of patients with ‘lean NASH’ who have a normal body mass index (BMI), even though the BMI cutoff for defining overweight in Asia (BMI > 23) is lower than in North America and Europe (BMI > 25)9.
NAFLD is an umbrella term that comprises a continuum of liver conditions varying in severity ofinjury and resulting fibrosis (Box 1). Among these, hepatic steatosis (fatty liver) alone is referred to as NAFL, and NASH is defined as a more serious process with inflammation and hepatocyte damage (steatohepatitis); typically, NASH is accompanied by percicellular fibrosis, which may progress to cirrhosis (Box 1). Although NAFL or NASH can be strongly suspected in an individual on the basis of imaging and clinical features (such as the presence of metabolic comorbidities and abnormal lab tests), NASH can only be definitely diagnosed by liver biopsy; additional subgroups of NASH have also been defined recently10. Patients with only NAFL carry a very low risk of adverse outcomes11, whereas the presence of NASH increases the risks of liver and possibly non-liver-related outcomes compared to those of patients with NAFL alone. Adverse hepatic outcomes related to NASH may include cirrhosis, liver failure and hepatocellular carcinoma, whereas non-liver-associated adverse outcomes are primarily related to increased cardiovascular disease and malignancy12,13.
Box 1 |. Diagnosis of NAFLD and NASH.
A diagnosis of NASH is typically considered when aminotransferases are elevated or when abdominal imaging incidentally detects hepatic fat. Less commonly, NAFLD is suspected when metabolic comorbidities are present. Hepatic steatosis can be reliably identified noninvasively through imaging tests, including ultrasound, computed tomography (CT) or magnetic resonance imaging (MRI). Of the available modalities, ultrasound and CT have comparable sensitivity and reliably detect steatosis when it comprises ≥20% of liver mass, whereas MRI can detect as little as 5% steatosis, but at a higher cost. Whereas the diagnosis of NAFLD is straightforward, the diagnosis of NASH, characterized by steatosis, hepatocellular ballooning and lobular inflammation with varying degrees of fibrosis, requires liver biopsy. Currently available biomarkers do not outperform serum ALT in identifying patients with NASH, which has poor sensitivity (< 50%). Promising serum biomarkers and several imaging techniques are undergoing evaluation in clinical trials and may ultimately obviate the need for liver biopsy to establish a diagnosis of NASH. Clinical prediction rules (CPRs), such as the NAFLD fibrosis score FIB-4 and elastography techniques, have acceptable performance characteristics to identify advanced fibrosis, with excellent negative predictive value.
The vast majority of patients with NAFLD across the disease spectrum, including those with compensated cirrhosis, are asymptomatic and often have normal laboratory profiles. Therefore, many patients can progress to cirrhosis undiagnosed.
Histologic features of human NASH.
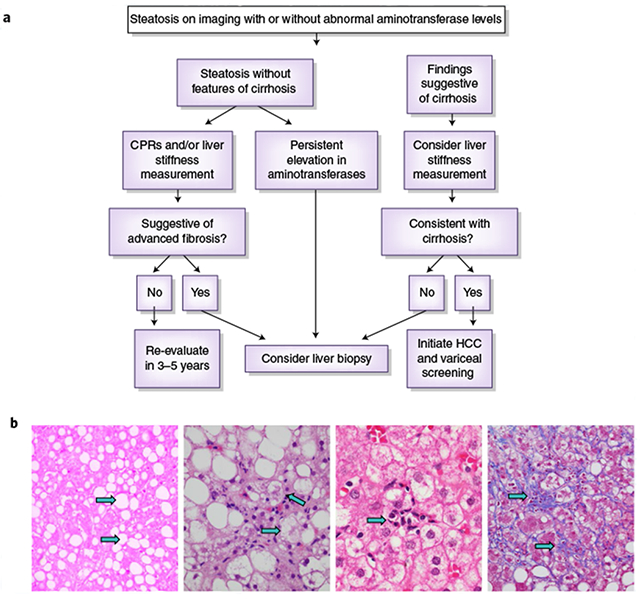
a, A schema for diagnosing NAFLD and NASH. Credit: Marina Corral Spence/Springer Nature. b, The panel of images from liver biopsies demonstrate the typical appearances of macrovesicular steatosis (fat), hepatocellular ballooning, lobular inflammation and pericellular fibrosis (arrows). As the disease progresses into cirrhosis (not shown), these features may also regress. H&E staining images were courtesy of Pierre Bedossa)
Although there has been steady progress in clarifying the pathogenesis of NAFLD, identifying therapeutic targets and advancing drug development, there are significant unmet challenges, and no agent is approved yet for this condition. The identification of those at risk for NAFLD and NASH is imprecise (Box 1), and there is inadequate knowledge of the natural history, key pathogenic drivers and optimal in vitro and animal models to mimic the disease and test new agents. Also, the validation of predictive biomarkers of disease risk and response to therapy are lacking and are urgently needed. Finally, although the field is moving rapidly toward combination therapies, there are also single drugs with multiple cellular or molecular targets of action that are under evaluation.
In view of both the rapid pace of progress as well as the unmet challenges, this Review highlights the current understanding of NAFLD, providing a critical assessment of the natural history, histologic features and clinical drivers of disease progression, pathogenesis, preclinical models, biomarkers, therapeutic targets and clinical trial designs. Further understanding of these key elements of NAFLD should ultimately help mitigate its global impact.
Clinical disease progression
NAFLD is a disease that has very different rates of progression among individuals and different clinical manifestations (Box 1). This highly variable natural history of NAFLD reflects the diverse but convergent impacts of the environment, the microbiome, metabolism, comorbidities and genetic risk factors. The importance of genetic and potentially microbiome-related risk factors in the development and severity of NASH have been underscored by studies identifying a higher risk of fibrosis among family members of those diagnosed with NASH14,15. Clarifying both genetic and other factors that result in different subtypes of NAFLD could lead to more accurate prediction of disease progression and more effective treatments based on individualized drivers of disease. Disease subclassification of this type has been pursued in pulmonary disease, for example, but has not yet yielded therapeutic advances16.
Most patients with NASH are asymptomatic without clinically relevant outcomes for decades, but some can progress rapidly. The diagnosis is often made serendipitously (Box 1), and even once identified, clinicians may not appreciate its potentially advanced stage. On average, those with a diagnosis of NASH progress one stage of fibrosis (based on the NASH Clinical Research Network fibrosis classification) every 7 years, whereas those with NAFL progress half as fast, as determined through a meta-analysis of randomized control trials (RCTs) and observational studies11. Overall, ~20% of patients rapidly progress to advanced fibrosis; however, methods to identify these rapid progressors remain elusive11. Further confounding natural history assessments, the histological lesions of NAFLD can progress, regress or remain stable over time11,17.
Risk factors for NAFLD and NASH
Presence of metabolic syndrome (MetS) in an individual is the strongest risk factor for NAFLD and NASH (Box 2). MetS is variably defined, but typically includes increased waist circumference (i.e., obesity), hyperglycemia, dyslipidemia and systemic hypertension (HTN)18. The association between NAFLD and features of MetS may be bidirectional, particularly with respect to diabetes and HTN, meaning that not only does MetS increase the risk of NAFLD, but also NAFLD may enhance several features and comorbidities of MetS. Thus, effective treatment of NASH could have the additional benefit of improving the features of MetS. MetS is also an important driver of adverse cardiovascular (CV) events and overall mortality in patients with NAFLD19,20. Among the features of MetS, diabetes mellitus has the clearest biologic link to the progression of NAFLD, and up to 75% of individuals with type 2 diabetes have NAFLD. Among individuals with diabetes and NAFLD, the prevalence of NASH and advanced fibrosis is also enriched compared to nondiabetics with NAFLD, irrespective of whether aminotransferases in the blood are elevated, which ordinarily indicates a higher likelihood of underlying liver injury (Box 1)21–23. Not surprisingly, those with NAFLD and diabetes are also at increased risk of developing liver-related complications24. Insulin resistance has been recognized for many years to be an integral component of NAFLD pathogenesis25 and worsens with disease progression. Although bettering insulin resistance improves NASH, doing so may not be sufficient by itself to attenuate NASH progression. Patients with NAFLD are also at increased risk of incident diabetes26.
Box 2 |. Comorbidities of NAFLD.
Patients with NAFLD are twice as likely to die of cardiovascular disease than liver disease, which is largely related to shared risk factors including diabetes mellitus, HTN and obesity12. In fact, liver disease is only the third leading cause of death in patients with NAFLD, following cardiovascular disease and malignancy36,175. Both increased age and metabolic comorbidities increase the occurrence and severity of NASH176,177. NAFLD and especially NASH also confer an independent risk of adverse cardiovascular events in affected individuals beyond that conferred by the shared risk factors, likely related to abnormalities including systemic vascular endothelial dysfunction, atherogenic dyslipidemia and the systemic and hepatic proinflammatory state associated with NAFLD and NASH; the magnitude of this risk has not yet been quantified, however. NAFLD and NASH are also associated with abnormalities of cardiac structure and function, which further contribute to its impact on CVD mortality28,178. In a recent meta-analysis of 34,000 patients with NAFLD diagnosed by imaging or histology over a median follow-up of 6.9 years, there was a 65% increased risk of developing both fatal and nonfatal cardiovascular events179.
Fifty percent of patients with HTN have NAFLD27, and NAFLD has been associated with changes in arterial stiffness, myocardial remodeling, kidney disease and heart failure28–30. HTN is strongly associated with fibrosis progression11; in a longitudinal cohort of Italian patients with NAFLD, those with HTN had a higher risk of fibrosis progression over the 6.2-year follow-up period31. There is also evidence that antagonism of the renin–angiotensin aldosterone system, a pathway that contributes to systemic HTN, may also improve NASH and hepatic fibrosis32–34.
Although NAFL alone has been conventionally thought to indicate that there is no risk of future fibrosis, recent studies refute this assumption. For example, in one longitudinal study, 22% of participants with NAFL developed fibrosis and 44% developed NASH over time17. In another, 13 of 16 patients with NAFLD and inflammation not sufficient to be diagnostic of NASH progressed to NASH or bridging fibrosis (i.e., fibrotic bands that extend from one portal triad to another)35.
The presence and stage of fibrosis is the clearest histologic determinant of outcomes in patients with NASH, but steatohepatitis is clearly the driving force for fibrosis development36–39. Although the link between fibrosis and outcomes is strong, there is substantial colinearity between the presence of NASH and fibrosis severity. For example, in one study, 94% ofthose with stage 4 fibrosis had NASH compared to only 35% of those with stage 0 fibrosis (P < 0.001)39; in this study, advanced fibrosis was present in 17% of those with NASH and 2% of those without NASH39.
Several genetic risk factors have been identified that increase the risk of NASH40–42. Among these, the best characterized is a single-nucleotide polymorphism in the PNPLA3 gene, which regulates lipolysis of hepatocyte lipid droplets and is the most strongly associated genetic variant linked to NASH43,44. The risk-associated PNPLA3-I148M variant is resistant to normal proteasomal degradation and accumulates on lipid droplets, which interferes with lipolysis45. Interestingly, the risk of NASH in patients with this variant is maximized only if it coexists with adiposity, illustrating the additive effects of genetic and environmental drivers in this disease46. More recently, a genome-wide association study revealed a splice variant (rs72613567:TA) in HSD17B13, a gene that encodes the hepatic lipid droplet protein 17β-hydroxysteroid dehydrogenase type 13, that was associated with reduced levels of alanine amino transferase (ALT) and aspartate amino transferase (AST), suggestive of less inflammation and liver injury (Box 1) in patients with fatty liver47. The splice variant yields a truncated, nonfunctional protein, implying that HSD17B13 normally generates a product, as yet unknown, that promotes hepatocellular damage. When the level of HSD17B13 protein is reduced, as is the case with the rs72613567:TA variant, attenuated hepatocyte injury is reflected in lower serum ALT and AST levels. Pharmaceutical efforts have been initiated to reduce expression of HSD17B13 by administering siRNA to silence HSD17B13 and mimic the phenotype of individuals who lack the protein (http://investors.alnylam.com/news-releases/news-release-details/regeneron-and-alnylam-pharmaceuticals-announce-collaboration).
Pathogenesis
A ‘two-hit’ theory was posited for several years to explain NASH pathogenesis. This theory suggests that in the setting of steatosis alone (i.e., NAFL), a second ‘hit’ from other factors (for example, oxidant stress) was required for the development of NASH; however, this view is now considered outdated. There are many molecular pathways that contribute to the development of NASH, and it is not even certain whether NASH is always preceded by NAFL. Moreover, pathogenic drivers are not likely to be identical among all patients. Thus, both the mechanisms leading to disease and their clinical manifestations are highly heterogeneous48.
In defining pathogenic drivers of NAFL and NASH, a useful conceptual framework is that the liver’s capacity to handle the primary metabolic energy substrates, carbohydrates and fatty acids, is overwhelmed, leading to accumulation of toxic lipid species49–53 (Fig. 1). These metabolites induce hepatocellular stress, injury and death, leading to fibrogenesis and genomic instability that predispose to cirrhosis and hepatocellular carcinoma. Thus, clarifying the sources and fates of fatty acids in hepatocytes is essential for understanding the metabolic underpinnings of NASH. When fatty acids are either supplied in excess or their disposal is impaired, they may serve as substrates for the generation of lipotoxic species that provoke ER stress and hepatocellular injury. Elucidating the pathways leading to lipotoxicity, ER stress and cell injury has led to rational therapeutic targeting (Fig. 2) (see ‘Treatment Targets’)54.
Fig. 1|. The substrate-overload liver injury model of NASH pathogenesis.
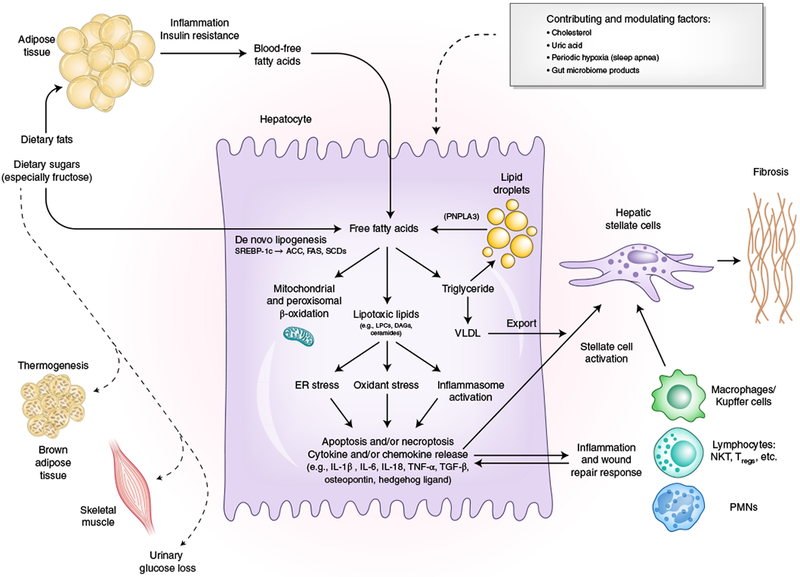
Free fatty adds are central to the pathogenesis of NASH. Free fatty adds that originate from lipolysis of triglyceride in adipose tissue are delivered through blood to the liver. The other major contributor to the free fatty acid flux through the liver is DNL, the process by which hepatocytes convert excess carbohydrates, especially fructose, to fatty acids. The two major fates of fatty acids in hepatocytes are mitochondrial beta-oxidation and re-esterification to form triglyceride. Triglyceride can be exported into the blood as VLDL or stored in lipid droplets. Lipid droplet triglyceride undergoes regulated lipolysis to release fatty acids back into the hepatocyte free fatty acid pool. PNPLA3 participates in this lipolytic process, and a single-nucleotide variant of PNPLA3 is strongly associated with NASH progression, underscoring the importance of the regulation of this lipolysis. When the disposal of fatty acids through beta-oxidation or formation of triglyceride is overwhelmed, fatty acids can contribute to the formation of lipotoxic species that lead to ER stress, oxidant stress and inflammasome activation. These processes are responsible for the phenotype of NASH with hepatocellular injury, inflammation, stellate cell activation and progressive accumulation of excess extracellular matrix. Lifestyle modifications that include healthy eating habits and regular exercise reduce the substrate overload through decreased intake and diversion of metabolic substrates to metabolically active tissues and can thereby prevent or reverse NASH. SCD, steroyl CoA-desaturase; FAS, fatty acid synthase; NKT, natural killer T cell; Tregs, regulatory T cells; PMNs, polymorphonuclear leukocytes. Credit: Marina Corral Spence/Springer Nature
Fig. 2|. Intrahepatic drug targets in phase 2 and 3 clinical trials for NASH.
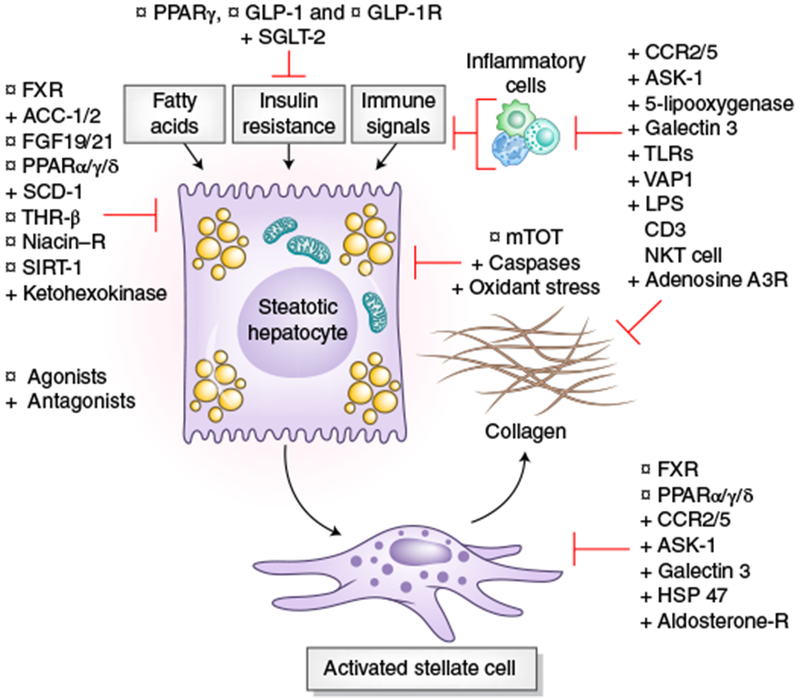
Depiction of the sites of action of drugs that are currently in phase 2 or 3 clinical trials, based on their primary locus of activity within the liver. Targets include those that regulate lipids and glucose homeostasis, and oxidant stress and mitochondrial targets in hepatocytes, inflammatory signals converge on hepatocytes, and those inflammatory signals and intracellular targets related to stellate cell activation and fibrogenesis. Gray boxes indicate disease drivers. Some targets (e.g, FXR agonists, CCR2 and CCR5 (CCR2/5) antagonist) have more than one action within the injury milieu. Agonists are indicated with a circle and antagonists with a cross. DGAT, Diacylglycerol O-acyltransferase; SCD, steroyl CoA-desaturase; THR, thyroid hormone receptor; SIRT, sirtuin; GLP, glucagon-like peptide; SGLT, sodium–glucose cotransporter; VAP, vascular adhesion protein; LPS, lipopolysaccharide; PPARα/δ/γ, peroxisome proliferator–activated receptors PPARα, PPARδ and PPARγ. Credit: Marina Corral Spence/Springer Nature
Sources of fatty acids in the liver
Fatty acids are primarily delivered to the liver from blood following lipolysis of triglyceride in adipose tissue, a process that is regulated by the actions of insulin on adipocytes. Impaired postreceptor signaling by insulin (i.e., insulin resistance) in adipose tissue contributes to NASH through dysregulated lipolysis resulting in excessive delivery of fatty acids to the liver55. Phosphorylation of c-Jun N-terminal kinases (JNKs) in adipocytes significantly impairs post-receptor insulin signaling during inflammation56. Thus, inflammatory and metabolic events in adipose tissue can drive the pathogenesis of NASH and may be appropriate therapeutic targets57,58. Modest weight reduction also improves adipose tissue insulin resistance and homeostasis in humans59, which may be beneficial in NASH60.
The second major source of fatty acids is their synthesis from glucose and fructose by de novo lipogenesis (DNL), and a stable isotope study demonstrated that the increase in hepatic lipid content in patients with NAFLD is largely attributable to DNL61. Whereas introduction of glucose into the DNL pathway is highly regulated, nearly all fructose is removed from portal blood by the liver, where it is phosphorylated and committed to DNL without regulation. How much ingested fructose reaches the hepatic portal circulation is uncertain, as studies in mice have shown gut enterocytes are involved in a component of fructose metabolism62, and human studies have shown limited absorptive capacity for fructose by the gut epithelium63,64. Following a large fructose load, the phosphorylation of fructose in hepatocytes commits it to DNL, resulting in the depletion of liver ATP in humans and animals65, which may enhance cellular stress. The consumption of sugar-sweetened beverages that contain either sucrose (which is converted to fructose and glucose in the gut) or a mixture of fructose and glucose is epidemiologically associated with fat accumulation in the liver and with NASH66,67.
DNL can be pharmacologically inhibited by targeting its synthetic enzymes, for example acetyl-CoA carboxylase (ACC)68. In addition, inducing downregulation of the expression of steroyl-CoA response element binding protein-1c (SREBP-1c), the major transcriptional regulator of the enzymes involved in DNL, could be beneficial in treating NASH because the nuclear transcription factor farnesoid X receptor (FXR) regulates the expression of enzymes of the DNL pathway. The carbohydrate response element–binding protein (ChREBP) also induces the expression of enzymes involved in DNL, but studies of human liver biopsies have suggested that ChREBP expression is diminished in patients with NASH69, so it is uncertain whether ChREBP would be a viable target for NASH therapeutics.
Diversion of fatty acids and carbohydrates away from the liver into other tissues, such as peripheral adipose tissue, brown adipose tissue and skeletal muscle, also reduces their delivery to the liver. For example, peroxisome proliferator–activated receptor gamma (PPARγ) ligands (for example, pioglitazone), a specialized type of nuclear receptor ligand, enhance adipocyte storage of fat and improve NASH, albeit at the expense of increased peripheral fat stores and weight gain, as shown in clinical trials of these drugs in patients with type 2 diabetes70.
Diversion of energy substrates to extrahepatic tissues can also be promoted by their consumption in active muscle and brown adipose tissue. Brown adipose tissue metabolizes energy substrates; this metabolism is accompanied by uncoupled mitochondrial respiration, resulting in thermogenesis, rather than storage, of triglyceride. Brown adipose tissue is targeted by bile acid–induced signaling, an effect in addition to the canonical function of bile acids as detergents that facilitate fat absorption from the gut71. Bile acids upregulate thermogenesis by binding to the TGR5 bile acid membrane receptor on brown adipose tissue72. Thus, one proposed therapeutic strategy for prevention or treatment of NASH is to augment brown adipose tissue abundance and function (see ‘Treatment Targets’)73.
Fates of fatty acids in the liver
Fatty acids in the liver are noncovalently bound to fatty acid–binding protein-1 (FABP-1, also known as liver-type FABP (L-FABP)) and are primarily metabolized either by mitochondrial β-oxidation or through esterification to form triglycerides. Some patients with NASH have mitochondrial dysfunction and impaired β-oxidation in the liver and perhaps throughout the body in other tissues74,75; however, it is unclear whether an underlying defect in mitochondrial function in the liver predisposes to NASH or if the mitochondrial dysfunction is a consequence of cellular stress in the disease. The disposal of fatty acids through formation of triglyceride is generally thought of as an adaptive, protective response to a supply of fatty acids that exceeds the capacity to metabolize them76; however, some studies suggest that excess triglyceride may also contribute to metabolic abnormalities, rather than just serving as a marker of those abnormalities58,77. Triglyceride not exported into the blood from the liver as very low-density lipoprotein (VLDL) forms lipid droplets in hepatocytes, a defining feature of NAFLD, and these droplets may contribute to the fatty acid pool within cells following their lipolysis. Following a meal, the timing and intracellular localization of this lipolysis is highly regulated.
The specific lipotoxic lipids that promote cell injury and the NASH phenotype are not yet identified, but preventing their generation would be a logical target of therapy. Candidate lipotoxic lipids include diacylglycerols78, ceramides79,80 and lysophosphatidyl choline species52,81. Cholesterol accumulation in the liver may also promote NASH82, and several mouse and rat models of NASH include a diet with high cholesterol content to increase disease severity83–85.
Response to lipotoxic lipids
Hepatocellular injury in NASH is characterized by endoplasmic reticulum (ER) stress86, a dysfunctional unfolded protein response87, inflammasome activation88, activation of apoptotic pathways, inflammation and an enhanced wound response89. External factors, including dysregulation of cytokines and adipokines90, ATP depletion65, uric acid toxicity91, periodic hypoxia in the setting of sleep apnea92 and products of the gut microbiome93–95, likely modulate the hepatocyte’s development of lipotoxic stress, injury and inflammation. The relative contribution of these defects in individuals with NASH is not clarified yet and likely varies among individuals.
The inflammasome
Recent studies in mice have revealed that hepatocyte inflammasome activation may be an important link between the initial metabolic stress and subsequent hepatocyte death and stimulation of fibrogenesis in NASH96. The inflammasome is a multiprotein cytoplasmic complex that responds to danger-associated molecular patterns (DAMPs), which in this context include saturated fatty acids96 that are the product of DNL, as well as pathogen-associated molecular protein (PAMPs), which are the products of gut microbiota and are delivered to the liver in the portal circulation. Mouse studies have demonstrated that hepatic inflammasome activation in the liver leads to the expression of pro-inflammatory cytokines, such as interleukin (IL)-1β and IL-18, and promotes apoptosis through caspase-1 activation. Although inhibition of inflammasome activation may be an attractive therapeutic target for NASH97,98, any therapy may need to be specifically targeted to the liver because inflammasome deficiency in the gut may promote an increase in proinflammatory toll-like receptor (TLR)-4 and TLR-9 ligands in the portal circulation that could aggravate NASH93.
Insulin resistance
Insulin resistance is a common feature of NAFLD that contributes to its pathogenesis99. Insulin resistance is characterized by reduced glucose disposal in nonhepatic tissues, including adipose tissue and muscle100. Insulin resistance in adipose tissue leads to inappropriate release of fatty acids through dysregulated lipolysis that further contributes to impaired insulin signaling throughout the body. Studies have shown metabolic crosstalk between adipose tissue and the liver. Adiponectin, IL-6 and other peptides released by adipose tissue have both protective and proinflammtory effects in the liver101,102. Another link between defects in liver and adipose tissue may be the enzyme dipeptidyl peptidase 4 (DPP-4)· which can promote insulin resistance. Circulating DPP-4 is secreted by hepatocytes and acts in concert with plasma factor Xa to stimulate inflammatory macrophages within visceral adipose tissue in mice. This effect is known to promote insulin resistance· underscoring the important cross talk between liver and other tissues underlying metabolic dysregulation in NAFLD103.
Microbiome
The precipitous increase in NAFLD in the past 25 years in the developed world has been largely attributed to both a diet that is rich in fructose, sucrose and saturated fats and an increasingly more sedentary lifestyle. An additional risk factor for NAFLD may be the evolution of the human microbiome, reflecting both a changing diet as well as the widespread use of antibiotics in farm animals and the indiscriminate prescription of antibiotics to humans. Regardless of the etiology, compelling mouse data demonstrate the transmissibility of a NASH phenotype through the microbiome93. Human studies document a gut microbiome among patients with NASH that is less complex than that of healthy subjects104,105 and indicate that weight loss also alters the microbiome. However, a cause–effect relationship between a change in the microbiome due to weight loss and the resolution of NASH has not been established94,106. Plausible mechanistic links between an altered microbiome and fatty liver are emerging and include the potential for bacterial proteins to function as ligands for G protein–coupled receptors107 as well as the gut bacteria’s activity that modulates the gut-liver axis through intestinal FXR signaling and release of fibroblast growth factor 19 (FGF19), a gut-derived hormone that regulates bile acid synthesis and also lipid and glucose metabolism108–110. An altered microbiome (i.e., ‘dysbiosis’) may also lead to increased intestinal permeability, which can amplify many of these gut-derived effects111. Studies of the microbiome are relatively nascent, and considerable progress in linking its role to NASH is anticipated.
Fibrogenesis
The accumulation of extracellular matrix in the liver, which leads to progressive fibrosis, cirrhosis, portal HTN and liver failure, is the major cause of liver-related death in patients with NAFLD. Fibrogenesis is driven by signaling from stressed or injured hepatocytes and activated macrophages (Kupffer cells in the liver), leading to activation of resident hepatic stellate cells into myofibroblasts to produce matrix proteins faster than they are degraded112. NASH-specific fibrogenic pathways are being increasingly identified113. For example, in mouse models of NASH, there is increased signaling by the transcriptional activator TAZ in NASH hepatocytes, which promotes secretion of Indian hedgehog ligand that results in paracrine fibrogenic signaling to stellate cells114. Another NASH-specific pathway is through the PNPLA3-I148M variant’s effects directly on stellate cell fibrogenesis44, suggesting that therapies directed at PNPLA3 function could also be antifibrotic115.
Preclinical models of NAFLD and NASH
The need for effective and safe therapy has spurred development of both in vitro and animal models of NASH to better elucidate disease pathogenesis, identify potential treatment targets and test the therapeutic potential of single drugs or drug combinations before human studies.
In vitro tissue engineering models
In vitro models of NAFLD have ranged from simple cell culture–based systems exposed to various lipids and cytokines to more sophisticated models based on 3D multicellular organoid cultures with or without perfusion mimicking sinusoidal blood flow116. Diverse approaches, including layered cocultures, spheroids, micropatterned surfaces, human precision-cut slices and bioprinting, have been used to develop such 3D cultures to replicate the architecture of the intact liver117 (Table 1). Although most of these have been used to detect toxicity of potential therapeutic compounds in various settings, some have been further modified to recreate NASH-like conditions. Human liver slices can be obtained for short-term (~3-day) cultures to assess transcriptomic responses to drugs while preserving the in vivo multicellular architecture118, but tissues from patients with NASH are very difficult to obtain. The Organovo bioprinting system using HepG2 cells develops steatosis when perfused with free fatty acids119; however, use of this system is limited because HepG2 cells alone fail to replicate the multicellular milieu of the intact liver.
Table 1 |.
Most commonly used in vitro and mouse and rat models of NASH
| Platform | Description | Phenotype and relevance to human disease |
|---|---|---|
| In vitro | ||
| Human liver slices118 | Small (250-μM thick) tissue disc generated from fresh human liver suitable for multiwell plating | Suitable for short-term (~3-day) transcriptomic studies of drug treatments, but direct analysis of NASH tissues is rarely possible. |
| Organova: bioprinted human liver119 | Scaffold-free assembly of human hepatocytes, stellate cells and umbilical vein cells in a 3D in vivo–relevant architecture | Primarily optimized for drug toxicity evaluation, but conditions that mimic NASH are being developed. |
| Hemoshear: in vitro–engineered liver model117 | Primary human hepatocyte, stellate cell and macrophage coculture that incorporates sinusoidal flow in a lipotoxic milieu | Designed primarily to test NASH therapeutics, with transcriptomic and metabolic pathways resembling human disease. |
| Rat and mouse models | ||
| Dietary | ||
| Methionine and choline deficiency (MCD)163 | High-fat and sucrose diet, deficient in methionine and choline | The histologic appearance in 2–4 weeks resembles human NASH, but animals lose weight and are not insulin resistant, with lack of concordance of the liver transcriptome with human NASH. |
| Choline-deficient, l-amino acid–defined supplemented, high-fat diet (CDAA)164 | High-fat, choline-deficient, reduced-methionine diet for 24 weeks | Like the MCD diet, animals do not gain weight or exhibit insulin resistance, but can develop fibrosis within 6–8 weeks. |
| High-fat, fructose and cholesterol (AMLN diet)165 | Diet high in fat (40%, of which 18% is trans-fat), plus fructose (22%) and cholesterol (2%) administered for 30 weeks | Animals develop both histologic and metabolic features of human NASH with diffuse fibrosis but no cirrhosis. |
| Genetic | ||
| ob/ob166 | Leptin-deficient mice | Animals develop severe insulin resistance and steatosis but are resistant to fibrosis without a second insult (e.g., LPS). Unlike this model, humans with NASH have normal or elevated serum leptin. |
| db/db167 | Leptin receptor–deficient mouse | Severe glucose intolerance, obesity and hepatic steatosis; susceptible to fibrosis with a second insult (e.g., MCD or high-fat diet). |
| Obese (fa/fa) Zucker rat168 | Nonfunctional leptin receptor due to mutation | Severe hyperphagia, insulin resistance and obesity on a high-fat diet, with hepatic steatosis but modest fibrosis. |
| foz/foz169 | Truncating mutation in Alstrom syndrome I (ALMS1) protein, a ciliary protein | Severe obesity, insulin resistance, hepatic steatosis and mild fibrosis on a high-fat diet for 300 days, with hypoadiponectinemia. Phenotype may be strain-dependent. |
| SREBP-1c transgenic170 | Hepatocyte-specific overexpression of SREBP-1c | Severe insulin resistance and spontaneous steatohepatitis with oxidant stress at 30 weeks, with some perivenular fibrosis. |
| MUP-uPA transgenic171 | Hepatocyte-specific overexpression of urokinase plasminogen activator | Severe ER stress, hepatic steatosis, mild fibrosis and HCC on high-fat diet (60% of calories), with TNF-α-driven inflammation. |
| LDLR knockout172 | LDL receptor global knockout | Marked steatosis on a diabetogenic diet for 24 weeks, made worse with addition of dietary cholesterol, and association with inflammation, oxidant stress, increased fatty acid production and mild fibrosis. |
| Phosphatase and tensin homolog (PTEN) knockout173 | Alb-Cre-mediated hepatocyte-specific knockout of PTEN | Spontaneous hepatomegaly, steatohepatitis and triglyceride accumulation with lipogenic gene induction at 40 weeks and high penetrance of ademomas, then carcinomas at 74–78 weeks. |
| Combinations | ||
| Streptozotocin plus high fat-diet (STAM) model174 | Streptozotocin administered at 2 days, followed by high-fat diet | Steatohepatitis and mild fibrosis with occasional HCCs at 20 weeks. However, animals have type 1 not type 2 diabetes, do not gain weight, and the liver transcriptome is discordant from human NASH. |
| DIAMOND mouse84 | Inbred isogenic strain of C57Bl6/J and S129S1/svlmj fed a high-fat, high-carbohydrate diet | Progressive steatohepatitis, fibrosis then cirrhosis and HCC at 1 year. High transcriptomic concordance to human NASH. |
| Western diet plus CCl4 addition121 | High-fat, high-fructose, high-cholesterol diet with weekly intraperitoneal CCl4 dosing | Progressive steatosis, insulin resistance and fibrosis at 12 weeks, with cirrhosis and frequent HCC at 24 weeks. High transcriptomic concordance to human NASH. |
| High-fat diet with thermoneutral housing123 | Animals housed at thermoneutral (30 °C) conditions, high-fat diet for 8 weeks | Greatly amplified steatohepatitis compared to standard (22 °C) housing, with altered microbiome, increased inflammatory signaling but modest fibrosis. |
Commonly used models are listed here with brief descriptions and relevance to human NASH. Rodent models refer to mice unless otherwise specified. This list is not intended to be all-inclusive and does not include other nonrodent animal models (e.g., pigs and nonhuman primates). LPS, lipopolysaccharide.
Another in vitro system, developed by Hemoshear, includes primary human hepatocytes, stellate cells and Kupffer cells in a microfluidic chamber which is perfused with free fatty acids (FFA), glucose, insulin and inflammatory cytokines to mimic a NASH-like system117. This system can mirror the transcriptomic, cell-signaling and pathophysiological changes seen in NASH, including increased DNL, gluconeogenesis, oxidative stress, production of inflammatory and fibrogenic cytokines and activation of stellate cells.
Animal models
Animal models of NASH can be broadly categorized as diet-induced, genetic or a combination of more than one intervention (Table 1)120. Although many models can lead to lipid accumulation in the liver, not all do so by pathways known to be relevant to human NASH. Moreover, while the histologic appearance of many models may resemble human NASH, the metabolic and transcriptomic features in the liver may not121, and models are increasingly evaluated on the basis of proteomic, lipidomic and transcriptomic features, rather than on histology alone, to assess their relevance to human disease. Some of these transcriptomic features include altered immune signaling and altered lipid and glucose metabolism121. A diet-induced obesity model with macronutrient composition resembling that of obese humans is a popular mouse model of NASH; however, C57Bl/6J mice do not consistently develop steatohepatitis or advanced fibrosis on a high-fat diet122. In mouse models of NASH, the use of a 1–2% cholesterol diet in mice may be necessary to achieve a level of hepatic cholesterol comparable to that in human liver114. On the other hand, an approach using streptozotocin to ablate pancreatic islet cells in mice does not fully recreate the systemic dysmetabolic insulin-resistant environment of type 2 diabetes seen in humans and therefore does not completely recreate the phenotype of human NASH120. Other factors, such as ambient temperature, may additionally impact the severity of NASH in animal models of the disease, as animals housed in thermoneutral conditions have more advanced human-like disease when fed a high-fat diet123.
The principal genetic animal models of NASH are the ob/ob (leptin deficient) and db/db (leptin receptor deficient) mice, the Zucker (fa/fa) rat, foz mice (deficient in the Alstrom syndrome 1 gene) and several transgenic or conditional knockout mice (Table 1)120. Normal mice (for example, C57/BL6) fed the methionine-choline deficient (MCD) and choline-deficient l-amino acid (CDAA)-defined diet develop a histological appearance similar to NASH with fat, inflammation and fibrosis; however, the defects underlying this model are not related to human disease pathogenesis in that the lack of choline prevents processing of hepatic lipids. Moreover, the mice lose rather than gain weight and are not insulin resistant, and conversely, patients with NASH are not choline-deficient124. This example highlights the potential to experimentally drive accumulation of hepatic triglycerides in the liver of an animal model, yet the pathophysiology of this model does not replicate human NASH.
Recently, the inbred isogenic offspring of a C57Bl/6J mouse crossed with a s129/SvlmJ mouse (a strain commonly used to create mice with targeted mutations) was shown to develop obesity, insulin resistance, hypoadiponectinemia, adipose tissue inflammation, dyslipidemia, NAFL followed by progressive NASH and fibrosis and HCC with cell signaling, transcriptomic and lipidomic changes similar to humans with NASH84,125. Similarly, a model in which animals are administered a Western diet with weekly CCl4, a hepatotoxin to amplify injury and fibrosis, also appears to closely resemble human NASH121. Both of these models replicate the progressive stages of NAFLD from steatosis alone (NAFL) through NASH to cirrhosis and hepatocellular carcinoma without the addition of a carcinogen (for example, diethylnitrosamine). Although other animal species, ranging from zebrafish126 and fruit flies127 to the Ossabaw pig128, have also been studied with respect to NASH, the translatability of results from these models (for example, zebrafish) to humans and logistical difficulties (for example, pig models) limit their utility.
Treatment targets
With regard to therapy for NASH, herein we focus primarily on targets associated with liver, gut, adipose and muscle, and we do not review those targets that solely affect the central nervous system by altering eating behavior or centrally regulated metabolism129. Furthermore, it is known that significant weight loss, regardless of the underlying mechanism, will have a salutary effect on NASH, with sustained weight loss of 10% associated with a reduction in liver fibrosis130, and so a long-term benefit of sustained weight loss on NASH severity needs to be investigated.
On the basis of the current pathophysiologic classification of NASH, metabolic and intrahepatic treatment targets can be organized using the scheme in Fig. 2, which defines the pathogenic drivers of hepatocyte dysregulation and the pathways of inflammation associated with fibrosis. In addition to these liver-specific targets, extrahepatic targets include the microbiome and gut–liver signaling axis as well as other organs that contribute to MetS, especially muscle and adipose tissue. Current phase 2 and 3 clinical trials are summarized in Table 2.
Table 2 |.
Ongoing major clinical trials of pharmacotherapies for the treatment of NASH1
| Agent (trial name) | Primary mechanism | Major inclusion criteria | Primary outcome(s) | Estimated completion | Trial number |
|---|---|---|---|---|---|
| Phase 3 | |||||
| Cenicriviroc (AURORA) | CCR2/5 inhibitor | NASH on biopsy with stage 2 or 3 fibrosis | Improvement in fibrosis without worsening NASH | Jul 2019 | NCT03028740 |
| Elafibranor (RESOLVE-IT) | PPARα/δ ligand | NASH on biopsy with stage 1–3 fibrosis | Resolution of NASH without worsening fibrosis | Dec 2021 | NCT02704403 |
| Obeticholic acid (REGENERATE) | FXR ligand | NASH on biopsy with stage 2 or 3 fibrosis or stage 1 fibrosis with risks for progression | Resolution of NASH without worsening fibrosis or improvement in fibrosis without worsening NASH | Oct 2022 | NCT02548351 |
| Obeticholic acid (REVERSE) | FXR agonist | NASH on biopsy with stage 4 fibrosis | Improvement in fibrosis without worsening NASH | Jul 2020 | NCT03439254 |
| Selonsertib (STELLAR 3) | ASK-1 inhibitor | NASH on biopsy with stage 3 fibrosis | Improvement in fibrosis without worsening NASH | Jan 2020 | NCT03053050 |
| Selonsertib (STELLAR 4) | ASK-1 inhibitor | NASH on biopsy with stage 4 fibrosis but no ascites, HE or variceal bleeding | Improvement in fibrosis without worsening NASH | Jan 2019 | NCT03053063 |
| Phase 2 | |||||
| Aramchol (Aramchol_005) | SCD-1 inhibitor | NASH on biopsy and >5% fat by MRS | Improvement in fat by MRS | Mar 2018 | NCT02279524 |
| BMS 130-045 (LIGHT-1) | Pegylated FGF-21 | NASH on biopsy and PDFF ≥10% | Improvement in liver fat by MRI | Jan 2017 | NCT02413372 |
| EDP-505 | FXR agonist | Histologic or phenotypic NASH | Improvement in ALT | Apr 2019 | NCT03421431 |
| Emricasan (ENCORE-NF) | Caspase inhibitor | NASH on biopsy with stage 2 or 3 fibrosis or stage 1 fibrosis with risks for progression | Improvement in fibrosis without worsening NASH | Dec 2018 | NCT02686762 |
| Emricasan (ENCORE-PH) | Caspase inhibitor | NASH cirrhosis and HVPG ≥12 mmHg | Improvement in HVPG | Oct 2018 | NCT02960204 |
| Emricasan (ENCORE-LF) | Caspase inhibitor | NASH cirrhosis with history of variceal bleeding and/or moderate or severe ascites | Event-free survival | Aug 2019 | NCT03205345 |
| GR-MD-02 (NASH-CX) | Galectin-3 inhibitor | NASH cirrhosis and HVPG ≥6 mmHg | Improvement in HVPG | Oct 2017 | NCT02462967 |
| IMM-124E | Anti-LPS | NASH on biopsy | Improvement in liver fat by MRI | Oct 2017 | NCT02316717 |
| IVA337 (NATIVE) | Pan-PPARγ agonist | NASH on biopsy and SAF >2 | SAF reduction ≥2 points without worsening fibrosis | Jan2019 | NCT03008070 |
| LIK066 | SGLT1/2 inhibitor | NASH on biopsy with stage 1–3 fibrosis or clinical risks for NASH | Improvement in ALT | Apr 2019 | NCT03205150 |
| LMB-763 | FXR agonist | Histologic or phenotypic NASH | Improvement in ALT | Aug 2019 | NCT02913105 |
| MGL-3196 | THR-β agonist | NASH on biopsy with stage 1–3 fibrosis | Improvement in liver fat by MRI | Oct 2017 | NCT02912260 |
| MSDC 0602K (EMMINENCE) | mTOT inhibitor | NASH on biopsy with stage 1–3 fibrosis | NAS reduction ≥2 points without worsening fibrosis | Jul 2019 | NCT02784444 |
| NGM282 | FGF19 analog | NASH on biopsy | Improvement in liver fat by MRI | Apr 2018 | NCT02443116 |
| PF-05221304 | ACC inhibitor | NAFLD and PDFF ≥8% | Improvement in liver fat by MRI | Feb 2019 | NCT03248882 |
| SAR425899 (RESTORE) | GLP-1 and glucagon receptor agonist | NASH on biopsy and type 2 diabetes | Resolution of ballooning without worsening of fibrosis | Jul 2020 | NCT03437720 |
| Saroglitazar (EVIDENCES II) | PPARα and PPARγ agonist | NAFLD and ALT ≥ 1.5 × ULN | Improvement in ALT | Dec 2018 | NCT03061721 |
| Selonsertib + GS-0976 + GS-9674 | ASK1 inhibitor, ACC inhibitor, FXR agonist | NASH on biopsy with stage 3 or 4 fibrosis or Fibroscan stiffness value ≥ 14.5 kPa orELF ≥ 9.8 | Improvement in fibrosis without worsening NASH | Mar 2020 | NCT03449446 |
| Semaglutide | GLP-1 mimetic | NASH on biopsy with stage 1–3 fibrosis | Resolution of NASH without worsening fibrosis | Nov 2019 | NCT02970942 |
| Tropifexor (FLIGHT-FXR) | FXR agonist | NASH on biopsy with stage 1–3 fibrosis or Clinical risks for NASH | Improvement in ALT and liver fat by MRI | Dec 2018 | NCT02855164 |
| Volixibat | ASBT inhibitor | NASH on biopsy with stage 0–3 fibrosis, >5% liver fat by MRI | NAS reduction ≥2 points without worsening fibrosis | Jul 2020 | NCT02787304 |
Trials registered on clinicaltrials.gov as of 16 March 2018 with the following criteria: (i) include a liver-related primary endpoint; (ii) enroll more than 50 patients; (iii) enroll at more than 3 study sites; (iv) results have not been published. CCR2/5, chemokine receptors 2 and 5; HE, hepatic encephalopathy; SCD, stearoyl-CoA desaturase; MRS, magnetic resonance spectroscopy; MRI, magnetic resonance imaging; HVPG, hepatic venous pressure gradient; SAF, steatosis, activity (inflammation and ballooning), fibrosis score; SGLT, sodium-dependent glucose cotransporter; NAS, NAFLD activity score; PDFF, proton density fat fraction by magnetic resonance imaging; ULN, upper limit of normal; ASBT, apical sodium-dependent bile acid transporter; ELF, Enhanced liver fibrosis score.
Although activity in drug development for NASH is intense and advancing rapidly, it is, in our opinion, too early to prioritize those drugs or mechanisms that are most promising, as clinical trials thus far have been relatively short (3–18 months), whereas any approved therapies will likely be administered long-term. Thus, although the efficacies of several agents in clinical trials, such as an FXR agonist (obeticholic acid), a PPARα and PPARδ agonist (elafibrinor) and a CCR2 and CCR5 antagonist (cencriviroc), are encouraging based on liver biopsy or noninvasive markers, these findings will need to translate into durable safety and efficacy. Moreover, the possibility of drug combinations as a future therapeutic option is increasingly likely because of concern that attacking a single target will not be sufficiently potent, but there is no data yet from long-term, controlled trials (see Challenges in Trial Design).
Metabolic targets
Metabolic targets can be used for treatment of NASH with the aim of either reduce metabolic substrate delivery to the liver or to facilitate its safe disposal. Diet- and lifestyle-induced weight loss decreases metabolic substrate delivery to the liver and can improve all of the features of NASH, including hepatic fibrosis130, providing proof of concept that attenuating metabolic drivers of NASH should be an effective therapy.
PPARγ ligands (for example, pioglitazone, a member of the family of thiazolidinedione class of drugs) have been studied in several trials and improve steatosis, inflammation and hepatocellular ballooning as well as hepatic fibrosis70,131. However, their appeal is limited by some side effects, such as weight gain, fluid retention, osteopenia and increased fracture risk, especially in older women. Interestingly, pioglitazone has poorer PPARγ agonism than the PPARγ agonist rosiglitazone but more profound effects on NASH70,132, which suggests that the activity of pioglitazone is mediated by additional mechanisms, such as the mitochondrial target of thiazolidinediones (mTOT), the mitochondrial pyruvate carrier; accordingly, specific agonists of mTOT are currently in phase 2B clinical trials.
Synthetic ligands that activate another nuclear receptor, the farnesoid X receptor (FXR), improve insulin sensitivity and exhibit direct anti-inflammatory and antifibrotic effects in mouse models of NASH and human tissues133,134. The prototype for this class of compounds is obeticholic acid (OCA), which improves insulin resistance, as revealed through euglycemic-hyperinsulinemic clamp studies in mice, a method that is considered to be the gold standard for the assessment of insulin resistance133. A positive phase 2B clinical trial of OCA (25 mg per day) was terminated early because of demonstration of efficacy during an interim analysis planned a priori135. However, OCA, in 25 mg per day doses, causes both pruritus and moderate increases in low-density lipoprotein (LDL) cholesterol in some individuals. The ongoing phase 3 Randomized Global Phase 3 Study to Evaluate the Impact on NASH With Fibrosis of Obeticholic Acid Treatment (REGENERATE) trial with OCA will determine whether a lower dose of OCA will render it more tolerable without losing efficacy (NCT02548351).
Several small-molecule agonists of FXR that do not have a bile acid structural backbone have been developed on the premise that they will not increase LDL cholesterol or cause pruritus, which is yet unproven.
Another potential pathway that potentiates FXR activity is the release of the growth factor FGF19 from the intestine upon bile acid binding to FXR136, which has beneficial effects on NASH in animal models, although results are conflicting137,138. The proliferative and protumorigenic effects of FGF19 have been obviated by the development of recombinant analogs in current trials that lack these detrimental activities but preserve its favorable metabolic effects. In a phase 2a study, this FGF19 analog yielded a dramatic reduction in hepatic fat and liver enzymes in patients with biopsy-confirmed NASH139. FGF21 is another member of the fibroblast growth factor family that is not mitogenic and has insulin-sensitizing and antifibrotic properties140. It too improved steatosis, insulin resistance and liver stiffness in short-term trials and is under active investigation as a treatment for NASH.
Glucagon-like peptide (GLP)-1 is an intestinal hormone generated through the proteolytic processing of proglucagon that stimulates insulin secretion and inhibits secretion of glucagon. GLP-1 is also an insulin sensitizer with additional metabolic effects that contribute to its anti-NASH activity141. Liraglutide, a GLP-1 agonist requiring daily injection, improved NASH histology in a small pilot study142. Studies of semaglutide, a GLP-1 agonist that requires only weekly dosing, are under way (NCT02970942). Therapy with GLP-1, in combination with either glucagon inhibitory peptide or GLP-2, which improves intestinal barrier function, is in early development.
Agents that accelerate the safe disposal of metabolic substrates are also under study, in particular PPARα/δ (i.e., possesses dual activity on both PPAR-α and PPAR-δ receptors) and PPARα/γ agonists. The PPARα component increases fatty acid oxidation, whereas the PPARδ component has anti-inflammatory effects. Elafibrinor is the prototypic PPARα/δ agonist that improves insulin resistance and improves inflammation143. In a post hoc analysis of a phase 2B trial, the drug improved the metabolic features of NASH, and it is currently being tested in a pivotal phase 3 trial, RESOLVE IT (NCT02704403). As described above, acyl-CoA carboxylase (ACC) inhibitors reduce DNL and potentially improve NASH. However, a possible limitation to this approach is that reduced DNL can promote a compensatory increase in SREBP-1 activity, which stimulates triglyceride generation from peripheral FFAs and thus promotes hypertriglyceridemia144. A thyroid hormone receptor (THR)-β-selective agonist is being evaluated as a means to reduce lipotoxic load, and its utilization has been shown to improve circulating lipids in healthy humans145 and rapidly ‘de-fat’ the liver in a phase 2A proof-of-concept trial in NAFLD (NCT02912260). This receptor agonist also has mitogenic properties146, and the longterm safety of targeting this pathway has not yet been established in humans, as with most drugs in phase 2 trials.
Cell stress and apoptosis
Oxidative stress and activation of the unfolded protein response are two well-characterized cell stress pathways that promote cell death in NASH. Vitamin E is a prototypic anti-oxidant and has the most profound antisteatohepatitic effect of any drug studied to date70. However, its appeal is limited by lack of efficacy in reducing hepatic fibrosis in a large randomized controlled trial70.
Programmed cell death (apoptosis) in NASH and other chronic liver diseases promotes tissue injury and fibrosis147, which establishes a rationale for inhibiting apotosis as a therapeutic strategy. Emricasan, a pancaspase inhibitor, reduces apoptosis that can attenuate inflammation and fibrosis148; it is also currently in phase 2B clinical trials for NASH (NCT02686762).
Immune targets
Inflammatory cells and cytokines are each implicated in NASH pathogenesis. Both the innate and adaptive immune systems are involved in the pathogenesis of NASH; however, most attention has focused on the innate immune system. The pro-inflammatory pathways involving apoptosis signaling kinase-1 (ASK-1)–JNK, MAP kinases, ERK and NFκB are potent mediators of inflammation and thus potential targets for therapy, although their ubiquitous expression in all cells and their vital role in maintaining tissue defenses against injury raise the potential specter of ‘off-target’ effects; fortunately, clinical trials in NASH have not revealed any such effects to date. In particular, the inhibition of ASK-1 ameliorated NASH and improved fibrosis in some patients in a short-term clinical trial149. These results await validation in larger more rigorously designed phase 3 trials, which are currently underway (NCT03053050).
The C–C motif chemokine receptor 2 (CCR2)–CCR5 chemokine axis amplifies the liver’s innate immune response and links inflammation to activation of hepatic stellate cells and fibrogenesis. Inhibition of CCR2–CCR5 reduced short-term fibrosis progression in a clinical trial of NASH150. After 2 years of treatment, however, the mean fibrosis stage in those on active therapy was not significantly different from that of patients on placebo. This molecule is also being evaluated in a phase 3 trial (NCT03028740).
Fibrosis as a target
Progressive accumulation of fibrosis is the hallmark of NASH disease progression, and therefore, reduced fibrosis is an important goal of therapy. Fibrosis reflects the net outcome of fibrogenesis (i.e., fibrosis production) and fibrolysis (i.e., fibrosis degradation). Both occur concurrently in ongoing liver injury, but over time, fibrogenesis exceeds the liver’s capacity to degrade the accumulating extracellular matrix. Therefore, an antifibrotic approach can either inhibit fibrogenesis and/or accelerate fibrolysis. To date, most studies have focused on inhibiting fibrogenesis as a target, but two large trials in patients with NASH tested an inhibitory antibody to lysyl oxidase-2 (LOXL2), an enzyme that chemically crosslinks fibrillary collagen, so that LOXL2 inhibition might render collagen more susceptible to degradation. Unfortunately, these trials were both negative; however, other efforts to inhibit LOXL2 using small molecules are still being pursued. The clinical trial of a carbohydrate molecule that inhibits galectin, a fibrogenic factor, yielded mixed results. In this trial, a subset of treated individuals with cirrhosis who lacked a key feature of advanced portal HTN, esophageal varices, demonstrated both reduced portal pressure and decreased hepatocyte ballooning.
Because activated stellate cells are the key producers of collagen, apoptosis of stellate cells is hypothesized to reduce fibrosis. In that context, a trial of a liposomal formulation that delivers siRNA silencing expression of heat shock protein 47 (HSP47) to promote stellate cell apoptosis is underway in patients with advanced fibrosis of different etiologies (NCT02227459). HSP47 is a chaperone protein that regulates the proper folding of fibrillary collagens; by inhibiting this molecule, collagen is misfolded within the cell and is accumulated rather than secreted, which leads to cell death.
Targets outside the liver
The intestinal microbiome has multiple physiological and pathophysiological effects that are relevant to NAFLD151. Probiotics and bovine colostrum that contains anti-bodies to endotoxin are currently under evaluation for NASH. Although not a direct target of current therapies, enhancement of brown adipose tissue function remains an appealing mechanism to attenuate MetS and NASH, as this specialized tissue burns glucose to produce heat and its enhanced activity can improve obesity and lipid and glucose abnormalities73,152. A molecule with dual activity as a ligand for FXR and TGR5 (a G protein–coupled receptor specific for bile acids) has shown the potential to browning of white adipose in an animal model of NASH, but further studies are needed153. Similarly, a strategy to inhibit mitochondrial uncoupling protein using a controlled release protonophore (i.e., a molecule that translocates proteins across the mitochondrial membrane) in a rodent model establishes proof of principle for therapeutically altering mitochondrial energetics. At present, however, there is a paucity of human data supporting the rationale to enhance energy utilization as a NASH treatment154, and historical use of such agents for weight loss resulted in toxicities and some deaths.
Combination therapies
Early evidence from clinical trials suggests that features of NASH are pharmacologically responsive. Nonetheless, no more than ~40% of patients in these trials have shown benefit from a single therapy, which may not be sufficiently efficacious to justify regulatory approval for long-term monotherapy. Thus, the field is rapidly moving toward combination therapies, as noted above. Combinations may also include conjugate therapies—for example, a fatty acid–bile acid conjugate, aramchol, that is undergoing clinical trials for NASH155; no drug–antibody conjugates have been developed thus far, however. Similarly, single drugs with multiple targets and modes of action have the potential to be efficacious, as NASH pathogenesis involves many disease drivers. As with drug combinations, single agents with more than one molecular or cellular target will be most valuable if all of their individual activities improve features of the disease and are not antagonistic. A regulatory pathway to approval of combination therapies has been outlined with a focus on the mechanistic plausibility of the drug combinations156.
Strategies to combine drugs are largely empiric at present, with most combinations seeking to include metabolic targets with one agent, combined with either antifibrotic and/or anti-inflammatory activity in another. At present, we lack a comprehensive understanding of critical drivers of NASH that might represent therapeutically vulnerable nodes of pathogenesis. As a result, efforts are directed at capturing as many potential pathogenic drivers in a single combination as possible. These combinations appear promising on the basis of results from animal studies, but they assume that all patients with the same histologic appearance or stage of NASH arise from the same pathogenesis. However, as noted above, different individuals could have variable contributions from the convergent pathways that are dysregulated in the disease. Although appealing in principle, such ‘personalized’ therapy for NASH remains elusive.
A different strategy for more data-driven, rational combination therapy could rely instead on big data approaches that assess gene expression changes in NASH liver tissue, as has already been employed to define new targets for liver cancer118,157. For example, consistent changes in liver transcriptomic pathways in response to one drug could also uncover unaffected pathways, which would benefit from a second, complementary drug. Moreover, such strategies could be validated using ex vivo approaches, including human liver slices118 or new multicellular platforms that contain human cells and replicate flow117, thereby streamlining the identification of optimal combinations and dosages and also uncovering any early toxicity signals.
Challenges in trial design
Principles of clinical trial design for NASH therapies are evolving rapidly as new targets and diagnostic technologies emerge and our understanding of the disease’s natural history is refined10,158. In particular, the selection of patients suitable for clinical trials has focused increasingly on those with intermediate or advanced fibrosis (stages 2–4), whereas patients with only NAFL (without inflammation, ballooning or fibrosis) are not trial candidates, primarily because they are at no demonstrable risk of short-term liver-related outcomes. Moreover, whereas the first major controlled trials included patients with intermediate NAFLD activity scores (for example, >3) and any amount of fibrosis, focus has shifted toward trials with fibrosis as the primary indicator of efficacy, as fibrosis is the histologic feature of NASH most clearly linked to risk of decompensation or liver-related events.
Trials intended to establish proof of target engagement and pharmacodynamic activity, typically phase 2A trials, rely increasingly on noninvasive readouts, primarily imaging to assess fat content and/or to quantify either stiffness or inflammatory activity, which suggest possible disease amelioration. In contrast, phase 2B and phase 3 trials to establish efficacy still require pre- and post-treatment liver biopsies in almost all cases, but this could change significantly as newer diagnostic methods that can supplant biopsy are validated. Joint efforts among all stakeholders to standardize approaches to diagnosis and clinical trial design have been initiated to accelerate clinical trial development and enable sharing of information and best practices159. It is anticipated that within 5–10 years, noninvasive diagnostic modalities, alone or in combination, will supplant liver biopsy as approvable endpoints. Such noninvasive tests, whether by assessing serum markers, examining liver structure through imaging tests or functionally testing liver reserve, offer the appeal of capturing information from the entire organ, rather than from a tiny fraction of the tissue obtained through a needle biopsy.
Drugs are approved on the basis of demonstration of clinically meaningful benefit160. This is broadly defined as an improvement in how affected individuals feel, function or survive. It is difficult to ascribe changes in functional status or in quality of life to a specific therapeutic intervention given the number of comorbidities present and concomitant therapies given to patients with NASH. Also, the capacity of patients to manage activities of daily living declines with progression to cirrhosis. Thus, the focus of therapeutic development has been on the demonstration of improved survival.
It is challenging to demonstrate survival benefit for patients who are in the precirrhotic stages of NASH, as they are not at short-term risk for liver-related outcomes. This has led to drug approval being sought under the Food and Drug Administration’s subpart H pathway in the United States, in which short-term improvement in histology, based on the mechanism of action of a drug, allows conditional approval with the requirement that additional studies are undertaken to demonstrate that short-term changes translate into reduced progression to cirrhosis and its complications. In individuals with cirrhosis, decreased fibrosis stage or preventing progression of model for end-stage liver disease (MELD) score to 15 or higher can serve as potential subpart H endpoints, but demonstration of survival or liver-related outcomes are needed in the long-term.
Future prospects
The public health and economic impacts of fatty liver disease have provoked intense interest in treatments for NASH among patients, regulators and the biotechnology and pharmaceutical industries161. Moreover, it is already clear that improvement in NASH-associated histology can be achieved pharmacologically, despite the limited number of clinical trials to date. Still, more robust and durable effects of pharmacotherapy must be demonstrated and linked to improvement in clinical outcomes, including reduced progression to cirrhosis, complications of end-stage liver disease, need for transplantation and hepatocellular carcinoma. Ideally, these therapies should additionally mitigate features of MetS, considering that, as discussed above, cardiovascular disease remains the most common cause of death in individuals with NAFLD even among those with NASH162. Standardization of clinical trial designs and validation of noninvasive technologies will greatly accelerate progress and are likely to emerge within 5 years, in part through all-stakeholder discussions that include academic leaders, regulators, drug developers and patients10. Nonetheless, success in discovering effective therapies for NASH will continue to rely upon fundamental biomedical investigation that links the microbiome, liver metabolism and response to injury, systemic consequences of obesity and MetS.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Estes C, Razavi H, Loomba R, Younossi Z & Sanyal AJ Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67, 123–133 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Younossi ZM et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 64, 1577–1586 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Goldberg D et al. Changes in the Prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology 152, 1090–1099 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong RJ et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 148, 547–555 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Mittal S et al. Hepatocellular carcinoma in the absence of cirrhosis in united states veterans is associated with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol 14, 124–131.e1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dyson J et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol 60, 110–117 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Piscaglia F et al. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: a multicenter prospective study. Hepatology 63, 827–838 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Younossi ZM et al. Global epidemiology of nonalcoholic fatty liver disease—meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Loomba R & Sanyal AJ The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol 10, 686–690 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Siddiqui MS et al. Case definitions for inclusion and analysis of endpoints in clinical trials for NASH through the lens of regulatory science. Hepatology 67, 2001–2012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh S et al. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol 13, 643–654.e1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindenmeyer CC & McCullough AJ The natural history of nonalcoholic fatty liver disease—an evolving view. Clin. Liver Dis 22, 11–21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rinella ME & Sanyal AJ Management of NAFLD: a stage-based approach. Nat. Rev. Gastroenterol. Hepatol 13, 196–205 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Caussy C et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J. Clin. Invest 127, 2697–2704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loomba R et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology 149, 1784–1793 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pouladi N, Bime C, Garcia JGN & Lussier YA Complex genetics of pulmonary diseases: lessons from genome-wide association studies and next-generation sequencing. Transl. Res 168, 22–39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McPherson S et al. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J. Hepatol 62, 1148–1155 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Huang PL A comprehensive definition for metabolic syndrome. Dis. Model. Mech 2, 231–237 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Käräjämäki AJ et al. Non-alcoholic fatty liver disease with and without metabolic syndrome: different long-term outcomes. Metabolism 66, 55–63 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Allen AM et al. Nonalcoholic fatty liver disease incidence and impact on metabolic burden and death: a 20 year-community study. Hepatology 67, 1726–1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bazick J et al. Clinical model for NASH and advanced fibrosis in adult patients with diabetes and NAFLD: guidelines for referral in NAFLD. Diabetes Care 38, 1347–1355 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Portillo-Sanchez P et al. High prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus and normal plasma aminotransferase levels. J. Clin. Endocrinol. Metab 100, 2231–2238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwok R et al. Screening diabetic patients for non-alcoholic fatty liver disease with controlled attenuation parameter and liver stiffness measurements: a prospective cohort study. Gut 65, 1359–1368 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Anstee QM, Targher G & Day CP Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol 10, 330–344 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Choudhury J & Sanyal AJ Insulin resistance and the pathogenesis of nonalcoholic fatty liver disease. Clin. Liver Dis 8, 575–594 (2004). ix. [DOI] [PubMed] [Google Scholar]
- 26.Ballestri S et al. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol 31, 936–944 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Lorbeer R et al. Association between MRI-derived hepatic fat fraction and blood pressure in participants without history of cardiovascular disease. J. Hypertens 35, 737–744 (2017). [DOI] [PubMed] [Google Scholar]
- 28.VanWagner LB et al. Association of nonalcoholic fatty liver disease with subclinical myocardial remodeling and dysfunction: a population-based study. Hepatology 62, 773–783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Musso G et al. Association of non-alcoholic fatty liver disease with chronic kidney disease: a systematic review and meta-analysis. PLoS Med. 11, e1001680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valbusa F et al. Nonalcoholic fatty liver disease and increased risk of 1-year all-cause and cardiac hospital readmissions in elderly patients admitted for acute heart failure. PLoS One 12, e0173398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sorrentino P et al. Predicting fibrosis worsening in obese patients with NASH through parenchymal fibronectin, HOMA-IR, and hypertension. Am. J. Gastroenterol 105, 336–344 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Pelusi S et al. Renin–angiotensin system inhibitors, type 2 diabetes and fibrosis progression: an observational study in patients with nonalcoholic fatty liver disease. PLoS One 11, e0163069 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Namisaki T et al. Beneficial effects of combined ursodeoxycholic acid and angiotensin-II type 1 receptor blocker on hepatic fibrogenesis in a rat model of nonalcoholic steatohepatitis. J. Gastroenterol 51, 162–172 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Noguchi R et al. Selective aldosterone blocker ameliorates the progression of non-alcoholic steatohepatitis in rats. Int. J. Mol. Med 26, 407–413 (2010). [PubMed] [Google Scholar]
- 35.Pais R et al. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J. Hepatol 59, 550–556 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Ekstedt M et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 44, 865–873 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Ekstedt M et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 61, 1547–1554 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Angulo P et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149, 389–397.10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hagström H et al. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol 67, 1265–1273 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Sookoian S & Pirola CJ Genetic predisposition in nonalcoholic fatty liver disease. Clin. Mol. Hepatol 23, 1–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anstee QM, Daly AK & Day CP Genetic modifiers of non-alcoholic fatty liver disease progression. Biochim. Biophys. Acta 1812, 1557–1566 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Eslam M, Valenti L & Romeo S Genetics and epigenetics of NAFLD and NASH: clinical impact. J. Hepatol 68, 268–279 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Romeo S et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet 40, 1461–1465 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bruschi FV et al. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 65, 1875–1890 (2017). [DOI] [PubMed] [Google Scholar]
- 45.BasuRay S, Smagris E, Cohen JC & Hobbs HH The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 66, 1111–1124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stender S et al. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat. Genet 49, 842–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abul-Husn NS et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N. Engl. J. Med 378, 1096–1106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alonso C et al. Metabolomic identification of subtypes of nonalcoholic steatohepatitis. Gastroenterology 152, 1449–1461.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neuschwander-Tetri BA Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 52, 774–788 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Cusi K Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology 142, 711–725.e6 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Boursier J et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63, 764–775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirsova P, Ibrahim SH, Gores GJ & Malhi H Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J. Lipid Res 57, 1758–1770 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mota M, Banini BA, Cazanave SC & Sanyal AJ Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 65, 1049–1061 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neuschwander-Tetri BA Non-alcoholic fatty liver disease. BMC Med. 15, 45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lomonaco R et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 55, 1389–1397 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Pal M, Febbraio MA & Lancaster GI The roles of c-Jun NH2-terminal kinases (JNKs) in obesity and insulin resistance. J. Physiol. (Lond.) 594, 267–279 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han MS et al. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339, 218–222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samuel VT & Shulman GI The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J. Clin. Invest 126, 12–22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Magkos F et al. Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab. 23, 591–601 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gastaldelli A et al. Importance of changes in adipose tissue insulin resistance to histological response during thiazolidinedione treatment of patients with nonalcoholic steatohepatitis. Hepatology 50, 1087–1093 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Donnelly KL et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest 115, 1343–1351 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jang C et al. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab. 27, 351–361.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Truswell AS, Seach JM & Thorburn AW Incomplete absorption of pure fructose in healthy subjects and the facilitating effect of glucose. Am. J. Clin. Nutr 48, 1424–1430 (1988). [DOI] [PubMed] [Google Scholar]
- 64.Rao SS, Attaluri A, Anderson L & Stumbo P Ability of the normal human small intestine to absorb fructose: evaluation by breath testing. Clin. Gastroenterol. Hepatol 5, 959–963 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abdelmalek MF et al. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 56, 952–960 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schwarz JM et al. Effects of dietary fructose restriction on liver fat, de novo lipogenesis, and insulin kinetics in children with obesity. Gastroenterology 153, 743–752 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Softic S, Cohen DE & Kahn CR Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci 61, 1282–1293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wakil SJ & Abu-Elheiga LA Fatty acid metabolism: target for metabolic syndrome. J. Lipid Res 50 Suppl, S138–S143 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benhamed F et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Invest 122, 2176–2194 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanyal AJ et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med 362, 1675–1685 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arab JP, Karpen SJ, Dawson PA, Arrese M & Trauner M Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology 65, 350–362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Nierop FS et al. Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes Endocrinol. 5, 224–233 (2017). [DOI] [PubMed] [Google Scholar]
- 73.Kajimura S, Spiegelman BM & Seale P Brown and beige fat: physiological roles beyond heat generation. Cell Metab. 22, 546–559 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sanyal AJ et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120, 1183–1192 (2001). [DOI] [PubMed] [Google Scholar]
- 75.Pessayre D & Fromenty B NASH: a mitochondrial disease. J. Hepatol 42, 928–940 (2005). [DOI] [PubMed] [Google Scholar]
- 76.Bril F et al. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology 65, 1132–1144 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Yki-Järvinen H Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2, 901–910 (2014). [DOI] [PubMed] [Google Scholar]
- 78.Perry RJ, Samuel VT, Petersen KF & Shulman GI The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510, 84–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luukkonen PK et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol 64, 1167–1175 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Mauer AS, Hirsova P, Maiers JL, Shah VH & Malhi H Inhibition of sphingosine 1-phosphate signaling ameliorates murine nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol 312, G300–G313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han MS et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J. Lipid Res 49, 84–97 (2008). [DOI] [PubMed] [Google Scholar]
- 82.Ioannou GN The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol. Metab 27, 84–95 (2016). [DOI] [PubMed] [Google Scholar]
- 83.Trevaskis JL et al. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol 302, G762–G772 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Asgharpour A et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol 65, 579–588 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Krishnan A et al. A longitudinal study of whole body, tissue, and cellular physiology in a mouse model of fibrosing NASH with high fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol 312, G666–G680 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han J & Kaufman RJ The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res 57, 1329–1338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Puri P et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 134, 568–576 (2008). [DOI] [PubMed] [Google Scholar]
- 88.Szabo G & Petrasek J Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol 12, 387–400 (2015). [DOI] [PubMed] [Google Scholar]
- 89.Guy CD et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 55, 1711–1721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marra F & Bertolani C Adipokines in liver diseases. Hepatology 50, 957–969 (2009). [DOI] [PubMed] [Google Scholar]
- 91.Lanaspa MA et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem 287, 40732–40744 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sookoian S & Pirola CJ Obstructive sleep apnea is associated with fatty liver and abnormal liver enzymes: a meta-analysis. Obes. Surg 23, 1815–1825 (2013). [DOI] [PubMed] [Google Scholar]
- 93.Henao-Mejia J et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu R et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med 23, 859–868 (2017). [DOI] [PubMed] [Google Scholar]
- 95.Horwath JA et al. Obesity-induced hepatic steatosis is mediated by endoplasmic reticulum stress in the subfornical organ of the brain. JCI Insight 2, 90170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Csak T et al. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 54, 133–144 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stienstra R et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl Acad. Sci. USA 108, 15324–15329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mridha AR et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol 66, 1037–1046 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Loomba R et al. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology 56, 943–951 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bugianesi E, McCullough AJ & Marchesini G Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology 42, 987–1000 (2005). [DOI] [PubMed] [Google Scholar]
- 101.Sabio G et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 322, 1539–1543 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tilg H The role of cytokines in non-alcoholic fatty liver disease. Dig. Dis 28, 179–185 (2010). [DOI] [PubMed] [Google Scholar]
- 103.Ghorpade DS et al. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 555, 673–677 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Betrapally NS, Gillevet PM & Bajaj JS Changes in the intestinal microbiome and alcoholic and nonalcoholic liver diseases: causes or effects? Gastroenterology 150, 1745–1755.e3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Loomba R et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 25, 1054–1062.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bashiardes S, Shapiro H, Rozin S, Shibolet O & Elinav E Non-alcoholic fatty liver and the gut microbiota. Mol. Metab 5, 782–794 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cohen LJ et al. Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature 549, 48–53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schubert K, Olde Damink SWM, von Bergen M & Schaap FG Interactions between bile salts, gut microbiota, and hepatic innate immunity. Immunol. Rev 279, 23–35 (2017). [DOI] [PubMed] [Google Scholar]
- 109.Marra F & Svegliati-Baroni G Lipotoxicity and the gut–liver axis in NASH pathogenesis. J. Hepatol 68, 280–295 (2018). [DOI] [PubMed] [Google Scholar]
- 110.Brandl K & Schnabl B Intestinal microbiota and nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol 33, 128–133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Leung C, Rivera L, Furness JB & Angus PW The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol 13, 412–425 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Tsuchida T & Friedman SL Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol 14, 397–411 (2017). [DOI] [PubMed] [Google Scholar]
- 113.Lade A, Noon LA & Friedman SL Contributions of metabolic dysregulation and inflammation to nonalcoholic steatohepatitis, hepatic fibrosis, and cancer. Curr. Opin. Oncol 26, 100–107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang X et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 24, 848–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Valenti L & Dongiovanni P Mutant PNPLA3 I148M protein as pharmacological target for liver disease. Hepatology 66, 1026–1028 (2017). [DOI] [PubMed] [Google Scholar]
- 116.Oseini AM, Cole BK, Issa D, Feaver RE & Sanyal AJ Translating scientific discovery: the need for preclinical models of nonalcoholic steatohepatitis. Hepatol. Int 12, 6–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Feaver RE et al. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI Insight 1, e90954 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nakagawa S et al. Molecular liver cancer prevention in cirrhosis by organ transcriptome analysis and lysophosphatidic acid pathway inhibition. Cancer Cell 30, 879–890 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nguyen DG et al. Bioprinted 3D primary liver tissues allow assessment of organ-level response to clinical drug induced toxicity in vitro. PLoS One 11, e0158674 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Santhekadur PK, Kumar DP & Sanyal AJ Preclinical models of non-alcoholic fatty liver disease. J. Hepatol 68, 230–237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tsuchida T et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol 10.1016/j.jhep.2018.03.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Charlton M et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol 301, G825–G834 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Giles DA et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. Nat. Med 23, 829–838 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Anstee QM & Goldin RD Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol 87, 1–16 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sanyal AJ & Pacana T A lipidomic readout of disease progression in a diet-induced mouse model of nonalcoholic fatty liver disease. Trans. Am. Clin. Climatol. Assoc 126, 271–288 (2015). [PMC free article] [PubMed] [Google Scholar]
- 126.Lai CY, Lin CY, Hsu CC, Yeh KY & Her GM Liver-directed microRNA-7a depletion induces nonalcoholic fatty liver disease by stabilizing YY1-mediated lipogenic pathways in zebrafish. Biochim. Biophys. Acta 1863, 844–856 (2018). [DOI] [PubMed] [Google Scholar]
- 127.Musselman LP et al. Role of fat body lipogenesis in protection against the effects of caloric overload in Drosophila. J. Biol. Chem 288, 8028–8042 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee L et al. Nutritional model of steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Hepatology 50, 56–67 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sternson SM & Eiselt AK three pillars for the neural control of appetite. Annu. Rev. Physiol 79, 401–423 (2017). [DOI] [PubMed] [Google Scholar]
- 130.Vilar-Gomez E et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 149, 367–378.e5 (2015). [DOI] [PubMed] [Google Scholar]
- 131.Mahady SE, Webster AC, Walker S, Sanyal A & George J The role of thiazolidinediones in non-alcoholic steatohepatitis—a systematic review and meta analysis. J. Hepatol 55, 1383–1390 (2011). [DOI] [PubMed] [Google Scholar]
- 132.Ratziu V et al. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 51, 445–453 (2010). [DOI] [PubMed] [Google Scholar]
- 133.Mudaliar S et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145, 574–582.e1 (2013). [DOI] [PubMed] [Google Scholar]
- 134.Kong B, Luyendyk JP, Tawfik O & Guo GL Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor–knockout mice fed a high-fat diet. J. Pharmacol. Exp. Ther 328, 116–122 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Neuschwander-Tetri BA et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385, 956–965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nies VJ et al. Fibroblast growth factor signaling in metabolic regulation. Front. Endocrinol. (Lausanne) 6, 193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hartmann P et al. Modulation of the intestinal bile acid/FXR/FGF15 axis improves alcoholic liver disease in mice. Hepatology 67, 2150–2166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jiang C et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Invest 125, 386–402 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Harrison SA et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 391, 1174–1185 (2018). [DOI] [PubMed] [Google Scholar]
- 140.Staiger H, Keuper M, Berti L, Hrabe de Angelis M & Häring HU Fibroblast growth factor 21-metabolic role in mice and men. Endocr. Rev 38, 468–488 (2017). [DOI] [PubMed] [Google Scholar]
- 141.Jinnouchi H et al. Liraglutide, a glucagon-like peptide-1 analog, increased insulin sensitivity assessed by hyperinsulinemic-euglycemic clamp examination in patients with uncontrolled type 2 diabetes mellitus. J. Diabetes Res 2015, 706416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Armstrong MJ et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 387, 679–690 (2016). [DOI] [PubMed] [Google Scholar]
- 143.Ratziu V et al. Elafibranor, an agonist of the peroxisome proliferator–activated receptor–α and –δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150, 1147–1159.e5 (2016). [DOI] [PubMed] [Google Scholar]
- 144.Kim CW et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 26, 394–406.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Taub R et al. Lipid lowering in healthy volunteers treated with multiple doses of MGL-3196, a liver-targeted thyroid hormone receptor-β agonist. Atherosclerosis 230, 373–380 (2013). [DOI] [PubMed] [Google Scholar]
- 146.Alvarado TF et al. Thyroid hormone receptor β agonist induces β-catenin-dependent hepatocyte proliferation in mice: implications in hepatic regeneration. Gene Expr. 17, 19–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Alkhouri N, Carter-Kent C & Feldstein AE Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol 5, 201–212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Barreyro FJ et al. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 35, 953–966 (2015). [DOI] [PubMed] [Google Scholar]
- 149.Loomba R et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Friedman SL et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 67, 1754–1767 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Puri P & Sanyal AJ The intestinal microbiome in nonalcoholic fatty liver disease. Clin. Liver Dis 22, 121–132 (2018). [DOI] [PubMed] [Google Scholar]
- 152.Ahmadian M et al. ERRγ preserves brown fat innate thermogenic activity. Cell Rep. 22, 2849–2859 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Carino A et al. BAR502, a dual FXR and GPBAR1 agonist, promotes browning of white adipose tissue and reverses liver steatosis and fibrosis. Sci. Rep 7, 42801 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Perry RJ, Zhang D, Zhang XM, Boyer JL & Shulman GI Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347, 1253–1256 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Safadi R et al. The fatty acid–bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol 12, 2085–2091.e1 (2014). [DOI] [PubMed] [Google Scholar]
- 156.Woodcock J, Griffin JP & Behrman RE Development of novel combination therapies. N. Engl. J. Med 364, 985–987 (2011). [DOI] [PubMed] [Google Scholar]
- 157.Wooden B, Goossens N, Hoshida Y & Friedman SL Using big data to discover diagnostics and therapeutics for gastrointestinal and liver diseases. Gastroenterology 152, 53–67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Konerman MA, Jones JC & Harrison SA Pharmacotherapy for NASH: current and emerging. J. Hepatol 68, 362–375 (2018). [DOI] [PubMed] [Google Scholar]
- 159.Patel YA et al. Baseline parameters in clinical trials for nonalcoholic steatohepatitis: recommendations from the liver forum. Gastroenterology 153, 621–625.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sanyal AJ et al. Challenges and opportunities in drug and biomarker development for nonalcoholic steatohepatitis: findings and recommendations from an American Association for the Study of Liver Diseases–U.S. Food and Drug Administration Joint Workshop. Hepatology 61, 1392–1405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Drew L Drug development: sprint finish. Nature 551 10.1038/d41586-017-06926-1 (2017). [DOI] [PubMed] [Google Scholar]
- 162.Haflidadottir S et al. Long-term follow-up and liver-related death rate in patients with non-alcoholic and alcoholic related fatty liver disease. BMC Gastroenterol 14, 166 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Weltman MD, Farrell GC & Liddle C Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 111, 1645–1653 (1996). [DOI] [PubMed] [Google Scholar]
- 164.Matsumoto M et al. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol 94, 93–103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Clapper JR et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol. Gastrointest. Liver Physiol 305, G483–G495 (2013). [DOI] [PubMed] [Google Scholar]
- 166.Zhang Y et al. Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432 (1994). [DOI] [PubMed] [Google Scholar]
- 167.Hummel KP, Dickie MM & Coleman DL Diabetes, a new mutation in the mouse. Science 153, 1127–1128 (1966). [DOI] [PubMed] [Google Scholar]
- 168.Oana F et al. Physiological difference between obese (fa/fa) Zucker rats and lean Zucker rats concerning adiponectin. Metabolism 54, 995–1001 (2005). [DOI] [PubMed] [Google Scholar]
- 169.Arsov T et al. Adaptive failure to high-fat diet characterizes steatohepatitis in Alms1 mutant mice. Biochem. Biophys. Res. Commun 342, 1152–1159 (2006). [DOI] [PubMed] [Google Scholar]
- 170.Nakayama H et al. Transgenic mice expressing nuclear sterol regulatory element–binding protein 1c in adipose tissue exhibit liver histology similar to nonalcoholic steatohepatitis. Metabolism 56, 470–475 (2007). [DOI] [PubMed] [Google Scholar]
- 171.Nakagawa H et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Subramanian S et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor–deficient mice. J. Lipid Res 52, 1626–1635 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Horie Y et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Invest 113, 1774–1783 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fujii M et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol 46, 141–152 (2013). [DOI] [PubMed] [Google Scholar]
- 175.Adams LA, Sanderson S, Lindor KD & Angulo P The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J. Hepatol 42, 132–138 (2005). [DOI] [PubMed] [Google Scholar]
- 176.Angulo P, Keach JC, Batts KP & Lindor KD Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 30, 1356–1362 (1999). [DOI] [PubMed] [Google Scholar]
- 177.Ratziu V et al. Liver fibrosis in overweight patients. Gastroenterology 118, 1117–1123 (2000). [DOI] [PubMed] [Google Scholar]
- 178.Mantovani A, Ballestri S, Lonardo A & Targher G Cardiovascular disease and myocardial abnormalities in nonalcoholic fatty liver disease. Dig. Dis. Sci 61, 1246–1267 (2016). [DOI] [PubMed] [Google Scholar]
- 179.Targher G, Byrne CD, Lonardo A, Zoppini G & Barbui C Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol 65, 589–600 (2016). [DOI] [PubMed] [Google Scholar]