

SUMMARY
Loss-of-function mutations in CNTNAP2 cause a syndromic form of autism spectrum disorder in humans and produce social deficits, repetitive behaviors, and seizures in mice. However, the functional effects of these mutations at cellular and circuit levels remain elusive. Using laser-scanning photostimulation, whole-cell recordings, and electron microscopy, we found a dramatic decrease in excitatory and inhibitory synaptic inputs onto L2/3 pyramidal neurons of the medial prefrontal cortex (mPFC) of Cntnap2 knockout (KO) mice, concurrent with reduced spines and synapses, despite normal dendritic complexity and intrinsic excitability. Moreover, recording of mPFC local field potentials (LFPs) and unit spiking in vivo revealed increased activity in inhibitory neurons, reduced phase-locking to delta and theta oscillations, and delayed phase preference during locomotion. Excitatory neurons showed similar phase modulation changes at delta frequencies. Finally, pairwise correlations increased during immobility in KO mice. Thus, reduced synaptic inputs can yield perturbed temporal coordination of neuronal firing in cortical ensembles.
In Brief
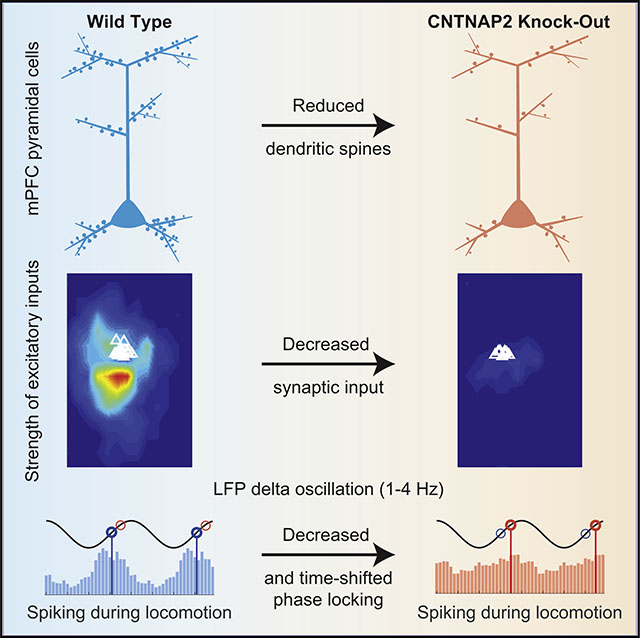
Lazaro et al. demonstrate a decrease in synaptic inputs onto mPFC L2/3 pyramidal neurons of Cntnap2 KO mice, concurrent with reduced spines and synapses. These lead to perturbed network activity, with mPFC cells exhibiting reduced phase locking and altered preferred phases to slow LFP oscillations, which may underlie autism-related phenotypes.
Graphical Abstract
INTRODUCTION
Autism spectrum disorder (ASD) is characterized by deficits in social communication and repetitive or restrictive behaviors (American Psychiatric Association, 2013). Genetic studies have revealed that the etiology of ASD is very heterogeneous, involving hundreds of genes (O’Roak et al., 2012; Sanders et al., 2012; Chen et al., 2015; Krishnan et al., 2016), a significant proportion of which appear as rare recessive or de novo dominant mutations (Geschwind, 2011; lossifov et al., 2014; Gilman et al., 2011; Leppa et al., 2016). One highly penetrant syndromic form of ASD is caused by loss-of-function mutations in the CNTNAP2 gene (Strauss et al., 2006), and CNTNAP2 polymorphisms have been associated with increased risk of ASD and other conditions (Poot et al., 2010; Scott-Van Zeeland et al., 2010; Arking et al., 2008).
CNTNAP2 encodes for contactin-associated protein-like 2 (Caspr2), a protein of the neurexin superfamily that has diverse cellular and circuit functions (Strauss et al., 2006; Poliak et al., 1999, 2001, 2003; Gdalyahu et al., 2015; Varea et al., 2015; Alarcon et al., 2008; Peñagarikano et al., 2011; Jurgensen and Castillo, 2015). Mice lacking the Cntnap2 gene recapitulate core behavioral deficits of ASD, including socialization and communication impairments, repetitive behaviors, and seizures (Peñagarikano et al., 2011). Recent in vivo evidence suggests that CNTNAP2 has a putative role in synapse formation and stabilization and that dendritic spine dynamics are affected in the Cntnap2 knockout (KO) mice, with reduced stability in newly formed spines (Gdalyahu et al., 2015). In addition, loss of CNTNAP2 leads to synaptic alterations in vitro, with decreased inhibition and axonal excitability deficits in acute hippocampal slices (Anderson et al., 2012; Jurgensen and Castillo, 2015; Scott et al., 2019). These results suggest that CNTNAP2 mutations may be linked to abnormal behavior by altering synaptic neurotransmission, functional connectivity, and neuronal network activity. However, the specific cellular and circuit mechanisms that lead to altered behavior in Cntnap2 KO mice remain unclear.
Here, we examined the neurophysiological consequences of Cntnap2 deletion in the mouse medial prefrontal cortex (mPFC), a brain region that is critically involved in social behavior (Yizhar et al., 2011; Grossmann, 2013) and notably affected in ASD (Voineagu et al., 2011; Redcay et al., 2013; Selimbeyoglu et al., 2017). mPFC cells can modulate social behavior, are critical for cortico-cortical communication, and have been considered a critical hub for autism-related gene expression (Yizhar et al., 2011; de la Torre-Ubieta et al., 2016; Selimbeyoglu et al., 2017; Parikshak et al., 2013). Using glutamate uncaging via laser-scanning photostimulation (LSPS) on layer 2/3 (L2/3) pyramidal neurons of the mPFC in combination with in vitro whole-cell patch-clamp recordings, we observed a reduction in both excitatory and inhibitory synaptic inputs onto excitatory neurons and decreased excitatory neurotransmission. Anatomical studies showed a concomitant decrease in dendritic spine and synapse densities. Using multichannel silicon microprobes to record in vivo local field potentials (LFPs) and activity from single neurons in the mPFC, we observed robust alterations in the phase locking of units to delta and theta oscillations during locomotion. These findings demonstrate that the loss of Cntnap2 results in decreased excitatory drive onto pyramidal cells, which further leads to alterations in circuit-level synchronous activity in the mPFC. Therefore, mutations in CNTNAP2 could be mechanistically linked to alterations in microcircuit connectivity and lead to abnormal population activity, providing a potential substrate for behavioral abnormalities in ASD.
RESULTS
Decreased Excitatory and Inhibitory Inputs in the mPFC of Cntnap2 KO Mice
To test how the loss of CNTNAP2 alters mPFC microcircuits, we used LSPS via glutamate uncaging to map and quantify local excitatory and inhibitory cortical inputs onto L2/3 mPFC pyramidal neurons. By voltage clamping patched pyramidal neurons at −70 and +5 mV, we recorded excitatory and inhibitory synaptic inputs, respectively, while uncaging glutamate and activating small clusters of surrounding neurons (Figures 1A, 1B, and S1). We observed that, similar to wild-type (WT), L2/3 pyramidal neurons in KO mice received most of their excitatory and inhibitory synaptic inputs from L2/3 and L5 in the mPFC (Figures 1C and 1D). However, L2/3 excitatory neurons in KO mice displayed a dramatic reduction in both excitatory and inhibitory local synaptic inputs compared to WT (Figures 1C–1E), while the balance of excitation to inhibition (E/I) in individual neurons was not significantly altered (Figure 1F). This reduction was not due to lower neuronal responsiveness to glutamate uncaging in KO mice, since both mouse groups showed equivalent responses to uncaging onto perisomatic regions (Figure S1).
Figure 1. Reduced Excitatory and Inhibitory Synaptic Inputs to L2/3 Pyramidal Neurons in the mPFC of Cntnap2 KO Mice.

(A) Schematic of laser-scanning photostimulation (LSPS) via glutamate uncaging paradigm, combined with whole-cell patch-clamp recordings of L2/3 pyramidal (Pyr) neurons in acute slices of the medial prefrontal cortex (mPFC). Patched neurons were clamped at −70 or +5 mV for the detection of local excitatory or inhibitory synaptic connections arising from photostimulated presynaptic glutamatergic (Excit.) and GABAergic (Inhib.) neurons.
(B) Example of an LSPS experiment in an mPFC slice, in which differential interference contrast imaging was used for tissue visualization. Photostimulation sites are superimposed (dots) and spaced within a 100-μm × 60-μm grid. The red circle indicates the location of the recorded glutamatergic neuron in L2/3, approached by the patch pipette.
(C and D) Group-averaged excitatory (C) and inhibitory (D) input maps of L2/3 excitatory neurons for WT (n = 20 cells from 3 mice in C; n = 11 cells from 3 mice in D) and KO (n = 9 cells from 3 mice in C; n = 13 cells from 3 mice in D) mice. Triangles indicate the location of individually recorded neurons.
(E) Average total synaptic excitatory and inhibitory input strength (log) measured for L2/3 excitatory cells depicting a robust decrease in the KO mice, compared to WT mice (WT: EPSC 2.51 ± 0.17, n = 20 cells; KO: EPSC 1.72 ± 0.17, n = 9 cells; WT: IPSC 2.83 ± 0.16, n = 11 cells; KO: IPSC 2.49 ± 0.15, n = 13 cells; EPSC: **p = 0.0051, IPSC: *p = 0.0218; Wilcoxon test).
(F) Average ratios of total EPSCs over IPSCs from individual cells (WT: n = 17 cells; KO: n = 8 cells). There is no significant difference in E/I ratio between WT and KO (p = 0.8873; unpaired t test). Scale bars: 200 μm.
All errors bars indicate the SEM.
Our input mapping findings could also be associated with alterations in the intrinsic excitability of cortical neurons. Caspr2 has a known role in the clustering of potassium channels in the juxtaparanodes of axons, which are important for the propagation of action potentials (Poliak et al., 1999, 2001, 2003). To examine whether the loss of Cntnap2 resulted in altered excitability and intrinsic properties in mPFC, we performed whole-cell current-clamp recordings on mPFC L2/3 pyramidal and parvalbumin-positive (PV+) inhibitory neurons (recorded in Cntnap2-PV-Cre × Ai9 animals) in KO and WT controls. We focused on PV+ interneurons, as these cells provide powerful perisomatic inhibition to cortical pyramidal neurons, and their dysfunction has been implicated in autism-associated deficits resulting from the loss of Cntnap2 (Scott et al., 2019; Peñagarikano et al., 2011). Input-output curves, showing the average number of action potentials elicited by increasing current injections in pyramidal and PV+ neurons, revealed no significant alterations in the action potential firing rate between the two groups (Figure S2). Action potential threshold, amplitude, half-width, afterhyperpolarization (AHP) potential, or time from peak to AHP were also not significantly different between WT and KO. The same was observed for resting membrane potential, input resistance, cell membrane capacitance, and membrane time constant (Table S1).
These results indicate that the loss of Cntnap2 does not affect the intrinsic excitability of L2/3 neurons of the mPFC, but leads to a robust reduction of local excitatory and inhibitory inputs onto these cells.
Decreased Excitatory Neurotransmission in Pyramidal Neurons of Cntnap2 KO Mice
To investigate the specific cellular processes that lead to reduced synaptic responses in KO mice, we performed whole-cell patch-clamp recordings of miniature excitatory and inhibitory postsynaptic currents (mEPSCs and mIPSCs, respectively) in mPFC L2/3 neurons. We measured mEPSC and mIPSC amplitude, frequency, and kinetics, as changes in amplitude are a reliable measure of the number of receptors at synapses (quantal size), while frequency correlates with the number of contacts or probability of release (Greer et al., 2010). In agreement with our LSPS findings, we observed a 2-fold decrease in the frequency of mEPSCs (Figures 2A and 2B) and a significant decrease in the average amplitude of mEPSCs in Cntnap2 KO pyramidal neurons (Figure 2C). We observed no statistically significant alterations in the frequency, amplitude, or kinetics of mIPSCs (Figures 2D–2F), despite the marked reduction in inhibition found with LSPS cortical input mapping (Figures 1D and 1E). This could reflect compensatory changes between synapse number and release probability or altered distribution of proximal and distal inhibitory inputs. In addition, we examined miniature postsynaptic currents in PV+ interneurons and found no significant differences in mEPSCs or mIPSCs (Figures S3A–S3F).
Figure 2. Reduced mEPSC Frequency and Long-Range Excitatory Inputs in Cntnap2 KO Pyramidal Neurons.

(A) Representative traces from recorded mEPSCs in Cntnap2 WT and KO pyramidal cells, voltage clamped at −70 mV, with corresponding average unitary events.
(B) Frequency of mEPSCs (WT: 2.42 ± 0.45 Hz, n = 24 cells, 6 mice; KO: 0.89 ± 0.12 Hz, n = 24 cells, 5 mice; Wilcoxon test, *p = 0.0410) is decreased in KO mice.
(C) Amplitude of mEPSCs (WT: 15.73 ± 0.63 pA, n = 24, 6 mice; KO: 13.88 ± 0.45 pA, n = 24 cells, 5 mice; Wilcoxon test, *p = 0.0172) is decreased in KO mice.
(D–F) Same as (A)–(C), but for mIPSCs. There is no significant decrease in frequency (WT: 5.76 ± 0.83 Hz, n = 28 cells, 8 mice; KO: 3.87 ± 0.45 Hz, n = 27 cells, 4 mice; p = 0.1327; Wilcoxon test) or amplitude of mIPSCs (WT: 24.43 ± 1.30 pA, n = 28 cells, 8 mice; KO: 27.21 ± 2.48 pA, n = 27 cells, 4 mice; p = 0.8740, Wilcoxon test) in KO mice compared to WT mice.
(G) A monopolar tungsten electrode was used to stimulate long-range axons (purple), which extend from the anterior forceps of the corpus callosum and project onto a patched excitatory neuron in L2/3 mPFC.
(H) Input-output curves of excitatory responses resulting from a range of increasing stimulus intensities in Cntnap2 WT and KO mice (WT: n = 7 cells, n = 6 mice; KO: n = 9 cells, n = 5 mice; *p < 0.0001, 2-way ANOVA).
(I) Representative current responses from paired pulses given at various interstimulus intervals (ISIs) in WT and KO mice.
(J) Ratio of second to first evoked synaptic response to paired-pulse stimulation at increasing ISIs suggests no significant deficits in the probability of synaptic vesicle release in Cntnap2 KO mice (WT: n = 10 cells, n = 6 mice; KO: n = 8 cells, n = 5 mice; p = 0.8926, 2-way ANOVA).
(K) Evoked AMPA (cells voltage clamped at −70 mV) and NMDA (cells voltage clamped at +40 mV) currents in WT and KO mice. Stimulus artifact was blanked for clarity. Dashed line indicates point where NMDA current amplitudes were measured, immediately after AMPA current decay.
(L) AMPA to NMDA ratios of Cntnap2 KO mice were not significantly altered, compared to WT mice, suggesting no significant changes in synaptic maturity (WT: 0.67 ± 0.10, n = 11 cells, 6 mice; KO: 0.53 ± 0.08, n = 8 cells, 5 mice; p = 0.3471, unpaired t test).
All errors bars indicate the SEM.
We then asked whether the observed decrease in mEPSC frequency on pyramidal neurons could be caused by a disruption in the probability of synaptic vesicle release (Toni et al., 1999; Sorra et al., 1998; Calverley and Jones, 1990). We tested this by stimulating long-range axonal projections to mPFC in slices and measuring evoked excitatory currents elicited in L2/3 pyramidal cells (Figure 2G). We observed reduced evoked EPSC amplitudes (Figure 2H) and significantly increased EPSC latencies (Figures S3G and S3H) in KO mice compared to controls, corroborating our previous findings of reduced excitatory neurotransmission. However, we found no significant differences in paired-pulse ratios of evoked currents between WT and KO mice (Figures 2I and 2J), indicating similar excitatory neurotransmitter release probabilities.
Finally, we tested whether KO mice had altered, immature, or silent synapses, characterized by the decreased ratio of α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate to N-methyl-D-aspartate (AMPA/NMDA) receptors (Toni et al., 1999; Calverley and Jones, 1990; Dani et al., 2005; Gibson et al., 2008). We recorded evoked AMPA and NMDA currents in the presence of the GABAA receptor blocker, picrotoxin, by holding the cells at −70 and +40 mV, respectively, in voltage clamp. We found no significant difference in the AMPA:NMDA ratios when comparing WT and KO mice (Figures 2K and 2L), indicating that KO mice do not have more immature or silent synapses.
These results indicate a reduction in the frequency and amplitude of excitatory drive onto single pyramidal cells, which cannot be explained by alterations in single synapse maturity or neurotransmitter vesicle release.
Decreased Dendritic Spine Density in Cntnap2 KO Mice
We next asked whether the decrease in excitatory neurotransmission was caused by a reduction in the total number of synaptic inputs, either through decreased dendritic branching or decreased spine density. We performed 3D anatomical reconstructions of L2/3 pyramidal neurons by filling cells with biocytin during in vitro slice recording experiments and imaged them with confocal microscopy. Sholl analysis did not reveal significant changes in total dendritic length, total number of dendritic branches, or dendritic complexity (Figures 3A and 3B), suggesting that L2/3 pyramidal neurons in Cntnap2 KO mice have normal dendritic arborization. In addition, the density of neuronal cell bodies in L2/3 and the density of immunolabeled PV+ neurons in the prelimbic cortex was similar in Cntnap2 KO mice and controls (Figures S4A and S4B).
Figure 3. Decreased Dendritic Excitatory and Inhibitory Synapses in Cntnap2 KO Mice.

(A) Representative z stack projection of biocytin-filled L2/3 neuron, visualized with a streptavidin-Alexa 488 antibody. Scale bar indicates 100-μm length.
(B) Sholl analysis showing number of intersections (p = 0.0632, 2-way ANOVA) and length (p = 0.9315, 2-way ANOVA) of dendrites is comparable between Cntnap2 WT (n = 8 cells) and KO (n = 9 cells).
(C) Confocal image of L2/3Thy1-GFP+ pyramidal neurons in mPFC, demonstrating representative apical and basal dendrites for WT (+/+) and Cntnap2 KO (−/−) mice.
(D) Summary graphs showing the quantification of average spine density in apical (WT: 0.86 ± 0.09 spines/μm, n = 21 dendrites; KO: 0.57 ± 0.07 spines/μm, n = 15 dendrites) and basal branches (WT: 0.70 ± 0.03 spines/mm, n = 34 dendrites; KO: 0.52 ± 0.02, n = 24 dendrites). *p < 0.05, unpaired t test; ****p < 0.0001, Welch’s t test.
(E) Representative electron micrographs showing neuropils of L2/3 mPFC in WT and Cntnap2 KO mice. Arrow indicates a multisynapse bouton (MSB). Arrowheads indicate perforated asymmetrical synapses. Spine profiles are pseudo-colored in orange. Scale bar: 500 nm.
(F) Graphs showing the quantification of asymmetric (putative excitatory; WT: 11.53 ± 0.35 synapses/100 μm2, n = 80 fields; KO: 8.59 ± 0.21 synapses/100 μm2, n = 90 fields; 3 mice per genotype; ***p < 0.0001, Wilcoxon test) and symmetric synapses (putative inhibitory; WT: 9.30 ± 0.46 synapses/100 μm2, n = 80 fields; KO: 5.87 ± 0.38 synapses/100 μm2, n = 90 fields; ***p < 0.0001, Wilcoxon test).
(G) Spine and postsynaptic density (PSD) area (WT: 0.1313 ± 0.006 μm2, n = 257 spines; KO: 0.1604 ± 0.010 μm2, n = 187 spines; p = 0.1142, Wilcoxon test) and length (WT: 281.10 ± 8.89 nm, n = 257 spines; KO: 309.90 ± 11.83 nm, n = 187 spines; p = 0.0709 Wilcoxon test).
(H) Density of MSBs for WT (1.33 ± 0.23 MSBs/100 μm2, n = 80 fields) and KO (0.41 ± 0.13 MSBs/100 μm2, n = 90 fields) mice; ***p = 0.0002, Wilcoxon test.
(I) Perforated synapses (PSs) for WT (3.38 ± 0.31 PS/100 μm2, n = 90 fields) and KO (4.52 ± 0.31 PS/100 μm2, n = 80 fields) mice; *p = 0.0122, Wilcoxon test.
All errors bars indicate the SEM.
To determine whether Cntnap2 KO neurons display a decrease in dendritic spine density, we crossed homozygous Cntnap2 KO (or WT) mice with Thy1-GFP mice, which express GFP in a subset of cortical pyramidal neurons, including sparsely labeled L2/3 mPFC pyramidal cells. The quantification of dendritic spines in these cells revealed that L2/3 pyramidal neurons in KO mice show a significant decrease in both basal and apical dendritic spine density (Figures 3C and 3D), which may underlie the reduction in functional synaptic inputs that we observed in our electrophysiology experiments.
To further validate this hypothesis, we used electron microscopy to examine L2/3 mPFC dendritic spines and synaptic contacts in WT (n = 3) and Cntnap2 KO mice (n = 3) (Figure 3E). Consistent with our previous findings, we observed a significant (~25%) reduction in the number of both asymmetric (excitatory) and symmetric (inhibitory) synapses in KO mice (Figure 3F). Furthermore, we found no significant changes in spine area or synapse length in the KO mice (Figure 3G). However, Cntnap2 KO mice had a markedly reduced density of multisynapse boutons (MSBs) (Figure 3H), a marker of synaptogenesis (Toni et al., 1999). We also found an increase in perforated synapses in these mice (Figure 3I), which are associated with increased synaptic turnover (Calverley and Jones, 1990; Sorra et al., 1998), supporting previous reports of increased dendritic spine turnover in Cntnap2 KO mice (Gdalyahu et al., 2015).
To determine how the loss of CNTNAP2 alters synaptic inputs to the distal apical tufts of excitatory neurons, we also counted asymmetric (putative excitatory) and symmetric (putative inhibitory) synapses in L1. Cntnap2 KO mice exhibited significantly decreased asymmetric and symmetric synapse numbers in L1 compared to controls, suggesting impairments in distal dendritic excitation and inhibition in these animals (Figures S4C and S4D).
These findings indicate that the loss of Cntnap2 leads to significant defects in both inhibitory and excitatory synaptic density, as well as alterations in markers of synapse plasticity and stability.
Altered In Vivo Network Activity in mPFC of Cntnap2 KO Mice
How does the observed decrease in cortical inputs onto L2/3 pyramidal neurons of Cntnap2 KO mice affect network activity in vivo? Such a robust decrease in functional synapses could affect the precise temporal coordination of neuronal firing during cortical network oscillations, which is critically dependent on the balance between excitation and inhibition (Sorra et al., 1998; Golomb and Hansel, 2000; Dani et al., 2005; Gibson et al., 2008).
To test this hypothesis, we recorded in vivo LFPs and single unit activity in the mPFC of KO and WT mice using multichannel silicon microprobes (Shobe et al., 2015) (Figures 4A and 4B). Head-fixed mice were free to rest or run on a spherical treadmill during the recordings (Polack et al., 2013), and locomotion was monitored. Both mouse groups exhibited similar locomotion characteristics, with a small but non-significant tendency for KO mice to have sparser but longer locomotion bouts (Figures S5A–S5F), in support of previous observations of hyperactivity in these animals (Peñagarikano et al., 2011). Activity during concatenated locomotion and immobility segments was analyzed separately (Figure 4A).
Figure 4. Increased Activity in In Vivo-Recorded Narrow-Spiking Units of Cntnap2 KO Mice, but Normal LFP Oscillations.

(A) Schematic of in vivo recordings and subsequent analysis of electrophysiological data. Mice were head fixed while free to run on a spherical treadmill. Extracellular signals were recorded from all layers of the mPFC of both WT (n = 8) and KO mice (n = 5) using multi-electrode silicon microprobes. Spikes were sorted into tentative single units methods(see Method Details). Motion on the treadmill was also recorded, and traces were separated into locomotion and immobility segments.
(B) Distribution and corresponding histograms of mean waveform peak-to-trough ratios versus peak-to-trough time distances for the two unit clusters consisting of WS and NS units. The inset depicts the average waveforms from all units in the two clusters.
(C) Unit spike waveforms averaged over all individual spikes (gray) from two example units from a WT mouse and a KO mouse.
(D and E) Distributions of average firing rates during locomotion (D) and immobility (E) for all WS and NS units in WT versus KO mice. Solid lines indicate the mean rates over all cells in the two groups. No significant difference for WS units. **p < 0.01; two-sample t test; Bonferroni corrected.
(F) Mean LFP bandpower during motion (top) and immobility (bottom) in WT and Cntnap2 KO mice, calculated from a single representative channel per mouse, located in the prelimbic mPFC. No significant differences were observed (p > 0.05, Wilcoxon test).
All errors bars indicate the SEM.
We recorded 249 single units from 8 WT mice and 145 units from 5 KO mice, which were clustered into wide-spiking (WS), putative excitatory units and narrow-spiking (NS), putative interneurons (Figures 4B and 4C). Firing rates of WS neurons had similar distributions between the WT and KO groups (Figures 4D, S5G, and S5H), but NS units from KO mice fired at a significantly higher rate (and consequently with lower inter-spike intervals), compared to the WT group, during both locomotion and immobility states (Figures 4E, S5G, and S5H). No differences between the two groups were observed in spiking variability or burst index in either unit type during either state (Figures S5G and S5H), suggesting unaltered intrinsic spiking characteristics in units of KO animals.
Since a decrease in synapse number could affect the coordinated synaptic activity that is thought to shape the LFP signal, particularly at low frequencies (Buzsáki et al., 2012), we first tested whether the power of low-frequency oscillations was altered in KO mice. We found no significant differences in the average power of the LFP between KO and WT mice in delta (1–4 Hz) or theta (5–11 Hz) oscillations, or even higher frequencies (beta 12–30 Hz, slow gamma 30–55 Hz, or high gamma 80–110 Hz), during either locomotion or immobility (Figure 4F), suggesting no major alterations in mPFC oscillatory activity on a broad neuronal population level.
Different neuronal populations are typically recruited to fire selectively at specific phases of ongoing oscillations, creating a dynamic circuit pattern (Klausberger and Somogyi, 2008). To assess how the observed synaptic alterations in KO mice reflect on the spiking modulation of individual units during LFP oscillations, we examined the preferred firing phase of each unit and its phase-locking strength to that phase, focusing again on delta and theta LFP oscillations during locomotion (Figures 5A and 5B) and immobility separately. We found a significant decrease in the strength of phase locking of WS units to delta oscillations and NS units to delta and theta oscillations in KO animals during locomotion, combined with significant shifts in both unit types to later phases in the respective oscillatory cycles (Figure 5C). Extending this analysis over the faster LFP oscillation rhythms mentioned above yielded fewer units that were significantly locked to such frequencies, particularly in KO animals (Figure S6). Notably, we found a decrease in phase locking to beta oscillations in NS units of KO mice, but over a small sample of phase-locked units.
Figure 5. Altered Phase Locking to Low-Frequency LFP Oscillations in In Vivo-Recorded Single Units of Cntnap2 KO Mice.

(A) Left: LFP delta frequency phase histograms of spikes (% over all spikes) of two WS units. Two cycles of the oscillation are shown together with a representation of the LFP oscillation for clarity. Solid lines indicate the mean (preferred) phase of each unit. Right: Same as the left, but for two example NS units.
(B) Similar to (A) for theta frequency phase distributions.
(C) Distributions and corresponding histograms of phase-locking strength (mean vector length) versus preferred phases of pooled WS units (left) or NS units (right) from WT and KO mice to delta (top) and theta (bottom) LFP oscillations during locomotion. Only significantly phase-locked units with sufficient spiking are included methods(see Method Details). Filled rectangles and solid lines depict the mean ± SE of the distributions and histograms, respectively (circular mean for phase distributions), for the two mouse groups. All of the distributions were significantly non-uniform (p < 0.05; Rayleigh test for non-uniformity; Bonferroni corrected over mouse groups), except for the theta frequency distribution of KO units (indicated by open rectangle and dashed lines accordingly). Asterisks indicate significant differences either in mean vector length (p < 0.05;t test if both distributions are normally distributed according to the Lilliefors test of normality with 5% significance level, or Wilcoxon test otherwise) or in mean preferred phase accordingly (p < 0.05; parametric Watson-Williams test).
(D) Same as (C) for immobility segments.
(E and F) Correlation coefficient distributions between pairs of wide-spiking units (left), narrow-spiking units (right), and wide-narrow spiking unit pairs (bottom), over all corresponding WT (blue) and KO (red) unit pairs in each mouse. Firing rates were computed during locomotion (E)or immobility segments (F) over500-ms-long time bins. Lines indicate the means of corresponding distributions. *p < 0.05, Wilcoxon test, Bonferroni corrected over the three unit-type combinations.
All errors bars indicate the SEM.
The observed reduction in phase locking in KO animals extended during immobility (Figure 5D), with significant reductions observed mainly for NS units in theta and gamma frequency ranges (Figure S6). During theta oscillations, we found a significant shift to later phases in WS units (Figure 5D). Finally, the number of significantly phase-locked units per animal to each frequency was on average comparable in both WT and KO groups (Table S2).
Decreased phase locking of individual units to LFP oscillations suggests a reduction in coordinated population activity during both motion and immobility in Cntnap2 KO animals. To test this, we compared correlations between the firing rates of all pairs of units in WT versus KO mice, during either locomotion or immobility, separately for WS-WS, NS-NS, and WS-NS pairs in each mouse (Figures 5E and 5F). Only units with adequate spiking (>200 spikes in each condition) were considered. Correlations between WS units exhibited a small but significant reduction in KO mice during locomotion and immobility. NS units exhibited no significant difference in locomotion, but they were significantly more correlated in KO animals during immobility, leading to increased WS-NS correlations as well. This finding was not affected by the firing rate binning since it was reproduced with firing rate time bins spanning from 500 (2 Hz, delta frequency; Figures 5E and 5F) down to 25 ms (40 Hz, slow gamma; Figure S7). Again, comparable numbers of units from each mouse group were included in each case (Table S2). Therefore, despite their reduction in phase locking to particular LFP oscillations, NS unit pairs remained more strongly correlated in immobile KO animals, despite a prominent desynchronization of WS units in each condition.
These results indicate a disrupted mPFC network in Cntnap2 KO mice, in which both excitatory and inhibitory neurons have less precise firing patterns that are also shifted relative to network activity and yield less coherent network dynamics. These alterations may lead to severely altered mPFC processing in Cntnap2 KO mice, potentially contributing to altered brain function and the previously described behavioral deficits observed in these mice.
DISCUSSION
Here, we find that the loss of Cntnap2, which causes a syndromic form of autism in humans, leads to reduced synaptic inputs onto L2/3 pyramidal neurons in the mPFC. LSPS mapping revealed a dramatic reduction of both excitatory and inhibitory inputs in this region, and mEPSCs occurred at a lower frequency in neurons. These findings suggest a decrease in the total number of excitatory synapses, which was confirmed with confocal microscopy as a decrease in spine density and the decrease in both excitatory and inhibitory synapses seen with electron microscopy. In vivo, these changes were associated with decreased phase-locking strength and shifted phase preference of putative excitatory neurons to delta oscillations and of inhibitory neurons to delta and theta oscillations during locomotion. We conclude that the loss of CNTNAP2 has a profound impact on synaptic connectivity and population dynamics of excitatory and inhibitory neurons in the mPFC.
The observed reduction in functional synaptic connectivity and in the density of synapses in the mPFC of Cntnap2 KO animals is consistent with recent studies showing reduced local and long-range functional connectivity in the prefrontal cortex of these mice (Liska et al., 2018), as well as work in somatosensory cortex, showing reductions in both excitatory and inhibitory input (Antoine et al., 2019). Moreover, work in dissociated neuronal cultures showed decreased mEPSC and mIPSC frequency after RNAi knockdown of CNTNAP2, which was linked to decreased dendritic arbor complexity and decreased spine head size (Anderson et al., 2012). While in general agreement with our findings, we found no changes in dendritic arborizations in the intact mPFC, despite a clear decrease in spine density. In addition, we found no significant changes in spine size or synapse length as assessed by electron microscopy in L2/3 of the mPFC. These inconsistencies between our findings and previous reports, specifically regarding dendritic morphology and spines, may arise from inherent differences between cultured neurons and in vivo preparations.
Our observed decrease in multisynapse boutons (MSBs) and increase in perforated postsynaptic densities (PSDs) further supports the notion that CNTNAP2 may have a complex role at the synaptic cleft. Since both MSBs and perforated PSDs are markers of well-developed synapses, this could also indicate that the loss of Cntnap2 disrupts synapse maturation dynamics via both pre- and postsynaptic mechanisms (Toni et al., 1999; Fiala et al., 2002; Nikonenko et al., 2002; Ganeshina et al., 2004; Geinisman, 1993). Accordingly, recent work in cultured cortical neurons from KO mice reported a decrease in spine density and in localization of the AMPA-subtype glutamate receptor GluA1 in the spines of Cntnap2 KO neurons (Varea et al., 2015), which is consistent with the small decrease in mEPSC amplitude that we observed. This is also concurrent with recent work, showing that Cntnap2 KO mice have reduced AMPA receptor expression and transmission in vivo (Kim et al., 2019). Our results are also in line with our previous work, reporting increased spine elimination and decreased spine density in apical dendrites of L5 neurons in the somatosensory cortex of Cntnap2 KO mice (Gdalyahu et al., 2015). Therefore, the effects of CNTNAP2 loss on spine density may generalize as decreased spine stability throughout the cortex.
Our observed lack of changes in intrinsic excitability of L2/3 pyramidal neurons or PV+ neurons appears surprising, given that Cntnap2 is important for potassium channel localization in axons (Poliak et al., 2003). Nonetheless, it is likely that Cntnap2 loss affects neurons in a cell type- and projection-specific manner, as supported by recent reports of decreased input resistance and intrinsic excitability of L5 subcortical projecting neurons of the mPFC (Brumback et al., 2018).
Such alterations in synaptic physiology and neurotransmission seem to be a common theme among mouse models of neurodevelopmental disorders. Loss-of-function mutations in Shank3, MECP2, and Ube3a (modeling Phelan-McDermid, Rett, and Angelman syndromes, respectively) result in decreased spine density and excitatory neurotransmission in the cortex (Dani et al., 2005; Belichenko et al., 2009; Wallace et al., 2012; Zhou et al., 2016). Moreover, spine maturation is impaired in fragile X model mice (Cruz-Martín et al., 2010), similar to what we find in Cntnap2 KO, and cortical inhibitory neurotransmission is similarly compromised in a number of these disorders (Gibson et al., 2008; Curia et al., 2009; Cea-Del Rio and Huntsman, 2014; Banerjee et al., 2016). This posits the notion that increasing or modulating excitatory and inhibitory synaptic connectivity, especially in a cell type- and projection-specific manner, may be therapeutically relevant.
Concurrently, we find that the loss of excitatory and inhibitory synaptic connectivity in Cntnap2 KO mice is associated with a decrease in the magnitude of phase-locked firing of inhibitory and excitatory neurons to delta oscillations in vivo. Inhibitory neurons were less phase locked to both theta oscillations, and they tended to fire later in the oscillatory cycle. These findings were more prominent during locomotion, suggesting that the effects from changes in connectivity can be more prominent during specific conditions or arousal states. Dysfunctional oscillations have often been reported in humans diagnosed with ASD and have been proposed as biomarkers (Rojas and Wilson, 2014; Simon and Wallace, 2016; Sidorov et al., 2017). Specifically, delta (4 Hz) oscillations in mPFC can entrain other brain regions, such as the amygdala during fear expression and the ventral tegmental area and hippocampus during working memory (Fujisawa and Buzsáki, 2011). Theta (4–8 Hz) oscillations in the mPFC have been associated with signaling safety under conditions of learned fear (Likhtik et al., 2014). Thus, the phase-locking alterations observed in mPFC neurons of Cntnap2 KO mice could be linked to some of the cognitive and affective behavioral disruptions displayed by this mouse model.
The mPFC electrophysiological alterations we observed in Cntnap2 KO mice could underlie some of the autism-related phenotypes in the model, such as deficits in social interactions and communication, as supported by observations that increasing the ratio of excitation to inhibition in the mPFC could disrupt social interactions in WT mice (Yizhar et al., 2011). Moreover, an opsin-mediated increase in PV+ cell excitability or a decrease in pyramidal neuron activity within the prelimbic mPFC can rescue social behavior and hyperactivity in Cntnap2 KO mice (Selimbeyoglu et al., 2017). Such disruptions in E/I balance could also reflect as broader-scale alterations in oscillatory power and synchrony and could be mechanistically linked to the altered representation of social stimuli in the mPFC of Cntnap2 KO mice (Levy et al., 2018).
Future studies need to dissect the inputs and outputs of the prefrontal cortex in a cell type- and projection-specific manner to uncover whether changes in excitatory and inhibitory connectivity are generalized or selectively impaired in specific circuits. This will require experiments in which Cntnap2 is conditionally deleted in specific cell types using Cre-Lox techniques. Also, it is not known whether the synaptic and population dynamic changes we found can be reversed or ameliorated by restoring Cntnap2 gene expression in adulthood or whether very early interventions will be needed. Finally, it will be important to understand how the delta and theta phase locking affects the recruitment of other connected brain regions, especially in the context of social engagement.
STAR⋆METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peyman Golshani (pgolshani@mednet.ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cntnap2 null mice were obtained from E. Peles and backcrossed to the C57BL/6J background for over 12 generations. For targeted electrophysiological recordings of parvalbumin-positive interneurons, Cntnap2 heterozygous mice were backcrossed to PV-Cre (Jackson labs number 008069) × Ai9 (Jackson labs number 007909) mice. For spine density analysis, Cntnap2 heterozygous mice were backcrossed with Thy1GFP (Jackson labs × 007788) mice. Experimental mice were obtained from heterozygous crossings and born with the expected Mendelian frequencies; both genders were used. The date of birth was designated at P0 and the three obtained genotypes (wild-type, heterozygous, homozygous knock-out) were housed together with three to four mice per same-sex cage. Mice were kept in a 12-hour light/12-hour dark cycle and had ad libitum access to food and water. All procedures involving animals were performed in accordance with the University of California, Los Angeles (UCLA) animal research committee, and the National Institutes of Health Guide for the Use and Care of Laboratory Animals.
METHOD DETAILS
Slice preparation
Acute coronal slices (300 μm thickness) containing the medial prefrontal cortex were prepared from 4 to 6-wk-old Cntnap2 knock-out mice and wild-type littermates. Mice were anaesthetized with isoflurane gas and beheaded after disappearance of toe-pinch reflex. The brain was removed and placed in ice-cold cutting solution consisting of (mM): 222 sucrose, 11 D-glucose, 26 NaHCO3, 1 NaH2PO4, 3 KCl, 7 MgCl2, 0.5 CaCl2, aerated with 95% O2, 5% CO2. The brain was cut in a Leica VT1000S Vibratome. Slices were allowed to recover for 30 minutes at 37°C in standard artificial cerebrospinal fluid (ACSF, in mM): 124 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4,10 D-glucose, 4 sucrose, 2.5 CaCl2,2 MgCl2, aerated with 95% O2,5%CO2, and kept at room temperature for at least 40 min until time of recording.
Electrophysiology
Whole-cell patch-clamp recordings of L2/3 neurons were obtained under visual guidance using infrared DIC video-microscopy and water-immersion 40x objective, with patch pipettes (3–5 MOhms) pulled from borosilicate capillary glass (Sutter) with a Sutter puller. TdTomato-expressing parvalbumin-positive inhibitory neurons were targeted under epifluorescence. All electrophysiological recordings were performed using Multiclamp 700B (Molecular Devices) patch clamp amplifiers and ACSF was maintained at 33–35°C. Signals were filtered at 4 kHz using Bessel filter and digitized at 10 kHz with WinWCP and WinEDR electrophysiology software interface for voltage-clamp recordings (Strathclyde). Current clamp recordings were digitized at 15 and Bessel filtered at 6 kHz. Series/access resistance was monitored in all recordings and compensated in current clamp mode. Recordings were discarded if series resistance changed significantly (> 20%) or exceeded 25 MOhms. Junction potential was not compensated.
Current-clamp recordings
For intrinsic excitability experiments, the internal pipette solution contained (in mM): 115 KGluc, 20 KCl, 10 HEPES, 10 phosphocreatine, 4 ATP–Mg2+, 0.3 GTP–Na+ (pH 7.2, 270–290 mOsm); in some recordings, 0.2% biocytin was added to the solution. Patched pyramidal excitatory neurons were identified and included in the analysis based on their action potential firing characteristics. Resting membrane potential (Vm) was measured after breaking into the cell (rupturing the patch) and applying zero current. Input resistance (Rin) was calculated as the slope of the linear fit of the voltage-current plot, generated from a family of negative and positive 500 ms current injections (−60 pA to +60 pA at 20 pA intervals, for pyramidal cells; −150 pA to +150 pAat 50 pA intervals, for parvalbumin-positive interneurons). The membrane decay constant (t) was calculated by fitting a single exponential curve to the current-voltage plot that resulted from a −20 pA current injection. Cell membrane capacitance (Cm) was given by Cm = τ/Rin. For assessment of intrinsic excitability, cells were clamped at −70 mV and injected a series of increasing current steps at 50 pA intervals. Action potential properties were determined from the first action potential elicited by minimum current injection. The spike adaptation ratio was calculated by dividing the last inter-spike interval to the first inter-spike interval in an action potential train elicited by a 500 ms pulse of 200 pA. All data was analyzed using custom-written MATLAB software. Unless specified otherwise, sample size n was defined as cell number and all statistical tests were performed based on the number of cells.
Voltage-clamp recordings
Miniature excitatory postsynaptic currents (mEPSCs) were isolated by applying (in mM): 0.5 tetrodotoxin (TTX) and 10 pictrotoxin to ACSF (described above). Pipette internal solution contained (in mM): 20 KCl, 10 Na-phosphocreatinine, 100 cesium methyl sulfonate, 3 QX-314, 10 HEPES, 4 ATP-Mg2+ and 0.3 GTP-Na+ (pH 7.2, 270–290 mOsm). Recordings were performed with cells clamped at −70 mV. Miniature inhibitory postsynaptic currents (mIPSCs) were isolated by applying (in mM): 0.5 tetrodotoxin (TTX), 10 CNQX, and 50 APV to ACSF. A high-chloride pipette internal solution was used, which contained (in mM): 120 KCl, 10 HEPES, 4 ATP- Mg2+, 0.3 GTP-Na+ and 10 Na-phosphocreatinine (pH 7.2, 270–290 mOsm). Recordings were performed with cells clamped at −50 mV. Miniature and spontaneous events were recorded for 2 min. MiniAnalysis software (Synaptosoft) was used to automatically identify synaptic events, based on template parameters. Events were then manually examined to exclude false positives. For voltage clamp recordings with a cesium-containing electrode, pyramidal cells were targeted based on soma shape and identity was manually verified based on EPSC decay, where cells with mean ESPC decay time constant ≤ 2 ms were considered to likely inhibitory and excluded. Events were excluded if the 10%−90% rise time was > 2 ms, as these events were likely recorded from synapses far from the soma and with poor space clamp. Inter-event intervals (event frequency), amplitude, decay time constant, area, 10%−90% rise time, and half-width, were analyzed and comparisons between groups were analyzed by Student’s t test. Grouped data are expressed as mean ± SEM. Unless specified otherwise, sample size n was defined as cell number and all statistical tests were performed based on the number of cells.
Evoked Excitatory Postsynaptic Currents
A tungsten concentric bipolar stimulating electrode (WPI) was placed in the white matter to stimulate axon fibers emerging from the anterior forceps of the corpus callosum, which project onto a whole-cell recorded L2/3 pyramidal neuron in PL-mPFC, voltage-clamped at −70 mV. Input-output curves were derived by increasing the stimulus duration (0.1 ms increments) and recording current responses in the recorded postsynaptic neurons. Short-term plasticity was assessed by measuring paired-pulse ratios, calculated as the peak amplitudes of 10 averaged episodes at various inter-stimulus intervals (25, 50, 100, 500ms). AMPA/NMDA ratios were measured by voltage-clamping the cells at a holding potential of −70 mV for AMPA currents and +40 mV for NMDA currents. Peak amplitude current responses were averaged over 10 episodes. Peak NMDA currents were measured after the offset of AMPA currents (25–30 ms post-stimulus) within the same cell. Data was analyzed manually using WinEDR software and plotted in MATLAB. Unless specified otherwise, sample size n was defined as cell number and all statistical tests were performed based on the number of cells.
Laser Scanning Photostimulation (LSPS)
Coronal sections of medial prefrontal cortex were cut 400 μm thick with a vibratome (VT1200S, Leica Systems) in sucrose-containing artificial cerebrospinal fluid (ACSF) (in mM: 85 NaCl, 75 sucrose, 2.5 KCl, 25 glucose, 1.25 NaH2PO4, 4 MgCl2, 0.5 CaCl2, and 24 NaHCO3). Slices were first incubated in sucrose-containing ACSF for 30 min to 1 h at 32°C, and then transferred to recording ACSF (in mM: 126 NaCI, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2,1.25 NaH2PO4, and 10 glucose) at room temperature. Throughout incubation and recording, the slices were continuously bubbled with 95% O2%−5% CO2.
The design of our laser scanning photostimulation system has been described previously(Xu et al., 2010). A laser unit (model 3501, DPSS Lasers, Santa Clara, CA) was used to generate a 355 nm UV laser for glutamate uncaging. Various laser stimulation positions were achieved through galvanometer-driven X-Y scanning mirrors (Cambridge Technology, Cambridge, MA), as the mirrors and the back aperture of the objective were in conjugate planes, thereby translating mirror positions into different scanning locations at the objective lens focal plane. Data were acquired with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA), data acquisition boards (models PCI MIO16E-4 and 6713, National Instruments, Austin, TX), and custom-modified version of Ephus software (Ephus, available at https://www.ephus.org/). Data were low-pass filtered at 2 kHz using a Bessel filter, digitized at 10 kHz, and stored on a computer.
Cortical slices were visualized with an upright microscope (BW51X, Olympus) with infrared differential interference contrast optics. Electrophysiological recordings, photostimulation, and imaging of the slice preparations were done in a slice perfusion chamber mounted on a motorized stage of the microscope at room temperature. An aliquot of MNI-caged-L-glutamate (4-methoxy-7-nitroindolinyl-caged L-glutamate, Tocris Bioscience, Ellisville, MO) was added to 20–25 mL of circulating ACSF for a concentration of 0.2 mM caged glutamate. To perform whole cell recording, cells were visualized at high magnification (60 × objective, 0.9 NA; LUMPlanFl/IR, Olympus). Excitatory neurons were selected based upon their pyramidal somata detected under differential interference contrast (DIC) microscopy. For experiments to assess photo-stimulation evoked spiking profiles of excitatory in mPFC (similar to our published studies(Shi et al., 2010; Xu et al., 2010)), the patch pipettes (4–6 MΩ resistance) were filled with an K+ internal solution containing (in mM) 126 K-gluconate, 4 KCl, 10 HEPES, 4 ATP-Mg, 0.3 GTP-Na, and 10 phosphocreatine (pH 7.2, 300 mOsm). For the photostimulation experiments to map synaptic inputs, we used a Cs+ internal solution containing (in mM) 6 CsCl, 130 CsOH, 130 D-Gluconic acid, 2 MgCl2, 0.2 EGTA, 10 HEPES, 2.5 ATP-Na, 0.5 GTP-Na, and 10 phosphocreatine-Na2 (pH 7.2, 300 mOsm). Because glutamate uncaging agnostically activates both excitatory and inhibitory neurons, we empirically determined the excitatory and inhibitory reversal potentials in L2/3 pyramidal cells to properly isolate EPSCs and IPSCs. Whole-cell voltage-clamp recordings were made from the recorded postsynaptic neurons with LSPS-evoked EPSCs and IPSCs measured at the holding potential of −70 mV and +5 mV, respectively, across photostimulation sites. The internal solution also contained 0.1% biocytin for cell labeling and morphological identification. The morphology of recorded pyramidal neuron was determined using post hoc staining with Cy3-conjugated streptavidin (1:500 dilution; Jackson ImmunoResearch). Once stable whole cell recordings were achieved with good access resistance (usually < 30 MΩ), the microscope objective was switched from 60 × to 4 ×; laser scanning photostimulation (LSPS) was performed through the 4× objective lens. At low magnification (4 × objective lens, 0.16 NA; UplanApo, Olympus), the slice images were acquired by a high-resolution digital CCD camera (Retiga 2000, Q-imaging, Austin, TX) and used for guiding and registering photostimulation sites in cortical slices.
Photostimulation (1.5 ms duration, 15 mW pulses) from a 350 nm UV laser generator (DPSS Lasers, Santa Clara, CA) was delivered to the sample, controlled via an electro-optical modulator and a mechanical shutter. Focal laser spots approximated a Gaussian profile with a diameter of ~50–100 μm. Under our experimental conditions, LSPS evoked action potentials were recorded from stimulation locations within 100 μm of targeted somata of excitatory neurons and occurred within 150 ms post photostimulation. Our calibration analysis indicates that LSPS allows for mapping direct synaptic inputs to recorded neurons. Synaptic currents in patched neurons were detected under voltage clamp. By systematically surveying synaptic inputs from hundreds of different sites across a large cortical region, aggregate synaptic input maps were generated for individual neurons. For our mapping experiments, a standard stimulus grid (16 × 16 stimulation sites, 100 × 60 μm2 spacing) was used to tessellate mPFC from pia to white matter. The LSPS site spacing was empirically determined to capture the smallest predicted distance in which photostimulation differentially activates adjacent neurons. Glutamate uncaging was delivered sequentially in a nonraster, nonrandom sequence, following a “shifting-X” pattern designed to avoid revisiting the vicinity of recently stimulated sites.
Photostimulation induces two forms of excitatory responses: (1) those that result from direct activation of the recorded neuron’s glutamate receptors, and (2) synaptically mediated responses (EPSCs) resulting from the suprathreshold activation of presynaptic excitatory neurons. Responses that occur within 10 ms of laser pulse onset were considered direct; these responses exhibited a distinct waveform and occurred immediately after glutamate uncaging. Synaptic currents with such short latencies are not possible because they would have to occur before the generation of action potentials in photostimulated neurons. Therefore, direct responses were excluded from local synaptic input analysis, but they were used to assess glutamate mediated excitability/responsiveness of recorded neurons. At some locations, synaptic responses were overriding on the relatively small direct responses, and these responses were identified and included in synaptic input analysis. The IPSC input was similarly analyzed as the EPSC input. For data map analysis, we implemented the approach for detection and extraction of photostimulation-evoked postsynaptic current responses as previously described(Shi et al., 2010). LSPS evoked EPSCs/IPSCs were quantified across the 16×16 mapping grid for each cell, and 1–2 individual maps were used per recorded cell. The PSC input from each stimulation site was the measurement of the sum of individual PSCs within the analysis window (> 10 ms to 160 ms post photostimulation), with the baseline spontaneous response subtracted from the photostimulation response of the same site. The value was normalized with the duration of the analysis window (i.e., 150 ms) and expressed as average integrated amplitudes in picoamperes (pA). The analysis window was chosen because photostimulated neurons fire most of their action potentials during this time. For the color-coded map display, data were plotted as the average integrated PSCs amplitude per pixel location (stimulation site), with the color scale coding input strength. For the group maps obtained across multiple cells, the individual cell maps were first aligned by their slice images using laminar cytoarchitectonic landmarks. Then a new map grid was created to re-sample and average input strength at each site location across cell maps; a smooth version of color-coded map was presented for overall assessments. To further quantitatively compare input strength across cell groups, we measured the total PSC inputs (total synaptic currents) across all map sites (total synaptic input strength) for individual cells. The total EPSC/IPSC input strength ratios were also measured for the cells when both EPSC and IPSC data were available from the same cells.
As virtually all Layer 1 neurons are inhibitory cells, and pyramidal neurons with apical dendritic tufts in layer 1 could fire action potentials when their tufts were stimulated in layer 1(Dantzker and Callaway, 2000), EPSCs detected after photostimulation in layer 1 were not included for analyses. However, because layer 1 neurons can provide inhibition to layer 2/3 neurons, we did analyze IPSCs detected after photostimulation in layer 1. All data are reported as mean ± standard error of the mean (SEM). When comparing two independent groups, a Wilcoxon rank sum test was used. Unless specified otherwise, sample size n was defined as cell number. A P value (≤0.05) was considered statistically significant.
Immunohistochemistry
For assessment of dendritic morphology and complexity, cells were filled during electrophysiological recordings via passive diffusion of internal pipette solution containing 0.2% biocytin. After recording for at least 10 min, slices were transferred to a 4% PFA solution for overnight fixation, washed for 10 min (×3) in 0.1 M phosphate buffered saline (PBS), blocked with 10% normal goat serum (NGS) containing 0.3% Triton-X in 0.1 M PBS for 1.5 hr, and incubated overnight with an Alexa 555 or Alexa 488-conjugated Streptavidin antibody (1:500, Invitrogen) in 0.1M PBS. Sections were finally washed 3× 10 min in 0.1M PBS and mounted on slides using DAPI Fluoromount-G (Invitrogen) for visualization. We assessed dendritic complexity of biocytin-filled cells by imaging at 20X magnification in an LSM 520 confocal microscope. Z stacks of optical sections (1 um) were compiled and images were processed in Neurolucida 10 (MFB Biosciences) for Sholl analysis.
For quantification of spine density, Cntnap2 WT and KO mice were crossed with a Thy1-GFP mouse line, which sparsely labels pyramidal neurons, including their dendritic projections and spines. Mice were perfused intracardially with 25mL0.1 M PBS, followed by 25 mL of 4% PFA in 0.1 M PBS (at 2 mL/min). The brains were dissected and fixed for at least 24 hr in the same solution. Brains were then sectioned at a thickness of 100 um, using a Leica vibratome. Sections containing the mPFC were mounted in slides using DAPI Fluoromount-G media. Apical and basal dendrites of GFP-expressing L2/3 mPFC neurons were imaged at high resolution using a 63X oil magnification objective on an LSM 520 confocal microscope (Zeiss). Optical sections of 0.32 um were acquired and maximum intensity projections of dendritic arbors were created in ImageJ (NIH). Dendritic segments were chosen using consistent criteria and spines were manually counted. Dendritic spine density was calculated by dividing the total number of spines over a given length of dendrite (spines/μm). Student’s t test was performed for statistical comparison between WT and KO mice.
For quantification of the density of parvalbumin-positive neurons in prelimbic cortex, wild-type and CNTNAP2 knockout mice were deeply anesthetized with 4% isoflurane and intracardially perfused with 4% paraformaldehyde 0.1M phosphate-buffered saline (freshly diluted from 32% stock, Electron Microscopy Sciences). Brains were subsequently removed and incubated in 0.1M phosphate-buffered solution containing 30% sucrose at 4°C for up to 2 days. Brains were then embedded in optimal cutting temperature solution (TissueTech) at −80°C, and cryosectioned at 50 μm thickness. Sections containing the prelimbic cortex were selected for immunostaining with mouse monoclonal anti-parvalbumin antibody (1:200, Sigma, P3088) and goat anti-mouse Alexa 488 secondary antibody (1:500). Confocal images were obtained at 10× magnification using a Zeiss 880 laser-scanning confocal microscope and analyzed using ImageJ (NIH). To establish counts, outlines over the prelimbic cortex were drawn with references to The Allen Mouse Brain Atlas (Allen Institute, http://mouse.brain-map.org/) then the number of parvalbumin-positive cells was quantified by hand by a blinded experimenter. Counts for each section were normalized to the size of selected area. Statistical comparisons were performed using Student’s t test with Prism 7 (Graphpad), where p values less than 0.05 were considered to be statistically significant.
Cell density measurements
The density of neurons in the prelimbic medial prefrontal cortex was quantified at the light microscopic level (Leica DM750) in toluidine blue-stained semithin sections (~300nm thin). Unbiased counting frames of known area (40.000 μm2) were superimposed on fields of these sections within the Layers 2/3 pyramidal cell layer using random sampling. The counting units were neuronal nuclei, and they were counted only if these did not contact the two exclusion lines of the counting frame. Within the frame, neuronal nuclei were counted. Cells with obvious glial characteristics were excluded from the analysis. Four sections were quantified per block in order to determine neuronal density in each animal. Our aim was not to make stereologically-rigorous estimates of the absolute values; instead we wanted to determine whether there are significant differences in neuronal density between WT and the CNTNAP2 KO animals.
Tissue preparation and electron microscopy
Animals from KO and WT groups (n = 3, respectively) were processed. Mice were deeply anesthetized with isoflurane were perfused transcardially with a mixture of 2% paraformaldehyde (PFA) and 2% glutaraldehyde in 0.1 M phosphate buffer (PB, pH 7.4). Brains were removed and post-fixed overnight at 4°C. 70 μm thick sections were cut with a Leica vibratome. Free-floating sections for electron microscopy were post-fixed with 1% OsO4, dehydrated in ascending ethanol series and embedded in epoxy resin (Durcupan; Sigma, Germany) within Aclar sheets (EMS, Hatfield, PA, USA). Uniform rectangular samples were cut from the prelimbic medial prefrontal cortices (mPFC, approx 1.98–1.94 AP, 3–3.75 DV, 0.25– 0.50 ML position) under a Leica S6E dissecting microscope, and mounted on plastic blocks. 60 nm ultrathin sections were cut on a Reichert ultramicrotome, mounted on 300 mesh copper grids, contrasted with lead citrate (Ultrostain II, Leica) and examined with a JEM-1011 transmission electron microscope (JEOL, Tokyo, Japan) equipped with a Mega-View-III digital camera and a Soft Imaging System (SIS, Münster, Germany) for the acquisition of the electron micrographs. Five to ten sections were analyzed per block, and two blocks per animal were used to collect micrographs. Sample areas (at least 50 μm2 per animal) were chosen in a pseudo-random fashion and photographed at a uniform magnification. Postsynaptic dendritic spines, axonal boutons, multi-synaptic boutons (MSB; a single presynaptic bouton that forms separate synapses with multiple spine heads) were identified on electron micrographs. Spine profile area were measured using the engine provided by NIH ImageJ v1.51j8 (Schneider et al., 2012); data were compiled using Excel (Microsoft) and Kaleidagraph (Synergy Software, Reading, PA, USA) software. The means and the effects of the loss of the Cntnap2 gene was determined by Wilcoxon rank sum test, with a p < 0.05 considered statistically significant. Data collection and quantification was performed blindly, to eliminate bias. We performed electron microscopy using random sampling from single sections to optimize sample size and to detect changes in synaptic features associated with loss of Cntnap2 in KO mice, compared to WT.
Surgery, behavioral habituation, and in vivo electrophysiology
Adult male and female Cntnap2 mutant and wild-type mice (2–5 months old) underwent an initial surgery for implantation of a stainless steel head restraint bar on their skull in preparation for in vivo electrophysiological recordings. All surgical procedures were performed under isoflurane anesthesia (3%–5% induction, 1.5% maintenance) in a stereotaxic apparatus. Mouse body temperature was monitored and kept at 37°C during surgery using a Harvard Apparatus feedback-controlled heating pad and were administered an subcutaneous injection of carprofen (5 mg/kg of body weight) for systemic analgesia. Mice were allowed to recover for 5 days, during which they were given antibiotic treatment (amoxicillin, 0.25 mg/mL in drinking water). After the recovery period, mice were habituated for at least three days for each of the following stages: human handling (5 min), headbar attachment (10 min), and head fixation on a spherical treadmill (10 min). The treadmill consisted of an 8-inch Styrofoam ball (Graham Sweet), tethered with a metal rod through the middle allowing only one axis of rotation. Air was blown, allowing the ball to float and the mouse to spin the ball and run in place and on top of it (Polack et al., 2013). After habituation, and one day prior to electrophysiological recordings, the mouse received a craniotomy above the medial prefrontal cortex on the right hemisphere (anterior 1.8 mm, lateral 0.5 mm to Bregma). The dura above the exposed brain area was carefully removed in order to facilitate electrode insertion. The exposed skull and brain were covered and sealed with a silicone elastomer sealant (Kwik-Sil, WPI). An additional craniotomy was performed over the posterior cerebellum for placement of a silver chloride electrical reference wire, which was glued into place with dental cement. The mouse was allowed to recover overnight. Mice were given a dose of carprofen on day of recording, to ameliorate any pain associated with the craniotomy surgery.
On the day of the recording, the mouse was head-fixed atop the spherical treadmill, the Kwik-Sil was removed and cortex buffer (135 mM NaCl, 5 mM KCl, 5 mM HEPES, 1.8 mM CaCl2 and 1 mM MgCl2) was immediately placed on top of the craniotomy in order to keep the exposed brain moist. The mouse skull was then stereotaxically aligned and the silicon microprobe coated with a fluorescent dye (DiI, Invitrogen), was stereotaxically lowered using a micromanipulator into the mPFC (relative to bregma: anterior 1.8 mm, lateral 0.5 mm, ventral 2.5 mm). This process was monitored using a surgical microscope (Zeiss STEMI 2000). The microprobes contained a total of 128 electrode recording sites that were densely distributed (hexagonal array geometry with 25 mm vertical spacing and 16–20 mm horizontal spacing) on two prongs (placed 0.4 mm apart), spanning L2/3 and L5 of the prelimbic (PL) and infralimbic (IL) medial prefrontal cortex. Only data from L2/3 prelimbic cortex was used. Once inserted, the probe was allowed to settle among the brain tissue for 1 hr. Recording of brain network activity was done for a total duration of 1 hr after that.
Data acquisition was performed using custom fabricated silicon probes and recorded with LabView Software (Du et al., 2011). Readout was achieved via a custom-built 128-channel detachable head stage module. Head stages contained commercial integrated electronic circuits (Intan Technologies RHA-2164B)(Harrison and Charles, 2003) providing signal multiplexing (32 electrodes per multiplexed output wire), amplification (gain 200), and filtering (0.1– 6500 Hz) functions. The head stage contained two 64-pin connectors (Molex, Slimstack 502426–6410) connecting to custom printed circuit boards wire bonded to the silicon microprobes. Analog signals were transmitted through thin flexible cables and subsequently digitized on 16-bit analog-to-digital conversion (ADC) cards (USB-6356, National Instruments). Multiplexed signals were recorded at 800 kHz and de-multiplexed with recording software into a sampling rate of 25 kHz per channel. All ADC cards were synchronized via a shared internal clock. All data acquisition, as well as control of stimulus timing, was performed with custom LabVIEW scripts. All data analysis was carried out with custom MATLAB scripts (Shobe et al., 2015).
After the recording session, mice were anaesthetized with isoflurane and sacrificed. The brain was extracted, sectioned (100 mm) on a Vibratome (Leica) and mounted on slides with DAPI Fluoromount-G (SouthernBiotech) mounting media. Confocal tiled images were taken to verify microprobe location (Zeiss LSM 800). Anatomical landmarks were used to determine anterior-posterior coordinates relative to bregma. Each of the 128 recording sites was then assigned an approximate coordinate in 3D Cartesian space and classified as belonging to prelimbic (PL) or infralimbic (IL) prefrontal cortex (Allen Brain Atlas).
Motion detection
Mouse treadmill rotation was recorded as an analog signal, using a custom printed circuit board based on a high sensitivity gaming mouse sensor (Avago ADNS-9500) connected to a microcontroller (Atmel Atmega328). The signal was initially recorded along with electrophysiology at 25 kHz, then down-sampled to 1 kHz, its sample mode value was subtracted, it was turned to absolute values and was smoothed by lowpass filtering < 1 Hz with a first order Butterworth filter. For one set of animals (n = 6) motion was detected when the smoothed treadmill motion-signal exceeded 0.8 × mean of recording and immobility was assumed when it dropped below 0.005 (a.u). For a second set (n = 7) these thresholds were changed to 2 × mean and 0.016 respectively due to increased recording noise. Motion segments shorter than 0.5 s long were discarded, and consecutive segments closer than 0.5 s were concatenated. This processed signal was treated as a proxy of velocity. Distance traveled on the ball per motion segment (motion bout) was approximated as the velocity integral over each segment.
QUANTIFICATION AND STATISTICAL ANALYSIS
Local Field Potential Analysis
To compare LFP bandpower, LFP recordings from the channel located closest to L2/3 were selected for each animal. LFPs were down-sampled to 1 kHz and all data points corresponding to either motion or immobility segments were concatenated. Bandpower over all frequency ranges (delta: 1–4 Hz, theta: 5–8 Hz, beta: 12–30 Hz, slow gamma: 30–55 Hz, fast gamma: 80–110 Hz) was computed using a periodogram with a Hamming window.
In vivo Unit Clustering and Analysis
Single units were isolated using custom spike-detection scripts and PyClust software. Raw data was initially background subtracted, bandpass filtered from 600–6500 Hz, and grouped into channel sets of neighboring electrodes for clustering. For each channel set, putative spikes were detected as any deviation greater than 4 standard deviations from the mean. The features of each spike were calculated (peak amplitude, valley, and trough, principle components) and individual clusters were isolated by outlining boundaries on each projection. Each unit was visually inspected and units that drifted outside recorded channels or were lost during the recording were eliminated. For each clustered unit, all peri-spike extracellular waveforms were collected from the channel that yielded the largest spike amplitude and were bandpass filtered at 600 – 6000 Hz with a first order Butterworth filter. The amplitudes and time-points of each unit’s mean waveform peak and trough were computed, together with the unit’s mean firing rate and burst index (percentage of consecutive spikes closer than 20 ms). Waveforms from identified units were visually inspected and units with low-quality individual and average waveforms were not included in the analysis (n = 25 rejected units).
To cluster units into broad and narrow spiking, the (i) peak-trough time distance, (ii) peak-trough amplitude ratio and (iii) burst index of all units of all animals were pooled together and z-scored. Principal component analysis was performed on the three variables and the scores from all three principal components were split into two clusters using k-means with squared Euclidean distance measure and 100 clustering repeats. This method yielded two well separated clusters with one containing ~20% of all units with smaller peak-trough distances and ratios and higher burst indexes compared to the other cluster. Units in that cluster are referred to as ‘narrow-spiking’ whereas those in the opposite cluster as ‘wide-spiking’.
Motion-related firing rates and inter-spike intervals were computed by time-binning each motion bout separately. Their average value and SD were computed after concatenating over all motion segments. Fano-factors were computed as mean/SD of firing rates using 100 ms non-overlapping time bins. Burst indexes were computed by concatenating spike times during motion or immobility accordingly and computing, for each unit, the percentage of consecutive spikes closer than 20 ms. Zero-lag Pearson correlations between firing rates of WS, NS or WS-NS pairs of units were computed per animal. Units with less than 200 spikes were excluded from correlation analysis.
Spike phases of each unit were computed using the LFP recording of the channel yielding the largest spike amplitude for that unit. LFPs were down-sampled at 1 kHz and bandpass filtered over the corresponding oscillation frequency range using a Butterworth bandpass filter matching the passband exactly. The phase of each spike was computed as the angle of the filtered LFP’s Hilbert transform at the spike peak. For producing preferred phase distributions of wide or narrow spiking units, only units with > 200 spikes over all motion, or immobility segments accordingly, and with significant phase locking at the corresponding frequency range (Rayleigh test, p value < 0.05, Bonferroni corrected over all WS or NS units accordingly) were included. Unless specified otherwise, sample size n was defined as cell number and all statistical tests were performed based on the number of cells.
DATA AND SOFTWARE AVAILABILITY
All data generated in the paper will be available upon request from the Lead Contact, Peyman Golshani, pgolshani@mednet.ucla.edu. MATLAB code used for all in-vivo electrophysiology analysis have been deposited in Github at https://github.com/jtaxidis/Lazaro-et-al.-Cell-Reports-2019/tree/master.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa-555 conjugated Streptavidin | Invitrogen/ ThermoFisher | Cat# S21381; RRID: AB_2307336 |
| Alexa 488 conjugated Streptavidin | Invitrogen/ ThermoFisher | Cat# S11223; RRID: AB_2336881 |
| Anti-Parvalbumin Antibody | Sigma | Cat# P3088; RRID: AB_477329 |
| Experimental Models: Organisms/Strains | ||
| CNTNAP2 KO Mice | Elior Peles | N/A |
| PV-Cre Mice | JAX | 008069 |
| Ai9 Mice | JAX | 007909 |
| Software and Algorithms | ||
| Ephus | Janelia Research Campus | N/A |
| WinWCP, WinEDR | Strathclyde | N/A |
| MiniAnalysis | Synaptosoft | N/A |
| MATLAB code for analysis of phase-locking and correlations | Golshani Lab | https://github.com/jtaxidis/Lazaro-et-al.-Cell-Reports-2019/tree/master |
| Prism | Graphpad | Prism 7 |
| PyClust | Mayank Mehta Lab | N/A |
| Custom Silicon Probe Data Acquisition Software (Labview) | Sotiris Masmanidis Lab | N/A |
Highlights.
Synaptic inputs onto mPFC L2/3 pyramidal neurons are reduced in Cntnap2 KO mice
The frequency and amplitude of mEPSCs are reduced in the mPFC of Cntnap2 KO neurons
Decreased density of dendritic excitatory and inhibitory synapses in Cntnap2 KO mice
Phase-modulated spiking to slow LFP oscillations is altered in Cntnap2 KO units
ACKNOWLEDGMENTS
P.G., J.T., and M.T.L. were supported by NIH grants 1R01MH101198, U01 NS094286, R01 MH105427, and U54 HD87101. M.T.L. was funded by the UCLA Eugene Cota-Robles Fellowship, NSF-GRFP DGE-0707424, NIMH T32MH073526, a UCLA Neurobehavioral Genetics Training Grant, the UCLA Dissertation Year Fellowship, and NIH grant 5U01NS094286. T.S. was supported by a postdoctoral fellowship from the Epilepsy Foundation. S.M. was supported by NIH DA034178. D.H.G. was supported by NIH grants P50 HD 055784-12 and U01 MH115746-02 and the Simons Foundation Grant 401457. We thank Tünde Magyar and Renáta Pop for excellent technical assistance. The project is supported by the European Union and co-financed by the European Social Fund (grant agreement no. EFOP-3.6.2-16-2017-0008: The Role of Neuro-inflammation in Neurodegeneration: From Molecules to Clinics). B.R. was supported by the János Bolyai Research Fellowship from the Hungarian Academy of Sciences and by ÚNKP-18-4 New National Excellence Program of the Ministry of Human Capacities. Silicon microprobes were supported by NSF Neuronex Award 1707408.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.006.
REFERENCES
- Alarcón M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Hum. Genet 82, 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association,. (2013). Diagnostic and Statistical Manual of Mental Disorders, 5th ed. DSM-5 (American Psychiatric Association; ). [Google Scholar]
- Anderson GR, Galfin T, Xu W, Aoto J, Malenka RC, and Sudhof TC (2012). Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc. Natl. Acad. Sci. USA 109, 18120–18125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine MW, Langberg T, Schnepel P, and Feldman DE (2019). Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron 101, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, Rea A, Guy M, Lin S, Cook EH, and Chakravarti A (2008). A Common Genetic Variant in the Neurexin Superfamily Member CNTNAP2 Increases Familial Risk of Autism. Am. J. Hum. Genet 82, 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Rikhye RV, Breton-Provencher V, Tang X, Li C, Li K, Runyan CA, Fu Z, Jaenisch R, and Sur M (2016). Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc. Natl. Acad. Sci. USA 113, E7287–E7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko NP, Belichenko PV, and Mobley WC (2009). Evidence for both neuronal cell autonomous and nonautonomous effects of methyl-CpG-binding protein 2 in the cerebral cortex of female mice with Mecp2 mutation. Neurobiol. Dis 34, 71–77. [DOI] [PubMed] [Google Scholar]
- Brumback AC, Ellwood IT, Kjaerby C, lafrati J, Robinson J, Robinson S, Lee AT, Patel T, Nagaraj S, Davatolhagh F, et al. (2018). Identifying specific prefrontal neurons that contribute to autism-associated abnormalities in physiology and social behavior. Mol. Psychiatry 23, 2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Anastassiou CA, and Koch C (2012). The origin of extracellular fields and currents-EEG, ECoG, LFP and spikes. Nat. Rev. Neurosci 13, 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calverley RKS, and Jones DG (1990). Contributions of dendritic spines and perforated synapses to synaptic plasticity. Brain Res. Brain Res. Rev 15, 215–249. [DOI] [PubMed] [Google Scholar]
- Cea-Del Rio CA, and Huntsman MM (2014). The contribution of inhibitory interneurons to circuit dysfunction in Fragile X Syndrome. Front. Cell. Neurosci 8, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JA, Peñagarikano O, Belgard TG, Swarup V, and Geschwind DH (2015). The emerging picture of autism spectrum disorder: genetics and pathology. Annu. Rev. Pathol 10, 111–144. [DOI] [PubMed] [Google Scholar]
- Cruz-Martín A, Crespo M, and Portera-Cailliau C (2010). Delayed Stabilization of Dendritic Spines in Fragile X Mice. J. Neurosci 30, 7793–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curia G, Papouin T, Séguéla P, and Avoli M (2009). Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb. Cortex 19, 1515–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, and Nelson SB (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 102, 12560–12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzker JL, and Callaway EM (2000). Laminar sources of synaptic input to cortical inhibitory interneurons and pyramidal neurons. Nat. Neurosci 3, 701–707. [DOI] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Won H, Stein JL, and Geschwind DH (2016). Advancing the understanding of autism disease mechanisms through genetics. Nat. Med 22, 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Blanche TJ, Harrison RR, Lester HA, and Masmanidis SC (2011). Multiplexed, high density electrophysiology with nanofabricated neural probes. PLoS One 6, e26204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Allwardt B, and Harris KM (2002). Dendritic spines do not split during hippocampal LTP or maturation. Nat. Neurosci 5, 297–298. [DOI] [PubMed] [Google Scholar]
- Fujisawa S, and Buzsáki G (2011). A 4 Hz oscillation adaptively synchronizes prefrontal, VTA, and hippocampal activities. Neuron 72, 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganeshina O, Berry RW, Petralia RS, Nicholson DA, and Geinisman Y (2004). Synapses with a segmented, completely partitioned postsynaptic density express more AMPA receptors than other axospinous synaptic junctions. Neuroscience 125, 615–623. [DOI] [PubMed] [Google Scholar]
- Gdalyahu A, Lazaro M, Penagarikano O, Golshani P, Trachtenberg JT, and Geschwind DH (2015). Correction: The Autism Related Protein Contactin-Associated Protein-Like 2 (CNTNAP2) Stabilizes New Spines: An In Vivo Mouse Study. PLoS One 10, e0129638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geinisman Y (1993). Perforated axospinous synapses with multiple, completely partitioned transmission zones: probable structural intermediates in synaptic plasticity. Hippocampus 3, 417–433. [DOI] [PubMed] [Google Scholar]
- Geschwind DH (2011). Genetics of autism spectrum disorders. Trends Cogn. Sci 15, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Bartley AF, Hays SA, and Huber KM (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol 100, 2615–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, and Vitkup D (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved information and function of synapses. Neuron 70, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golomb D, and Hansel D (2000). The number of synaptic inputs and the synchrony of large, sparse neuronal networks. Neural Comput. 12, 1095–1139. [DOI] [PubMed] [Google Scholar]
- Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, Kim TK, Griffith EC, Waldon Z, Maehr R, et al. (2010). The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 140, 704–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann T (2013). The role of medial prefrontal cortex in early social cognition. Front. Hum. Neurosci. 7, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RR, and Charles C (2003). A low-power low-noise CMOS amplifier for neural recording applications. IEEE J. Solid-State Circuits 38, 958–965. [Google Scholar]
- lossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515,216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgensen S, and Castillo PE (2015). Selective Dysregulation of Hippocampal Inhibition in the Mouse Lacking Autism Candidate Gene CNTNAP2. J. Neurosci 35, 14681–14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-W, Park K, Kang RJ, Gonzalez ELT, Kim DG, Oh HA, Seung H, Ko MJ, Kwon KJ, Kim KC, et al. (2019). Pharmacological modulation of AMPA receptor rescues social impairments in animal models of autism. Neuropsychopharmacology 44, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, and Somogyi P (2008). Neuronal Diversity and Temporal Dynamics: The Unity of Hippocampal Circuit Operations. Science 321, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A, Zhang R, Yao V, Theesfeld CL, Wong AK, Tadych A, Volfovsky N, Packer A, Lash A, and Troyanskaya OG (2016). Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci 19, 1454–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppa VM, Kravitz SN, Martin CL, Andrieux J, Le Caignec C, Martin-Coignard D, DyBuncio C, Sanders SJ, Lowe JK, Cantor RM, et al. (2016). Rare Inherited and De Novo CNVs Reveal Complex Contributions to ASD Risk in Multiplex Families. Am. J. Hum. Genet 99, 540–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DR, Tamir T, Kaufman M, Weissbrod A, Schneidman E, and Yizhar O (2018). Dynamics of social representation in the mouse prefrontal cortex. bioRxiv. 10.1101/321182. [DOI] [PubMed]
- Likhtik E, Stujenske JM, Topiwala MA, Harris AZ, and Gordon JA (2014). Prefrontal entrainment of amygdala activity signals safety in learned fear and innate anxiety. Nat. Neurosci 17, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liska A, Bertero A, Gomolka R, Sabbioni M, Galbusera A, Barsotti N, Panzeri S, Scattoni ML, Pasqualetti M, and Gozzi A (2018). Homozygous Loss of Autism-Risk Gene CNTNAP2 Results in Reduced Local and Long-Range Prefrontal Functional Connectivity. Cereb. Cortex 28, 1141–1153. [DOI] [PubMed] [Google Scholar]
- Nikonenko I, Jourdain P, Alberi S, Toni N, and Muller D (2002). Activity-induced changes of spine morphology. Hippocampus 12, 585–591. [DOI] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, et al. (2012). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, and Geschwind DH (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, Sonnenblick LI, Gruver R, Almajano J, Bragin A, et al. (2011). Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack P-O, Friedman J, and Golshani P (2013). Cellular mechanisms of brain state-dependent gain modulation in visual cortex. Nat. Neurosci 16, 1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, Trimmer JS, Shrager P, and Peles E (1999). Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 24, 1037–1047. [DOI] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Salomon D, Berglund EO, Ohara R, Ranscht B, and Peles E (2001). Localization of Caspr2 in Myelinated Nerves Depends on Axon-Glia Interactions and the Generation of Barriers along the Axon. J. Neurosci 21, 7568–7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL,Xu X, Chiu SY, Shrager P, et al. (2003). Juxtaparanodal clustering ofShaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J. Cell Biol 162, 1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poot M, Beyer V, Schwaab I, Damatova N, Van’t Slot R, Prothero J, Holder SE, and Haaf T (2010). Disruption of CNTNAP2 and additional structural genome changes in a boy with speech delay and autism spectrum disorder. Neurogenetics 11, 81–89. [DOI] [PubMed] [Google Scholar]
- Redcay E, Moran JM, Mavros PL, Tager-Flusberg H, Gabrieli JD, and Whitfield-Gabrieli S (2013). Intrinsic functional network organization in high-functioning adolescents with autism spectrum disorder. Front. Hum. Neurosci 7, 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas DC, and Wilson LB (2014). γ-band abnormalities as markers of autism spectrum disorders. Biomarkers Med. 8, 353–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R, Sánchez-Aguilera A, van Elst K, Lim L, Dehorter N, Bae SE, Bartolini G, Peles E, Kas MJH, Bruining H, and Marín O (2019). Loss of Cntnap2 Causes Axonal Excitability Deficits, Developmental Delay in Cortical Myelination, and Abnormal Stereotyped Motor Behavior. Cereb. Cortex 29, 586–597. [DOI] [PubMed] [Google Scholar]
- Scott-Van Zeeland AA, Abrahams BS, Alvarez-Retuerto AI, Sonnenblick LI, Rudie JD, Ghahremani D, Mumford JA, Poldrack RA, Dapretto M, Geschwind DH, and Bookheimer SY (2010).Altered Functional Connectivity in Frontal Lobe Circuits Is Associated with Variation in the Autism Risk Gene CNTNAP2. Sci. Transl. Med 2, 56ra80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimbeyoglu A, Kim CK, Inoue M, Lee SY, Hong ASO, Kauvar I, Ramakrishnan C, Fenno LE, Davidson TJ, Wright M, and Deisseroth K (2017). Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2 -deficient mice. Sci. Transl. Med 9, eaah6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Nenadic Z, and Xu X (2010). Novel use of matched filtering for synaptic event detection and extraction. PLoS One 5, e15517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shobe JL, Claar LD, Parhami S, Bakhurin KI, and Masmanidis SC (2015). Brain activity mapping at multiple scales with silicon microprobes containing 1,024 electrodes. J. Neurophysiol 114, 2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorov MS, Deck GM, Dolatshahi M, Thibert RL, Bird LM, Chu CJ, and Philpot BD (2017). Delta rhythmicity is a reliable EEG biomarker in Angel-man syndrome: a parallel mouse and human analysis. J. Neurodev. Disord 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DM, and Wallace MT (2016). Dysfunction of sensory oscillations in Autism Spectrum Disorder. Neurosci. Biobehav. Rev 68, 848–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorra KE, Fiala JC, and Harris KM (1998). Critical assessment of the involvement of perforations, spinules, and spine branching in hippocampal synapse formation. J. Comp. Neurol 398, 225–240. [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, and Morton DH (2006). Recessive Symptomatic Focal Epilepsy and Mutant Contactin-Associated Protein-like 2. N. Engl. J. Med 354, 1370–1377. [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, and Muller D (1999). LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 402, 421–425. [DOI] [PubMed] [Google Scholar]
- Varea O, Martin-de-Saavedra MD, Kopeikina KJ, Schürmann B, Fleming HJ, Fawcett-Patel JM, Bach A, Jang S, Peles E, Kim E, and Penzes P (2015). Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like Caspr2 knockout neurons. Proc. Natl. Acad. Sci. USA 112, 6176–6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, and Geschwind DH (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace ML, Burette AC, Weinberg RJ, and Philpot BD (2012). Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron 74, 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Olivas ND, Levi R, Ikrar T, and Nenadic Z (2010). High precision and fast functional mapping of cortical circuitry through a novel combination of voltage sensitive dye imaging and laser scanning photostimulation. J. Neurophysiol 103, 2301–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, et al. (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Kaiser T, Monteiro P, Zhang X, Van der Goes MS, Wang D, Barak B, Zeng M, Li C, Lu C, et al. (2016). Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron 89, 147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in the paper will be available upon request from the Lead Contact, Peyman Golshani, pgolshani@mednet.ucla.edu. MATLAB code used for all in-vivo electrophysiology analysis have been deposited in Github at https://github.com/jtaxidis/Lazaro-et-al.-Cell-Reports-2019/tree/master.