

Abstract
Interfering with microtubule dynamics is a well-established strategy in cancer treatment; however, many microtubule-targeting agents are associated with drug resistance and adverse effects. Substantial evidence points to ATP-binding cassette (ABC) transporters as critical players in the development of resistance. Herein, we demonstrate the efficacy of DJ95 (2-(1H-indol-6-yl)-4-(3,4,5-trimethoxyphenyl)-1H-imidazo[4,5-c]pyridine), a novel tubulin inhibitor, in a variety of cancer cell lines, including malignant melanomas, drug-selected resistant cell lines, specific ABC transporter–overexpressing cell lines, and the National Cancer Institute 60 cell line panel. DJ95 treatment inhibited cancer cell migration, caused morphologic changes to the microtubule network foundation, and severely disrupted mitotic spindle formation of mitotic cells. The high-resolution crystal structure of DJ95 in complex with tubulin protein and the detailed molecular interactions confirmed its direct binding to the colchicine site. In vitro pharmacological screening of DJ95 using SafetyScreen44 (Eurofins Cerep-Panlabs) revealed no significant off-target interactions, and pharmacokinetic analysis showed that DJ95 was maintained at therapeutically relevant plasma concentrations for up to 24 hours in mice. In an A375 xenograft model in nude mice, DJ95 inhibited tumor growth and disrupted tumor vasculature in xenograft tumors. These results demonstrate that DJ95 is potent against a variety of cell lines, demonstrated greater potency to ABC transporter–overexpressing cell lines than existing tubulin inhibitors, directly targets the colchicine binding domain, exhibits significant antitumor efficacy, and demonstrates vascular-disrupting properties. Collectively, these data suggest that DJ95 has great potential as a cancer therapeutic, particularly for multidrug resistance phenotypes, and warrants further development.
Significance Statement
Paclitaxel is a widely used tubulin inhibitor for cancer therapy, but its clinical efficacy is often limited by the development of multidrug resistance. In this study, we reported the preclinical characterization of a new tubulin inhibitor DJ95, and demonstrated its abilities to overcome paclitaxel resistance, disrupt tumor vasculature, and exhibit significant antitumor efficacy.
Introduction
Microtubules are cytoskeletal structures that are essential for a variety of cellular events including movement, intracellular transport, cell signaling, and mitosis (Jordan, 2002; Pasquier and Kavallaris, 2008; Perez, 2009; Kavallaris, 2010). Microtubules are composed of α- and β-tubulin proteins, which readily undergo polymerization and depolymerization in a phenomenon known as dynamic instability (Mitchison and Kirschner, 1984). They also form the highly dynamic mitotic spindles that are responsible for the alignment and segregation of chromosomes in the cell during mitosis (Kline-Smith and Walczak, 2004). Interference with microtubule dynamics consequently disrupts mitotic progression and ultimately leads to apoptosis and cell death (Schmidt and Bastians, 2007). Therefore, interfering with microtubule dynamics by targeting tubulin with small molecules is a validated anticancer strategy, and many agents are already used clinically or are undergoing development (Ravelli et al., 2004; Gigant et al., 2005; Dorléans et al., 2009; Perez, 2009).
Tubulin inhibitors can be broadly divided into two categories: microtubule stabilizing agents (e.g., taxanes) or destabilizing agents (e.g., vinca alkaloids and colchicine binding site agents). Currently, all approved tubulin inhibitors for cancer therapy target the taxane or vinca alkaloid binding site. However, many tubulin inhibitors, including paclitaxel (Taxol), are associated with multidrug resistance (MDR) mechanisms such as overexpression of drug efflux pumps or the βIII tubulin isoform (Orr et al., 2003; Kamath et al., 2005; Morris and Fornier, 2008; Lu et al., 2012). Drug efflux mediated by ATP-binding cassette (ABC) transporters is the most commonly observed mechanism responsible for inhibiting the intracellular accumulation of therapeutic agents in resistant cell lines (Szakács et al., 2006). Of the known human ABC transporters, resistant melanoma cells have been shown to overexpress ABCB1 (MDR1, P-glycoprotein), ABCC1 (MRP1), ABCC2 (MRP2), and ABCB5, among others (Schadendorf et al., 1995; Frank et al., 2005; Chen et al., 2009; Luo et al., 2012). ABCG2 (breast cancer resistant protein) is also well-characterized and its overexpression causes resistance to a variety of anticancer drugs including paclitaxel and docetaxel (Vlaming et al., 2009; Wu et al., 2011). Additionally, studies have demonstrated that alterations in β-tubulin isotypes can lead to resistance to taxanes in melanoma and other cancers (Ranganathan et al., 1998; Hari et al., 2003; Kamath et al., 2005; Mhaidat et al., 2008). The colchicine binding site is located at the interface between the α- and β-tubulin monomers, and agents that bind to this site may have important advantages over other tubulin inhibitors targeting the taxane or vina alkaloid binding domains. Extensive research efforts have addressed the issue of MDR, and numerous studies have demonstrated that colchicine binding agents can overcome ABCB1 (also known as P-glycoprotein) overexpression and βIII tubulin-mediated drug resistance (Gangjee et al., 2010;, 2013; Stengel et al., 2010; Wang et al., 2012; Dong et al., 2016; Wu et al., 2016; Devambatla et al., 2017; Li et al., 2017; Arnst et al., 2018). While colchicine is not employed as an anticancer agent due to its toxic side effects, other colchicine binding inhibitors have demonstrated promising potential and some are currently being investigated as anticancer candidates (Stanton et al., 2011; Lu et al., 2012).
More recently, there has been increasing interest in the vascular-disrupting capabilities possessed by some microtubule binding agents (Schwartz, 2009; Ji et al., 2015; Canela et al., 2017). It is well-known that tumor progression is dependent on blood vessels to supply oxygen, essential nutrients, and growth factors. Vascular-disrupting agents act on the tumor endothelium and induce destructive changes that decrease blood flow, induce vascular collapse, initiate hypoxia within the tumor, and cause necrosis (Canela et al., 2017). There is accumulating evidence that microtubule binding agents can act selectively on tumor endothelial cells, and this supports the pursuit of targeting tumor vasculature (Schwartz, 2009).
We previously reported a variety of compounds that inhibit tubulin polymerization through interactions with the colchicine binding site (Ahn et al., 2010; Lu et al., 2011; Wang et al., 2012; Hwang et al., 2015; Banerjee et al., 2018). This led to the development of a novel class of indolyl-imidazopyridines, and several of these compounds demonstrated superior potency in vitro and strong tubulin depolymerizing effects (Hwang et al., 2015; Arnst et al., 2018). Herein, we describe one of the most potent of the imidazopyridines, DJ95 (2-(1H-indol-6-yl)-4-(3,4,5-trimethoxyphenyl)-1H-imidazo[4,5-c]pyridine), and evaluate its potential as a small-molecule chemotherapeutic agent.
DJ95 is highly active against our melanoma panel, as well as many cancer types in the National Cancer Institute (NCI) 60 panel. Additionally, it presents a low-resistance index against ABC transporter–overexpressing cell lines and outperformed other tubulin-targeting agents such as paclitaxel, colchicine, and vincristine. We also evaluated its effect on cancer cell migration, clonogenic potential, and endothelial cell tube formation in vitro. Its depolymerization effects were demonstrated through visualization of the microtubule fragmentation as well as the distortion of mitotic spindles. The binding of DJ95 to the colchicine site was confidently confirmed through X-ray crystallographic analyses. Finally, DJ95 inhibited tumor growth in vivo in a melanoma xenograft model and did not reveal significant off-target effects in pharmacological screening.
Materials and Methods
Cell Culture and Reagents
Human melanoma cell lines, A375, RPMI-7951, WM-164, WM115, and SK-MEL-1 (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium (Corning, Manassas, VA) supplemented with 10% (v/v) FBS (Atlanta Biologicals, Lawrenceville, GA) and 1% antibiotic/antimycotic mixture (Sigma-Aldrich, St. Louis, MO). Human umbilical vein endothelial cells (HUVECs) were cultured in endothelial cell growth medium containing growth supplement (Promocell, Heidelberg, Germany) and 1% antibiotic/antimycotic mixture (Sigma-Aldrich). The human epidermoid carcinoma cell line KB-3-1, its drug-selected ABCB1-overexpressing KB-C2 cell line (maintained in medium with 2 μg/ml colchicine), and ABCC1-overexpressing KB-CV60 cell line (maintained in medium with 1 mg/ml of cepharanthine and 60 ng/ml of vincristine) (Wang et al., 2018) were kindly provided by Dr. Shinchi Akiyama at Kagoshima University (Kagoshima, Japan). The human nonsmall cell lung cancer cell line NCI-H460 and its mitoxantrone-selected ABCG2-overexpressing NCI-H460/MX20 cells (maintained in medium with 20 nM of mitoxantrone) (Robey et al., 2001), and the transfected cell lines human embryonic kidney 293 (HEK293)/pcDNA 3.1, HEK293/ABCB1, HEK293/ABCC1, and HEK293/ABCG2-R482 were kindly provided by Dr. Susan E. Bates (Columbia University, New York, NY) and Dr. Robert W. Robey [National Institutes of Health (NIH), Bethesda, MD]; these cell lines were established by transfecting HEK293 cells with either the empty pcDNA3.1 vector or vector containing full-length ABCB1, ABCC1, and wild-type ABCG2, respectively (Patel et al., 2017). These cell lines were cultured in Dulbecco’s modified Eagle’s medium, supplemented with penicillin/streptomycin (Corning) and FBS or Hyclone bovine calf serum (GE Healthcare Life Science, Pittsburgh, PA). All cell lines were authenticated by American Type Culture Collection by short tandem repeat profiling. Cultures were maintained to 80%–90% confluency at 37°C in a humidified atmosphere containing 5% CO2. Compounds were dissolved in DMSO (Sigma-Aldrich) to make a stock solution of 20 mM. Compound solutions were freshly prepared by diluting stocks with cell culture medium before use.
Cytotoxicity Assays
The cytotoxic effect against melanoma cell lines has been previously described (Arnst et al., 2018). Briefly, A375, RPMI-7951, WM-164, WM115, or SK-MEL-1 cells were seeded in 96-well plates at a density of 1000–3500 cells per well, depending on the growth rate of the cell line. After overnight incubation, test compounds were added to the wells at 10 concentrations ranging from 0.03 nM to 1 µM plus a media-only control for 72 hours in four replicates. Following treatment, MTS Reagent (Promega, Madison, WI) was added to the cells and incubated in the dark at 37°C for at least 1 hour. Absorbance at 490 nm was measured using a plate reader (BioTek Instruments Inc., Winooski, VT).
The cytotoxic effects of DJ95 to KB-3-1, KB-C2, KB-CV60, NCI-H460, and NCIH460/MX20 cell lines, and the transfected cell lines HEK293/pcDNA3.1, HEK293/ABCB1, HEK293/ABCC1, and HEK293/ABCG2-R482, were determined using MTT Reagent (Thermo Fisher Scientific Inc., Haverhill, MA) as previously described (Fan et al., 2018). Known tubulin inhibitors with different mechanisms of actions, including paclitaxel (microtubule-stabilizing agent, targeting the taxane-binding site in tubulin), colchicine (microtubule-destabilizing agent, targeting the colchicine-binding site in tubulin), and vincristine (microtubule-destabilizing agent, targeting the vinca alkaloid-binding site in tubulin) (Perez, 2009; Li et al., 2017), were selected as positive controls for comparison with DJ59. In this study, paclitaxel, colchicine, and vincristine, which are existing substrates of ABCB1 and ABCC1 (Chen et al., 2006; Deeley and Cole, 2006; Hodges et al., 2011), also served as positive substrate controls in the experiments involving ABCB1- or ABCC1-overexpressing cell lines. Mitoxantrone, a known substrate of ABCG2 (Homolya et al., 2011), was used as a positive substrate control in ABCG2-overexpressing cells. Cisplatin was used as a negative control since it is not a substrate of ABCB1, ABCC1, or ABCG2 (Fan et al., 2018).
The IC50 values were calculated by nonlinear regression analysis using GraphPad Prism (GraphPad Software, San Diego, CA). In addition, DJ95 was evaluated in one- and five-dose assays against the NCI-60 cell line panel by the National Cancer Institute Developmental Therapeutics Program.
ATPase Assay
The vanadate-sensitive ATPase activity of ABCB1 and ABCG2 using crude membranes of High Five insect cells was determined with the presence of DJ95 (0–10,000 nM) or positive substrate drug by PREDEASY ATPase Kits with modified protocols as previously described (Ambudkar, 1998; Wang et al., 2017b). Paclitaxel and topotecan were selected as positive substrate drugs for ABCB1 and ABCG2, respectively.
Colony-Forming Assay
A375 cells were seeded in six-well plates (500 cells per well) in replicates of four and incubated at 37°C overnight. Cells were treated with the compound or media-only control and incubated for 10 days. Cells were then fixed with methanol and stained with 0.5% crystal violet. Images were taken, and the colony area was quantified with ImageJ software (NIH).
Scratch Migration Assay
A375 and RPMI-7951 cells were seeded in 24-well plates (200,000 cells per well) in replicates of four and incubated overnight. A 200 µl pipette tip was used to scratch a straight line through the cell monolayer to remove an area of cells, and then washed several times to remove any debris and uprooted cells. Media were replaced, containing equivalent vehicle (DMSO) control or DJ95 at 10 or 25 nM concentrations. Images were obtained at the start of the experiment and after 24 hours with the Evos Fl Imaging System (LifeTechnologies, Carlsbad, CA). The analysis was performed with ImageJ software (NIH).
Endothelial Cell Tube Formation Assay
Matrigel (Corning) was thawed on ice overnight, and then diluted with serum-free media for a final concentration of 10 mg/ml. Matrigel was plated in 48-well plates and incubated at 37°C for 1 hour. Low passage number HUVECs (<5) in the logarithmic growth phase were trypsinized and suspended in endothelial cell growth media. Cells (7 × 104) were plated on the matrigel plates in quadruplicate containing the desired drug concentrations. Images were captured after 6-hour incubation with the Evos Fl Imaging System (LifeTechnologies). Analysis was performed with the angiogenesis tool plug-in with ImageJ software (NIH).
Immunofluorescent Staining
WM-164 cells were seeded 2.5–5 × 105 on glass coverslips in six-well plates and incubated overnight. Media were changed, and cells were treated with DJ95, paclitaxel, or the media-only control for 18 hours. Cells were then fixed with 4% paraformaldehyde, permeabilized in 0.1% Triton X (Sigma-Aldrich) in PBS (Thermo Fisher Scientific Inc.), and blocked with 3% bovine serum albumin (Cell Signaling Technology, Danvers, MA) prior to staining. Microtubules and mitotic spindles were visualized after incubating with anti-α-tubulin antibody (Thermo Scientific, Rockford, IL) and Alexa Fluor 647 goat anti-mouse IgG (Molecular Probes, Eugene, OR). The coverslips were mounted with Prolong Diamond Antifade mounting media containing 4′,6-diamidino-2-phenylindole (Invitrogen, Eugene, OR) and images were acquired with a Keyence BZ-X700 fluorescence microscope and BZ-X analyzer software (Keyence, Osaka, Japan).
X-Ray Crystallography
Protein Expression and Purification.
The stathmin-like domain (SLD) of RB3 from rat was transformed into and overexpressed in Escherichia coli. The protein was purified by anion-exchange chromatography (QFF; GE Healthcare), eluted with a 0–200 mM NaCl linear gradient in 20 mM Tris-HCl and 1 mM EGTA (pH 8.0), and gel filtration chromatography (Superdex 75; GE Healthcare), eluted with 10 mM HEPES (pH 7.2), 150 mM NaCl, and 2 mM dithiothreitol. The peak fractions from the gel filtration column were concentrated to 10 mg/ml and stored at −80°C (Charbaut et al., 2001; Dorléans et al., 2009; Wang et al., 2016). The tubulin tyrosine ligase (TTL) protein from chicken was expressed and purified from an E. coli expression system as described previously (Prota et al., 2013). Briefly, the protein was expressed in E. coli using Lysogeny broth, purified through nickel-nitrilotriacetic acid affinity chromatography and gel filtration chromatography [buffer: Bis-Tris propane (pH 6.5), 200 mM NaCl, 2.5 mM MgCl2, 5 mM βMe, and 1% glycerol]. The peak fractions were concentrated to 20 mg/ml and saved at −80°C. Sodium dodecyl sulfate polyacrylamide gel electrophoresis was performed to check the purity of RB3 and TTL. Porcine brain tubulin (Cytoskeleton Inc., Denver, CO) was supplied at 10 mg/ml in general tubulin buffer [80 mM 1,4-piperazinediethanesulfonic acid (pH 6.9), 2 mM MgCl2, 0.5 mM EGTA, and 1 mM GTP] as a frozen liquid and saved at −80°C until use.
Crystallization and Crystal Soaking
The previously published process of obtaining crystals of the tubulin-RB3_SLD-TTL (T2R-TTL) complex was applied (Prota et al., 2013; Wang et al., 2017a). In brief, tubulin (10 mg/ml), TTL (20 mg/ml), and RB3 (10 mg/ml) were mixed at the molar ratio of 2:1.3:1.2 (tubulin:RB3_SLD:TTL) and incubated on ice with 1 mM adenylylmethylenediphosphonate disodium salt acid adenylate ester, 5 mM tyrosinol, and 10 mM DDT, and the mixture was concentrated to 20 mg/ml at 4°C. The crystallization of the T2R-TTL complex was carried out at 20°C using the sitting-drop vapor diffusion method by mixing an equal volume of protein complex and crystallization buffer containing 6% polyethylene glycol 4000, 5% glycerol, 0.1 M 4-morpholineethanesulfonic acid, 30 mM CaCl2, and 30 mM MgCl2 (pH 6.7). Seeding was used to obtain the well diffracting crystals. Initial crystals were observed after 2 days of incubation and reached a final length of 200–300 μm within 3–5 days. Morphologically superior crystals were selected, cryoprotected with crystallization buffer containing 20% glycerol, and flash frozen in liquid nitrogen.
X-Ray Data Collection and Structure Determination
Diffraction data were collected at 100K on the BL19U1 beamline at the Shanghai Synchrotron Radiation Facility in Shanghai, China. Data were indexed, integrated, and scaled using HKL2000 (Otwinowski and Minor, 1997). The structure of T2R-TTL/DJ95 was determined by molecular replacement using the previously published T2R-TTL structure [Protein Data Bank identification (PDB ID): 4I55] as a search model. The rotation and translation function searches were performed by the program PHASER (McCoy et al., 2007). The model was further built with Coot (Emsley and Cowtan, 2004) and refined using the phenix.refine module Phenix (Adams et al., 2002). The model quality was checked with PROCHECK (Laskowski et al., 1993) and showed good stereochemistry according to the Ramachandran plot.
Surface Plasmon Resonance for Tubulin Binding Affinity Analyses
To evaluate the binding affinities of DJ95 with tubulin protein, we performed surface plasmon resonance analyses using a Biacore T200 system (GE Healthcare Life Sciences). A Series S Sensor Chip CM5 (GE Healthcare Life Sciences) was preconditioned with three consecutive 1-minute injections of 70% (w/w) BIA normalizing solution (GE Healthcare Life Sciences). Then, 20 μg/ml tubulin (Cytoskeleton Inc.) was immobilized to the sensor chip surface to attain 17,000 Resonance Units (1000 Resonance Units corresponds to an angle change of ∼0.1°). One of the four flow cells on the chip was left free as a negative control. DJ95, colchicine, or Combretastatin A4 (CA-4) (positive controls) was injected over the sensor chip surface for association analysis, followed by dissociation analysis. We adjusted the concentration gradients for each of the three compounds based on their different affinities to tubulin and different solubilities. The experiment data were obtained at 25°C with running a buffer PBS (10 mM phosphate, 2.7 mM KCl, and 137 mM NaCl), and 0.01% (v/v) Surfactant P20 (GE Healthcare Life Sciences) (pH 7.4). The flow rate was 30 µl/min. The analytes bound on the sensor chips were connected for 120 seconds and dissociated for 120 seconds. Regeneration of the sensor chips was performed for 30 seconds by 10 mM glycine-HCl buffer (pH 1.5). The equilibrium dissociation constant (Kd) was calculated by a steady-state fitting mode with Biacore T200 Evaluation Software, version 2.
In Vitro Pharmacological Profiling to Assess Potential Off-Target Effects
Screening of potentially significant off-target effects to DJ95 binding and enzyme targets was performed via SafetyScreen44 offered by Eurofins Cerep-Panlabs. DJ95 was tested at 100 nM. Compound binding was calculated as the percentage of inhibition of the binding of a radioactively labeled ligand specific for each target. The compound enzyme inhibition effect was calculated as the percentage of inhibition of control enzyme activity. Results showing inhibition (or stimulation for assays run in basal conditions) higher than 50% were considered to represent significant effects of the test compounds. Results showing inhibition (or stimulation) between 25% and 50% were indicative of weak effects. Results showing inhibition (or stimulation) lower than 25% were not considered significant and were mostly attributable to variability of the signal around the control level. In each experiment, the respective reference compound was tested concurrently with DJ95, and the data were compared with historical values determined at Eurofins. The experiment was accepted in accordance with the Eurofins validation standard operating procedure.
Liquid Chromatography–Tandem Mass Spectrometry Plasma Concentration Analysis
Liquid Chromatography–Tandem Mass Spectrometry Parameters.
The liquid chromatography–tandem mass spectrometry system comprised a Sciex (Framingham, MA) 5500 triple quadrupole mass spectrometer, equipped with a Turboionspray ionization interface and Analyst software version 1.6.3. Chromatographic separation was carried out using a ZORBAX SB-C18 column of 150 × 4.6 mm (i.d.) and 3.5 µm particle size (Agilent Technologies, Santa Clara, CA) maintained at 35°C using the Shimadzu (Columbia, MD) Nexera XR HPLC system and SIL-20ACXR autosampler. The mobile phase (A: Milli-Q water; B: methanol) was eluted at a flow rate of 0.5 ml/min. The gradient started at 50% of mobile phase B, was maintained for 0.5 minutes, and was then linearly increased to 100% B over 1 minute. Subsequently, the eluent composition was maintained at 100% B from 1.5 to 6 minutes before it was decreased to the initial condition of 50% mobile phase B for re-equilibration in 0.1 minutes. The total run time was 6.5 minutes plus pre-equilibration of 0.5 minutes. A switching valve directed the mobile phase to the mass spectrometry system between 4.5 and 6.1 minutes. The electrospray ion source was operated in positive ionization mode for all of the experiments. The typical parameters were as follows: capillary, 5.5 kV; entrance potential, 10 V; channel electron multiplier, 1800 V; and source temperature, 600°C. Other compound-specific parameters are listed in Supplemental Table 1.
Liquid Chromatography–Tandem Mass Spectrometry Sample Preparation
Protein precipitation was used to extract DJ95 from plasma. Plasma (50 μl) was added in 150 μl precipitation solution methanol, including 26.5 nM ABI-231 as the internal standard, vortexed for 15 seconds, and then centrifuged for 15 minutes at 4°C (∼12,000 rpm). Supernatant (120 μl) was then transferred to a 96-well plate and a 1 μl sample was injected into the liquid chromatography–tandem mass spectrometry system.
Calibration standards were prepared by mixing DJ95 stock solution in pooled human plasma, resulting in matrix concentrations of 1, 10, 100, 200, 1000, 2000, and 10,000 nM. Blank samples were prepared using blank plasma. All samples were stored at −20°C prior to use. The lowest standard of 1 nM was not detected. The lower limit of quantification was determined to be 10 nM.
In Vivo Mouse Models and Treatments
All protocols and methods, including methods of anesthesia, administration of drugs, blood collection, and endpoints requiring euthanasia, were approved by the University of Tennessee Health Science Center Animal Care and Use Committee, consistent with the Guide for the Care and Use of Laboratory Animals, eighth edition, as published by the National Academy of Sciences (National Research Council, 2011). All animals were maintained in a room with a 12-hour light/dark cycle and provided food and water ad libitum.
Pharmacokinetic Studies
Thirty CD-1 ICR mice from Charles River Laboratories (Wilmington, MA), approximately 6 weeks of age, were used for the study. A mix of males and females were used, and at least one animal of each sex was collected at each time point. For each time point cohort, animals (n = 3 per time point) were dosed with 15 mg/kg DJ95 via intraperitoneal injection and the drug vehicle was polyethylene glycol 300 (Sigma-Aldrich) and PBS at a 1:1 ratio.
Blood was collected using heparinized syringes from mice deeply anesthetized with isoflurane during the terminal blood collection via cardiac venipuncture into lithium heparinized tubes. At each time point, blood (∼0.6 ml) was collected from a separate cohort of three mice at the following time points: 0, 15, 30, 60, 90, 180, 260, 480, 720, and 1440 minutes. Samples were centrifuged at 3000 rpm for 10 minutes. Plasma was collected into 1.5 ml centrifuge tubes and frozen at −80°C until analysis by liquid chromatography mass spectrometry. Pharmacokinetic parameters were determined by noncompartmental analysis using Phoenix WinNonlin 8.1 (Certara, Princeton, NJ). These parameters included the area under the concentration-time profile curve, half-life, clearance, volume of distribution, and Cmax.
Maximum Tolerable Dose and Xenograft Study
Nude mice, 50 male and 50 female (6–8 weeks old), were purchased from Evigo Laboratories (Indianapolis, IN). A maximum tolerable dose study was performed by subjecting nude mice to a dose of up to 30 mg/kg of DJ95 formulated in the vehicle (equal parts polyethylene glycol 300 and PBS) by intraperitoneal injection for five consecutive days. Because mice began to show signs of toxicity beyond 30 mg/kg for the 5-day treatment, we scaled the dose back to 15 mg/kg for the xenograft study to ensure an adequate safety margin. Logarithmic growth phase A375 cells were prepared in phenyl red–free, FBS-free media and mixed with thawed matrigel prior to injecting into mice. Tumors were established by subcutaneously injecting 100 μl of matrigel/cell suspension containing 2.5 × 106 cells in the hind flank of each mouse. After tumors were established, mice were ranked on tumor size and randomized into control or treatment groups, with each group receiving one-half males and one-half females. Then, 100 µl of the drug treatment or vehicle control solution was administered via intraperitoneal injection five times a week for the duration of the studies.
Tumor volume was measured three times a week with a caliper and calculated by using the formula a × b2 × 0.5, where a and b represent the larger and smaller diameters, respectively. Tumor growth inhibition at the conclusion of the experiments was calculated as 100 − 100 × [(T − T0)/(C − C0)], where T, T0, C, and C0 are the mean tumor volume for the specific group on the last day of treatment, mean tumor volume of the same group on the first day of treatment, mean tumor volume for the vehicle control group on the last day of treatment, and mean tumor volume for the vehicle control group on the first day of treatment, respectively (Wang et al., 2014). Animal activity was monitored, and body weights were recorded throughout the study to assess potential acute toxicity. At the end of the experiment, mice were sacrificed, and the tumors were dissected out, weighed, and fixed in 10% neutral buffered formalin solution prior to pathology staining analysis.
Histology and Immunohistochemistry
The fixed tumor xenograft tissues were embedded in paraffin. Serial sections were obtained for immunohistochemistry analysis. Staining was performed with rabbit anti-CD31 (Cell Signaling Technology Inc.) following Avidin Biotin Complex methods. Antigen retrieval was performed with H-3300 antigen unmasking solution (Vector Laboratories, Burlingame, CA). Images were captured with a Keyence BZ-X fluorescent microscope (Keyence Corporation, Itasca, IL) at 10× and 20× magnifications. Five representative fields of view from three tumors per group at 20× magnification were analyzed in ImageJ to calculate the positive stained area.
Statistical Analysis
In all studies, it was presumed that the null hypothesis would be that the DJ95 treatment had no effect on the biologic phenotype measured compared with the control; therefore, significance was based on two-tailed statistics tests. The sample sizes for all in vitro and in vivo studies were predetermined before the studies were initiated. The In vitro studies included multiple technical replicates per assay and were repeated at least three times (biologic replicates). The IC50 values were calculated by nonlinear regression, and the S.D. was determined from at least three independent experiments. For the in vivo studies, sample sizes were based on our prior experience from studies testing similar drug compounds in xenograft animal models using the subcutaneous injection method. For the in vitro experiments comparing multiple doses of DJ95 (Figs. 2 and 8), the statistical significance (P < 0.05) was calculated by one-way ANOVA followed by Dunnett’s multiple comparisons test. For the in vivo experiments (Fig. 7), two cohorts were compared with each other, the control mice (vehicle, n = 7) and the DJ95-treated mice (15 mg/kg, n = 6), using the unpaired Student’s t test. Data for endpoint tumor volume and tumor wet weight were shown as a scatter plot presenting the mean with 1 S.D. All data were analyzed using GraphPad Prism Software 5.0 (GraphPad Software, Inc.).
Fig. 2.

DJ95 inhibits cell proliferation and migration of melanoma. (A) Colony formation assay of A375 cells treated with DJ95 in six-well plates (n = 3). Cells were treated with indicated concentrations of DJ95 and medium only was used as the control. (B) Quantification of colony area using ImageJ software. Graph is represented as area ± S.D. (C) Representative images of A375 (top) and RPMI-7951 cells (bottom) in a wound healing assay (n = 3). A scratch was created through a monolayer of confluent cells, which were then treated with DJ95 or control. The migrating ability was calculated from images taken at the start of the treatment and after 24-hour incubation with compound. The yellow outline represents the leading edge of the area boundary as determined by ImageJ software. (D) Quantification of A375 and (E) RPMI-7951 migration represented as a percentage of the initial scratch area ± S.D. Statistical significance was determined by one-way ANOVA followed by Dunnett’s multiple comparisons test, comparing each treatment group to the control group for the aforementioned experiments (**P< 0.01; ***P< 0.001; ****P< 0.0001).
Fig. 8.

DJ95 targets tumor vascular and capillary network formation. (A) Representative images of HUVEC capillary formation on matrigel. Images were taken after 6-hour incubation with indicated concentrations of DJ95 or colchicine. (B) Quantification of total tube length using ImageJ analysis expressed as mean ± S.D. (n = 4). Statistical analysis was performed using one-way ANOVA comparing each group to the control. (C) Representative immunohistochemistry images of CD31-stained tumor sections taken at 10× magnification. (D) Positive stained area calculated for tumor sections at 20× magnification (five images per tumor; three separate tumors per group) represented as mean pixels ± S.D. Statistical significance was determined by student’s t test without correction for multiple comparisons (**P< 0.01; ***P< 0.001; ****P< 0.0001).
Fig. 7.

DJ95 inhibits melanoma tumor growth in vivo. (A) Plasma concentrations at each time point following intraperitoneal injection of DJ95 at 15 mg/kg concentration shown as individual replicates connected by mean (n = 3 per time point). (B) A375 melanoma xenograft model in nude mice. Mice were dosed by intraperitoneal injection five times per week for 2 weeks with vehicle solution only or 15 mg/kg DJ95. Mean tumor volume is expressed as percentage of growth compared with tumor volume at the beginning of the experiment ± S.E.M. (n = 6 for the treated group; n = 7 for the control group). (C) Individual tumor volumes at the end of the experiment shown as mean ± S.D. (D) Mouse weight change throughout the study shown as mean ± S.D. (E) Resected tumor weight at the end of the experiment expressed as mean ± S.D. Statistical significance was determined by student’s t test comparing the treatment group to the vehicle control group (*P< 0.05; **P< 0.01).
Results
DJ95 Is Potent against Cancer Cell Lines and Overcomes ABC Transporter–Mediated Resistance.
We previously reported that DJ95 is active against a variety of melanoma and prostate cancer cell lines (Hwang et al., 2015). To further explore these initial findings, we tested DJ95 against a panel of malignant melanoma cell lines representing genomic complexity and heterogeneity, including A375, RPMI-7951, WM-164, WM115, and SK-MEL-1 (Supplemental Table 2). We discovered that DJ95 was highly potent in melanoma cell lines and had IC50 values of less than 100 nM (Table 1). It was the most potent against A375, with an average IC50 value of 24.7 ± 4.9 nM, and was similar to colchicine (10.6 ± 1.8 nM). Paclitaxel was generally the most potent against the parental melanoma cell lines.
TABLE 1.
Cytotoxic effects of DJ95 against malignant melanoma cancer cell lines
The IC50 values represent the mean ± S.D. of three independent experiments performed in quadruplicate.
| Treatment |
IC50 ± S.D. |
||||
|---|---|---|---|---|---|
| A375 |
RPMI-7951 |
WM-164 |
WM115 |
SK-MEL-1 |
|
| nM | nM | nM | nM | nM | |
| DJ95 | 24.7 ± 4.9 | 58.9 ± 13.3 | 68.2 ± 12.4 | 99.8 ± 16.8 | 78.7 ± 16.5 |
| Colchicine | 10.6 ± 1.8 | 11.1 ± 0.7 | 10.1 ± 0.7 | 9.3 ± 1.0 | 11.5 ± 2.5 |
| Paclitaxel | 2.5 ± 1.0 | 3.4 ± 0.8 | 8.0 ± 1.0 | 3.3 ± 1.6 | 1.8 ± 0.8 |
Because we were particularly interested in the ability of DJ95 to overcome transporter-mediated drug resistance, we tested DJ95 against genetically engineered HEK293 cell lines that stably transfected full-length ABCB1, ABCC1, wild-type ABCG2, or the pcDNA3.1 blank vector (Supplemental Fig. 1). Cisplatin, which is not a substrate for any of these ABC transporters, was used as the negative control. Greater than 100-fold resistance was observed for the ABCB1-overexpressing HEK293 cells against paclitaxel, colchicine, and vincristine (Table 2). On the other hand, HEK293/ABCB1-resistant cells showed virtually no resistance to DJ95. A slight resistance index (3.7) was revealed for the ABCC1-overexpressing HEK293-transfected cells against DJ95 compared with the vector-only cells, but all other microtubule-targeting drugs that were tested (paclitaxel, colchicine, and vincristine) demonstrated significantly increased resistance, with IC50 values greater than 10,000 nM against HEK293/ABCC1. Similarly, against the HEK293/ABCG2-R482 cells, resistance was evident for all tubulin inhibitors, although less resistance was developed against the DJ95 compound (about 8.7-fold). This was still lower than mitoxantrone, a known substrate of ABCG2, showing about 11.2-fold decrease in potency in the ABCG2 overexpressing cell line.
TABLE 2.
Cytotoxic effects of DJ95 against parental and gene-transfected ABCB1-, ABCC1-, and ABCG2-overexpressing cell lines
The IC50 values represent the mean ± S.D. of three independent experiments performed in triplicate.
—, data not available.
| Treatment |
IC50 ± S.D. |
|||
|---|---|---|---|---|
| HEK293/pcDNA3.1 |
HEK293/ABCB1 |
HEK293/ABCC1 |
HEK293/ABCG2-R482 |
|
| nM | nM | nM | nM | |
| DJ95 | 330.0 ± 44.1 | 314.7 ± 73.1 | 1213.9 ± 241.6 | 2869.2 ± 123.3 |
| Paclitaxel | 34.6 ± 6.3 | 3496.0 ± 657.8 | >10,000 | >10,000 |
| Colchicine | 112.1 ± 41.5 | >10,000 | >10,000 | >10,000 |
| Vincristine | 99.4 ± 23.1 | >10,000 | >10,000 | >10,000 |
| Mitoxantrone | 108.0 ± 27.5 | — | — | 1209.7 ± 111.4 |
| Cisplatin | 2510.1 ± 362.6 | 2583.1 ± 474.5 | 2584.1 ± 151.8 | 2475.3 ± 127.4 |
We also wanted to assess the effects of DJ95 against drug-selected, resistant cell lines. From the parental epidermoid carcinoma cell line KB-3-1, the colchicine resistant/ABCB1-overexpressing (KB-C2) and vincristine resistant/ABCC1-overexpressing (KB-CV60) cell lines were developed. The mitoxantrone-selected ABCG2-overexpressing cell line NCI-H460/MX20 generated from the parental nonsmall cell lung cancer cell line NCI-H460 was used to further evaluate the effects against ABCG2 upregulation. Overexpression of ABCB1, ABCC1, or ABCG2 for each of these cell lines is shown in Supplemental Fig. 1.
Compared with the existing tubulin inhibitors paclitaxel, colchicine, and vincristine, DJ95 exhibited higher cytotoxic levels in drug-selected ABCB1-overexpressing KB-C2 and ABCC1-overexpressing KB-CV60 cell lines, with IC50 values of 719.54 ± 181.48 and 35.90 ± 4.50 nM, respectively (Table 3). Although there was still some increase in resistance against the KB-C2 (colchicine selected/ABCB1-overexpressing) cells compared with the parental KB-3-1 cells, both KB-C2 and KB-CV60 were much less resistant to DJ95 than they were against the three other tubulin inhibitors tested. Since no resistance to DJ95 was revealed from the ABCB1-transfected cell line, the increase in resistance observed for DJ95 against the colchicine-selected KB-C2 may be related to mechanisms affiliated with the colchicine binding site and not solely with ABCB1 overexpression.
TABLE 3.
Cytotoxic effects of DJ95 against parental and drug-selected ABCB1-, ABCC1-, and ABCG2-overexpressing cancer cell lines
The IC50 values represent the mean ± S.D. of three independent experiments performed in triplicate.
—, data not available.
| Treatment |
IC50 ± S.D. |
||||
|---|---|---|---|---|---|
| KB-3-1 |
KB-C2 (ABCB1) |
KB-CV60 (ABCC1) |
NCI-H460 |
NCI-H460/MX20 (ABCG2) |
|
| nM | nM | nM | nM | nM | |
| DJ95 | 23.5 ± 2.2 | 719.5 ± 181.5 | 35.9 ± 4.5 | 673.0 ± 111.7 | 5361.5 ± 645.5 |
| Paclitaxel | 9.2 ± 2.1 | 1918.5 ± 106.3 | 120.0 ± 14.6 | >10,000 | >10,000 |
| Colchicine | 23.9 ± 4.9 | 7445.3 ± 446.3 | 91.7 ± 7.7 | >10,000 | >10,000 |
| Vincristine | 1.3 ± 0.2 | 748.8 ± 99.8 | 218.6 ± 1.9 | >10,000 | >10,000 |
| Mitoxantrone | — | — | — | 118.7 ± 40.2 | 2456.5 ± 420.4 |
| Cisplatin | 2024.5 ± 463.7 | 2871.2 ± 115.3 | 1796.5 ± 198.8 | 2226.6 ± 179.5 | 2870.6 ± 281.7 |
In both NCI-H460 and its mitoxantrone-selected ABCG2-overexpressing cell line, DJ95 demonstrated better anticancer activities than paclitaxel, colchicine, and vincristine, with IC50 values of 673.04 ± 111.71 and 5361.50 ± 645.45 nM, respectively, whereas the IC50 values of the three other tubulin inhibitors were higher than 10,000 nM in both cell lines. However, low resistance against DJ95 was observed in the NCI-H460/MX20 cell line compared with its parental cell line (about 8-fold resistance), although the resistance index was not as high as that of the substrate control mitoxantrone (about 20-fold resistance). The resistance index is the ratio of the IC50 value of the drug-resistant cell (or transfected cell overexpressing the ABC transporter) over the IC50 value of the parental cell (or vector control for the transfected cell). The results from the drug-selected resistant cells are consistent with those obtained from the transfected ABCG2-overexpressing cell line HEK293/ABCG2-R482 and its vector control, which indicated that DJ95 might be a weak substrate of ABCG2.
To follow up on these results, the ABCB1 or ABCG2 ATPase activity with the presence of DJ95 were investigated to assess the interaction between DJ95 and these ABC transporters. DJ95 inhibited ABCB1-mediated ATP hydrolysis, while paclitaxel, which is a substrate of ABCB1, stimulated the vanadate-sensitive ATPase activity of ABCB1 (Supplemental Fig. 2A). This suggests that DJ95 may not be a substrate of ABCB1 and may be able to circumvent ABCB1-medited MDR, which is consistent with the cytotoxicity results from HEK293/ABCB1 and HEK293/pcDNA3.1 cells. On the contrary, DJ95 showed stimulating effects on ABCG2 ATPase activity, similar to the positive substrate drug topotecan (Supplemental Fig. 2B). This is in accordance with the resistance to DJ95 shown by ABCG2-overexpressing cells, which further supports the hypothesis that DJ95 may be a substrate of ABCG2.
Finally, DJ95 was tested in the NCI-60 screening, assaying a diverse selection of cancer types. DJ95 was evaluated in a single-dose assay (Supplemental Fig. 3, A and B) as well as a five-concentration assay to see if it was sensitive in other kinds of cancer (Fig. 1). DJ95 was highly active and demonstrated low Growth Inhibition of 50% (GI50) values for the majority of melanoma cell lines, and was especially effective against leukemia, colon, central nervous system, and prostate cancers. The average total growth inhibition and Lethal Concentration of 50% (LC50) values were greatest for colon cancer and melanoma. Additionally, we assessed the dose-response curves of each individual melanoma cell line tested and determined that the cell lines most sensitive to DJ95 treatment were LOX IMVI and SK-MEL-5 (Supplemental Fig. 3C; Supplemental Table 3). Together, these results indicate that DJ95 is effective against a multitude of cancer cell lines representing genomic heterogeneity, drug-resistant profiles, and cancer types.
Fig. 1.

DJ95 is potent against NCI-60 cell lines. National Cancer Institute Developmental Therapeutics Program 5-dose assay mean graphs. Positive values indicate net cell growth, zero represents no net growth, and negative values indicate cell lethality.
DJ95 Impedes Cancer Cell Colony Formation and Migration.
After verifying the potency of DJ95, it was further evaluated for functional inhibition of cell proliferation and migration in vitro. We first determined its ability to inhibit colony formation in a concentration-dependent manner compared with untreated A375 control cells (Fig. 2A). A significant decrease was observed for all treated cells, and concentrations as low as 10 nM caused 26.78% ± 1.9% inhibition of colony area compared with control (P < 0.001) and higher concentrations of 25 and 50 nM inhibited colony formation by 44.73% ± 1.9% and 65.01% ± 3.08%, respectively (P < 0.0001) (Fig. 2B). At 100 nM, DJ95 almost completely eliminated colony formation and only 1.25% ± 0.07% of the area compared with the control remained. DJ95 was then tested in both A375 and RPMI-7951 melanoma cell lines to determine its effect on migration ability of the cells in a scratch assay (Fig. 2C). After 24 hours, the untreated cells migrated into 80.73% ± 4.48% of the wound channel, nearly closing the gap. Treatment to the A375 cells with 10 and 25 nM of DJ95 decreased the cell migration and led to cells only occupying 58.23% ± 2.15% (P < 0.01) and 35.60% ± 1.35% (P < 0.0001) of the scratch area, respectively, which was significantly less than the control (Fig. 2D). A similar phenomenon was also observed in the RPMI-7951 cell line, where the control cells reclaimed 77.43% ± 3.91% of the scratch area (Fig. 2E). Treatments of 10 and 25 nM of DJ95 caused the cells to only migrate into 34.37% ± 6.09% (P < 0.001) and 23.10% ± 2.92% (P < 0.0001) of the scratch area, respectively. There was no significant difference in the scratch channel area for each group at the beginning of the experiment (time 0) as determined by two-way ANOVA analysis factoring in both time and treatment group (Supplemental Fig. 4). To this end, it can be interpreted that DJ95 inhibits cell proliferation and migration at low concentrations for these melanoma cell lines.
DJ95 Disrupts Microtubule Networks and Mitotic Spindle Formation.
Previously, we demonstrated that DJ95 is able to potently inhibit polymerization of purified tubulin protein in a cell-free assay (Hwang et al., 2015). Here, we visually present the effect DJ95 has on the microtubule networks of WM-164 melanoma cells (Fig. 3A). The stabilizing agent paclitaxel, a potent enhancer of polymerization, was also used for comparison. After 18 hours of treatment at a 50 nM concentration with DJ95, cells showed dramatically disrupted microtubule networks and an increase in soluble, cytoplasmic tubulin. Furthermore, at 200 nM there appears to be very little polymeric tubulin framework left intact. In contrast, paclitaxel-treated cells demonstrated highly condensed, polymeric tubulin. For cells treated at the higher concentration of 200 nM paclitaxel, the fluorescent signal was amplified and the aggregation of the filaments was more pronounced, consistent with increased stabilization of microtubules.
Fig. 3.

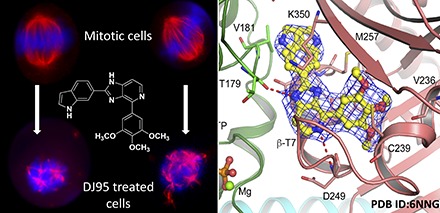
DJ95 interferes with microtubule networks and mitotic spindle organization. (A) Microtubule networks of WM-164 cells after 18-hour treatment with paclitaxel or DJ95 at indicated concentrations compared with untreated cells. The fragmentation and destabilization of the microtubules is evident from DJ95 treatment, whereas paclitaxel increased polymerization and stabilization. (B) The effect on mitotic spindle organization after 18-hour treatment with 100 and 200 nM concentrations of DJ95. Mitotic spindle suppression is evidenced by multiple asters and disorganized spindle morphology. Confocal images were obtained from a Keyence BZX microscope. Tubulin (red) is visualized by α-tubulin primary antibody and Alexa Fluor 647 secondary antibody and DNA (blue) is stained with 4′,6-diamidino-2-phenylindole.
Microtubules also make up the mitotic spindle and use the dynamic properties for chromosome segregation during mitosis (Kline-Smith and Walczak, 2004). Therefore, tubulin-targeting agents also interfere with the formation and organization of mitotic spindles (Weiderhold et al., 2006; Rai et al., 2012; Rohena et al., 2016; Sakchaisri et al., 2017). We investigated the effects of DJ95 in a concentration-dependent manner compared with untreated control cells (Fig. 3B). While control cells exhibited normal, bilateral spindle formation extruding from centrosomes, DJ95 treatment led to aberrant spindle development as evidenced by multipolar and disorganized spindle morphology. These abnormalities were exacerbated with increasing drug concentrations. This is further proof that DJ95 inhibits the progression of mitosis by interfering with microtubule dynamics.
X-Ray Crystallography Confirms that DJ95 Occupies the Colchicine Binding Site.
We determined the crystal structure of the T2R-TTL complex bound with compound DJ95 at 2.4 Å resolution (Fig. 4A), and the coordinates and structure factors have been deposited in the Protein Data Bank with the PDB ID: 6NNG. The refinement statistics and overall geometric parameters confirm that the structure quality is excellent; this information, together with the data collection parameters, is presented in Table 4. DJ95 binds at the interface of the α/β-tubulin heterodimer and occupies the same locale as colchicine in the β-tubulin protomer (PDB ID: 4O2B) directly opposite the GTP that binds within the α-tubulin. Unlike colchicine that binds both available α/β interfaces in the T2R-TTL complex, DJ95 only binds to the interface proximal to the TTL subunit. DJ95 is slightly more extended than colchicine, and it appears that the single occupancy is not biologically relevant but rather the result of small conformational changes in the assembly that cannot easily be accommodated by the packing within the crystal lattice. The 3-methoxyphenyl group of DJ95 occupies a deep pocket that is lined with residues Tyr200, Val236, Cys239, Leu240, Leu250, Leu253, Ala314, Ile316, Ala352, and Ile368 (Fig. 4B). The central imidazopyridine ring and the distal indole ring are linearly connected and flanked on one side by residues from the short α-helix β-H8, specifically Lys252, Leu253, Asn256, and Met257, and residues Lys350 and Leu246 on the other side. The DJ95 binding interactions are almost exclusively van der Waals in nature but there are several specific interactions (Fig. 4B). The central imidazopyridine ring forms two hydrogen bonds: a nitrogen atom with the main chain amide of Asp249 and an NH group with the main chain carbonyl oxygen of Thr179 of α-tubulin across the interface. The 3-methoxyphenyl moiety has three interactions; the central methoxy group interacts via a water molecule with the main chain NH of Cys239, and the side chains of Leu253 and Cys239 make flanking π-H interactions with the phenyl ring.
Fig. 4.

The Crystal structure of the T2R-TTL-DJ95 complex. (A) Quaternary structure of the T2R-TTL-DJ95 complex showing the arrangement of the protein components, the bound nucleotides, and the bound DJ95. Note that DJ95 only occupies one of the available α/β-tubulin interfaces. (B) Close-up view of the bound DJ95 (yellow carbons) and its interactions with surrounding residues from β-tubulin (cyan). Note the hydrogen bond to the backbone carbonyl oxygen of α-tubulin Thr179 (green) across the α/β interface. (C) Close-up view of the superimposed DJ95 complex and the colchicine complex (PDB ID: 4O2B, gray). Note that DJ95 is slightly more extended than colchicine. (D) Close-up view of the curved DJ95 complex superimposed on the straight Taxol complex (PDB ID: 1JFF, wheat). The red arrows indicate the extent of movement of b-tubulin with respect to a-tubulin upon the binding of DJ95. Note that the binding of DJ95 is not compatible with the straight complex.
TABLE 4 .
X-ray data collection and refinement statistics Tubulin-RB3_SLD-TTL complex bound with DJ95 (PDB ID: 6NNG). Values in parentheses indicate the highest-resolution shell.
| Data Collection | Value |
|---|---|
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 105.42, 157.98, 182.84 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 50–2.4 (2.44–2.4) |
| Rmeas | 0.092 (0.922) |
| I/σ(I) | 20.89 (2.14) |
| Completeness (%) | 99.2 (99.5) |
| Redundancy | 6.6 (6.4) |
| Refinement | |
| Resolution (Å) | 48.25–2.4 (2.48–2.4) |
| Number of reflections | 117,909 (10,915) |
| Rwork/Rfree | 0.1866/0.2253 |
| Number of atoms | 17,724 |
| Protein | 16,956 |
| Ligand/ion | 215 |
| Water | 553 |
| B factors | |
| Protein | 49.92 |
| Ligand/ion | 52.57 |
| Water | 43.07 |
| Root-mean-square deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.05 |
| Ramachandran Plot | |
| Favored (%) | 98.04 |
| Allowed (%) | 1.91 |
| Outliers (%) | 0.05 |
Comparison with the colchicine complex reveals that the binding interactions are very similar, with most of the residues described previously in equivalent conformations (Fig. 4C). There are two noticeable differences. First, the equivalent 3-methoxyphenyl moiety of colchicine does not penetrate the pocket as deeply (by some 2.5 Å), which precludes the water-mediated hydrogen bond to the central methoxy group. Second, in the unliganded (PDB ID: 4I55) and colchicine-liganded T2R-TTL complexes, the side chain of Lys350 is extended and interacts across the α/β interface with Ser178 and Thr179, respectively, in the flexible α-T5 loop of α-tubulin. However, in the DJ95 complex steric interference with the distal indole ring and favorable van der Waals interactions with the molecule cause Lys350 to swing around, which in turn allows Thr179 to form the main chain hydrogen bond described previously. Finally, in a similar fashion to colchicine, the binding of DJ95 elicits substantial conformational changes at the α/β interface, including within loops β-T7 of β-tubulin and α-Τ5 of α-tubulin (Fig. 4D). These are incompatible with the straight structure of the tubulin filament (PDB ID: 1JFF), and therefore is consistent with DJ95 acting as a destabilizer of filament formation. The electron density map is demonstrated in Supplemental Fig. 5.
DJ95 Shows Lower Binding Affinity to Tubulin than that of Colchicine and CA-4.
To quantitatively determine the binding affinity of DJ95 with tubulin protein, we performed surface plasmon resonance experiments using the Biacore T200 system (GE Healthcare Life Sciences). A CM5 sensor chip with glucan on the surface was used to immobilize tubulin. DJ95, colchicine, or CA-4 was injected over the sensor chip surface for binding detection. As determined by a 1:1 kinetics fitting model, the equilibrium dissociation constant Kd values of DJ95, colchicine, and CA-4 were 59.4, 5.7 and 7.7 μM, respectively (Fig. 5). These data reveal that DJ95 has approximately 10-fold lower binding affinity than colchicine and CA-4, and further modifications will be required to increase the affinity to tubulin and avoid potential binding to additional molecular targets.
Fig. 5.

Biacore surface plasmon resonance kinetic analyses of DJ95, colchicine, and CA-4 interacting with tubulin. Binding kinetics of DJ95 (32, 16, 8, 4, and 2 μM), Colchicine (20, 10, 5, 2.5, and 1.25 μM), and CA-4 (20, 10, 5, 2.5, and 1.25 μM) to tubulin (17,000 Resonance Units) determined by surface plasmon resonance technology. The calculated equilibrium dissociation constant Kd values for DJ95, colchicine, and CA-4 are 59.4, 5.7, and 7.7 μM, respectively.
DJ95 Has Negligible Interactions with 44 Physiologically Important Targets.
Prior to in vivo studies, we wanted to determine if DJ95 would exhibit a relatively safe profile based on interactions with off-targets and predict if there would be any potentially detrimental clinical effects. In vitro pharmacological profiling involves screening the compound of interest against a wide range of targets such as receptors, ion channels, transporters, and enzymes other than the intended therapeutic target to identify specific interactions that may elicit adverse drug-related side effects (Bowes et al., 2012). Here, DJ95 was evaluated in binding and enzyme uptake assays performed by Eurofins Cerep Panlabs. DJ95 demonstrated minimal specific binding inhibition or stimulation against 37 radioactive labeled ligand targets (Fig. 6A). Additionally, DJ95 did not induce significant alterations in enzyme function for COX1, COX2, PDE3A, PDE4D2, Lck kinase, or ACHE (Fig. 6B). Results showing inhibition or stimulation higher than 50% were considered significant for the test compounds, none of which were observed at any of the targets studied in this screening. Specific binding assay targets and enzyme-based assays, along with the corresponding reference compound, are summarized in Supplemental Table 4.
Fig. 6.

DJ95 in binding and enzyme and uptake assays. (A) DJ95 control-specific binding as a percentage of inhibition to the binding to a radioactively labeled ligand specific for each target. (B) DJ95 enzyme-based functional percentage of control enzyme activity. Results showing inhibition or stimulation higher than 50% are considered to represent significant effects of the test compounds. Graphs are represented as the mean of duplicate assays ± S.D. Screening was performed by Eurofins Cerep-Panlabs.
DJ95 Pharmacokinetics and Antitumor Activity In Vivo.
Based on the preliminary in vitro data suggesting that DJ95 is potently active against melanoma cell lines and minimally interferes with off-targets, we sought to test the activity of DJ95 in vivo. We determined that concentrations up to 30 mg/kg administered by intraperitoneal injection daily were well tolerated for at least 5 days in nude mice; however, additional treatments caused a decrease in mouse weight and decline in behavioral activity. Therefore, we scaled back the dose to one-half of that (15 mg/kg) for the pharmacokinetic and xenograft study. To determine if DJ95 could reach therapeutically relevant biologic concentrations at doses of 15 mg/kg, we collected blood samples from 15 minutes to 24 hours following intraperitoneal injection of the drug and analyzed the plasma concentrations by liquid chromatography mass spectrometry methods. The Cmax value for DJ95 was 13.65 µM and the detected concentrations stayed above 13 µM for at least 1.5 hours in mouse plasma (Fig. 7A). While the concentration of DJ95 gradually decline over the course of the 24 hours when samples were collected, there was still an average of 126.5 nM at 12 hours and 8.1 nM at 24 hours. These data, along with the area under the concentration-time profile curve (50,500 hour*nM), suggest acceptable exposure for DJ95 over the course of a day. We also determined additional pharmacokinetic parameters including the half-life (3.28 hours), volume of distribution (3.51 l/kg), and clearance (0.744 l/h per kilogram). We reasoned that these results supported a dosing regimen of five treatments per week, allowing for two recovery days to avoid accumulating toxicities. To test the anticancer efficacy of DJ95 in vivo, xenografts were established by subcutaneous inoculation of A375 cells, and treatment began after viable tumors developed. Groups were dosed by intraperitoneal injection with either 15 mg/kg treatments of DJ95 or vehicle solution only. After 2 weeks and a total of 10 treatments, tumor growth for the DJ95-treated group was significantly inhibited compared with the control group, with a tumor growth inhibition of 61.4% (Fig. 7B). A student’s t test gave an overall P value of 0.0081 based on the final percentage change and 0.0382 based on the final tumor volume (Fig. 7C), compared with the control group. Animal behavior and mouse body weights were measured and recorded throughout the course of the experiment to assess for acute toxicities, and major deviations were not observed (Fig. 7D). At the end of the study, tumors were resected and weighed, and there was a significant decrease in tumor weight for the DJ95 group (P = 0.0406) (Fig. 7E). This in vivo study demonstrates the antitumor efficacy of DJ95 in a melanoma xenograft model and supports its continued investigation as an anticancer agent.
DJ95 Shows Vascular-Disrupting Capabilities.
In recent years, there has been extensive research on the vascular-disrupting properties of tubulin-targeting agents and their ability to selectively target tumor vasculature (Wang et al., 2012; Ji et al., 2015; Martel-Frachet et al., 2015; Banerjee et al., 2016; Su et al., 2016). To see whether DJ95 would exhibit these characteristics, we tested the in vitro effect on the formation of capillary-like networks of HUVECs plated on matrigel. The matrigel basement membrane allows endothelial cells to form tubules with tight cell-to-cell and cell-to-membrane contacts (Benton et al., 2014). After 6 hours of incubation, it was clearly observed that both DJ95 and colchicine disrupted the tube cell formation in a concentration-dependent manner (Fig. 8A). At concentrations of 100 and 200 nM, DJ95 disrupted networks by 35.71% ± 5.62% and 51.81% ± 3.25%, respectively (P < 0.0001) (Fig. 8B). This was similar to the effect observed upon colchicine treatment at the same concentrations, inhibiting HUVEC tube cell formation by 60.43% ± 9.04% and 81.59% ± 8.01%, respectively (P < 0.0001). Since this result was observed in a short time frame, we expect that the drug action on tube cell formation was not a result of antiproliferative activity.
We also wanted to assess the effect on tumor vasculature from the DJ95-treated xenografts. CD31 staining of tumor sections revealed the change in microvessels of the DJ95-treated group compared with the control group (Fig. 8C.) It is apparent that the control group has more abundant and in-tact microvessels throughout the tumors, whereas DJ95 treatment induced vessel fragmentation and decreased overall density and occupied area. Quantification of the positive CD31-stained area revealed that there was a 50.47% decrease in total microvessel area, representing a significant difference (P = 0.0038) (Fig. 8D). Detailed images used for quantification can be found in Supplemental Fig. 6. This finding corroborates the in vitro results and suggests that the antivascular capacities portrayed by DJ95 may contribute to its anticancer efficacy.
Discussion
Importance of Overcoming Drug Resistance.
Melanoma is the most aggressive form of skin cancer and is one of the most rapidly increasing cancers worldwide (Linos et al., 2009). Malignant melanoma is characterized by resistance to chemotherapy and is incurable in most affected patients (Wu and Singh, 2011; Kalal et al., 2017). Despite recent advances in treating melanoma with targeted therapies and immunotherapies, significant obstacles still exist for finding satisfactory treatments. Intrinsic and acquired resistance are the major causes of treatment failure, and it is of utmost importance to discover and develop agents that can overcome drug resistance, improve response rates, and extend survival for melanoma patients.
We previously identified DJ95, a small molecule synthesized from a series of indolyl-imidazopyridines, that targets the colchicine binding site and has shown promising activity in vitro (Hwang et al., 2015). Genetic heterogeneity among cancer cell lines of the same cancer type has emphasized the necessity to study multiple cell lines in a panel (Gillet et al., 2011). To expand on its cytotoxic potential, DJ95 was tested against our malignant melanoma cell line panel and demonstrated IC50 values ranging from 25 to 100 nM, confirming its strong activity against a variety of melanoma cell lines. We also tested DJ95 against the NCI-60 cell panel to assess its efficacy against other cancers. The NCI-60 cell panel includes numerous cell lines from nine different tumor types, which are extensively characterized. DJ95 had an exceptionally low GI50 value in the majority of cell lines for all cancer types. In addition to melanoma, DJ95 was particularly active against colon cancers based on the total growth inhibition and LC50 values.
One of the most significant factors that limits the efficacy of chemotherapeutics is the development of MDR. One of the major culprits responsible for contributing to MDR is the overexpression of ABC transporters such as ABCB1 (P-glycoprotein/MDR1), ABCC1 (MRP1), and ABCG2 (breast cancer resistant protein) (Gottesman et al., 2002). Agents that target the taxane and vinca binding sites of tubulin are particularly susceptible to resistance from the overexpression ABCB1 and are effectively effluxed from the cell (Yusuf et al., 2003; Fojo and Menefee, 2007; Xia and Smith, 2012; Callaghan et al., 2014). These agents are also substrates to several ABCC/MRP members that decrease their intracellular concentrations (Dumontet and Jordan, 2010). We discovered that DJ95 had a lower resistance index than the other tested tubulin inhibitors in both the drug-selected and gene-transfected cell lines overexpressing ABCC1. While it did not show resistance to the transfected ABCB1-overexpressing cells, it did show some resistance to the KB-C2 drug-selected cell line overexpressing ABCB1. Since DJ95 was considered not to be a substrate of ABCB1 based on the data indicating that it did not stimulate ABCB1 ATPase activity, the resistance showed from KB-C2 might be contributed by the more complex MDR mechanisms in drug-selected MDR cells in comparison with gene-transfected cells. Since KB-C2 cells were established by continuous selection with colchicine (2 μg/ml) they may have incurred additional drug-resistance mechanisms besides solely ABCB1 upregulation. For example, mutations to the colchicine binding domain on tubulin could account for the crossresistance to DJ95 or other agents targeting this site. Taken together, we infer that DJ95 may be able to circumvent ABCB1- or ABCC1-mediated MDR but may have reduced efficacy in colchicine-selected drug-resistant cases.
Targeting Mitotic Machinery.
The main function of microtubule-targeting agents is to inhibit mitosis through disruption of microtubule dynamics (Mukhtar et al., 2014). While stabilizing agents such as paclitaxel promote polymerization by blocking the disassembly of GDP-bound tubulin and form stable tubulin polymers, destabilizing agents inhibit this assembly of tubulin into microtubules and promote depolymerization (Field et al., 2014). This phenomenon was observed in DJ95-treated cells, which displayed fragmented microtubules and a dramatic decrease of visible tubulin filaments. On the contrary, paclitaxel-treated cells demonstrated rigid and condensed polymeric microtubules. Furthermore, microtubules constitute the mitotic spindle in cells undergoing mitosis. Disruption of the mitotic spindle through suppression of treadmilling and dynamic instability are the primary means by which microtubule inhibitors thwart cellular functions and induce apoptosis (Loong and Yeo, 2014). Typical features of mitotic spindle suppression and aberrant formation such as multiple asters in mitotic cells were evident in the DJ95-treated cells, whereas the control cells demonstrated polar spindle formation and centrally aligned chromosomes.
Optimizing Scaffolds through X-Ray Crystallography.
The crystal structure of the T2R-TTL complex bound with DJ95 provides further evidence to the mechanism of action for the anticancer activity of this compound. It binds at the colchicine binding pocket at the interface of α- and β-tubulin heterodimers. Colchicine targets the β subunit of tubulin and keeps it from adopting a straight conformation, thus inhibiting microtubule assembly. The binding of DJ95 also blocked the curve-to-straight conformational change of tubulin by the steric clashes between DJ95 and surrounding secondary structure elements (Fig. 4C). Therefore, DJ95 likely shares the same inhibition mechanism as that of colchicine. The crystal structure not only provides the structural basis for the anticancer activity of DJ95, it also presents opportunities for designing better analogs of this compound that may show improved potency. For example, replacing a carbon atom that faces residue Asn347 by a hydrogen bond donor (NH) could pick up an additional hydrogen bond with the main chain C=O of Asn347.
Minimizing Failure in Drug Development.
Reducing the attrition rate of potential pharmaceutical drug candidates is an important step in drug discovery and development. Identification of off-target, adverse drug reactions to important pharmacological targets such as G protein–coupled receptors, drug transporters, ion channels, nuclear receptors, kinases, and nonkinase enzymes can aid in determining if a drug may elicit side effects that would prevent its further advancement. Use of predictive safety panels that implement assays to identify significant off-target interactions of drug candidates is supported by major pharmaceutical companies such as AstraZeneca, GlaxoSmithKline, Novartis, and Pfizer (Bowes et al., 2012). DJ95 underwent this pharmacological profiling in Eurofins SafetyScreen44 Panel. DJ95 did not show significant inhibition or activation to any of the 44 physiologically important targets tested in the screening. This suggests that DJ95 minimally interacts with or activates some of the key targets that potentially cause adverse drug reactions. Therefore, we proceeded with in vivo studies and found that DJ95 effectively inhibited melanoma tumor growth at a 15 mg/kg dose. While colchicine is known to cause significant toxicity, no toxic side effects based on animal body weight and behavior were observed in the DJ95-treated groups.
DJ95 Disrupts Tumor Vasculature.
It is well known that tumor growth and metastasis require a reliable system of blood vessels to support the tumor microenvironment, and interfering with this process is an attractive strategy for inhibiting tumor growth (Matter, 2001; Banerjee et al., 2016). This has prompted research in the development of antiangiogenic and vascular-disrupting agents, and many tubulin inhibitors that target the colchicine binding site demonstrate this capability (Wang et al., 2012; Ji et al., 2015; Banerjee et al., 2016; Arnst et al., 2018; Galmarini et al., 2018). Microtubule stabilizing agents such as paclitaxel, peloruside A, and laulimalide also have reported vascular-disrupting properties, but they have other shortcomings, such as development of resistance and dose-limiting toxicities (Akiyama et al., 2012; Bocci et al., 2013; Kanakkanthara et al., 2014; Chan et al., 2015). Here, we demonstrate the ability of DJ95 in suppressing capillary-like networks in vitro in HUVECs in a concentration-dependent manner. We also discovered vascular-disrupting action evident in mouse xenografts after DJ95 treatment, where there was significant disturbance of endothelial vasculature and an overall decrease in microvessel area. These in vitro and in vivo data also support the notion of DJ95 as a potential dual inhibitor that may exert its anticancer effects through multiple mechanisms.
Conclusions
In summary, we report the preclinical evaluation of DJ95. DJ95 is effective against a variety of melanoma cell lines and other cancer types, significantly reduces tubulin polymerization, binds to the colchicine binding site on tubulin, reduces melanoma tumor growth in vivo without causing toxicity, and has vascular-disrupting properties. DJ95 incurred less resistance in ABC transporter–overexpressing cell lines and could prove to be an effective alternative treatment when other tubulin-targeting agents fail to show efficacy due to MDR. There are currently no colchicine binding site agents approved for chemotherapy, and DJ95 shows great potential for further development as an anticancer agent.
Acknowledgments
The authors thank Dr. Benoît Gigant [Institute for Integrative Biology of the Cell (I2BC), Université Paris-Sud, Orsay, and Université Paris-Saclay, Paris, France] and Dr. Michel O. Steinmetz (Paul Scherrer Institute, Villigen, Switzerland) for kindly providing the plasmids of RB3-SLD and TTL. We thank Dr. Shinichi Akiyama (Kagoshima University, Kagoshima, Japan) for kindly providing the KB-3-1, KB-C2, and KB-CV60 cell lines. We thank Dr. Susan E. Bates (Columbia University, New York, NY) and Dr. Robert W. Robey (National Institutes of Health, Bethesda, MD) for generously providing the NCI-H460, NCI-H460/MX20, and HEK293/pcDNA 3.1 cell lines, and the transfected cell lines HEK293/ABCB1, HEK293/ABCC1, and HEK293/ABCG2-482R. We also thank Dr. Yi Xue at the University of Tennessee Health Science Center for helping with the immunohistochemical analyses and Dr. Pradeep Lukka for help with the liquid chromatography mass spectrometry analysis of the plasma samples.
Abbreviations
- ABC
ATP-binding cassette
- CA-4
Combretastatin A-4
- DJ95
(2-(1H-indol-6-yl)-4-(3,4,5-trimethoxyphenyl)-1H-imidazo[4,5-c]pyridine)
- GI50
Growth Inhibition of 50%
- HEK293
human embryonic kidney 293
- HUVEC
human umbilical vein endothelial cell
- LC50
Lethal Concentration of 50%
- MDR
multidrug resistance
- NCI
National Cancer Institute
- NIH
National Institutes of Health
- PDB ID
Protein Data Bank identification
- SLD
stathmin-like domain
- T2R-TTL
tubulin stathmin-like domain of RB3/tubulin tyrosine ligase (tubulin-RB3_SLD-TTL)
- TTL
tubulin tyrosine ligase
Authorship Contributions
Participated in research design: Arnst, Miller, Li.
Conducted experiments: Arnst, Wang, Lei, Ma, Parke.
Contributed new reagents or analytic tools: Arnst, Wang, Lei, Hwang, Seagroves, Q. Chen, Yang, Z-S Chen, Li.
Wrote or contributed to the writing of the manuscript: Arnst, Lei, Kumar, White, Li.
Footnotes
This work is supported by the National Institutes of Health National Cancer Institute [Grant R01CA148706 (to W.L. and D.D.M.), Grants 1S10OD010678-01 and 1S10RR026377-01 (to W.L.), and other support from Grant 1S10OD016226-01A1]. Additional support was received from the University of Tennessee College of Pharmacy Drug Discovery Center. The X-ray work was partially supported by the National Natural Science Foundation of China [Grant S81703553 (to Y.W., Q.C., and J.Y.)]. G.K. and S.W.W. acknowledge support from American Lebanese Syrian Associated Charities.
The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. No National Institutes of Health funds were used for any experiments contributed by Y.W., Q.C. and J.Y. to this work.
Dr. W.L. started in January 2019 to serve as a consultant to Veru Inc., who licensed the patent portfolios covering DJ95 and its related analogs for commercial development. Veru Inc. did not provide any financial support or have any influence on research design, experiments, data collection, data analyses, or the writing of this manuscript.
All intellectual properties are owned by the University of Tennessee Health Science Center.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58:1948–1954. [DOI] [PubMed] [Google Scholar]
- Ahn S, Duke CB, III, Barrett CM, Hwang DJ, Li CM, Miller DD, Dalton JT. (2010) I-387, a novel antimitotic indole, displays a potent in vitro and in vivo antitumor activity with less neurotoxicity. Mol Cancer Ther 9:2859–2868. [DOI] [PubMed] [Google Scholar]
- Akiyama K, Ohga N, Hida Y, Kawamoto T, Sadamoto Y, Ishikawa S, Maishi N, Akino T, Kondoh M, Matsuda A, et al. (2012) Tumor endothelial cells acquire drug resistance by MDR1 up-regulation via VEGF signaling in tumor microenvironment. Am J Pathol 180:1283–1293. [DOI] [PubMed] [Google Scholar]
- Ambudkar SV. (1998) Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol 292:504–514. [DOI] [PubMed] [Google Scholar]
- Arnst KE, Wang Y, Hwang DJ, Xue Y, Costello T, Hamilton D, Chen Q, Yang J, Park F, Dalton JT, et al. (2018) A potent, metabolically stable tubulin inhibitor targets the colchicine binding site and overcomes taxane resistance. Cancer Res 78:265–277. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Arnst KE, Wang Y, Kumar G, Deng S, Yang L, Li GB, Yang J, White SW, Li W, et al. (2018) Heterocyclic-fused pyrimidines as novel tubulin polymerization inhibitors targeting the colchicine binding site: structural basis and antitumor efficacy. J Med Chem 61:1704–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Hwang DJ, Li W, Miller DD. (2016) Current advances of tubulin inhibitors in nanoparticle drug delivery and vascular disruption/angiogenesis. Molecules 21:1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton G, Arnaoutova I, George J, Kleinman HK, Koblinski J. (2014) Matrigel: from discovery and ECM mimicry to assays and models for cancer research. Adv Drug Deliv Rev 79–80:3–18. [DOI] [PubMed] [Google Scholar]
- Bocci G, Di Paolo A, Danesi R. (2013) The pharmacological bases of the antiangiogenic activity of paclitaxel. Angiogenesis 16:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes J, Brown AJ, Hamon J, Jarolimek W, Sridhar A, Waldron G, Whitebread S. (2012) Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov 11:909–922. [DOI] [PubMed] [Google Scholar]
- Callaghan R, Luk F, Bebawy M. (2014) Inhibition of the multidrug resistance P-glycoprotein: time for a change of strategy? Drug Metab Dispos 42:623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela MD, Noppen S, Bueno O, Prota AE, Bargsten K, Sáez-Calvo G, Jimeno ML, Benkheil M, Ribatti D, Velázquez S, et al. (2017) Antivascular and antitumor properties of the tubulin-binding chalcone TUB091. Oncotarget 8:14325–14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A, Singh AJ, Northcote PT, Miller JH. (2015) Inhibition of human vascular endothelial cell migration and capillary-like tube formation by the microtubule-stabilizing agent peloruside A. Invest New Drugs 33:564–574. [DOI] [PubMed] [Google Scholar]
- Charbaut E, Curmi PA, Ozon S, Lachkar S, Redeker V, Sobel A. (2001) Stathmin family proteins display specific molecular and tubulin binding properties. J Biol Chem 276:16146–16154. [DOI] [PubMed] [Google Scholar]
- Chen KG, Valencia JC, Gillet JP, Hearing VJ, Gottesman MM. (2009) Involvement of ABC transporters in melanogenesis and the development of multidrug resistance of melanoma. Pigment Cell Melanoma Res 22:740–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Yang Y, Li L, Zhang JT. (2006) The amino terminus of the human multidrug resistance transporter ABCC1 has a U-shaped folding with a gating function. J Biol Chem 281:31152–31163. [DOI] [PubMed] [Google Scholar]
- Deeley RG, Cole SPC. (2006) Substrate recognition and transport by multidrug resistance protein 1 (ABCC1). FEBS Lett 580:1103–1111. [DOI] [PubMed] [Google Scholar]
- Devambatla RKV, Li W, Zaware N, Choudhary S, Hamel E, Mooberry SL, Gangjee A. (2017) Design, synthesis, and structure-activity relationships of pyrimido[4,5-b]indole-4-amines as microtubule depolymerizing agents that are effective against multidrug resistant cells. Bioorg Med Chem Lett 27:3423–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong M, Liu F, Zhou H, Zhai S, Yan B. (2016) Novel natural product- and privileged scaffold-based tubulin inhibitors targeting the colchicine binding site. Molecules 21:1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorléans A, Gigant B, Ravelli RB, Mailliet P, Mikol V, Knossow M. (2009) Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc Natl Acad Sci USA 106:13775–13779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumontet C, Jordan MA. (2010) Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 9:790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- Fan YF, Zhang W, Zeng L, Lei ZN, Cai CY, Gupta P, Yang DH, Cui Q, Qin ZD, Chen ZS, et al. (2018) Dacomitinib antagonizes multidrug resistance (MDR) in cancer cells by inhibiting the efflux activity of ABCB1 and ABCG2 transporters. Cancer Lett 421:186–198. [DOI] [PubMed] [Google Scholar]
- Field JJ, Kanakkanthara A, Miller JH. (2014) Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg Med Chem 22:5050–5059. [DOI] [PubMed] [Google Scholar]
- Fojo T, Menefee M. (2007) Mechanisms of multidrug resistance: the potential role of microtubule-stabilizing agents. Ann Oncol 18 (Suppl 5):v3–v8. [DOI] [PubMed] [Google Scholar]
- Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, Sayegh MH, Sadee W, Frank MH. (2005) ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res 65:4320–4333. [DOI] [PubMed] [Google Scholar]
- Galmarini CM, Martin M, Bouchet BP, Guillen-Navarro MJ, Martínez-Diez M, Martinez-Leal JF, Akhmanova A, Aviles P. (2018) Plocabulin, a novel tubulin-binding agent, inhibits angiogenesis by modulation of microtubule dynamics in endothelial cells. BMC Cancer 18:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangjee A, Zaware N, Devambatla RK, Raghavan S, Westbrook CD, Dybdal-Hargreaves NF, Hamel E, Mooberry SL. (2013) Synthesis of N4-(substituted phenyl)-N4-alkyl/desalkyl-9H-pyrimido[4,5-b]indole-2,4-diamines and identification of new microtubule disrupting compounds that are effective against multidrug resistant cells. Bioorg Med Chem 21:891–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangjee A, Zhao Y, Lin L, Raghavan S, Roberts EG, Risinger AL, Hamel E, Mooberry SL. (2010) Synthesis and discovery of water-soluble microtubule targeting agents that bind to the colchicine site on tubulin and circumvent Pgp mediated resistance. J Med Chem 53:8116–8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigant B, Wang C, Ravelli RB, Roussi F, Steinmetz MO, Curmi PA, Sobel A, Knossow M. (2005) Structural basis for the regulation of tubulin by vinblastine. Nature 435:519–522. [DOI] [PubMed] [Google Scholar]
- Gillet JP, Calcagno AM, Varma S, Marino M, Green LJ, Vora MI, Patel C, Orina JN, Eliseeva TA, Singal V, et al. (2011) Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc Natl Acad Sci USA 108:18708–18713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2:48–58. [DOI] [PubMed] [Google Scholar]
- Hari M, Yang H, Zeng C, Canizales M, Cabral F. (2003) Expression of class III β-tubulin reduces microtubule assembly and confers resistance to paclitaxel. Cell Motil Cytoskeleton 56:45–56. [DOI] [PubMed] [Google Scholar]
- Hodges LM, Markova SM, Chinn LW, Gow JM, Kroetz DL, Klein TE, Altman RB. (2011) Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharmacogenet Genomics 21:152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homolya L, Orbán TI, Csanády L, Sarkadi B. (2011) Mitoxantrone is expelled by the ABCG2 multidrug transporter directly from the plasma membrane. Biochim Biophys Acta 1808:154–163. [DOI] [PubMed] [Google Scholar]
- Hwang DJ, Wang J, Li W, Miller DD. (2015) Structural optimization of indole derivatives acting at colchicine binding site as potential anticancer agents. ACS Med Chem Lett 6:993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji YT, Liu YN, Liu ZP. (2015) Tubulin colchicine binding site inhibitors as vascular disrupting agents in clinical developments. Curr Med Chem 22:1348–1360. [DOI] [PubMed] [Google Scholar]
- Jordan MA. (2002) Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr Med Chem Anticancer Agents 2:1–17. [DOI] [PubMed] [Google Scholar]
- Kalal BS, Upadhya D, Pai VR. (2017) Chemotherapy resistance mechanisms in advanced skin cancer. Oncol Rev 11:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath K, Wilson L, Cabral F, Jordan MA. (2005) βIII-tubulin induces paclitaxel resistance in association with reduced effects on microtubule dynamic instability. J Biol Chem 280:12902–12907. [DOI] [PubMed] [Google Scholar]
- Kanakkanthara A, Eras J, Northcote PT, Cabral F, Miller JH. (2014) Resistance to peloruside A and laulimalide: functional significance of acquired βI-tubulin mutations at sites important for drug-tubulin binding. Curr Cancer Drug Targets 14:79–90. [DOI] [PubMed] [Google Scholar]
- Kavallaris M. (2010) Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer 10:194–204. [DOI] [PubMed] [Google Scholar]
- Kline-Smith SL, Walczak CE. (2004) Mitotic spindle assembly and chromosome segregation: refocusing on microtubule dynamics. Mol Cell 15:317–327. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26 (2):283–291. [Google Scholar]
- Li W, Sun H, Xu S, Zhu Z, Xu J. (2017) Tubulin inhibitors targeting the colchicine binding site: a perspective of privileged structures. Future Med Chem 9:1765–1794. [DOI] [PubMed] [Google Scholar]
- Linos E, Swetter SM, Cockburn MG, Colditz GA, Clarke CA. (2009) Increasing burden of melanoma in the United States. J Invest Dermatol 129:1666–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loong HH, Yeo W. (2014) Microtubule-targeting agents in oncology and therapeutic potential in hepatocellular carcinoma. OncoTargets Ther 7:575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Chen J, Xiao M, Li W, Miller DD. (2012) An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res 29:2943–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Li CM, Wang Z, Chen J, Mohler ML, Li W, Dalton JT, Miller DD. (2011) Design, synthesis, and SAR studies of 4-substituted methoxylbenzoyl-aryl-thiazoles analogues as potent and orally bioavailable anticancer agents. J Med Chem 54:4678–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Ellis LZ, Dallaglio K, Takeda M, Robinson WA, Robinson SE, Liu W, Lewis KD, McCarter MD, Gonzalez R, et al. (2012) Side population cells from human melanoma tumors reveal diverse mechanisms for chemoresistance. J Invest Dermatol 132:2440–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel-Frachet V, Keramidas M, Nurisso A, DeBonis S, Rome C, Coll JL, Boumendjel A, Skoufias DA, Ronot X. (2015) IPP51, a chalcone acting as a microtubule inhibitor with in vivo antitumor activity against bladder carcinoma. Oncotarget 6:14669–14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter A. (2001) Tumor angiogenesis as a therapeutic target. Drug Discov Today 6:1005–1024. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. (2007) Phaser crystallographic software. J Appl Cryst 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhaidat NM, Thorne RF, de Bock CE, Zhang XD, Hersey P. (2008) Melanoma cell sensitivity to Docetaxel-induced apoptosis is determined by class III β-tubulin levels. FEBS Lett 582:267–272. [DOI] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M. (1984) Dynamic instability of microtubule growth. Nature 312:237–242. [DOI] [PubMed] [Google Scholar]
- Morris PG, Fornier MN. (2008) Microtubule active agents: beyond the taxane frontier. Clin Cancer Res 14:7167–7172. [DOI] [PubMed] [Google Scholar]
- Mukhtar E, Adhami VM, Mukhtar H. (2014) Targeting microtubules by natural agents for cancer therapy. Mol Cancer Ther 13:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council (2011) Guide for the Care and Use of Laboratory Animals, 8th ed, Washington, DC, The National Academies Press.
- Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. (2003) Mechanisms of Taxol resistance related to microtubules. Oncogene 22:7280–7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. (1997) [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- Pasquier E, Kavallaris M. (2008) Microtubules: a dynamic target in cancer therapy. IUBMB Life 60:165–170. [DOI] [PubMed] [Google Scholar]
- Patel A, Li TW, Anreddy N, Wang DS, Sodani K, Gadhia S, Kathawala R, Yang DH, Cheng C, Chen ZS. (2017) Suppression of ABCG2 mediated MDR in vitro and in vivo by a novel inhibitor of ABCG2 drug transport. Pharmacol Res 121:184–193. [DOI] [PubMed] [Google Scholar]
- Perez EA. (2009) Microtubule inhibitors: differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance [published correction appears in Mol Cancer Ther (2012) 11:1381]. Mol Cancer Ther 8:2086–2095. [DOI] [PubMed] [Google Scholar]
- Prota AE, Bargsten K, Zurwerra D, Field JJ, Díaz JF, Altmann KH, Steinmetz MO. (2013) Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 339:587–590. [DOI] [PubMed] [Google Scholar]
- Rai A, Surolia A, Panda D. (2012) An antitubulin agent BCFMT inhibits proliferation of cancer cells and induces cell death by inhibiting microtubule dynamics. PLoS One 7:e44311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan S, Benetatos CA, Colarusso PJ, Dexter DW, Hudes GR. (1998) Altered beta-tubulin isotype expression in paclitaxel-resistant human prostate carcinoma cells. Br J Cancer 77:562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. (2004) Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 428:198–202. [DOI] [PubMed] [Google Scholar]
- Robey RW, Honjo Y, van de Laar A, Miyake K, Regis JT, Litman T, Bates SE. (2001) A functional assay for detection of the mitoxantrone resistance protein, MXR (ABCG2). Biochim Biophys Acta 1512:171–182. [DOI] [PubMed] [Google Scholar]
- Rohena CC, Telang NS, Da C, Risinger AL, Sikorski JA, Kellogg GE, Gupton JT, Mooberry SL. (2016) Biological characterization of an improved pyrrole-based colchicine site agent identified through structure-based design. Mol Pharmacol 89:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakchaisri K, Kim SO, Hwang J, Soung NK, Lee KH, Choi TW, Lee Y, Park CM, Thimmegowda NR, Lee PY, et al. (2017) Anticancer activity of a novel small molecule tubulin inhibitor STK899704. PLoS One 12:e0173311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadendorf D, Herfordt R, Czarnetzki BM. (1995) P-glycoprotein expression in primary and metastatic malignant melanoma. Br J Dermatol 132:551–555. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Bastians H. (2007) Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Updat 10:162–181. [DOI] [PubMed] [Google Scholar]
- Schwartz EL. (2009) Antivascular actions of microtubule-binding drugs. Clin Cancer Res 15:2594–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton RA, Gernert KM, Nettles JH, Aneja R. (2011) Drugs that target dynamic microtubules: a new molecular perspective. Med Res Rev 31:443–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel C, Newman SP, Leese MP, Potter BV, Reed MJ, Purohit A. (2010) Class III β-tubulin expression and in vitro resistance to microtubule targeting agents. Br J Cancer 102:316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M, Huang J, Liu S, Xiao Y, Qin X, Liu J, Pi C, Luo T, Li J, Chen X, et al. (2016) The anti-angiogenic effect and novel mechanisms of action of Combretastatin A-4. Sci Rep 6:28139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5:219–234. [DOI] [PubMed] [Google Scholar]
- Vlaming ML, Lagas JS, Schinkel AH. (2009) Physiological and pharmacological roles of ABCG2 (BCRP): recent findings in Abcg2 knockout mice. Adv Drug Deliv Rev 61:14–25. [DOI] [PubMed] [Google Scholar]
- Wang B, Ma LY, Wang JQ, Lei ZN, Gupta P, Zhao YD, Li ZH, Liu Y, Zhang XH, Li YN, et al. (2018) Discovery of 5-cyano-6-phenylpyrimidin derivatives containing an acylurea moiety as orally bioavailable reversal agents against P-glycoprotein-mediated mutidrug resistance. J Med Chem 61:5988–6001. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen J, Miller DD, Li W. (2014) Synergistic combination of novel tubulin inhibitor ABI-274 and vemurafenib overcome vemurafenib acquired resistance in BRAFV600E melanoma. Mol Cancer Ther 13:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yu Y, Li GB, Li SA, Wu C, Gigant B, Qin W, Chen H, Wu Y, Chen Q, et al. (2017a) Mechanism of microtubule stabilization by taccalonolide AJ. Nat Commun 8:15787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang H, Gigant B, Yu Y, Wu Y, Chen X, Lai Q, Yang Z, Chen Q, Yang J. (2016) Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J 283:102–111. [DOI] [PubMed] [Google Scholar]
- Wang YJ, Zhang YK, Zhang GN, Al Rihani SB, Wei MN, Gupta P, Zhang XY, Shukla S, Ambudkar SV, Kaddoumi A, et al. (2017b) Regorafenib overcomes chemotherapeutic multidrug resistance mediated by ABCB1 transporter in colorectal cancer: in vitro and in vivo study. Cancer Lett 396:145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Chen J, Wang J, Ahn S, Li CM, Lu Y, Loveless VS, Dalton JT, Miller DD, Li W. (2012) Novel tubulin polymerization inhibitors overcome multidrug resistance and reduce melanoma lung metastasis. Pharm Res 29:3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiderhold KN, Randall-Hlubek DA, Polin LA, Hamel E, Mooberry SL. (2006) CB694, a novel antimitotic with antitumor activities. Int J Cancer 118:1032–1040. [DOI] [PubMed] [Google Scholar]
- Wu CP, Hsieh CH, Wu YS. (2011) The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol Pharm 8:1996–2011. [DOI] [PubMed] [Google Scholar]
- Wu S, Singh RK. (2011) Resistance to chemotherapy and molecularly targeted therapies: rationale for combination therapy in malignant melanoma. Curr Mol Med 11:553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Wang Q, Li W. (2016) Recent advances in heterocyclic tubulin inhibitors targeting the colchicine binding site. Anticancer Agents Med Chem 16:1325–1338. [DOI] [PubMed] [Google Scholar]
- Xia CQ, Smith PG. (2012) Drug efflux transporters and multidrug resistance in acute leukemia: therapeutic impact and novel approaches to mediation. Mol Pharmacol 82:1008–1021. [DOI] [PubMed] [Google Scholar]
- Yusuf RZ, Duan Z, Lamendola DE, Penson RT, Seiden MV. (2003) Paclitaxel resistance: molecular mechanisms and pharmacologic manipulation. Curr Cancer Drug Targets 3:1–19. [DOI] [PubMed] [Google Scholar]