

Abstract
The cyanobacterial marine natural product honaucin A inhibits mammalian innate inflammation in vitro and in vivo. To decipher its mechanism of action, RNA sequencing was used to evaluate differences in gene expression of cultured macrophages following honaucin A treatment. This analysis led to the hypothesis that honaucin A exerts its anti-inflammatory activity through activation of the cytoprotective nuclear erythroid 2-related factor 2 (Nrf2)-antioxidant response element/electrophile response element (ARE/EpRE) signaling pathway. Activation of this pathway by honaucin A in cultured human MCF7 cells was confirmed using an Nrf2 luciferase reporter assay. In vitro alkylation experiments with the natural product and N-acetyl-L-cysteine suggest that honaucin A activates this pathway through covalent interaction with the sulfhydryl residues of the cytosolic repressor protein Keapl. Honaucin A presents a potential therapeutic lead for diseases with an inflammatory component modulated by Nrf2-ARE.
Graphical Abstract

Natural products produced by cyanobacteria have therapeutic potential in the treatment of many human diseases.1,2 The broadly bioactive nature of these compounds may be attributed, in part, to their impressive structural diversity. In addition, these specialized metabolites are thought to serve important ecological functions, including cell-cell communication and chemical defense.3 It is therefore likely that bioactive cyanobacterial natural products have been subject to evolutionary selection for increased binding specificity to conserved biological targets across phyla.4
Advancement of a bioactive compound with therapeutic potential is greatly facilitated by knowledge of its cellular target(s) and mechanism(s) of action (MOA). Natural products are most often identified as entities of interest by means of phenotypic cell, tissue, or whole organism screens. As such, at the stage of discovery, they lack clearly defined MOAs. Identification of the MOA(s) of a natural product can be difficult and time-consuming and, for some compounds, has not yet been possible.5 Fortunately, methods for MOA determination are constantly evolving. In addition to classical approaches, such as biochemical affinity assays, methods arising from technological advances in DNA/RNA sequencing, high-resolution/high-throughput microscopy, digital computing, and proteomics have helped expand the tool kits used for this task.5–12
Honaucin A is one bioactive small molecule for which there was a lack of mechanistic understanding. Originally isolated from the marine filamentous cyanobacterium Leptolyngbya crossbyana found overgrowing a Hawaiian coral reef, it was previously reported to inhibit both mammalian innate inflammation and bacterial quorum sensing in vitro.13 Given the promising bioactivity of honaucin A in mammalian cells and the ongoing need to develop novel and effective therapeutics to treat chronic inflammation and/or diseases with an inflammatory component, we further evaluated the molecule’s antiinflammatory properties in vivo as well as the molecular basis of its activity. Following transcriptomic analysis, we tested the hypothesis that honaucin A activates the cytoprotective nuclear erythroid 2-related factor 2 (Nrf2)-antioxidant response element/electrophile response element (ARE/EpRE) pathway.
The Nrf2-ARE/EpRE pathway serves as an environmental sensor for the cell, allowing it to monitor for the presence of oxidants, electrophiles, and xenobiotic compounds that may react with and thereby damage cellular components.14 In the presence of these insults, pathway activation results in translocation of transcription factor Nrf2 into the nucleus, triggering a detoxifying antioxidant response aimed at neutralizing the threat. Some weak electrophilic compounds, by virtue of their chemical structures, trigger this pathway in the absence of appreciable cellular damage and thus induce a net cytoprotective effect.
RESULTS AND DISCUSSION
Honaucin A Modulates in Vivo Inflammation.
The in vivo activity of honaucin A was assayed in a generalized hypersensitivity mouse ear edema model.15 The ability of the natural product to attenuate innate inflammation and subsequent ear edema in the presence of the irritant phorbol 12-myristate 13-acetate (PMA) was dependent upon the amount of natural product applied and displayed a biphasic profile (Figure 1).
Figure 1.

Honaucin A dose response in the mouse ear edema model. Low doses of honaucin A (A) caused a significant reduction in PMA-induced mouse ear edema, whereas the highest dose tested increased swelling significantly (B). Ears treated with honaucin A and PMA were compared to the paired PMA-treated ear, and the honaucin A-free, PMA positive control was compared to the paired vehicle-treated ear. All mean values between treated and untreated ears were significantly different when compared using the Wilcoxon’s signed ranks test (p ≤ 0.05).
Exposure to 1 μg of PMA per ear (1.62 nmol/ear) in the absence of the natural product caused a significant increase in swelling with the mean increase equal to 32.5 ± 7.5% of the average punch weight of the vehicle-treated ears (p = 0.02). Treatment with the greatest amount of honaucin A (3 μmol/ ear) also caused significant swelling (p = 0.02; mean increase in swelling = 51.4 ± 14.4%). Lower concentrations of honaucin A caused a significant reduction in PMA-induced ear edema; application of 1 μmol/ear resulted in a 27.6 ± 11.0% reduction (p = 0.03); application of 0.3 μmol/ear resulted in a 10.0 ± 4.1% reduction (p = 0.03), and application of 0.1 μmol/ear resulted in a 17.1 ± 3.5% reduction (p = 0.02).
The biphasic dose response for honaucin A was not wholly unexpected. The finding mimicked previous in vitro observations of cytotoxicity at higher compound concentrations when honaucin A was tested for its ability to diminish LPS-induced nitric oxide production in murine RAW 264.7 cells.13 Biphasic dose response curves have been reported for other chemical compounds with anti-inflammatory properties, including the natural products chloroquine and curcumin.16,17 Despite the augmented inflammation at its highest dose, the significant inhibition of inflammation induced by lower doses of honaucin A justified further exploration of its mechanism of action.
Treatment of Murine Macrophages with Honaucin A Results in Differential Regulation of Genes Involved in Inflammation and Redox Balance in a Manner Suggestive of Nrf2-ARE Activation.
Transcriptomic methods have proven to be valuable tools for generating hypotheses regarding the mechanisms of small molecules.8,11 RNA sequencing of the transcriptome of murine macrophages exposed to honaucin A allowed us to pinpoint differences in gene expression that resulted from treatment. Exposure of the murine macrophage cell line RAW 264.7 to honaucin A in the presence of proinflammatory lipopolysaccharide (LPS) induced the differential regulation of several genes as compared that of the LPS-only control (Figure 2).
Figure 2.
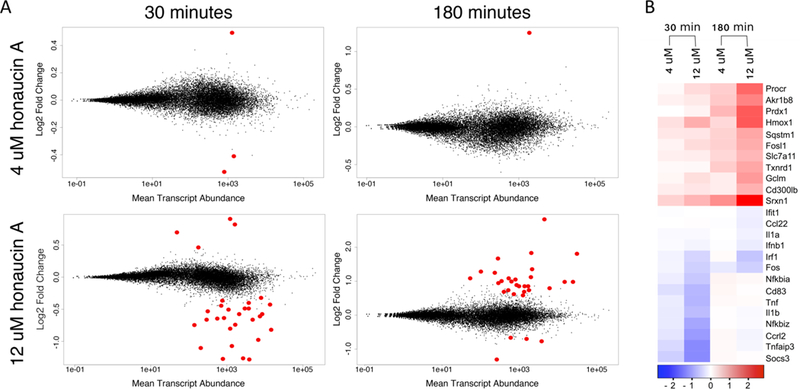
Differential gene expression in murine macrophages exposed to honaucin A. Significant differences in transcript abundance (red points) were observed in cells treated with a low or high concentration of honaucin A at two time points (A). Relative to LPS, RAW 264.7 cells treated with honaucin A and LPS displayed a general decrease in the abundance of transcripts involved in the inflammatory response and an increase in the abundance of transcripts related to phase II enzymes (B). Scale depicts the log2 fold change in abundance. See also Table S1.
The concentration of honaucin A was a greater determinant of differential gene expression than was incubation time. Exposure of cells to 4 μM honaucin A, the half-maximal inhibitory concentration (IC50) for nitric oxide production in LPS-stimulated RAW 264.7 cells,13 resulted in the differential expression of a small number of genes at both the earlier and later time points. In contrast, exposure of cells to 12 μM honaucin A elicited a greater degree of differential gene expression.
Time did influence the character of differential gene expression. At 30 min, the majority of differentially expressed genes in the 12 μM treatment were downregulated. At 180 min, the trend was reversed, and the majority of differentially expressed genes in the 12 μM treatment were upregulated.
Few statistically significant, differentially expressed genes were shared between treatments. Among shared genes with increased expression, only two genes, sulfiredoxin 1 (Srxnl) and heme oxygenase 1 (Hmoxl), were shared (Table S1). Srxnl, an enzyme involved in antioxidant metabolism, which may be expressed under conditions of oxidative stress to regenerate peroxiredoxins, was upregulated in the 4 treatment at both early and late time points as well as in the 12 μM treatment at the early time point. Although Srxnl also displayed increased expression in the 12 μM late time point treatment, the regulation did not meet the threshold of significance as determined by the R statistical package DESeq2. Hmoxl, an inducible regulator of inflammatory processes whose activation results in production of the antioxidant bilirubin, was upregulated in RAW 264.7 cells exposed to the higher concentration of honaucin A at both time points.
Of the downregulated genes, only two were shared among treatments. These two genes, suppressor of cytokine signaling 3 (Socs3), a regulator of STAT3 activation for cytokines belonging to the IL-6 family, and RasGEF domain family, member 1B (Rasgefl), a signaling network regulator that is induced via TLR activation by LPS, were downregulated at 30 min for both high and low honaucin A concentrations (Table S1).18,19 The other downregulated genes were all observed from the 12 μM treatment.
Overall, honaucin A treatment resulted in a trend of decreased abundance of transcripts of proinflammatory genes, including cytokines and chemokines (Figure 2B, Table S1). The finding mirrored previous in vitro findings in which honaucin A treatment of RAW 264.7 cells resulted in a dampened response to LPS. This included downregulation of nitric oxide production as well as decreased transcription of tumor necrosis factor alpha (Tnfa), interleukin-1-beta (Illfi), interleukin-6 (Il6), and inducible nitric oxide synthase (Nos2).13
An increase in the abundance of transcripts involved in redox balance was somewhat at odds with the anti-inflammatory profile observed from the diminished transcription of immune genes. It implied that honaucin A acts as an oxidant, inducing oxidative stress in macrophages (Figure 2B, Table S1). Transcriptional activation of many of the genes whose transcripts were observed in greater abundances, such as Hmoxl, Srxnl, Txrdnl, Prdxl, Sqstml, Gclm, Slc7all, and Akrlb8, is known to result from Nrf2 binding to the antioxidant response element (ARE/EpRE).20–22 One plausible explan ation that reconciles the apparent incongruity between honaucin A’s oxidant and anti-inflammatory effects is that the natural product activates the Nrf2-ARE/EpRE pathway.
Nrf2 is a transcription factor that, under basal conditions, is sequestered in the cell cytoplasm by its repressor protein Kelch-like ECH-associated protein 1 (Keapl).23 Keapl additionally binds Cul3, serving as an adapter protein between Nrf2 and the E3 ubiquitin ligase.24 The complex is ubiquitinated and degraded by the 26S proteasome during the normal turnover of these proteins in the cell (Figure 3A).
Figure 3.
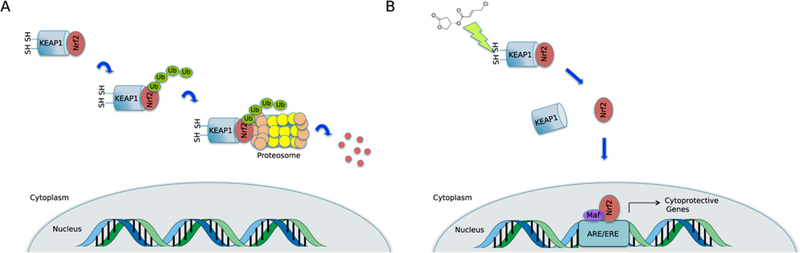
Nrf2-ARE signaling pathway. Under normoxic conditions, cytoplasmic Keap1 is complexed to the transcription factor Nrf2. The protein complex is ubiquitinated and degraded by the proteasome (A). If an oxidant or electrophile such as honaucin A is present, it may interact with cysteine residues on Keap1 leading to Nrf2 stabilization and translocation into the nucleus where it can bind to the antioxidant response element/electrophile response element (ARE/EpRE) initiating the transcription of cytoprotective genes (B).
Keapl contains either 25 (murine) or 27 (human) cysteine residues that act as environmental sensors.25 If oxidants or electrophiles are present in the cytoplasm, they are able to interact with the sulfhydryl groups of these cysteines. The number and identities of the residues to which they bind depend on the architecture of the interacting molecule and its strength as a Michael acceptor. Moreover, a molecule’s pattern of cysteine residue engagement may be referred to as its “cysteine code”.26 Dynamics may differ depending on the character of this interaction and other factors (see refs 27 and 28), but interaction with these cysteine residues ultimately promotes dissociation of Nrf2 from Keapl. Nrf2 then enters the nucleus where, in concert with small Maf proteins, it forms a complex with the ARE/EpRE promoter and initiates the transcription of cytoprotective genes (Figure 3B).
Ultimately, transcription of Nrf2-activated genes results in the increased presence of detoxifying and antioxidant phase II enzymes. For this reason, they have been termed “indirect antioxidants”.20‘29 Classical antioxidants administered through diet or otherwise may not reach the site at which they are needed and, in the absence of regenerating enzymes, are consumed when they react with an electrophile restricting their usefulness to a single chemical reaction. These factors limit the therapeutic utility of classical antioxidants. Indirect antioxidants, conversely, prompt the production of antioxidants such as glutathione and bilirubin in situ. They also prompt the production of enzymes able to regenerate these antioxidants so that they may react multiple times to neutralize chemical threats. This renders indirect antioxidants much more potent than classical antioxidants. Thus, although the pathway is triggered by oxidant species and electrophiles, it ultimately has a protective outcome; this is consistent with the reduction of inflammation observed upon honaucin A exposure.
Because of the generalized response that results from Nrf2-ARE/EpRE activation, activating molecules exert pleiotropic effects. Neuroprotective, chemoprotective, and anti-inflammatory effects have been reported for these compounds.30 There is also evidence of increased proteasomal activity as well as antibacterial and antifungal properties.31,32 One example of a synthetic Nrf2-ARE activator, dimethyl fumarate, is prescribed to treat relapsing multiple sclerosis under the trade name Tecfidera. Besides honaucin A, other marine natural product activators of Nrf2-ARE include halomadurones C and D from an Actinomadura sp. cultured from an ascidian, zonarol from the brown alga Dictyopteris undulate, and strongylophorine-8 from the sponge Petrosia (Strongylophora) corticata, all of which were investigated for their potential neuroprotective properties.33–35
Confirmation of ARE/EpRE Engagement by Nrf2 in the Presence of Honaucin A.
For evaluating our prediction of Nrf2-ARE/EpRE involvement in the MOA of honaucin A, the ability of the natural product to stabilize the transcription factor Nrf2 was assessed using an in vitro luciferase reporter assay. When honaucin A was added to the human breast cancer cell line MCF7 into which an Nrf2-responsive luciferase construct had been stably integrated, an increase in luminescence in the presence of firefly luciferase substrate was observed relative to that of untreated cells (Figure 4A). The level of Nrf2 activation was compared to the known Nrf2 activators tert-butylhydroquinone (tBHQ) and dimethyl fumarate (DMF).
Figure 4.

Activation of the Nrf2-ARE pathway in MCF7 cells in the presence of honaucin A. Honaucin A displayed a robust and dose-dependent activation of Nrf2-ARE relative to untreated cells (A). Less activation at higher concentrations of the natural product coincides with a cytotoxic effect at these doses (B).
Honaucin A, at all but the highest concentration (15 μM), caused Nrf2 stabilization and subsequent binding to the ARE/ EpRE comparable to that caused by tBHQ and DMF. The contribution of some cytotoxicity at higher compound concentrations confounded the interpretation of honaucin A-induced Nrf2 activity levels in the reporter assay at these doses (Figure 4B). tBHQ and DMF were exempt from major cytotoxicity and displayed a linear increase in activation with the greatest activation occurring at the highest concentrations.
ARE/EpRE promoter binding confirmed that honaucin A activates the Nrf2-ARE pathway. More robust pathway activation at lower concentrations was in contrast to higher concentrations of honaucin A at which cytotoxicity prevailed. These findings mimicked prior results from in vitro assays of nitric oxide inhibition13 and the in vivo ear edema assay results reported here. Bell-shaped dose-response curves are not uncommon for Nrf2 activators.35,36 It appears that an Nrf2 stabilizing effect predominates at low honaucin A concentrations, whereas at higher concentrations, other factors contribute to a shift toward cytotoxicity.
Although the precise cause of honaucin A cytotoxicity at higher concentrations is unknown, it has generally been recognized that electrophiles can exert either cytoprotective or cytotoxic effects. When cytoprotective effects predominate and cells detoxify alkylating agents through Nrf2 stabilization, this has been termed “electrophile counterattack”.37 In contrast, some electrophiles decrease the overall reductive capacity of the cell and thereby induce apoptosis.37 This can occur if the electrophile reacts with the sulfhydryl groups of glutathione, compromising the cell’s response to oxidants and alkylating agents. Other mechanisms through which this type of electrophile response can occur include macromolecule damage, reactive oxygen species production, dysregulation of signaling pathways, and mitochondrial impairment.37,38
Gloire et al. have proposed a hierarchical model of oxidative stress, wherein the cellular response to a stressor is dependent upon the intensity of the stressor.39 Low stress initiates signaling through Nrf2, intermediate stress engages NFκB, AP-1, and MAP kinase pathways, and high levels of oxidative stress overwhelm the electrophile counterattack response and culminate in cell death. This high stress type of response is typified by strong electrophiles or by persistent electrophile bombardment. Electrophile counterattack is generally elicited by weak electrophiles such as tBHQ and DMF and, as shown here, honaucin A.
Covalent Interaction of Honaucin A and N-Acetylcys-teine and a Proposed Structural Basis for Honaucin A Nrf2 Activation.
One mechanism of Nrf2 stabilization by small molecules involves their covalent interaction with the cysteine residues of the Kelch-like ECH-associated protein 1 (Keapl) repressor.23 We hypothesized that honaucin A might also interact with Keap1 in this way. For this hypothesis to be tested, honaucin A was incubated with the cysteine-bearing small molecules N-acetylcysteine (NAC) and reduced glutathione (GSH) (Figure S1). Addition products were interrogated using high-resolution mass spectrometry.
Honaucin A reaction with NAC formed the predicted adduct and its derivative (Figure 5). This likely occurred through 1,4-conjugate addition in which the electron-withdrawing carbonyl oxygen and chlorine atom of honaucin A contributed to make the 4-position carbon of the Michael acceptor highly electropositive and thus very reactive toward the nucleophilic thiolate of NAC (Figure S2). Interestingly, there was no evidence that honaucin A reacted with glutathione (data not shown).
Figure 5.
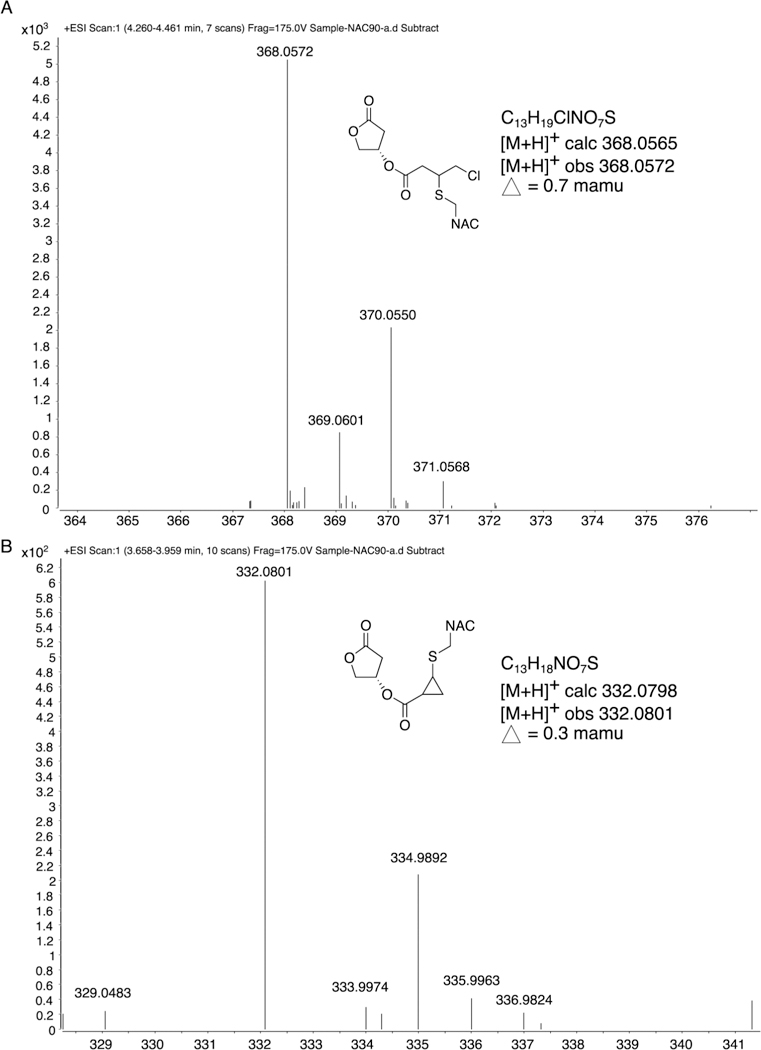
Reaction of honaucin A with the cysteine-bearing small molecule N-acetylcysteine (NAC) through 1,4-conjugate addition to produce two products. The later eluting peak was centered at 4.3 min (A) and had an [M + H]+ m/z 368.0572 (C13H19ClNO7S) for the SNAC addition product, whereas the earlier eluting peak was centered at 3.8 min (B) and showed an [M + H]+ m/z 332.0801 (C13H18NO7S) for the SNAC addition plus loss of HCl to produce the proposed cyclopropyl ring compound. This indicates that honaucin A may stabilize Nrf2 by engaging sulfhydryl residues of the repressor protein Keap1. See also Figures S1 and S2.
Nrf2-ARE/EpRE pathway activators often share the common structural feature of an electrophilic Michael acceptor motif. This motif enables electrophilic Nrf2-ARE activators to react covalently with cysteine residues on Keapl through S-alkylation. The sulfhydryl group of cysteine is ionizable and can be deprotonated to yield a reactive thiolate group; in addition, the local protein environment around cysteine sulfhydryl residues can stabilize nucleophilic thiolates.40 Cysteine-mediated reactivity and post-translational modification of Keapl make it an ideal environmental sensor able to detect reactive electrophile species (RES) (and also oxidants such as reactive oxygen species and reactive nitrogen species) in the reducing cellular environment. Potential threats may then be neutralized to avoid damage to the cell.
Honaucin A is composed of one Michael acceptor system, an unsaturated ester motif with an electron-withdrawing group (Figure S2). We hypothesize that honaucin A’s structural architecture and electronic configuration facilitate interaction with sulfhydryl groups on Keapl, thereby liberating Nrf2 to enter the nucleus and initiate transcription of cytoprotective genes. Mass spectrometric (MS) analysis of the reaction products that resulted from the interaction of honaucin A and NAC confirmed the ability of the marine natural product to interact with cysteine through 1,4-conjugate addition in the manner predicted. This indicates that honaucin A might also alkylate the cysteine thiols of Keapl to activate Nrf2. Preparation of a fluorescent honaucin A analogue and observation of its cellular localization in murine macrophages demonstrated that the compound was extranuclear, supporting its activation of Nrf2 through Keapl engagement (Supplementary Experimental Procedures, Figures S3–S5). The concept that honaucin A operates through this mechanism is also supported by the results of a previous structure-activity relationship study in which structural alterations to the molecule’s Michael acceptor system diminished its ability to attenuate LPS-induced nitric oxide production in murine macrophages13
The lack of in vitro reaction between glutathione and honaucin A in our hands suggests that honaucin A does not exert toxic effects through depletion of the cell’s reductive capacity. Rather, its initiation of Nrf2-ARE signaling as well as its induction of phase II enzymes and glutathione synthesis signify that at low doses it is a weak electrophile that elicits an electrophile counterattack from cells. Cytotoxicity at higher concentrations may arise from an independent mechanism.
Broader Implications of Nrf2-ARE Activation for Honaucin A Bioactivity.
The Nrf2-ARE signaling pathway is highly conserved among eukaryotes.41 Bacteria also possess an oxidative stress system that relies on sulfur chemistry.42,43 It has been shown that, in addition to maintaining cellular redox balance, reactive thiols in bacteria are used in signaling to initiate expression of genes not directly involved in redox homeostasis. Vibrio cholerae employs one such thiol switch to activate expression of virulence-related genes.44 Perhaps of even greater interest, redox sensing and the quorum sensing accessory gene regulator (agr) signaling system are linked in Staphylococcus aureus.45 Oxidation of Cys-199 on the quorum sensing response regulator AgrA induces transcription of genes associated with oxidative stress resistance. Also related to quorum sensing, halogenated furanone natural products from the red alga Delisea pulchra were shown to alkylate the cysteine residues on LuxS and thereby inactivate AI-2 quorum sensing.46 We have previously observed quorum sensing inhibition in Vibrio harveyi by honaucin A.13 It may be that this activity as well is regulated through modification of cysteine thiols, thereby providing a structural and chemical basis for interkingdom signaling by honaucin A.47
In conclusion, honaucin A has been shown to inhibit inflammation both in vitro and in vivo. Experimental evidence indicates that, at low doses, honaucin A targets the NRF2-ARE/ EpRE pathway and activates cytoprotective genes, generating an anti-inflammatory response. Further investigation of honaucin A as an anti-inflammatory agent would be of great interest.
EXPERIMENTAL SECTION
General Experimental Procedures.
Synthetic honaucin A was used in all assays and measurements. For bioassays, lipopolysacchar-ides from Escherichia coli (O26:B6) as well as phorbol 12-myristate 13-acetate and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide were purchased from Sigma-Aldrich (St. Louis, MO). NRF2 luciferase reporter MCF7 stable cells were purchased from Signosis (Santa Clara, CA). RNA sequencing was carried out using an Illumina Hiseq2000 (San Diego, CA) following RNA quality assessment via Bioanalyzer 2100 (Agilent, Santa Clara, CA). For in vitro alkylation experiments, reaction products were separated via liquid chromatography using a Phenomenex Kinetex 5 μm EVO C-18 column. Highresolution mass analysis was accomplished with an Agilent 6230 ESI-TOF-MS operating in positive mode.
Dose Response Ear Edema Assay.
Eight-week old female CD1 mice were divided into treatment groups of six animals. Animals were anesthetized with isoflurane, and the underside of the right ear was treated with 10 of either water/ethanol/polysorbate 20 vehicle or honaucin A at 3.0, 1.0, 0.3, or 0.1 μmol/ear. The underside of the left ear was treated with vehicle only. Ten minutes after application of the compound and/or vehicle, 1 μg of PMA in 10 μL was applied to the underside of both ears except in the case of a PMA-only control. For this control, no test compounds were used, and the irritant was applied to the right ear and vehicle was applied to the left ear. At 6 h, the animals were euthanized, and 6 mm biopsy punches were collected from both ears and weighed. The percent inhibition of edema was calculated as (vehicle — treatment/vehicle) × 100. Statistical significance was evaluated with GraphPad Prism 6 software using the one-tailed Wilcoxon signed rank test. The significance level was set at 0.05, and p-values were rounded to two decimals. The experiment was carried out under a protocol approved by the Animal Subjects Committee at the University of California, San Diego.
Treatment of RAW 264.7 Cells with Honaucin A and RNA Isolation.
RAW 264.7 murine macrophages were grown in Dulbecco’s modified Eagle medium (Gibco) supplemented with 10% fetal bovine serum, penicillin, and streptomycin. Cells were seeded in 6-well plates and grown to near confluency. EtOH or honaucin A (4 or 12 μM) in EtOH were added to the appropriate wells, and plates were incubated for 1 h at 37 °C with 5% CO2. Lipopolysaccharide (0.5 μg/mL) was then added to all treatments. Three replicates comprised each treatment. The treated cells were incubated for 30 or 180 min after which total RNA was isolated and purified with TRIzol (Life Technologies) according to the manufacturer’s protocol.
RNA Sequencing.
RNA quality was determined using an Agilent Technologies Bioanalyzer 2100. Following rRNA depletion and library preparation using standard Illumina reagents and protocols, Illumina Hiseq2000 sequencing was completed to generate 100 base pair singleend reads. RNA quality assessment, library preparation, and sequencing were completed at the Next Generation Sequencing Core at The Scripps Research Institute in La Jolla, California. Data were deposited to the National Center for Biotechnology Information Gene Expression Omnibus database (GEO: GSE93558).
Read Processing and Differential Expression Analysis.
Quality of the sequenced reads was inspected using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Sequencing adapters were removed from reads using the fastx toolkit clipper tool (http://hannonlab.cshl.edu/fastx_toolkit/index.html). Quality trimming was done using Prinseq (http://prinseq.sourceforge.net/). All reads with a phred quality score less than 20 and all reads shorter than 55 base pairs after polyA trimming were excluded from subsequent analysis. The ultrafast universal RNA-seq aligner STAR was used to map reads to the Ensembl Mus musculus GRCm38.75 reference genome.48 The Python package HTSeq (http://wwwhuber.embl.de/HTSeq/doc/overview.html), specifically htseq-count, was used to quantify the number of reads that mapped to each feature in the genome.49 Once counts were obtained for the different experimental treatments, the R statistical package DESeq2 (https://bioconductor.org/packages/release/bioc/html/DESeq2. html) was used to perform differential expression analysis.50 This was accomplished by performing pairwise comparisons of each treatment to its appropriate LPS control.
NRF2 Luciferase Reporter MCF7 Stable Cell Line Assay.
NRF2 luciferase reporter MCF7 stable cells (Signosis) were grown in Dulbecco’s modified Eagle medium (Gibco) supplemented with 10% fetal bovine serum, 10,000 units/mL of penicillin, 100 μg/mL of streptomycin, and 75 μg/mL of G418 (Life Technologies). The day before the assay, cells were seeded into white 96-well flat-bottomed plates at a density of 5 × 104 cells/220 μL/well and allowed to adhere and grow overnight at 37 °C with 5% CO2. Honaucin A and controls were solubilized in DMSO and added to the plate the following day in a volume of 2 μL. Concentrations tested ranged from 1.98 to 15.0 μM. The known Nrf2-ARE/EpRE activators tBHQ and DMF served as positive controls and DMSO as the negative control. After compound addition, the plates were again incubated overnight at 37 °C with 5% CO2. After incubation, the media were removed, and the cells were washed in phosphate buffered saline. Twenty-five microliters of passive lysis buffer (Promega) was added to cells, which were then incubated at room temperature for 15 min. Firefly luciferase substrate (Signosis) was added via injector, and the relative luminescence of each well was read using a GloMax Microplate Luminometer (Promega). Three biological replicates of honaucin A and tBHQ and two biological replicates of DMF, each consisting of three technical replicates, were assayed.
Relative luminescence units (RLU) for the technical replicates were averaged and compared to the average RLU of the untreated cells to arrive at the percentage of Nrf2-ARE activation over background via the formula (treatment — cell background/cell background) × 100. Biological replicates were then averaged, and the values for DMSO vehicle-treated cells were subtracted from the overall percentage for each treatment. Standard error for each treatment was calculated.
MTT Assay for Cell Viability.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma, St. Louis, MI) was used to measure the viability of the treated MCF7 cells. NRF2 luciferase reporter MCF7 stable cells (Signosis) were grown, seeded, and treated in an identical manner as for the luminescent assay with the exception that clear plates were used. The day after cell treatment, medium was aspirated, and the cells were washed with PBS. Then, 60 μL of 1 mg/ mL MTT in serum-free DMEM was added to each well, and the plates were incubated for 25 min at 37 °C. After the incubation, the medium with MTT was aspirated, and the plates were dried. Then, 100 μL of DMSO was added to each well, and the absorbance was measured at 630 and 570 nm. Background absorbance was subtracted, and viability for each treatment was calculated as a percentage of cell survival compared to that of an untreated control. Data are expressed as mean ± standard error.
In Vitro Alkylation of Honaucin A.
N-Acetyl-L-cysteine (NAC, Spectrum Chemical) was dissolved in milli-Q water, and synthetic honaucin A was solubilized in DMSO. Honaucin A was incubated with NAC in either 2-or 50-fold excess with stirring under argon for 2 h at room temperature (after Wang et al., 2013).22 At the conclusion of the reaction, vial contents were passed over a Bond Elut-C18 SPE column (Agilent) that had been washed with 3 column volumes of methanol and then equilibrated with 3 column volumes of water. The reaction products were subsequently eluted with increasing percentages of methanol. Elutions were dried using rotary evaporation and then reconstituted at a concentration of 1 mg/mL in 50:50 methanol/water. This preparation was analyzed for the presence of the hypothesized addition products via high-resolution MS at the Molecular Mass Spectrometry Facility at the University of California, San Diego. Briefly, reaction products were separated via liquid chromatography using a Phenomenex Kinetex 5 μm EVO C-18 column prior to being introduced to an Agilent 6230 ESI-TOF-MS running in positive mode for high-resolution mass measurement.
Supplementary Material
ACKNOWLEDGMENTS
RNA sequencing and subsequent data analyses were supported by NIEHS/NIH R21ES024105 and NEI/NIH R01EY022306 to T.G. NIH CA100851 and NIH ES024105 to L.G. Additional research support was provided to S.J.M. through NIH Marine Biotechnology Training Fellowship NIH GM067550, P.E.O. Scholar Award, Claude E. Zobell Fellowship, Robert Scripps Fellowship, SIO Cheng An Lun Fellowship, SIO Haymet First Year Fellowship, and Wyer Family Fellowship.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnat-prod.7b00734.
Reaction scheme for honaucin A and N-acetyl-L-cysteine, honaucin A structure with Michael acceptor motif highlighted, synthetic scheme for generation of the fluorescent probe, bioactivity data for the fluorescent probe, confocal images of the fluorescent probe and cells, and supporting experimental procedures (PDF)
Accession Codes
All data used in the study may be accessed through the National Center for Biotechnology Information Gene Expression Omnibus database (GEO: GSE93558).
The authors declare no competing financial interest.
DEDICATION
Dedicated to Dr. Susan Band Horwitz, of Albert Einstein College of Medicine, Bronx, NY, for her pioneering work on bioactive natural products.
REFERENCES
- (1).Gerwick WH; Moore BS Chem. Biol. 2012, 19, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).McKerrow JH Nat. Prod. Rep. 2015, 32, 1610–1611. [DOI] [PubMed] [Google Scholar]
- (3).Puglisi MP; Sneed JM; Sharp KH; Ritson-Williams R; Paul VJ Nat. Prod. Rep. 2014, 31, 1510–1553. [DOI] [PubMed] [Google Scholar]
- (4).Koehn FE; Carter GT Nat. Rev. Drug Discovery 2005, 4, 206–220. [DOI] [PubMed] [Google Scholar]
- (5).Gregori-Puigjané E; Setola V; Hert J; Crews BA; Irwin JJ; Lounkine E; Marnett L; Roth BL; Shoichet BK Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11178–11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Alves MJ; Froufe HJ; Costa AF; Santos AF; Oliveira LG; Osório SR; Abreu RM; Pintado M; Ferreira IC Molecules 2014, 19, 1672–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Flannery EL; Fidock DA; Winzeler EA J. Med. Chem. 2013, 56, 7761–7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kibble M; Saarinen N; Tang J; Wennerberg K; Mäkelä S; Aittokallio T Nat. Prod. Rep. 2015, 32, 1249–1266. [DOI] [PubMed] [Google Scholar]
- (9).Ong SE; Li X; Schenone M; Schreiber SL; Carr S A Methods Mol. Biol. 2012, 803, 129–140. [DOI] [PubMed] [Google Scholar]
- (10).Parsons AB; Brost RL; Ding H; Li Z; Zhang C; Sheikh B; Brown GW; Kane PM; Hughes TR; Boone C Nat. Biotechnol. 2004, 22, 62–69. [DOI] [PubMed] [Google Scholar]
- (11).Schenone M; Danak V; Wagner BK; Clemons PA Nat. Chem. Biol. 2013, 9, 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Schulze CJ; Bray WM; Woerhmann MH; Stuart J; Lokey RS; Linington RG Chem. Biol. 2013, 20, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Choi H; Mascuch SJ; Villa FA; Byrum T; Teasdale ME; Smith JE; Preskitt LB; Rowley DC; Gerwick L; Gerwick WH Chem. Biol. 2012, 195, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dinkova-Kostova AT; Holtzclaw WD; Kensler TW Chem. Res. Toxicol. 2005, 18, 1779–1791. [DOI] [PubMed] [Google Scholar]
- (15).Gábor MJ Toxicol., Cutaneous Ocul. Toxicol. 2002, 213, 191–202. [Google Scholar]
- (16).Ali RE; Rattan SI Ann. N. Y. Acad. Sci. 2006, 1067, 394–399. [DOI] [PubMed] [Google Scholar]
- (17).Park D; Lee Y Dev. Reprod 2014, 18, 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Andrade WA; Silva AM; Alves VS; Salgado AP; Melo MB; Andrade HM; Dall’Orto FV; Garcia SA; Silveira TN; Gazzinelli RT Genes Immun. 2010, 116, 447–457. [DOI] [PubMed] [Google Scholar]
- (19).Carow B; Rottenberg ME Front. Immunol. 2014, 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Jung KA; Kwak MK Molecules 2010, 15, 7266–7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liu Y; Kern JT; Walker JR; Johnson JA; Schultz PG; Luesch H Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 5205–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang R; Mason DE; Choe KP; Lewin AS; Peters EC; Luesch H ACS Chem. Biol. 2013, 8, 1764–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Itoh K; Wakabayashi N; Katoh Y; Ishii T; Igarashi K; Engel JD; Yamamoto M Genes Dev. 1999, 13, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kobayashi A; Kang MI; Okawa H; Ohtsuji M; Zenke Y; Chiba T; Igarashi K; Yamamoto M Mol. Cell. Biol. 2004, 24, 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dinkova-Kostova AT; Holtzclaw WD; Cole RN; Itoh K; Wakabayashi N; Katoh Y; Yamamoto M; Talalay P Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 11908–11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kobayashi M; Li L; Iwamoto N; Nakajima-Takagi Y; Kaneko H; Nakayama Y; Eguchi M; Wada Y; Kumagai Y; Yamamoto M Mol. Cell. Biol. 2009, 29, 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sun Z; Chin YE; Zhang DD Mol. Cell. Biol. 2009, 29, 2658–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Sun Z; Huang Z; Zhang DD PLoS One 2009, 4, No. e6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dinkova-Kostova AT; Talalay P Mol. Nutr. Food Res. 2008, 52 (Suppl 1), S128–138. [DOI] [PubMed] [Google Scholar]
- (30).Singh S; Vrishni S; Singh BK; Rahman I; Kakkar P Free Radical Res. 2010, 44, 1267–1288. [DOI] [PubMed] [Google Scholar]
- (31).Kwak MK; Huang B; Chang H; Kim JA; Kensler TW Life Sci. 2007, 80, 2411–2420. [DOI] [PubMed] [Google Scholar]
- (32).Wang HH; Sun DW; Kuang R Int. J. Food Microbiol. 2001, 65, 125–130. [DOI] [PubMed] [Google Scholar]
- (33).Sasaki S; Tozawa T; Van Wagoner RM; Ireland CM; Harper MK; Satoh T Biochem. Biophys. Res. Commun. 2011, 415, 6–10. [DOI] [PubMed] [Google Scholar]
- (34).Shimizu H; Koyama T; Yamada S; Lipton SA; Satoh T Biochem. Biophys. Res. Commun. 2015, 457, 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wyche TP; Standiford M; Hou Y; Braun D; Johnson DA; Johnson JA; Bugni TS Mar. Drugs 2013, 11, 5089–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hybertson BM; Gao B; Bose SK; McCord JM Mol. Aspects Med. 2011, 32, 234–246. [DOI] [PubMed] [Google Scholar]
- (37).Satoh T; Lipton SA Trends Neurosci. 2007, 30, 37–45. [DOI] [PubMed] [Google Scholar]
- (38).Itoh K; Tong KI; Yamamoto M Free Radical Biol. Med. 2004, 36, 1208–1213. [DOI] [PubMed] [Google Scholar]
- (39).Gloire G; Legrand-Poels S; Piette J Biochem. Pharmacol. 2006, 72, 1493–1505. [DOI] [PubMed] [Google Scholar]
- (40).Poole LB Free Radical Biol. Med. 2015, 80, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fuse Y; Kobayashi M Molecules 2017, 223, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Antelmann H; Helmann JD Antioxid. Redox Signaling 2011, 14, 1049–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lushchak VI Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol. 2011, 153, 175–190. [DOI] [PubMed] [Google Scholar]
- (44).Liu Z; Yang M; Peterfreund GL; Tsou AM; Selamoglu N; Daldal F; Zhong Z; Kan B; Zhu J Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 810–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sun F; Liang H; Kong X; Xie S; Cho H; Deng X; Ji Q; Zhang H; Alvarez S; Hicks LM; Bae T; Luo C; Jiang H; He C Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9095–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zang T; Lee BW; Cannon LM; Ritter KA; Dai S; Ren D; Wood TK; Zhou ZS Bioorg. Med. Chem. Lett. 2009, 19, 6200–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Gerwick L; Boudreau P; Choi H; Mascuch SJ; Villa FA; Balunas MJ; Malloy KL; Teasdale ME; Rowley DC; Gerwick WH Phytochem. Rev. 2013, 12, 459–465. [Google Scholar]
- (48).Dobin A; Davis CA; Schlesinger F; Drenkow J; Zaleski C; Jha S; Batut P; Chaisson M; Gingeras TR Bioinformatics 2013, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Anders S; Pyl PT; Huber W Bioinformatics 2015, 312, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Love MI; Huber W; Anders S Genome Biol. 2014, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.