

Abstract
Glycogen synthase kinase 3 (GSK3), a key regulatory kinase in the WNT pathway, remains a therapeutic target of interest in many diseases. While dual GSK3α/β inhibitors have entered clinical trials, none has successfully translated to clinical application. Mechanism-based toxicities, driven in part by the inhibition of both GSK3 paralogs and subsequent β-catenin stabilization, are a concern in the translation of this target class to cancer therapy, particularly for the treatment of acute myeloid leukemia (AML). Knockdown of GSK3α or GSK3β individually does not increase β-catenin in certain cellular subtypes and offers a conceptual resolution to targeting GSK3: paralog-selective inhibition. However, inadequate chemical tools exist. The design of selective ATP competitive inhibitors poses a drug discovery challenge due to the high homology (95% identity, 100% similarity) in their ATP binding domains. Taking advantage of an Asp133→Glu196 “switch” in their hinge binding domains, we present a rational design strategy towards the discovery of a set of paralog selective GSK3 inhibitors. These first-in-class GSK3α and GSK3β selective inhibitors provide insights into GSK3 targeting in AML where GSK3α has been identified as a therapeutic target using genetic approaches. Our GSK3α selective compound (BRD0705) inhibits kinase function and does not stabilize β-catenin, mitigating potential neoplastic concerns. BRD0705 induces myeloid differentiation and impairs colony formation in AML cells while no effect is observed on normal hematopoietic cells. Moreover, BRD0705 impairs leukemia initiation and prolongs survival in AML mouse models. These studies validate feasibility of paralog selective GSK3α inhibition offering a promising therapeutic approach in AML.
One Sentence Summary:
Exploiting a single amino acid difference in GSK3 kinases’ ATP binding domains to design a first-in-class set of paralog selective inhibitors for AML.
Introduction
The development of selective, targeted small molecules to understand the mechanisms of human diseases and improve their treatment remains a challenge. This challenge is pronounced when selective inhibitors of highly similar and ubiquitously expressed proteins are desirable. One exemplary target class, relevant to numerous biological pathways and human diseases, is glycogen synthase kinase 3 (GSK3).
Multiple eukaryotic kinases have undergone gene duplication events resulting in two or more paralog genes. Duplicate genes are thought to diverge either in their regulation or their biochemical functions. GSK3 represents an example of such an evolutionary selection system, resulting in two paralog genes 1, GSK3A on chromosome 19 and GSK3B on chromosome 3. The encoded paralog proteins, GSK3α and GSK3β, have a high degree of similarity (Figure 1A, 67% overall amino acid identity and 95% identity in the ATP binding site 2). While some functional overlap is clear, the GSK3α and GSK3β paralogs also have distinct, albeit uncharacterized, functions 3,4. Both GSK3 paralogs are ubiquitously expressed and implicated in the pathophysiology of a number of human disorders 5,6, including non-insulin-dependent diabetes mellitus 7, cardiac hypertrophy 8,9, neurological and neurodevelopmental disorders (e.g., bipolar disorder and Alzheimer’s disease) 10 and cancer 11–16. The precise mechanistic role of each paralog in disease pathogenesis, however, has been difficult to determine given the lack of paralog selective chemical probes.
Figure 1. A single amino acid difference in the ATP binding domain of GSK3α and GSK3β leads to structural and topological differences.
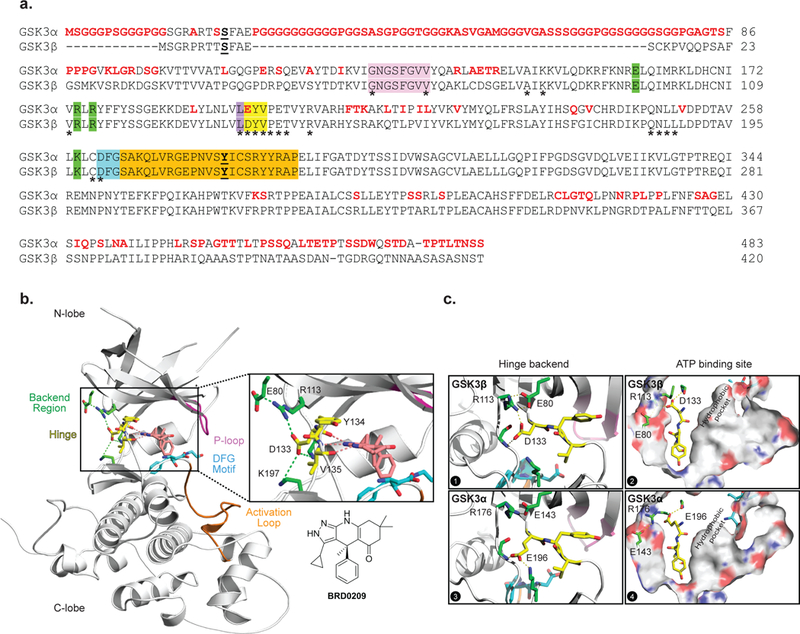
(a) Aligned primary amino acid sequences of GSK3α and GSK3β. Residue differences between the two paralogs are in red. The main regions of the kinases are color-coded: hinge (yellow), backend region (green), P-loop (pink), DFG motif (blue) and activation loop (orange). (b) 2.4 Å resolution X-ray crystal structure of hGSK3β bound to BRD0209, a dual GSK3α/β inhibitor. In the inset, a previously undescribed hydrogen bond network, centered on the Asp133 side chain, on the backend region of the hinge connecting the N-and C-lobes of GSK3. The main regions of the kinases are again color-coded. (c) A comparison between the hinge backend and ATP binding site of apo GSK3β and a MD simulation of apo GSK3α highlight a size difference between their hydrophobic pockets.
A large number of dual GSK3α/β inhibitors from varied chemotypes have been reported: the natural product-based indirubins (e.g., 6-bromoindirubin-3’-oxime, BIO), paullones (e.g., alsterpaullone), bisarylmaleimides (e.g., SB-216763, LY2090314), indolocarbazoles (e.g., staurosporine), synthetically derived aminopyrimidines (e.g., CHIR99021), ruthenium complexes (e.g., complex 5), thiadiazolidinones (e.g., TDZD-8, tideglusib), and imino-thiadiazoles (e.g., VP1.14) 17–20. Few of these inhibitors possess good selectivity versus the human kinome, a property paramount to the unambiguous interpretation of in vitro and in vivo effects as solely driven by GSK3 inhibition 21. Furthermore, as might be expected, paralog selective inhibitors of GSK3α or GSK3β with demonstrated functional selectivity in vitro and activity in vivo are non-existent. Oxadiazole-based selective GSK3α inhibitors have been described, but their functional selectivity or activity in mammalian models in vivo, has not been characterized. 22–24 In addition, although a small molecule preferentially inhibiting GSK3β has been recently reported, its degree of paralog selectivity and off-target inhibition against other kinases complicates the use of these molecules to study the biology of GSK3 paralogs.25
One concern in the clinical application of dual GSK3α/β inhibitors is the predicted stabilization and nuclear translocation of β-catenin19–21, a direct GSK3 phosphorylation substrate in the APC/AXIN/GSK3 complex. Indeed, this is likely a problem in the translation of dual GSK3α/β inhibitors in the treatment of acute myeloid leukemia (AML), a poorly treated form of cancer in need of new therapies. Numerous studies support the targeting of GSK3 in AML 26–30, however, the well-established oncogenic role of β-catenin activation in the context of myeloid leukemia, results in a clear biological challenge to targeting GSK3 in this disease 31–33. We recently suggested a more parsimonious approach to targeting GSK3 in AML. We found that selective genetic suppression of the GSK3α paralog promotes AML differentiation and impairs leukemia progression in mouse models of AML without increasing β-catenin 34. Additionally, Woodgett et al. reported that selective genetic suppression of either GSK3 paralog in embryonic stem cells and whole body KO of GSK3α in mice does not increase β-catenin 22. A gene-dosage effect resulting in β-catenin pathway activation was observed only in cells lacking three or all four of the GSK3 alleles. Interestingly, in an adult hematopoietic stem cell (HSC) context, GSK3β paralog KO or dual GSK3α/β KO, respectively, induced a pre-neoplasic disease and overt AML, whereas HSC GSK3α KO did not result in myeloid transformation 31. Therefore, paralog selective inhibition of GSK3α should avoid this mechanism-based safety concern.
In this study, we aimed to develop small molecule, paralog selective inhibitors of GSK3. Additionally, we intended to determine if the pharmacological inhibition of the GSK3α paralog would mimic its paralog selective genetic suppression in the context of AML differentiation and leukemia progression. Starting from BRD0209, a highly kinome selective GSK3 inhibitor recently published by our group 21, a set of chemically “matched” or isochemogenic inhibitors, with paralog selectivity, were rationally designed, synthesized and characterized. We applied this set of selective GSK3α and GSK3β inhibitors to a series of cellular assays to refine our understanding of the individual paralogs in driving mechanism-based β-catenin toxicities. BRD0705, our selective GSK3α inhibitor, induced differentiation, reduced transcriptional programs of stemness and impaired colony formation in AML cell lines and primary patient samples without affecting normal hematopoietic cell growth. Importantly, BRD0705 did not induce β-catenin stabilization or nuclear translocation at concentrations efficacious in multiple mouse models of AML, resulting in leukemia initiation impairment and prolonged survival. Taken together, our findings suggest that paralog selective, small molecule inhibition of GSK3α offers a safe and novel therapeutic strategy for the treatment of AML.
Results
Structure-based design considerations.
GSK3α and GSK3β, which are highly similar in terms of sequence and domain architecture, are highly conserved across species (Figure S1). The primary sequence of human GSK3α is 67% identical to human GSK3β 2 with the least homology in the N-and C-terminal domains (difference in amino acid sequence in red, Figure 1a). Inspection of the adenosine triphosphate (ATP) binding domain (* in Figure 1a) reveals a 95% amino acid sequence identity (100% similarity) with a single amino acid difference in the hinge binding region (amino acids in yellow): a glutamic acid (Glu(E)196) in GSK3α to an aspartic acid (Asp(D)133) in GSK3β.
Our group recently published the discovery of kinome selective, dual inhibitors of the GSK3 kinases, such as BRD0209 (Figure 1b) 21. A high-resolution (2.4 Å) X-ray crystal structure of hGSK3Β bound to BRD0209 was obtained (PDB: 5KPK, detailed experimental conditions and crystallographic parameters in supporting information). Consistent with previous X-ray crystal structures of hGSK3β (53 unique structures with more than 20 amino acids, in Protein Data Bank (PDB) based on a UniProt search of P49841), the general structural motifs consist of 1) an N-lobe characterized by six β-sheets and a flexible P-loop (pink in Figures 1a-b); 2) a kinase hinge region (D/E-Y-V, yellow in Figures 1a-b); and 3) a C-lobe, consisting primarily of α-helices and featuring the activation loop (orange in Figures 1a-b) and the DFG motif (blue). Within the ATP binding domain (Figure 1b, inset), the crystal structure revealed a tridentate hydrogen bond interaction (pink dotted lines) established by the constrained tricyclic pyrazolo-tetrahydroquinolinone structure of BRD0209. This hydrogen bond donor-acceptor-donor motif serves not only as a critical binding motif but also a geometric anchor which orients substituents from the pyrazole ring towards the kinase hydrophobic pocket 35 adjacent to the gatekeeper residue (Leu (L), purple in Figure 1a). Notably, an H-bond donor interaction, through the pyrazolo-NH-, engages the backbone carbonyl of Asp133, the unique and key residue difference between GSK3β and GSKα (Glu196), within the ATP binding domain (Figure 1b-c). Intriguingly, we observed a previously undescribed hydrogen bond network on the backend region of the hinge connecting the N-and C-lobes centered on the Asp133 side chain (Figure 1b). The Asp133 carboxylate forms one H-bond with Agr113 in the N-lobe and a second with Lys197 in the C-lobe. Inspection of all GSK3β structures in the PDB confirmed that this backend hydrogen network was indeed conserved across structures independent of the bound ligand. We wondered whether this hydrogen bond network was unique to the Asp133 in GSK3β or would be conserved in GSK3α where the corresponding residue, Glu196, contains one additional side chain carbon atom.
Figure 2. Design and characterization of first paralog selective inhibitors of GSK3α and GSK3β.
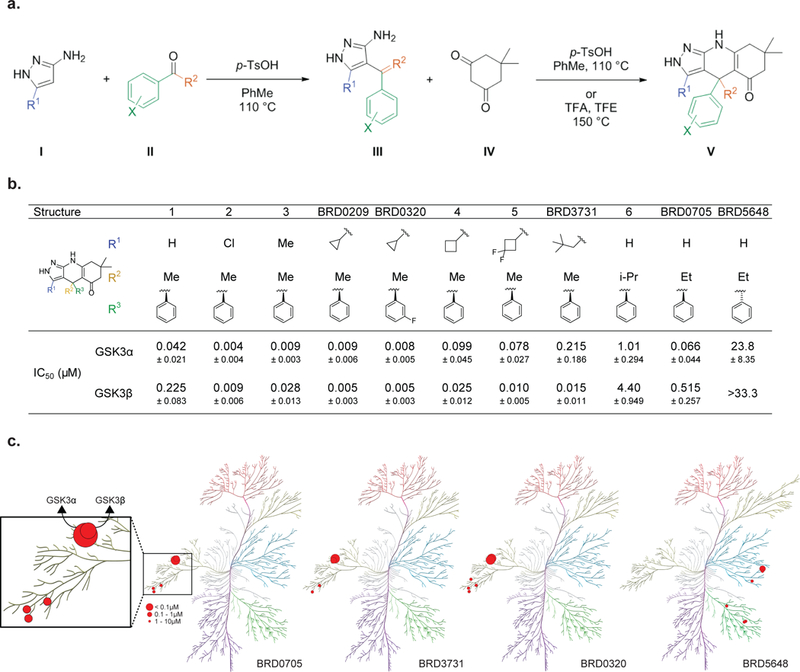
(a) General synthetic scheme for synthesis of the pyrazolo-tetrahydroquinolinone scaffold. (b) IC50 values for the inhibition of GSK3α and GSK3β were determined at Km of ATP in a motility-shift microfluidic assay (Caliper, MA) measuring phosphorylation of a synthetic substrate. Values are average of three or more experiments. Data are shown as IC50 values in μM ± standard deviation. Compounds were tested in duplicate in a 12-point dose curve with 3-fold dilution starting at 33.3 μM. (c) Kinome-wide selectivity for BRD0705 and BRD3731 represented on a kinome phylogenetic tree. Each inhibitor was screened against 311 kinases at a 10 μM concentration. Kinases with >50% inhibition are depicted (percentage inhibition proportional to size of red dot).
Figure 3. An Asp (D) –to-Glu (E) “switch” in the enzymatic hinge backend underlies paralog selectivity of GSK3α and GSK3β inhibitors.
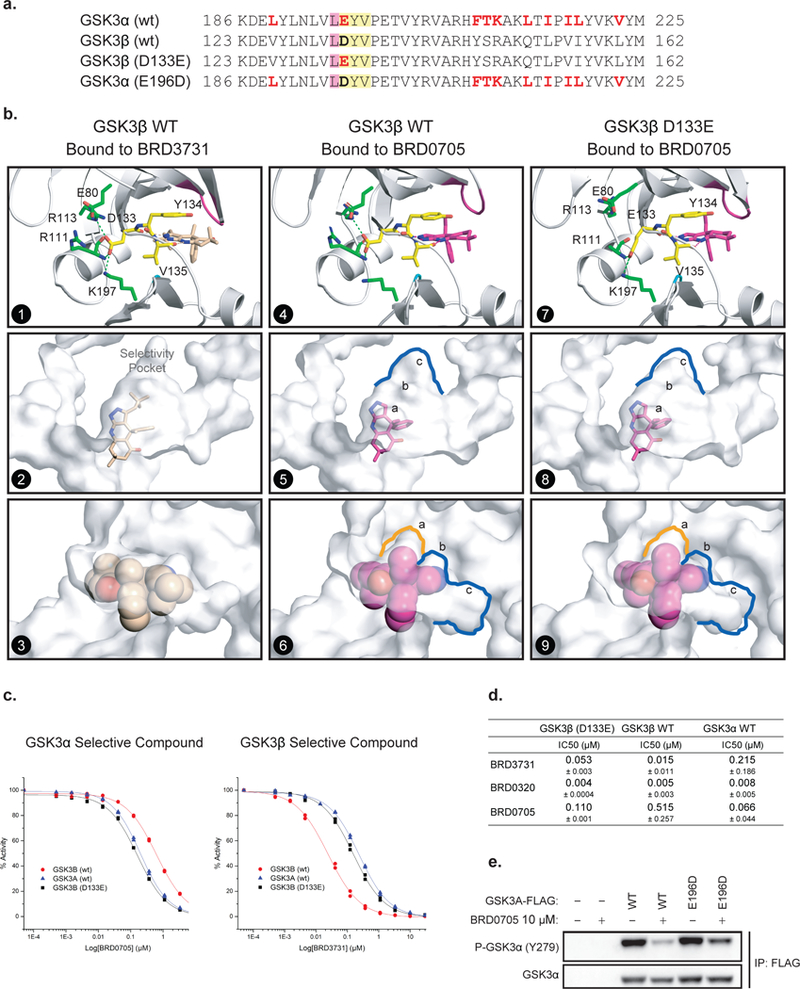
(a) Primary sequence comparison of GSK3α, GSK3β and GSK3β (D133E) and GSK3α (E196D) mutants. (b) X-ray structures of hGSK3β bound to BRD3731 (panels 1–3) and BRD0705 (panels 4–6) and hGSK3β (D133E) bound to BRD0705 (panels 7–9). The side chain of Asp133 in GSK3β forms a complex hydrogen bond network at the back of the kinase hinge and controls the shape and size of the adjacent selectivity pocket. (c-d) GSK3β (D133E) mutation “switches” compound selectivity: IC50 values for the inhibition GSK3β (D133E) were determined at Km of ATP in a motility-shift microfluidic assay (Caliper, MA) measuring phosphorylation of a synthetic substrate. Representative IC50 curves comparison in enzymatic assay using GSK3α (WT, blue), GSK3β (WT, red) and GSK3β (D133E, black) protein constructs (c). Values are the average of three or more experiments. Data are shown as IC50 values in μM ± standard deviation (d). Compounds were tested in duplicate in a 12-point dose curve with 3-fold dilution starting at 33.3 μM. (e) FLAG tagged WT and E196D mutant GSK3α are overexpressed in HEK-293T cells. Western immunoblot for p-GSK3α (Tyr279) and T-GSK3α after FLAG-GSK3α immunoprecipitation and treatment with DMSO or BRD0705 at 10 μM.
While numerous structures of hGSK3β are reported in the PDB, no X-ray crystal structures of GSK3α have been reported preventing a direct structural comparison. Several attempts to generate a GSK3α structure utilizing a variety of constructs were also unsuccessful (unpublished data). To understand the impact of a glutamic acid substitution at this residue on the backend hydrogen bond network a Basic Local Alignment Search Tool (BLAST) search was performed of GSK3α to identify a kinase with high similarity to GSK3α, including a hinge glutamic acid. Our search identified a fungal GSK3 orthologue found in Ustilago maydis (PDB 4E7W), which possesses 49% and 58% overall identity to GSK3α and GSK3β respectively 36. Analysis of the fungal structure revealed a different Glu126 sidechain conformation compared to the analogous Asp133 sidechain in hGSK3β (Figure S2). The GSK3β Asp133 hydrogen bonds to the sidechain of Arg131 within the N-lobe, and Arg131 in turn hydrogen bonds with the sidechain of Glu80 within the N-lobe forming a network of hydrogen bonds (Figure 1b). However, the Glu126 in the fungal structure adopts a downward conformation and does not engage the corresponding Lys106, and there is no Lys106 to Asp73 hydrogen bonding interaction, indicating that a single carbon difference at this hinge residue may indeed impact the overall architecture of this backend hydrogen bond network (Figure S2).
This observation led us to investigate whether similar structural differences between the Asp-Glu “switch” in the human GSK3 paralogs were present by running long time-scale molecular dynamics (MD) simulations of the GSK3α and GSK3β apo forms (see supporting information for details). Analysis of the two sets of kinase simulations were consistent with our initial observations and revealed two different geometrically preferred paralog conformations of the hinge Asp133/Glu196 sidechains (in GSK3β and GSK3α respectively) (Figure 1c, panels 1 and 3). The sidechain of Asp133 in GSK3β hydrogen bonds to the sidechain of Arg113 within the N-lobe (Figure 1c, panel 1, Figure S3, distance A, and Figure S4A), which in turn stabilizes an Arg113:Glu80 sidechain-to-sidechain hydrogen bonding interaction (Figure 1c, panel 1, Figure S3, distance B and Figure S4A). Additionally, Asp133 also forms a hydrogen bond interaction to Lys197 within the C-lobe, effectively bridging the two major structural components within the kinase, the N-and C-lobes. Conversely, the corresponding amino acid at the hinge of GSK3α, Glu196, does not strongly interact with Arg176 (Figure 1c, panel 3, and Figure S3, distance A) due to the extra side chain methylene which in turn destabilizes the corresponding Arg176:Glu143 sidechain-to-sidechain hydrogen bond interconnections within the N-lobe (Figure 1c, panel 3, and Figure S3, distance B). This difference in the stability of the network of hydrogen bonds on the backend of the ATP binding pocket for GSK3α and GSK3β was quantified by measuring these distances across time as well as plotting the free energy contour plots (Figure S3, S4; see supporting information for simulation protocol). The ability of the carboxylate sidechain of Asp133 in GSK3β to hydrogen bond with Arg113 stabilizes the backend region driving it into a single preferred conformation. In comparison, the carboxylate sidechain of Glu196 in GSK3α has a fleeting interaction with the analogous Arg176, which in turn does not favor the Arg176 to Glu143 interaction as all three residues sample distances outside typical hydrogen bond distances (Figure S4C-D). This subtle, but quantifiable, conformational difference between the two kinase hinges triggers an indirect effect on the size and topology of the ATP binding domain and the adjacent hydrophobic pocket (Figure 1c, panels 2 and 4).
Recognizing that these topological differences are driven via the Asp→Glu “switch” within the hinge domain and that our pyrazolo-tetrahydroquinolinone-based hinge binders formed a direct H-bond to the analogous hinge position (Asp133 or Glu196 residue of GSK3β or GSK3α respectively), we envisioned a rational approach to exploit this hypothesis and generate paralog selective inhibitors. The tridentate binding mode of our core scaffold provided a rigid molecular platform within the ATP binding domain well suited to explore apparent differences within the hydrophobic selectivity pockets. Therefore, we set out to systematically probe these differences by designing inhibitors predicted to be preferential binders for either GSK3α or GSK3β.
Exploiting an Asp (D) -to-Glu (E) “switch” to design paralog selective GSK3 inhibitors.
In order to map the topography of the selectivity pocket adjacent to the hinge region, our initial structure-activity relationship (SAR) exploration probed substituents from the pyrazole ring at R1 which vectors into the hydrophobic selectivity pocket (see V, Figure 2a). Structural modifications to the pyrazolo-tetrahydroquinolinone scaffolds are easily obtained via an efficient three component, two-step reaction sequence (Figure 2a). An electrophilic aromatic substitution between aminopyrazoles (I) and acetophenones (II) generate C-substituted pyrazolo-intermediates such as III. Intermediate III reacts with dimedones (IV) to form vinylogous amides which cyclize in situ to provide the fully assembled tricycles, such as (V) (see supporting information for detailed synthetic protocols and compound characterization).
Starting with minimal structural requirements at R1, we installed a hydrogen, leaving the selectivity pocket unoccupied (Figure 2b). Compound 1 displayed 5-fold selectivity for GSK3α versus GSK3β (IC50 = 42 nM and 225 nM, respectively; measured using a mobility shift microfluidic assay (Caliper, assays were run at Km, ATP of each enzyme). However, the chloro-substituted (~50% increase in atomic radius versus hydrogen) compound 2, displays equipotency for two paralogs (IC50 = 4 nM and 9 nM respectively). A similar increase in potency for both paralogs was observed with electronically neutral, but increasingly bulkier substituents: a methyl in compound 3 and a cyclopropyl in BRD0209, implicating hydrophobic rather than electronic effects. These modifications and increased binding affinities were tolerant of simple substitutions on the phenyl ring (cf. BRD0320). Increasing the ring size at R1 to cyclobutyl in compound 4 we observed our first hints of preferential binding (4-fold selectivity) for GSK3β (IC50 = 25 nM) over GSK3α (IC50 = 100 nM). To further exploit these differential interactions and steric requirements in this highly hydrophobic selectivity pocket, we synthesized the difluorocyclobutyl compound 5 which increased our preferential binding towards GSK3β, exhibiting an 8-fold selectivity window. Further structure-based design aimed towards maximizing steric requirement at the R1 position and the corresponding differences within the selectivity pockets of GSK3α and β led to the neopentyl substituted compound BRD3731, which displayed 14-fold selectivity for GSK3β (GSK3β IC50 = 15 nM; GSK3α IC50 = 215 nM).
With the successful achievement of >10 fold selectivity for GSK3β utilizing large substituents at R1, we turned our attention toward the development of more selective GSK3α inhibitors by revisiting the smallest substituent at R1, hydrogen. Recognizing subtle differences within the ATP binding domains between GSK3α and β, in addition to the now validated differences in the hydrophobic selectivity pockets, we explored combinations between R1 = H and the quaternary center R2. While branched alkyl (cf. compound 6) and long chain alkyl substituents at R2 led to losses in potency, an ethyl substitution (BRD0705) maintained potency and displayed increased selectivity for GSK3α (8-fold) versus GSK3β (GSK3α IC50 = 66 nM; GSK3β IC50 = 515 nM). The inverted quaternary center stereochemistry, the enantiomeric BRD5648, is relatively inactive against the GSK3s, demonstrating the limited three-dimensional space in the ATP binding domain. BRD0705 represents one of the most selective GSK3α reported to date.
With the successful design of a chemically matched set of GSK3α (BRD0705) and GSK3β (BRD3731) selective inhibitors, as well as a corresponding negative control compound (BRD5648), we turned our attention towards the larger kinome as a final test of specificity. While minor chemical modifications provided a robust selectivity and SAR within the GSK3 kinases our compounds were untested against the larger human kinome. Testing all four compounds against a panel of human kinases (311 kinases at 10 μM compound concentration, Carna Biosciences) revealed exquisite overall kinase selectivity (Figure 2c, Tables S5–6). For BRD0705, the CDK family of kinases (CDK2, 3 and 5) were next most potently inhibited at values of 6.87 μM, 9.74 μM and 9.20 μM (87-fold, 123-fold and 116-fold selectivity relative to GSK3α, Table S6). This set of small molecule inhibitors represents the most selective and fully characterized set of chemical tools to probe the biological function of the GSK3 kinases.
Site-directed mutagenesis and crystallographic studies reveal a key role for the hinge D-to-E “switch”.
With the successful demonstration of GSK3α and GSK3β selective inhibitors, and a working hypothesis based on computational and structural data for the observed paralog selectivity of BRD0705 and BRD3731, we set out to further validate our structural findings and computational predictions through additional crystallographic structure-based approaches. Since our efforts to obtain ligand bound or apo structures of GSK3α were not successful, presumably due to its unorganized N-terminus, we chose to pursue mutagenesis studies in GSK3β to test our Asp→Glu “switch” hypothesis. We surmised that if this one amino acid residue change (Asp133→Glu196) within the hinge -the net result being the addition of a single carbon atom in GSK3α -were the driver of the observed structure activity relationships, then mutating Asp133 to Glu133 in GSK3β would abrogate our observed selectivity patterns (see Figure 3a for construct primary sequence, GSK3β (D133E)). In addition, this design directly tests the significance of the hinge residue on the nature of the backend hydrogen bond network, the corresponding topological effects on the ATP binding domain and adjacent hydrophobic selectivity pocket. Towards this end, a single point GSK3β (D133E) mutant protein was designed, expressed, purified and fully characterized (PBD: 5T31, detailed experimental conditions and crystallographic parameters in supporting information). This GSK3β (D133E) mutant exhibited comparable kinase activity when compared to recombinant GSK3α (wt) or GSK3β (wt) (data not reported). For comparison, we also obtained additional high resolution X-ray GSK3β (wt) ligand bound structures with our GSK3β selective inhibitor BRD3731 (PDB: 5KPM, Figure 3b, panels 1–3) and our GSK3α selective inhibitor BRD0705 (PDB: 5KPL, Figure 3b, panels 4–6).
As in previous ligand bound GSK3β (wt) structures, for both GSK3 (wt) BRD3731 or BRD0705 bound crystal structures, we observed the hydrogen bond network on the backside of the hinge binding domain centered on Asp133 (Figure 3b, panels 1 and 4). In both GSK3β (wt) structures, Asp133 formed a hydrogen bond with Arg113 in the N-lobe and Lys197 in the C-lobe as previously described. However, in the corresponding BRD0705 ligand bound structure with the GSK3β (D133E) construct, the increase of a single carbon unit in the GSK3β mutant protein (Glu133) disrupted the described H-bond interaction. This observation was predicted from our earlier comparison to the fungal GSK3β orthologue with a Glu at this position and from our MD simulation studies of GSK3α. In addition, this Asp133→Glu133 “switch” on the kinase hinge has indirect topological effects on the hinge binding domain and adjacent selectivity pocket (Figure 3b, panels 7 and 9). The volume of the hydrophobic selectivity pocket adjacent to the hinge, specifically pocket “c”, is much larger in the BRD0705-GSK3β (wt) structure than in the BRD0705-mutant GSK3β (D133E) structure (Figure 3b; blue contour in panels 5 vs 8 and pocket c in panels 6 vs 9; and Figure S5). This observation is consistent with our observed SAR that the larger R1 neopentyl substituent in BRD3731 provides the most preferential binding for GSK3β. The smaller predicted pocket topology in regions “b” and “c” in GSK3α is less able to accommodate such large sterically demanding substituents. Conversely, smaller R1 substituents, such as a hydrogen in compound 1 and BRD0705, do not benefit from hydrophobic interactions in the hydrophobic selectivity pocket of GSK3β and showed preferred binding to GSK3α. Additionally, pocket “a” in GSK3β (D133E) was slightly larger (Figure 3b, orange contour in panel 6 vs 9) and tightly binds the R2 ethyl substituent of BRD0705, providing additional selectivity for GSK3α (overall 8-fold selectivity).
As further validation of our Asp→Glu “switch” hypothesis and SAR we tested our GSK3α selective (BRD0705) and GSK3β selective (BRD3731) inhibitors in the same enzymatic assay using the mutant GSK3β (D133E) construct. Remarkably, both compounds displayed “GSK3α-like” activity against this GSK3β mutant construct (Figure 3c). BRD0705 displayed similar potency towards the GSK3β (D133E) and GSK3α (wt) constructs with an IC50s of 110 nM and 66 nM, respectively, while BRD3731 displayed reduced potency towards the GSK3β (D133E) with an IC50 = 53 nM compared to GSK3β (wt) (cf. IC50 = 15 nM).
Finally, we sought to validate in cellulo the suggested biochemical explanation for the GSK3α paralog selectivity of BRD0705 using GSK3α 279 tyrosine auto-phosphorylation as a surrogate for GSK3α enzymatic activity 37,38. As predicted, overexpression of the E196D mutant form of GSK3α in 293T cells led to an attenuated repression of GSK3α active site phosphorylation after BRD0705 treatment compared to overexpression of GSK3α (wt) in this same cellular context (Figure 3e).
Through a variety of structural and functional approaches, we believe that the observed inhibitory profiles for our GSK3α (BRD0705) and GSK3β (BRD3731) selective inhibitors are driven in large part through their direct ligand interactions with Asp133 in GSK3β and the corresponding Glu196 in GSK3α. This interaction exploits a previously undescribed network of hydrogen bonds on the backside of the hinge domain which impacts the overall topology of the ATP binding domain and the adjacent hydrophobic selectivity pocket in each of these kinase paralogs. Having further validated our Asp→Glu “switch” hypothesis and increased our confidence in a bona fide selectivity profile for this series of compounds, we turned our attention towards the translation of these binding differences into cell-based systems. We therefore explored the biological impact of both GSK3α and GSK3β selective inhibition in a variety of cell-based systems and extended our investigation into the downstream effects of paralog inhibition on β-catenin stabilization.
Cell-based functional selectivity of GSK3 paralog selective inhibitors and cell context dependent effects on β-catenin stabilization.
With the ultimate goal of using this set of compounds in a cellular and in vivo context, it was crucial to demonstrate that a ~10-fold selectivity in a biochemical assay using recombinant enzymes would translate to sufficient selectivity in a human cellular context. Paralog selective target engagement for GSK3α and GSK3β was quantitatively assessed using a bioluminescence resonance energy transfer reporter system (NanoBRET) in live, non-permeabilized 293 cells to match physiological drug delivery conditions 39. N-terminal NanoLuc fusion constructs of the individual GSK3α and GSK3β paralogs were designed to serve as luminescent energy transfer donors and utilized a fluorescent non-selective kinase BRET tracer serving as an energy acceptor. Compound binding is evident from a loss of BRET signal owing to competitive displacement of the BRET tracer. In this functional cell-based assay, the 10-fold selectivity for each paralog selective GSK3 inhibitor translated to a >4–6-fold selectivity in intact live 293 cells. BRD0705 inhibits GSK3α with a Kd of 4.8 μM, and BRD3731 impairs GSK3β with a Kd of 3.3 μM in this cellular context (Figure 4a, and sample curves in Supplementary Figure 6).
Figure 4: GSK3α and GSK3β selective inhibitors play a differential role on β-catenin stabilization in a context dependent manner.
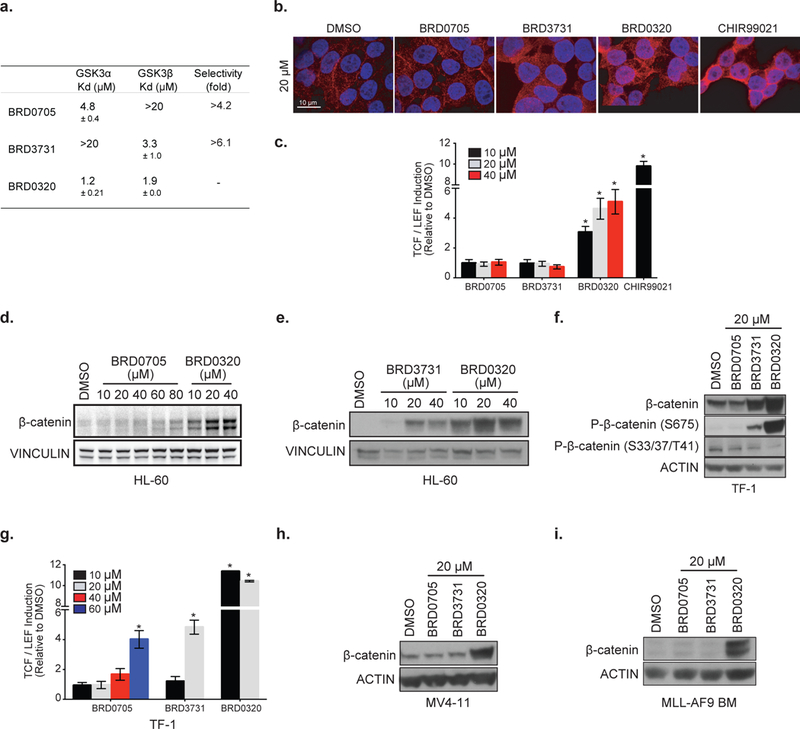
(a) Biophysical measure of GSK3 target engagement in HEK 293 by BRET signal between a NanoLuc fused protein target and a small molecule labeled with the NanoBRET acceptor dye. Kds for each inhibitor are reported as mean ± SEM of two replicates. (b) β-catenin immunofluorescence staining in HEK 293T after treatment with the indicated inhibitor (β-catenin in red and DAPI in blue). (c) β-catenin TCF/LEF luciferase reporter assay in HEK 293T after treatment with the indicated inhibitor. * p-value < 0.05 calculated using a Mann-Whitney test in comparison with control conditions. Error bars represent mean ± SEM of ten replicates. (d-e) Western immunoblot for β-catenin and vinculin after treatment with the indicated inhibitors in HL-60. (f) Western immunoblot for β-catenin, p-β-catenin (S675), p-β-catenin (S33/37/T41) and actin after treatment with the indicated inhibitors in TF-1. (g) β-catenin TCF/LEF luciferase reporter assay in TF-1 after 24 hours of treatment with the indicated inhibitor. * p-value < 0.05 is calculated using a Welch’s t test in comparison with control conditions. Error bars represent mean ± SEM of three replicates. (h-i) Western immunoblot for β-catenin and actin after treatment with the indicated inhibitors in MV4–11 (h) and MLL-AF9 murine leukemic cells (i).
Because genetic KO or KD of the GSK3α or GSK3β individually does not induce β-catenin accumulation in an embryonic stem cells context 22, we set out to define whether a ~10 and >4–6-fold selectivity in a biochemical and cellular context would translate to sufficient selectivity to mitigate the effect of dual inhibition on total β-catenin protein stabilization and nuclear translocation. Immunofluorescence staining of β-catenin was performed in 293T (Figure 4b), demonstrating an absence of nuclear β-catenin accumulation after selective inhibition of either GSK3α (BRD0705) or GSK3β (BRD3731) for 24 hours (20 μM). In contrast, dual paralog inhibition, with BRD0320 or CHIR99021, a commercially available pan-GSK3 inhibitor 40, led to total β-catenin stabilization and nuclear translocation. Using a β-catenin dependent TCF/LEF luciferase reporter assay, we confirmed the absence of β-catenin induced target activation after treatment with BRD0705 or BRD3731 (Figure 4c). In contrast, the dual GSK3 inhibitor BRD0320 or CHIR99021 activated the reporter.
Given the increasing interest in targeting GSK3 in cancer and the potential oncogenic risk of β-catenin stabilization, we set out to validate safety of our GSK3 inhibitors in a neoplastic context using multiple AML models. The effect of paralog GSK3 inhibition using our selective inhibitors was evaluated in three human AML cell lines and one murine primary AML model. As observed by β-catenin western immunoblotting in Figures 4d and 4e, total protein amounts remain largely below dual GSK3 inhibition induced β-catenin stabilization up to 80 μM of BRD0705 treatment whereas BRD3731 induced β-catenin stabilization starting at 20 μM in the HL-60 AML cell line. To further assess the threshold of β-catenin stabilization and nuclear translocation in AML, we transduced the TF-1 AML cell line with a TCF/LEF reporter construct. We first evaluated total protein amounts and phosphorylation of β-catenin after GSK3 inhibition (Figure 4f). Consistent with our observation in the HL-60 cell line, GSK3β inhibition with BRD3731 at 20 μM decreased β-catenin S33/37/T41 phosphorylation and induced β-catenin S675 phosphorylation resulting in increased β-catenin. GSK3α inhibition with BRD0705 at 20 μM resulted in unchanged β-catenin total protein and phosphorylation. Indeed, TCF/LEF induction was only observed at 60 μM and 20 μM of BRD0705 and BRD3731 treatment, respectively, whereas 10 μM of BRD0320 was sufficient to promote β-catenin nuclear translocation and subsequent transcriptional effects in the TF-1 context. Because the effects of the genetic suppression of GSK3β on β-catenin stabilization have been inconsistent,22,31,34 we decided to further evaluate the effects of BRD3731 in two additional AML models. As observed in Figures 4h and 4i, no β-catenin stabilization was observed after GSK3β inhibition in both the MV4–11 AML cell line and an MLL-AF9 induced murine AML at 20 μM, in contrast to our observation in TF-1 and HL-60 cells.
Taken together, this data demonstrates an absence of β-catenin stabilization after GSK3α inhibition in all tested AML cell lines tested, while GSK3β inhibition appears to drive β-catenin stabilization in a context-dependent manner, consistent with previous data obtained with genetic suppression of GSK3β. 22,31,34 Importantly from a toxicity perspective, we were able to demonstrate for the first time that ~10-fold selectivity for GSK3α versus GSK3β is sufficient to achieve potent GSK3α inhibition with no effect on β-catenin stabilization. With BRD0705, we have successfully decoupled GSK3α kinase inhibitory effects from WNT/β-catenin pathway activation, mitigating a potential mechanism-based toxicity. In light of this finding and prior studies supporting the targeting of GSK3α in AML and raising concerns over targeting GSK3β, 22,31,34 we have focused our attention on the GSK3α selective compound BRD0705 in the AML cellular context.
BRD0705 selectively targets GSK3α and induces differentiation in AML cell lines and primary patient samples.
To further assess GSK3α selective target engagement in AML, we performed time-(Figure 5a) and concentration-response (Figure 5b) immunoblotting studies of GSK3α/β Tyr279/216 phosphorylation after treatment with BRD0705 or BRD0320. GSK3α/β Tyr279/216 autophosphorylation sites are used as a surrogate for GSK3α/β enzymatic activity and are reflective of proximal-based substrate inhibition 37,38. As expected, the GSK3α selective inhibitor BRD0705 impaired GSK3α Tyr279 phosphorylation in a time-and concentration-dependent manner without affecting GSK3β Tyr216 phosphorylation. In contrast, the dual inhibitor BRD0320 decreased active site phosphorylation for both GSK3α and GSK3β. A decrease in the phosphorylation of glycogen synthase, a known direct target of GSK3 41 was also observed (Figure 5b). As expected, BRD5648, the inactive enantiomer of BRD0705, did not induce changes in enzyme phosphorylation or total β-catenin protein stabilization, supporting the on-target GSK3 activity of these selective inhibitors (Figure 5b).
Figure 5: BRD0705 induces differentiation in AML cell lines and primary patient samples through GSK3α selective inhibition.
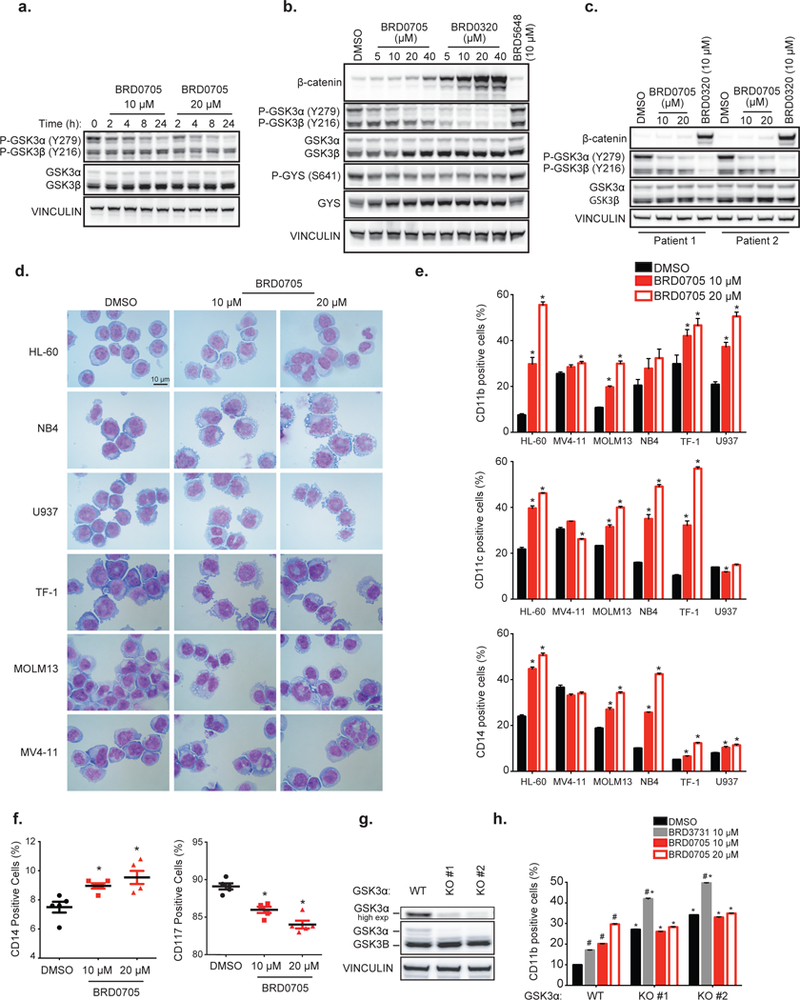
Time-(a) and (b) dose-response western immunoblots for β-catenin, p-GSK3α/β (Tyr279/216), T-GSK3α/β, GYS, p-GYS (S641) and vinculin after treatment with the indicated inhibitors in U937. (c) Dose-response western immunoblot for β-catenin, p-GSK3α/β(Tyr279/216), T-GSK3α/β, GYS and vinculin after treatment with the indicated inhibitors in primary AML patient samples.(d) May-Grunwald Giemsa staining after BRD0705 treatment for 6 days. (e) FACS analysis of the expression of CD11b, CD11c, and CD14 cell surface markers after BRD0705 treatment. *p-value < 0.05 calculated using a Welch’s t test in comparison with the control condition. Error bars represent mean ± SEM of three replicates. (f) FACS analysis of the expression of CD14 and CD117 cell surface markers in 5 primary AML samples treated with BRD0705. *p-value < 0.05 calculated using non-parametric Mann-Whitney test in comparison with control condition. (g) Western immunoblot for T-GSK3α/β and vinculin in U937 after GSK3α CRISPR KO. (h) FACS analysis of the expression of CD11b cell surface marker in U937 GSK3α-WT, GSK3α-KO#1 or GSK3α-KO#2, treated with the indicated inhibitor. P-value < 0.05 calculated using a Welch’s t test in comparison with the DMSO condition for each clone (#) or for the DMSO condition in the GSK3α-WT clone (*). Error bars represent mean ± SEM of three replicates.
Finally, to further evaluate BRD0705’s clinical potential, the functional selectivity of BRD0705 towards direct selective inhibition of GSK3α (P-GSK3α, Y279) and for an absence of downstream stabilization of β-catenin was evaluated in primary blasts from five independent patients with AML. Selective GSK3α active site inhibition and absence of β-catenin stabilization in patient samples highlights the clinical translational potential of BRD0705 in achieving on target activity (Figure 5c, data shown for 2 representative patient samples).
Taken together, these data demonstrate the functional selectivity of BRD0705 in targeting endogenous GSK3α kinase activity in multiple cell-based systems, including patient-derived AML cell lines and primary AML cells. This functional selectivity translated to proximal substrates including GSK3α, glycogen synthase and β-catenin.
Given the implication of GSK3α as a differentiation target in AML34, we treated a panel of six AML cell lines (HL-60, NB-4, U937, TF-1, MOLM13 and MV4–11) with the GSK3α selective inhibitor BRD0705 to evaluate potential phenotypic changes. Selective GSK3α inhibition induced morphological (Figure 5d) and cell surface markers (Figure 5e: CD11b, CD11c, CD14) changes consistent with AML differentiation. We then tested whether these findings were relevant to primary patient AML cells through cell surface marker staining (CD14 and CD117) in a panel of five primary AML patient blast samples (Figure 5f). Increased differentiation marker (CD14) expression and decreased immature marker (CD117) expression was confirmed in all five patient samples in a dose-responsive manner after BRD0705 treatment. Conversely, BRD3731 effects were inconsistent, with a modest differentiation phenotype in some cell lines but a decrease in cell surface differentiation markers in a subset of the AML cell lines (Figure S8a-c). These results support our previously reported GSK3α genetic suppression-induced phenotype in AML 34 and confirm that enzymatic inhibition recapitulates the phenotype observed with genetic suppression.
To provide more definitive evidence supporting the selectivity of BRD0705 for GSK3α, we generated GSK3α isogenic KO U937 clones (Figure 5g and 5h), characterized the phenotypic effect of KO on differentiation, and tested BRD0705 and BRD3731 treatment of two different KO clones. GSK3α KO recapitulated the GSK3α inhibition phenotype as evidenced by increased expression of differentiation cell surface markers with DMSO treatment. These cells show no additional increase in expression of the differentiation cell surface marker CD11b upon treatment with BRD0705, in contrast to the GSK3α expressing clones (Figure 5h), providing strong validation of in vitro paralog selectivity and on-target activity. Moreover, GSK3β inhibition using BRD3731 had a similar increase in CD11b marker on GSK3α WT and GSK3α KO clones (Figure 5h).
GSK3α inhibition triggers differentiation and reduced stemness transcriptional programs without increasing β-catenin related signatures.
To characterize the effects of GSK3 inhibitor treatment on genome-wide transcriptional programs, we profiled the U937 AML cell line after 24 hours of treatment with either BRD0705, BRD3731 or BRD0320 at 10 μM using RNA sequencing. Dual GSK3 inhibition induced a more dynamic change in transcriptional programs (556 upregulated and 975 downregulated genes) compared with selective GSK3α inhibition (55 upregulated and 193 downregulated genes) or selective GSK3β inhibition (187 upregulated and 203 downregulated genes) as illustrated in figures 6a and 6b. This finding suggests a partially, but not fully, redundant role of GSK3 paralogs. The top 30 differentially up-and down-regulated genes upon selective GSK3α, selective GSK3β or dual GSK3 inhibition are depicted in figure 6c. All differentially expressed genes based on a permutation p<0.05 and FDR<0.05 are listed in Supplementary Tables S7, S8 and S9.
Figure 6: GSK3α and GSK3β selective inhibitors trigger differential transcriptional programs.
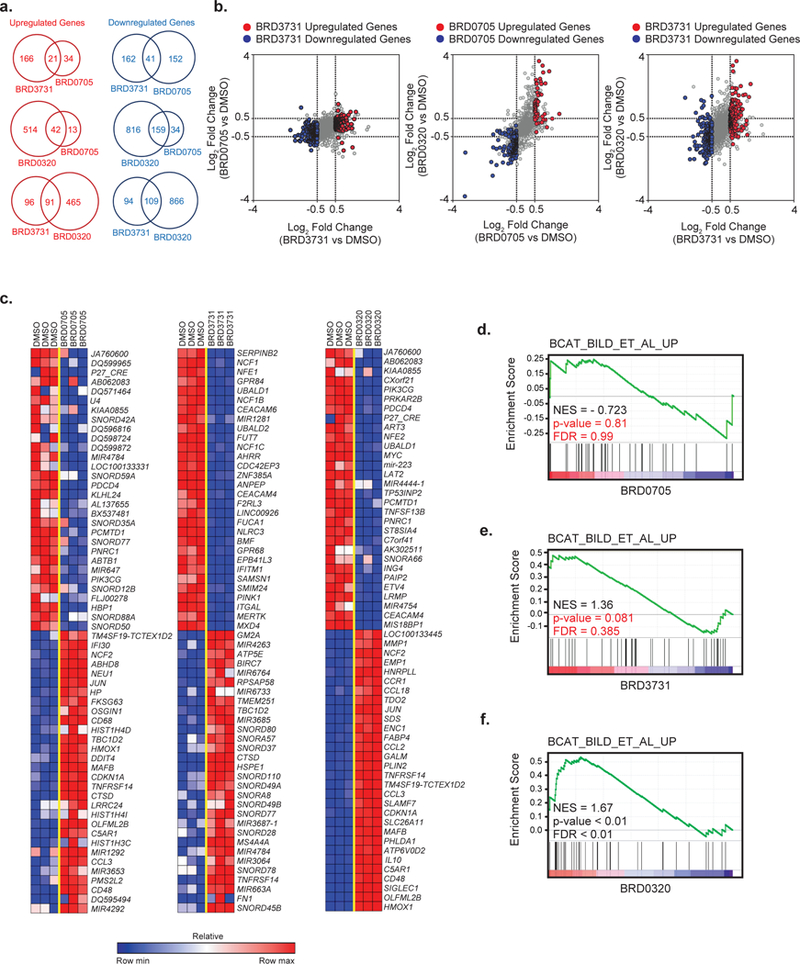
(a) Venn diagrams and (b) scatterplots of hits modulated after BRD0705, BRD3731 or BRD0320 treatment. Significantly depleted or enriched genes after BRD3731 or BRD0705 treatment are highlighted respectively in blue and red (Log2FC < −0.5 or > 0.5 and p-value ≤ 0.05). (c) Heatmaps of the top 30 genes differentially expressed by genomic profiling upon BRD0705, BRD3731 or BRD0320 treatment in the U937 cell line. Depleted and enriched genes are respectively in blue and red. Data is presented as row normalized. The BRD0705, BRD3731 and BRD0320 signatures identified by RNAseq were then interrogated in a functional enrichment analysis across the MsigDB database. GSEA plots for β-catenin signaling pathway induction after treatment with BRD0705 (d), BRD3731 (e) or BRD0320 (f).
To assess more specifically BRD0705’s functional effects in AML, we interrogated our RNA sequencing data with published gene signatures for statistically significant enrichment by gene set enrichment analysis (GSEA). We generated a functional network illustrating GSK3α inhibition transcriptional effects in AML (Figure S9a, Table S10). This analysis first confirmed that GSK3 inhibition gene sets were statistically enriched among genes downregulated by BRD0705 treatment (Figure S9b). Similarly, GSK3α inhibition by BRD0705 induced differentiation transcriptional programs and downregulated stemness signatures (Figure S9c-d). Interestingly, as reported by Guezguez et al.,31 GSK3α inhibition upregulated multiple mitochondria metabolism transcriptional programs (Figure S9e). Overall, this genomic profiling allows independent validation through an unbiased approach of the previously established on-target, phenotypic effects of BRD0705. Finally, a similar enrichment analysis performed on BRD0320 induced transcriptional signatures revealed a strong induction of β-catenin signaling pathway genes, whereas BRD0705 treatment did not induce enrichment for β-catenin-related gene sets. BRD3731 showed a trend toward induction of β-catenin transcriptional programs (Figure 6d-f).
BRD0705 impairs colony formation in both AML cell lines and patient samples and demonstrates in vivo efficacy in multiple xenograft and syngeneic AML mouse models.
To further characterize the phenotypic consequences of BRD0705 treatment, we tested its ability to impair AML colony formation in methylcellulose. We found that BRD0705 impaired AML colony formation in all six tested cell lines, MOLM13, TF-1, U937, MV4–11, HL-60 and NB4, in a concentration-dependent manner (Figure 7a), as opposed to BRD3731 which impaired colony formation in TF-1 while increasing colony forming ability in the MV4–11 cell line (Figure S8d). We then tested whether these findings were relevant to primary AML patient samples through a methylcellulose colony forming assays in AML samples from five patients previously selected for colony formation ability. Reduced ability to form colonies (Figure 7b) was confirmed in all five patient AML samples, again in a concentration-dependent manner, after BRD0705 treatment. No effect of BRD0705 treatment was observed on normal CD34 cell colony formation, thus offering a potential therapeutic window for selectively targeting leukemic cells (Figure 7c). Detailed patient characteristics are provided in Table S11. In accordance with these findings and the RNA sequencing data suggesting down regulation of stemness related gene expression signatures with BRD0705 treatment, we evaluated the capacity of our selective GSK3α inhibitor to eradicate leukemic initiating cells (LIC) in a syngeneic mouse model of AML driven by the oncogene MLL-AF9. BRD0705 treatment significantly impaired development of MLL-AF9 leukemia in secondary recipient mice in comparison to the vehicle-treated group (Figure 7d, p value<0.05). Interestingly, dual GSK3 inhibition, with BRD0320, only induced a mild delay in leukemia onset, whereas GSK3β selective inhibition with BRD3731 did not affect leukemia development. A limiting dilution experiment performed in this mouse model revealed a 3.79-fold decrease in LIC frequency after BRD0705 pre-treatment (Figure 7e). No significant difference in LIC frequency was observed after BRD3731 or BRD0320 pre-treatments.
Figure 7: BRD0705 impairs colony formation in AML cell lines and patient cells and shows in vivo efficacy in multiple AML mice models.
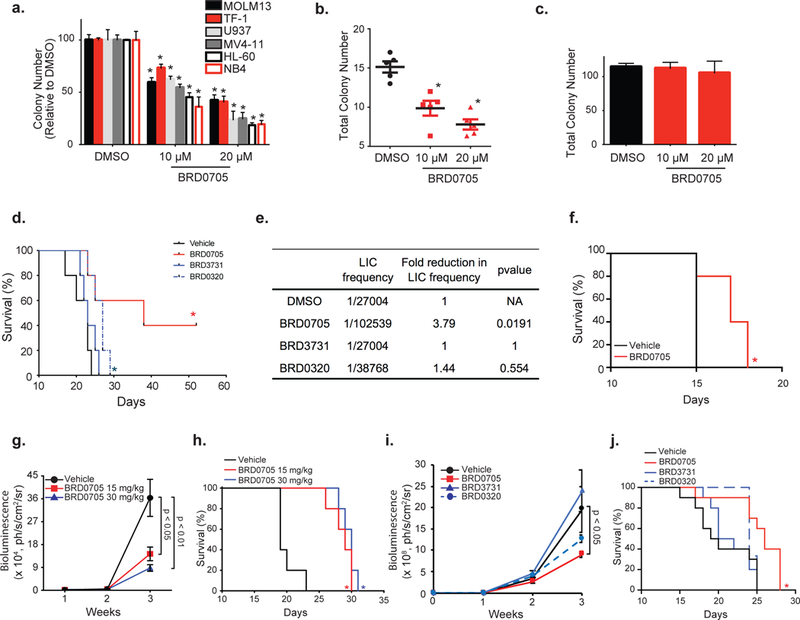
Colony formation assay of the indicated AML cell lines (a), 5 primary AML samples (b) and human CD34 cells (c) after BRD0705 treatment. Data represented as mean ± SEM of three replicates. (d) MLL-AF9 cells injected into secondary recipient mice after pretreatment with DMSO, BRD0705, BRD3731 or BRD0320 in a serially diluted manner. .Kaplan-Meier curves for the 250 000 cells dilution (n=5 per group). (e) Frequency of Leukemia Initiating Cells (LIC) calculated using ELDA software. (f) MLL-AF9 AML cells injected into secondary recipient mice before treatment with vehicle (black) or BRD0705 at 30 mg/kg (red). Kaplan-Meier curves for each group of mice (n=5 per group). (g) HL-60-Luc cells injected into recipient mice and treated with vehicle (black), BRD0705 at 15 mg/kg (red) or 30 mg/kg (blue). Bioluminescence was quantified. (h) Kaplan-Meier curves for each group of mice (n=5 per group). (i) MV4–11-Luc cells injected into recipient mice and treated with vehicle (black), BRD0705 (red), BRD3731 (blue) or BRD0320 (dashed blue). Bioluminescence was quantified. Data represented as mean ± SEM. P-value calculated on the latest time point. (j) Kaplan-Meier curves for each group of mice (n=10 per group). * p-value < 0.05 determined by log-rank (Mantel-Cox) test in comparison with vehicle.
To better enable in vivo studies, we next characterized the peripheral exposure of BRD0705 in mice. Following a single oral dose administration of BRD0705, plasma concentrations were quantifiable up to 24 hours with Tmax of 0.25 h and AUC of 67.6 μmol/L.h (Figure S10a). BRD0705 has 100% oral bioavailability in mice. To determine whether selective inhibition of GSK3α would translate to a decreased AML burden in vivo, we tested the efficacy of BRD0705 in the MLL-AF9 syngeneic mouse model. Mice were separated into two groups and treated with either vehicle or BRD0705 at 30 mg/kg through oral gavage twice daily (BID). Mice survival was significantly improved under BRD0705 treatment (Figure 7f, p value<0.05).
We then evaluated BRD0705 efficacy in an orthotopic HL–60 xenograft AML model generated through injection of luciferized HL-60 cells into the tail veins of NSG mice. Mice were separated into three groups and received either vehicle or BRD0705 at 15 or 30 mg/kg through oral gavage once a day (QD). BRD0705 treatment induced a decrease in leukemia progression in a dose-dependent manner as evidenced by the decrease of both bioluminescence measurement of disease burden (Figure 7g, p<0.05 and p<0.01 at 15 and 30 mg/kg, respectively) and circulating human CD45 expressing AML cells (Figure S10b) resulting in a prolonged overall survival (Figure 7h). No major weight loss and no significant blood toxicities were observed upon in vivo administration of BRD0705 (Figure S10c-g) at this dose and schedule. GSK3 inhibition has been previously shown to decrease glycogen synthase phosphorylation in peripheral blood mononuclear cells leading to an increased cellular glycogen content 25. Thus, we first validated glycogen accumulation (PAS staining and quantitative measurement) as an on-target effect of GSK3α inhibition after BRD0705 treatment in vitro in AML cell lines including HL-60 (Figure S7). We then confirmed the presence of increased glycogen in peripheral mononuclear cells obtained from BRD0705 treated mice as compared to vehicle treated (Figure S10h). Glycogen measure can therefore serve as a potential biomarker of BRD0705 on target activity in future clinical trials.
Finally, to evaluate the differential effects of paralog selective inhibition in vivo, we generated an MV4–11 orthotopic AML mouse model through tail vein injection of a luciferized MV4–11 cell line into irradiated NSG recipient mice. Mice were separated into four groups and treated either with vehicle, BRD0705, BRD3731 or BRD0320 at 30 mg/kg QD by oral gavage. BRD0705 treatment resulted in a significant disease burden decrease as measured by bioluminescence and prolonged mice survival (Figures 7i and 7j, p value<0.05). Interestingly, although no significant disease burden or survival effect was detected under BRD3731 or BRD0320 treatments, a trend towards increased disease burden after GSK3β inhibition was observed, matching the increased methylcellulose colony formation effect observed in MV4–11 cell line (Figure S8d). BRD0320 treatment resulted in a non-significant disease burden decrease. Together, these results suggest a potential oncogenic effect of GSK3β selective paralog inhibition, in part independent from β-catenin stabilization because BRD3731 did not increase β-catenin in MV4–11 cells. This undesirable effect could potentially counteract the beneficial effect of GSK3α inhibition while using a dual GSK3 inhibitor, thus offering an additional argument for selective GSK3α targeting in AML.
Discussion
Reported GSK3 inhibitors generally demonstrate poor to moderate selectivity versus the broader human kinome. To date, only one reported GSK3 inhibitor has demonstrated preferential inhibition of GSK3α over GSK3β 23,24,42, but this compound has not been characterized for cell-based, functional selectivity, or studied in mammalian models in vivo. Dual GSK3α/β inhibitors, however, have entered clinical testing for both neurodegenerative disorders and cancer at tolerated doses but the general lack of clinical activity was disappointing 43–48.
From a cancer therapy perspective, the compound LY2090314, a dual GSK3 inhibitor, was initially tested in a Phase I study in combination with pemetrexed and carboplatin in patients with advanced solid tumors where the primary toxicities included visual disturbances and peri-infusional chest pain that was relieved with IV H2 blockers 44. Next, a phase II study was performed testing LY2090314 in patients with relapsed/refractory AML or patients with newly diagnosed AML who could not tolerate standard induction chemotherapy 47. In this study, primary toxicities included decreased appetite and nausea; hematologic toxicities included rare febrile neutropenia, thrombocytopenia and anemia. While 5 patients had a decrease in peripheral blood blasts, there were no complete or partial remissions. Moreover, there was evidence of target inhibition based on β-catenin stabilization in peripheral mononuclear cells and in patient leukemia blasts.
While increased β-catenin provided confirmation of GSK3α/β target inhibition in the testing of LY2090314 in patients with AML, the notable lack of clinically meaningful responses does raise the question of the potential role of β-catenin stabilization in attenuating response to this drug in patients with leukemia. Indeed, the concurrent inhibition of GSK3α and GSK3β is well known to induce β-catenin stabilization and therefore increase self-renewal of leukemia initiating cells raising potential efficacy concerns 22,32,33,49. In support of these studies, more recently, Guezguez et al. reported that dual GSK3α/β KO led to an overt, aggressive AML, while GSK3β KO drove a pre-neoplastic mixed myeloproliferative/myelodysplastic state 31. Paralog selective GSK3α or GSK3β suppression with genetic approaches did not stabilize β-catenin in embryonic stem cell lines 22. In our own prior work, GSK3α or β directed shRNA did not stabilize β-catenin in the MOLM14 AML cell line, and GSK3α suppression promoted differentiation in AML cells 34. In the Guezguez study, however, it was reported that GSK3β KO drives a β-catenin signature in the context of adult hematopoietic stem/progenitor cells as a mechanism of inducing the pre-neoplastic phenotype, lending caution to the application of GSK3β selective inhibitors. These results are consistent with the data reported here highlighting the importance of cellular context on β-catenin stabilization induced by GSK3β inhibition. In line with our observations, where we did not see β-catenin stabilization with GSK3α inhibition, Guezguez et al. had shown that GSK3α KO did not stabilize β-catenin and did not lead to a leukemic nor pre-leukemic phenotype, offering additional support for the exploration of GSK3α selective inhibitors in AML treatment. Hence, in our own studies of paralog selective GSK3 molecules in AML, we have focused on GSK3α inhibitors which display no functional effects on β-catenin.
Our GSK3 inhibitors were designed from a single core pyrazolo-tetrahydroquinolinone scaffold, which presented a unique tridentate binding mode within the ATP binding domain. To design these highly selective competitive inhibitors, we employed a rational, structure-based approach exploiting a previously undescribed hydrogen bond network on the backside of the hinge binding domain centered on an Asp→Glu “switch” hypothesis. The difference of a single carbon atom between these two highly similar kinases (GSK3β-Asp133 → GSK3α-Glu196) translated into topological differences within the ATP binding and the adjacent hydrophobic selectivity pocket. A new secondary selectivity pocket unique to the ATP binding site of GSK3α is also described. These differences were engaged by distinct small molecule ligands to generate the first chemically matched, paralog selective and highly kinome selective GSK3 inhibitors. A robust structural rationale for the observed selectivity was developed and subsequently validated through site-directed mutagenesis and crystallographic studies. Further medicinal chemistry rational design, taking full advantage of residue and volume differences in these selectivity pockets, will undoubtedly lead to even greater paralog selectivity for second generation inhibitors. Most importantly, we were able to demonstrate that it is feasible to inhibit the kinase function of either GSK3α or GSK3β with sufficiently paralog selective inhibitors (10-fold) that functionally decouple paralog selective effects on β-catenin and further downstream activation of the WNT pathway in a cellular context. These first-in-class paralog selective inhibitors should provide an increased therapeutic index and allow for clinical doses and exposures which may have not been previously explored.
With an eye towards developing paralog selective GSK3 inhibitors as a therapeutically tractable approach to cancers, such as AML, we explored the effect of selective GSK3α inhibition on differentiation, stemness and colony formation. Small-molecule inhibition of GSK3α by BRD0705 induced myeloid differentiation through morphological and surface marker changes and impaired colony formation in AML cell lines and primary AML samples. Moreover, BRD0705 treatment reduced leukemia initiating cells frequency resulting in delayed disease onset in an MLL-AF9 syngeneic mice model, supporting the in vivo relevance of the observed repression in stemness transcriptional programs observed in AML cells with BRD0705 treatment. BRD0705 showed a favorable tolerability profile in mice treated with 15 and 30 mg/kg and BRD0705 decreased leukemia burden and prolonged survival in HL-60, MV4–11 and MLL-AF9 AML mouse models. No significant disease burden or survival effect was detected with BRD3731 (GSK3β inhibition) treatment. Future studies of this paralog series will extend testing to additional mouse models of AML and seek to identify novel drug-drug combinations with GSK3α inhibitors in AML. This manuscript provides a proof-of-concept roadmap toward the development of paralogue selective GSK3α inhibitors. However, additional optimization of theses molecular probes will be needed to develop this chemical series toward a drug candidate, setting the stage to test a paralog selective GSK3α inhibitor in phase I/II clinical trials for the treatment AML.
Beyond AML, this set of highly selective isochemogenic inhibitors will be useful chemical probes to refine our understanding of fundamental GSK3 biology and unravel substrate and functional selectivity of each GSK3 paralog, leading to potential therapeutic options in human diseases where the hyperactivity of the GSK3 kinases has been implicated, such as diabetes, cardiovascular and central nervous system disorders.
While we have pursued the study of an Asp→Glu switch within the GSK3 paralogs, it is tempting to speculate on the generality of this structural phenomenon in other paralog pairs of kinases. Interestingly, from an evolutionary perspective, studying the biological relevance of this newly unraveled backend hinge hydrogen bond network and single amino-acid switch will be key to decipher functional differences between such highly similar kinases and lead to the identification of preferential substrates of each paralog kinase. Indeed, our future investigations will seek to determine whether this Asp→Glu switch is conserved in other kinases and whether the chemistry discovered here can be exploited toward the development of other paralog selective compounds.
Materials and Methods
Study Design
This study sought to design a set of first-in-class isochemogenic and paralog selective GSK3 inhibitors and validate the use of our GSK3α inhibitor as a targeted therapy in AML. Exploiting a single carbon-atom difference between GSK3α and GSK3β and combining in silico and in vitro approaches, we were able to design paralog selective GSK3 inhibitors (BRD0705, BRD3731 and BRD0320). Selectivity and on target effects were validated using in vitro and in cellulo assays (kinome assays evaluating selectivity against >300 kinases; BRET-luminescence assays; ß-catenin accumulation through protein levels, phosphorylation modifications and nuclear translocation evaluated both by transcriptional reporter assays and immunofluorescence, Y279-GSK3α selective phosphorylation impairment, GSK3α KO single clones and selectivity pocket mutant forms of GSK3). Effects of BRD0705 in AML were studied in vitro in 6 AML cell lines and 5 primary patient AML blast samples and in vivo in 3 mouse models, including human orthotopic xenograft and syngeneic mouse models with a wide range of AML genetic backgrounds. Primary patient AML blasts were collected from bone marrow aspirates after obtaining patient informed consent under a Dana-Farber Cancer Institute Internal Review Board-approved protocol. Dana-Farber Cancer Institute Committee on Animal Care reviewed and approved the HL-60, MV4–11 and MLL-AF9 mouse experiments described in this study. Sample size was chosen in light of the fact that these in vivo models were historically highly penetrant, aggressive, and consistent. Blinded observers visually inspected mice for obvious signs of illness, such as loss of appetite, hunched posture, and lethargy. Mice were randomly assigned to each treatment group. The number of experimental replicates is specified in each figure legend.
Chemistry
All final compounds were confirmed to be of ≥95% purity based on HPLC LC-MS and 1H NMR analyses. General procedures for the synthesis of pyrazolo-tetrahydroquinolinone are described in Supporting Information along with analytical data for all final compounds.
Mobility Shift Microfluidics Assay
Purified GSK3β or GSK3α was incubated with tested compounds in the presence of 4.3 μM of ATP (at or just below Km to study competitive inhibitors) and 1.5 μM peptide substrate (Peptide 15, Caliper, MA) for 60 minutes at room temperature in 384-well plates (Seahorse Bioscience, MA) in assay buffer that contained 100 mM HEPES (pH 7.5), 10 mM MgCl2, 2.5 mM DTT, 0.004% Tween-20 and 0.003% Briji-35. Reactions were terminated by the addition of 10 mM EDTA. Substrate and product were separated electrophoretically, and fluorescence intensity of the substrate and product was determined by Labchip EZ Reader II (Caliper Life Sciences, MA). The kinase activity was measured as percent conversion. The reactions were performed in duplicate for each sample. The positive control, CHIR99021, was included in each plate and used to scale the data in conjunction with in-plate DMSO controls. The results were analyzed by Genedata Assay Analyzer. The percent inhibition was plotted against the compound concentration and the IC50 value was determined from the logistic dose-response curve fitting. Values are the average of at least three experiments. Compounds were tested using a 12-point dose curve with 3-fold serial dilution starting from 33 μM.
Functional Profiling
Briefly, a selection of compounds was screened against a panel of kinases at a single concentration of 10 μM. The kinases were selected from all families of the kinome and in all represented 60% of the entire kinome for a total of 311 kinases screened. This was completed utilizing one of two assays depending on the kinase being examined: 1) IMAP Assay. A solution of 4X inhibitor, 4X substrate/ATP/Metal solution and 2X kinase solution was prepared with assay buffer (20 mM HEPES, 0.01% Tween-20, 2 mM DTT, pH 7.4) and mixed/incubated in 384-well black plates for 1 hour at room temperature. A solution of IMAP binding reagent (IMAP Screening Express kit; Molecular Devices) was added to each well and incubated for 30 minutes. Kinase activity was then evaluated by fluorescence polarization at 485 nM (exc) and 530 nM (emm) of each well. 2) Off-Chip Mobility Shift Assay (MSA). A solution of 4X inhibitor, 4X substrate/ATP-Metal solution and 2X Kinase solution was prepared with assay buffer (20 mM HEPES, 0.01% Triton X-100, 2 mM DTT, pH 7.5) and mixed/incubated in 384-well plates for 1 or 5 hours depending on the kinase, at room temperature. A solution of termination buffer (QuickScout Screening assist MSA; Carna Biosciences) was added to each well. The entire reaction mixture was then applied to a LabChip3000 system (Caliper Life Science) and the product and substrate peptide peaks were separated and quantified. Evaluation of kinase activity was then determined based on the ratio of calculated peak heights of product (P) and substrate (S) peptides (P/(P+S)). For dose response IC50 determination: A solution of 4X inhibitor, 4X substrate/ATP Metal solution and 2X Kinase solution was prepared with assay buffer (20 mM HEPES, 0.01% Triton X-100, 2 mM DTT, pH 7.5) and mixed/incubated in 384-well plates for 1 or 5 hours, depending on the kinase, at room temperature. A solution of termination buffer (QuickScout Screening assist MSA; Carna Biosciences) was added to each well. The entire reaction mixture was then applied to a LabChip3000 system (Caliper Life Science) and the product and substrate peptide peaks were separated and quantified. Evaluation of kinase activity was then determined based on ratio of calculated peak heights of product (P) and substrate (S) peptides (P/(P+S)).
Cell transfection, treatments, and BRET measurements
To lower intracellular expression of the reporter fusion, GSK3α and GSK3β/NanoLuc fusion constructs (Promega) were diluted into promoterless Transfection Carrier DNA (Promega) at a mass ratio of 1:10 (mass/mass), prior to forming FuGENE HD complexes according to the manufacturer’s protocol (Promega). DNA:FuGENE complexes were formed at a ratio of 1:3 (mg DNA/mL FuGENE). One part of the transfection complexes was then mixed with 20 parts (v/v) of HEK293 cells (ATCC) suspended at a density of 2 × 105 in DMEM (Gibco) + 10% FBS (GE Healthcare), seeded into white, tissue culture treated 96-well plates at 2 × 104 cells per well, followed by incubation in a humidified, 37°C/5% CO2 incubator for 20 hr. Cells medium was removed and cells were resuspended in Opti-MEM without phenol red (Life Technologies). For target engagement, NanoBRET Kinase Tracer-06 (Promega) was added to the cells at 100 nM (below the observed EC50 for GSK3α and GSK3β) prior to test compound addition. Serially-diluted test compounds were then added to the cells and allowed to equilibrate for 2 h prior to BRET measurements. To measure BRET, NanoBRET NanoGlo Substrate and Extracellular NanoLuc Inhibitor (Promega) was added, and filtered luminescence was measure on GloMax Discover luminometer equipped with 450 nm BP filter (donor) and 600 nm LP filter (acceptor), using 0.5 s integration time. Milli-BRET units (mBU) are the BRET values x 1000. Competitive displacement data were then graphed with GraphPad Prism software using a 3-parameter curve fit with the following equation (Equation);
Crystal Structures
Detailed experimental conditions and parameters for X-ray co-crystal structures are provided in Supporting Information.
Cell Culture
HL-60, U937 and TF-1 cell lines were purchased from the American Type Culture Collection. NB-4 and MV4–11 cells were provided by Dr. Ross Levine (Memorial Sloan Kettering Cancer Center, NY), and MOLM-13 by Dr. Benjamin Ebert (Dana Farber Cancer Institute, MA). TF-1 cells were maintained in RPMI 1640 supplemented with 1% penicillin-streptomycin and 10% FBS with 20 ng per ml GM-CSF (Peprotech). MOLM-13 were maintained in RPMI 1640 supplemented with 1% penicillin-streptomycin and 20% FBS. All other cell lines were maintained in RPMI 1640 (Cellgro) supplemented with 1% penicillin-streptomycin (PS, Invitrogen) and 10% fetal bovine serum (FBS, Sigma-Aldrich) at 37 °C with 5% CO2, confirmed by STR genotyping and mycoplasma negative. The 293T cells were maintained in Dulbecco’s modified Eagle’s medium (Cellgro) supplemented with 10% FBS and 1% PS.
Mononuclear cells were isolated from primary patient AML bone marrow aspirates using Ficoll-Paque Plus (Amersham Biosciences) and red blood cells were lysed (eBioscience). These cells were maintained in StemSpan SFEM (StemCell Technologies Inc.) medium supplemented with 20 ng/ml IL-3 (Peprotech), 20 ng/ml IL-6 (Peprotech), 20 ng/ml GM-CSF (Peprotech), 100 ng/ml FLT3-Ligand (Peprotech) and 100 ng/ml SCF (Peprotech).
Plasmids and Cell Infection
The β-catenin reporter 7xTCF-luc-mCherry was a kind gift from Dr. William C. Hahn (Dana-Farber Cancer Institute, MA). LentiCRISPR v2 was a gift from Dr. Feng Zhang (Addgene plasmid # 52961). We generated lentiCRISPR v2-NT, lentiCRISPR v2-GSK3α#12, lentiCRISPR v2-GSK3α#8 and lentiCRISPR v2-GSK3α#9 vectors following the GeCKO Lentiviral CRISPR toolbox cloning protocol 50 and using the sgRNA guide sequences listed below. The GSK3α pWZL overexpression vector was generated as previously described 34. The pMMP LUC-NEO vector was a kind gift from Dr. Andrew Kung (Columbia University, New York).
For virus production, 12 μg of the above plasmids and 6 μg pCMV-GAG/POL and pCMV-VSVG (for retroviral infection), or 6 μg pCMV8.9 or psPAX2 and pCMV-VSVG (for lentiviral infection) packaging vectors were transfected into the 293T packaging cell line using X-tremeGENE 9 (Roche), and the resulting viral supernatants were harvested as previously described 34. The lentiviral and retroviral viruses were then concentrated using PEG-it™ Virus Precipitation Solution or Retro-concentin (SBI System Biosciences) respectively. Cells were infected using RetroNectin (Clontech) following the manufacturer’s recommendations for retroviral vectors and as previously described for lentiviral plasmids 34.
| Designation | sgRNA sequence |
|---|---|
| NT | GTAGCGAACGTGTCCGGCGT |
| GSK3α#12 | TACACGTGAGGCAAGGGTTG |
Site-directed Mutagenesis
The point mutation in the GSK3α sequence was obtained using a QuikChange XL Site-directed mutagenesis kit (Stratagene). The primers used for this mutagenesis reaction are listed below.
| Target | Mutant | Primer | Sequence |
|---|---|---|---|
| GSK3α | E196D (A588C) | Forward | CCTAAATCTGGTGCTGGACTATGTGCCCGAGACAGTG |
| Reverse | CACTGTCTCGGGCACATAGTCCAGCACCAGATTTAGG | ||
TCF/LEF β-catenin Reporter Assay
HEK 293T cells were transiently transfected with the β-catenin reporter 7xTCF-luc-mCherry and then treated with BRD0705, BRD3731, BRD0320 and CHIR99021 at the indicated concentrations for 48 hours. Cells were lysed in 1X Passive Lysis Buffer (Promega) for 15 minutes, Luciferase Assay Substrate (Promega) was added, and luminescence was measured using a FLUOstar Omega from BMG Labtech.
TF-1 cells were stably infected with the GFP β-catenin reporter (CLS-018G, Qiagen) and then treated with BRD0705, BRD3731 and BRD0320 at the indicated concentrations for 24 hours. GFP expression was measured using flow cytometry. 10,000 cells for each condition were analyzed with BD a FACSCanto II HTS.
Glycogen Quantitative Assay
Two million cells were harvested after 3 days of vehicle or BRD0705 treatment. Murine peripheral blood was collected and lysed for 10 minutes with red blood cell lysis buffer (Sigma). Samples were then lysed in dH2O. Glycogen was quantified using a Glycogen Colorimetric/Fluorometric Assay Kit per the manufacturer’s protocol (BioVision). The quantity of glycogen was normalized to total protein using Bradford colorimetric dosage (Bio-Rad). Data were normalized to cell number.
Western Immunoblotting
Cells were lysed in Cell Signaling Lysis Buffer (Cell Signaling Technology), EDTA-free Protease Inhibitor Cocktail tablets (Roche Diagnostics), and PhosSTOP Phosphatase Inhibitor (Roche Diagnostics), resolved by gel electrophoresis using Novex 4%–12% Bis-Tris Gel (Invitrogen), transferred to nitrocellulose membranes (Bio-Rad), and blocked for 1 hour in 5% BSA (Sigma-Aldrich). Blots were incubated with primary antibodies to p-GSK-3α/β(Y279/216) (Thermo Scientific #OPA1–03083), total GSK-3α (Cell Signaling Technology #9338), total GSK-3β (Cell Signaling Technology #9315), β-catenin (Cell Signaling Technology #8480S and BDB610154), p-β-catenin-S675 (Cell Signaling Technology #9567S), p-β-catenin-S33/37/T41 (Cell Signaling Technology #9561S), Actin (Thermo Scientific, MS1295P1) or Vinculin (Abcam #ab18058), followed by secondary antibodies anti-rabbit HRP (Amersham #NA9340V) or anti-mouse HRP (Amersham #NA9310V). Bound antibody was detected using Western Lightning Chemiluminescence Reagent (Perkin Elmer).
Morphology Assessment
Cytospin preparations and May-Grunwald Giemsa (Sigma) staining were performed at day 6 after treatment. Periodic Acid-Schiff staining was performed per the manufacturer’s protocol (Sigma). Representative images of three independent experiments were acquired by light microscopy (OLYMPUS BX40) under oil immersion at 100x magnification using SPOT software 5.1.
Immunofluorescence Staining
After 24 hours of treatment with DMSO, BRD0705, BRD3731, BRD0320 or CHR99021 at 20 μM, 30,000 to 50,000 cells were washed in PBS/2%FBS and then cytospun onto poly-L-Lysine coated slides (EMS). Cells were fixed in 4% paraformaldehyde (EMS) for 20 minutes, permeabilized in 0.2% Triton X-100 (RPI) for 30 minutes at 4°C, blocked in 1% BSA/0.05% triton for 1 hour at room temperature and then incubated for 1 hour at room temperature with primary anti-β-catenin (1/500) and then secondary antibody (Alexa Fluor 568 goat anti-rabbit, 1/1000). Three 5 minute washes were performed after each antibody incubation. Slides were mounted with Prolong (R) Gold Antifade with DAPI (Cell Signaling) and imaged with Yokogawa Spinning Disk Confocal / TIRF system. Representative images of three independent experiments were analyzed with Image-J software.
Flow Cytometry
Surface marker staining for human CD11b (PE-Cy7, Ebioscience), CD11c (APC, BD Pharmingen), CD14 (APC, Ebioscience) and CD117 (PE-Cy7, Biolegend) was performed at day 6 after treatment. Cell lines were washed in PBS containing 0.1% BSA and 2 mM EDTA before a 35 minutes incubation with anti-human conjugated antibodies. Cells were then washed twice with PBS containing 0.1%BSA and 2 mM EDTA before analysis of 10,000 cells for each condition with a BD FACSCanto II HTS.
Murine peripheral blood was collected and lysed for 10 minutes with red blood cell lysis buffer (Sigma). After two washing steps with PBS containing 0.1% BSA and 2 mM EDTA, cells were stained for 30 minutes at 4°C with anti-human CD45 antibody (APC, Ebioscience). Cells were then washed twice with PBS containing 0.1% BSA and 2 mM EDTA before analysis of 20,000 cells for each condition with BD a FACSCanto II HTS.
Methylcellulose Colony Formation Assay
Cell lines:
250 or 500 cells were seeded in methylcellulose-based medium (ClonaCell-TCS Medium) with DMSO, BRD0705 or BRD3731. The number of colonies was counted after MTT staining after 7–10 days in methylcellulose and analyzed through ImageQuant TL software. All experiments were performed in triplicate. PRISM GraphPad software was used for statistical analysis.
Primary patient samples and CD34 primary cells
Primary patient AML blasts were collected from bone marrow aspirates after obtaining patient informed consent under a Dana-Farber Cancer Institute Internal Review Board-approved protocol. Mononuclear cells were isolated using Ficoll-Paque Plus (Amersham Biosciences) and red blood cells were lysed. Normal human CD34 cells were purchased from Lonza. 2000–20000 cells were plated into semisolid methylcellulose medium (MethoCult H4236, StemCell Technologies Inc.) supplemented with 20 ng/ml IL-3 (Peprotech), 20 ng/ml IL-6 (Peprotech), 20 ng/ml GM-CSF (Peprotech), and 100 ng/ml SCF (Peprotech). After at least 10 days, colony number was evaluated after MTT staining of the colonies (R&D Systems). PRISM GraphPad software was used for statistical analysis.
RNA Sequencing
Genome expression profiling
U937 cells were treated in triplicate with either vehicle or 10 μM BRD0705, BRD3731 or BRD0320 for 24 hours. Total RNA was extracted with an RNeasy Kit (Qiagen) and profiled by RNA sequencing (HiSeq, Illumina) at the BioMicroCenter from the Massachusetts Institute of Technology (Cambridge, MA, USA). The total number of reads for individual samples ranged from 21 to 24 Million base pairs. Quality control tests for the unmapped reads were performed using the FASTQC software (www.bioinformatics.babraham.ac.uk/projects/fastqc/). The reads were aligned to the GRCh37/hg19 human genes by using Bowtie v2 51. The average percentage of uniquely mapped reads in the aligned data was 84.90%, with a standard deviation of 2.7%.
FPKM scores for genes were computed based on the RSEM R software package 52. Expression data was estimated as log2(FPKM). The significance of the differential expression between the control and treatment phenotypes was estimated based on the EBSeq method implemented in the EBSeq R library 53 by using the significance cutoff P ≤ 0.05 for the posterior probability. The aligned RNASeq data for this experiment have been deposited in the GEO database (GSE109987).
Comparative Marker Analysis
The 15 samples available in the data were separated into four groups: one corresponding to vehicle, the second to BRD0705, the third to BRD3731 and the fourth to BRD0320 treatment. The Comparative Marker Selection module from GenePattern v3.9.6 54 was employed to identify individual genes that were differentially expressed between treated and vehicle conditions. The analysis was performed on the log2(FPKM) expression data by applying a 2-sided Signal-to-Noise Ratio (SNR) test followed by 1000 permutations of phenotype labels. The settings for the SNR parameters were log-transformed-data:yes, complete:no, balanced:no, smooth p-values: yes.
Molecular signatures for genes down-regulated or up-regulated by each of the BRD0705 and BRD0320 compounds vs vehicle were defined based on the cut-offs SNR permutation p-value ≤ 0.05, Benjamini-Hochberg false discovery rate (FDR) ≤ 0.05, and absolute fold change [FC, log2(FPKM)] ≥ 1.5. The BRD0705 molecular signature consists of 193 genes down-regulated and 55 genes up-regulated between treated samples vs vehicle (Table S7), while the BRD0320 molecular signature consists of 975 genes down-regulated and 556 genes up-regulated between treated samples vs vehicle (Table S8). The BRD0705 and BRD0320 molecular signatures were visualized as highlighted dots in the volcano plots of fold changes of log2(RPKM) expression vs –log10(P-value) scores.
GSEA analysis
The Gene Set Enrichment Analysis (GSEA v2.1.0) software 55,56 was used to identify functional associations of the molecular phenotypes induced by BRD0705, BRD3731 and BRD0320 with a compendium of gene signatures including the MSigDB v5.0 56 collections c2 of 4,725 curated gene sets and c6 of 189 oncogenic signatures, the DMAP collection of hematopoietic lineage differentiation signatures 57 and with the GSK3 inhibitor CHIR99021 gene set signature (GSE54056 58). Gene sets with less than 15 genes or more than 500 genes were excluded from the analysis. Gene sets with an FDR ≤ 0.25 and a nominal P-value ≤ 0.05 were considered significant hits. The results were visualized on GSEA plots and heatmaps for selected gene signatures, and with the Enrichment Map software, which organizes the significant gene sets into a network called an “enrichment map.” In the enrichment map, the nodes correspond to gene sets and the edges reflect significant overlap between the nodes according to a two-tailed Fisher exact test. The size of the nodes is correlated with the number of genes in the gene set. The hubs correspond to collections of gene sets with a unifying class label according to gene ontology (GO) biologic processes.
In vivo studies
HL-60 and MV4–11 cells were transduced with the pMMP-LucNeo retrovirus and selected with neomycin at 1 mg/mL. 2×106 cells were subsequently injected via tail vein into 8-week-old male NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, The Jackson Laboratory). Three (HL-60) and four (MV4–11) days post cell injection, mice were randomized to be treated once daily by oral gavage with vehicle (7.5% NMP, 10% Kolliphor HS15, 30% PEG 400 and 52.5% normal saline; 5 μL/g), BRD0705 (15 or 30 mg/kg), BRD3731 (30 mg/kg) or BRD0320 (30 mg/kg). All mice received daily subcutaneous hydration with 5% dextrose. Mice were imaged at the indicated time points on an IVIS Spectrum (Caliper Life Sciences) to measure the bioluminescence signal. Whole blood was collected by sub-mandibular bleeding at the indicated time point.
MLL-AF9-DsRed L-GMP cells were treated for 24h with either DMSO, 10 μM BRD0705, 10 μM BRD3731 or 10 μM BRD0320 before tail vein injection of 500 000, 250 000, 100 000 or 50 000 cells into sub-lethally irradiated 6-week-old male C57BL/6 recipient mice. Mice were then followed for survival.
Statistical analysis
Statistical analysis was done using Microsoft Excel, PRISM 5.03 (GraphPad) or indicated software for more dedicated analysis. Data were analyzed using a nonparametric Mann-Whitney test (with the assumption of no Gaussian distribution of the group) unless otherwise specified, and the threshold of significance (alpha) was always set at 0.05.
Supplementary Material
Acknowledgments:
We thank Drs. Suto and White (Xtal Biosciences) for helpful discussions.
Funding: This work was supported by the Stanley Medical Research Institute and grants from the National Cancer Institute R01 CA140292 and R35CA210030 (KS) and the Children’s Leukemia Research Association (KS). KS is a Leukemia and Lymphoma Society Scholar. LB is an MD-PhD candidate of “Ecole de l’INSERM Liliane Bettencourt” and a recipient of Philippe Foundation fellowship. AP is a recipient of the ATIP-AVENIR and the ERC research programs.
Footnotes
Competing Financial Interests Statement: None. The following patent applications were filed US-2014–0107141 or granted US Patent 9,096,594.
Data and materials availability: The authors declare that data supporting the findings of this study are available within the paper and its supplementary information document. Structure factors and coordinates have been deposited in the Protein Data Bank under accession code: 5KPK (hGSK3β bound to BRD0209); 5KPM (hGSK3Β bound to BRD3731); 5KPL (hGSK3β bound to BRD0705); 5T31 (hGSK3β mutant bound to BRD0705). BRD0705, BRD3731, BRD0320 and BRD5648 can be obtained through an MTA from the Broad Institute of Harvard and MIT. The aligned RNASeq data have been deposited in the GEO database (GSE109987).
References:
- 1.Innan H & Kondrashov F The evolution of gene duplications: classifying and distinguishing between models. Nat Rev Genet 11, 97–108, doi:10.1038/nrg2689 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Woodgett JR cDNA cloning and properties of glycogen synthase kinase-3. Methods Enzymol 200, 564–577 (1991). [DOI] [PubMed] [Google Scholar]
- 3.Matsuda T et al. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc Natl Acad Sci U S A 105, 20900–20905, doi:10.1073/pnas.0808315106 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phiel CJ, Wilson CA, Lee VM & Klein PS GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423, 435–439, doi:10.1038/nature01640 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Beurel E, Grieco SF & Jope RS Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 148, 114–131, doi:10.1016/j.pharmthera.2014.11.016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen P & Goedert M GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov 3, 479–487, doi:10.1038/nrd1415 (2004). [DOI] [PubMed] [Google Scholar]
- 7.MacAulay K et al. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab 6, 329–337, doi:10.1016/j.cmet.2007.08.013 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Hall JL Glycogen synthase kinase-3 and the heart. J Am Coll Cardiol 64, 707–709, doi:10.1016/j.jacc.2014.06.1142 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Lal H, Ahmad F, Woodgett J & Force T The GSK-3 family as therapeutic target for myocardial diseases. Circ Res 116, 138–149, doi:10.1161/CIRCRESAHA.116.303613 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kramer T, Schmidt B & Lo Monte F Small-Molecule Inhibitors of GSK-3: Structural Insights and Their Application to Alzheimer’s Disease Models. Int J Alzheimers Dis 2012, 381029, doi:10.1155/2012/381029 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osolodkin DI, Palyulin VA & Zefirov NS Glycogen synthase kinase 3 as an anticancer drug target: novel experimental findings and trends in the design of inhibitors. Curr Pharm Des 19, 665–679 (2013). [PubMed] [Google Scholar]
- 12.Bang D, Wilson W, Ryan M, Yeh JJ & Baldwin AS GSK-3alpha promotes oncogenic KRAS function in pancreatic cancer via TAK1-TAB stabilization and regulation of noncanonical NF-kappaB. Cancer Discov 3, 690–703, doi:10.1158/2159–8290.CD-12–0541 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo J Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett 273, 194–200, doi:10.1016/j.canlet.2008.05.045 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ougolkov AV & Billadeau DD Targeting GSK-3: a promising approach for cancer therapy? Future Oncol 2, 91–100, doi:10.2217/14796694.2.1.91 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Patel S & Woodgett J Glycogen synthase kinase-3 and cancer: good cop, bad cop? Cancer Cell 14, 351–353, doi:10.1016/j.ccr.2008.10.013 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polakis P The many ways of Wnt in cancer. Curr Opin Genet Dev 17, 45–51, doi:10.1016/j.gde.2006.12.007 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Atilla-Gokcumen GE, Williams DS, Bregman H, Pagano N & Meggers E Organometallic compounds with biological activity: a very selective and highly potent cellular inhibitor for glycogen synthase kinase 3. Chembiochem 7, 1443–1450, doi:10.1002/cbic.200600117 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Forde JE & Dale TC Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci 64, 1930–1944, doi:10.1007/s00018–007-7045–7 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meijer L, Flajolet M & Greengard P Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci 25, 471–480, doi:10.1016/j.tips.2004.07.006 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Palomo V et al. 5-imino-1,2,4-thiadiazoles: first small molecules as substrate competitive inhibitors of glycogen synthase kinase 3. J Med Chem 55, 1645–1661, doi:10.1021/jm201463v (2012). [DOI] [PubMed] [Google Scholar]
- 21.Wagner FF et al. Inhibitors of Glycogen Synthase Kinase 3 with Exquisite Kinome-Wide Selectivity and Their Functional Effects. ACS Chem Biol, doi:10.1021/acschembio.6b00306 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Doble BW, Patel S, Wood GA, Kockeritz LK & Woodgett JR Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Developmental cell 12, 957–971, doi:10.1016/j.devcel.2007.04.001 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo Monte F et al. Identification of glycogen synthase kinase-3 inhibitors with a selective sting for glycogen synthase kinase-3alpha. J Med Chem 55, 4407–4424, doi:10.1021/jm300309a (2012). [DOI] [PubMed] [Google Scholar]
- 24.Lo Monte F et al. Structure-based optimization of oxadiazole-based GSK-3 inhibitors. Eur J Med Chem 61, 26–40, doi:10.1016/j.ejmech.2012.06.006 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Georgievska B et al. AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J Neurochem 125, 446–456, doi:10.1111/jnc.12203 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Gupta K et al. GSK3 is a regulator of RAR-mediated differentiation. Leukemia 26, 1277–1285, doi:10.1038/leu.2012.2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCubrey JA et al. Multifaceted roles of GSK-3 and Wnt/beta-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia 28, 15–33, doi:10.1038/leu.2013.184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Si J, Mueller L & Collins SJ GSK3 inhibitors enhance retinoic acid receptor activity and induce the differentiation of retinoic acid-sensitive myeloid leukemia cells. Leukemia 25, 1914–1918, doi:10.1038/leu.2011.171 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Song EY et al. Glycogen synthase kinase−−3beta inhibitors suppress leukemia cell growth. Exp Hematol 38, 908–921 e901, doi:10.1016/j.exphem.2010.06.001 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Wang Z et al. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature 455, 1205–1209, doi:10.1038/nature07284 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guezguez B et al. GSK3 Deficiencies in Hematopoietic Stem Cells Initiate Pre-neoplastic State that Is Predictive of Clinical Outcomes of Human Acute Leukemia. Cancer Cell 29, 61–74, doi:10.1016/j.ccell.2015.11.012 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Wang Y et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 327, 1650–1653, doi:10.1126/science.1186624 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeung J et al. beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 18, 606–618, doi:10.1016/j.ccr.2010.10.032 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Banerji V et al. The intersection of genetic and chemical genomic screens identifies GSK-3alpha as a target in human acute myeloid leukemia. J Clin Invest 122, 935–947, doi:10.1172/JCI46465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Licht-Murava A & Eldar-Finkelman H Exploiting substrate recognition for selective inhibition of protein kinases. Curr Pharm Des 18, 2914–2920 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Grutter C et al. Targeting GSK3 from Ustilago maydis: type-II kinase inhibitors as potential antifungals. ACS Chem Biol 7, 1257–1267, doi:10.1021/cb300128b (2012). [DOI] [PubMed] [Google Scholar]
- 37.Cole A, Frame S & Cohen P Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J 377, 249–255, doi:10.1042/BJ20031259 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lochhead PA et al. A chaperone-dependent GSK3beta transitional intermediate mediates activation-loop autophosphorylation. Mol Cell 24, 627–633, doi:10.1016/j.molcel.2006.10.009 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Robers MB et al. Target engagement and drug residence time can be observed in living cells with BRET. Nat Commun 6, 10091, doi:10.1038/ncomms10091 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennett CN et al. Regulation of Wnt signaling during adipogenesis. J Biol Chem 277, 30998–31004, doi:10.1074/jbc.M204527200 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Patel S et al. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol 28, 6314–6328, doi:10.1128/MCB.00763–08 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neumann T, Benajiba L, Goring S, Stegmaier K & Schmidt B Evaluation of Improved Glycogen Synthase Kinase-3alpha Inhibitors in Models of Acute Myeloid Leukemia. J Med Chem 58, 8907–8919, doi:10.1021/acs.jmedchem.5b01200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.del Ser T et al. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J Alzheimers Dis 33, 205–215, doi:10.3233/JAD-2012–120805 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Gray JE et al. A first-in-human phase I dose-escalation, pharmacokinetic, and pharmacodynamic evaluation of intravenous LY2090314, a glycogen synthase kinase 3 inhibitor, administered in combination with pemetrexed and carboplatin. Investigational new drugs 33, 1187–1196, doi:10.1007/s10637–015-0278–7 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Hoglinger GU et al. Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov Disord 29, 479–487, doi:10.1002/mds.25815 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Lovestone S et al. A phase II trial of tideglusib in Alzheimer’s disease. J Alzheimers Dis 45, 75–88, doi:10.3233/JAD-141959 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Rizzieri DA et al. An open-label phase 2 study of glycogen synthase kinase-3 inhibitor LY2090314 in patients with acute leukemia. Leuk Lymphoma, 1–7, doi:10.3109/10428194.2015.1122781 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Tolosa E et al. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord 29, 470–478, doi:10.1002/mds.25824 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Jiang Y et al. Activation of the Wnt pathway through AR79, a GSK3beta inhibitor, promotes prostate cancer growth in soft tissue and bone. Mol Cancer Res 11, 1597–1610, doi:10.1158/1541–7786.MCR-13–0332-T (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87, doi:10.1126/science.1247005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, doi:10.1038/nmeth.1923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323, doi:10.1186/1471–2105-12–323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leng N et al. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29, 1035–1043, doi:10.1093/bioinformatics/btt087 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reich M et al. GenePattern 2.0. Nat Genet 38, 500–501, doi:10.1038/ng0506–500 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Mootha VK et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273, doi:10.1038/ng1180 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550, doi:10.1073/pnas.0506580102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Novershtern N et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144, 296–309, doi:10.1016/j.cell.2011.01.004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suga A, Sadamoto K, Fujii M, Mandai M & Takahashi M Proliferation potential of Muller glia after retinal damage varies between mouse strains. PLoS One 9, e94556, doi:10.1371/journal.pone.0094556 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.