

Abstract
PURPOSE
Phosphatidylinositol 3-kinase (PI3K) signaling is highly active in glioblastomas. We assessed pharmacokinetics, pharmacodynamics, and efficacy of the pan-PI3K inhibitor buparlisib in patients with recurrent glioblastoma with PI3K pathway activation.
METHODS
This study was a multicenter, open-label, multi-arm, phase II trial in patients with PI3K pathway–activated glioblastoma at first or second recurrence. In cohort 1, patients scheduled for re-operation after progression received buparlisib for 7 to 13 days before surgery to evaluate brain penetration and modulation of the PI3K pathway in resected tumor tissue. In cohort 2, patients not eligible for re-operation received buparlisib until progression or unacceptable toxicity. Once daily oral buparlisib 100 mg was administered on a continuous 28-day schedule. Primary end points were PI3K pathway inhibition in tumor tissue and buparlisib pharmacokinetics in cohort 1 and 6-month progression-free survival (PFS6) in cohort 2.
RESULTS
Sixty-five patients were treated (cohort 1, n = 15; cohort 2, n = 50). In cohort 1, reduction of phosphorylated AKTS473 immunohistochemistry score was achieved in six (42.8%) of 14 patients, but effects on phosphoribosomal protein S6S235/236 and proliferation were not significant. Tumor-to-plasma drug level was 1.0. In cohort 2, four (8%) of 50 patients reached 6-month PFS6, and the median PFS was 1.7 months (95% CI, 1.4 to 1.8 months). The most common grade 3 or greater adverse events related to treatment were lipase elevation (n = 7 [10.8%]), fatigue (n = 4 [6.2%]), hyperglycemia (n = 3 [4.6%]), and elevated ALT (n = 3 [4.6%]).
CONCLUSION
Buparlisib had minimal single-agent efficacy in patients with PI3K-activated recurrent glioblastoma. Although buparlisib achieved significant brain penetration, the lack of clinical efficacy was explained by incomplete blockade of the PI3K pathway in tumor tissue. Integrative results suggest that additional study of PI3K inhibitors that achieve more-complete pathway inhibition may still be warranted.
INTRODUCTION
Glioblastoma is the most common malignant primary brain tumor.1 Despite treatment with surgery, radiation therapy (RT), and chemotherapy, outcomes have not substantially improved over the past two decades, with median overall survival (OS) of only 14 to 18 months.2-4 Limited drug delivery as a result of the blood-brain barrier (BBB) represents one of the most significant challenges and partly explains why many agents that target oncogenic pathways of glioblastoma but whose chemical properties do not allow significant BBB penetration have minimal efficacy.5 However, few studies directly examined tumor tissue during treatment,6,7 which prevents reliable conclusions about drug effectiveness with regard to level of target inhibition and effects on cell death and proliferation. Studies designed to confirm drug penetration and target engagement therefore may be critical to understanding trial results and improving outcomes in glioblastoma.
The PI3K pathway is activated in most glioblastomas.8 PTEN loss and PIK3CA or PIK3R1 mutations represent potential therapeutic targets that are found in approximately 45% of glioblastomas.8,9 Previous trials of mechanistic target of rapamycin (mTOR) complex 1 inhibitors did not show significant efficacy.6,10,11 More recently, PI3K inhibitors have been evaluated. In a trial of the pan-PI3K inhibitor PX-866 in 32 molecularly unselected patients with recurrent glioblastoma, one patient achieved a partial response (PR), and the 6-month progression-free survival (PFS6) rate was 17%.12 However, this study did not evaluate whether adequate brain penetration and target engagement was achieved.
Buparlisib (NVP-BKM120) is an oral pan-PI3K inhibitor that targets all four isoforms of class 1 PI3K (α, β, γ, and δ).13 Buparlisib has high penetration across the BBB. In preclinical studies, buparlisib enters the brain at therapeutic concentrations demonstrated to inhibit the PI3K pathway in normal brain and glioma models in vitro and in vivo.14-16 The Ivy Foundation Early Phase Clinical Trials Consortium conducted a phase II trial of buparlisib in patients with recurrent glioblastoma with evidence of PI3K pathway activation to assess the pharmacokinetics, pharmacodynamics, and efficacy of buparlisib in this population.
METHODS
Study Design and Participants
This study was a multicenter, open-label, and multi-arm phase II trial in patients with recurrent glioblastoma at first or second relapse. Written informed consent was obtained from all participants. The study was approved by the local institutional review board of each participating institution and consisted of two cohorts: a surgery plus treatment arm (cohort 1) and a treatment-only arm (cohort 2; Appendix Fig A1, online only). Eligible participants were age 18 years or older with a centrally confirmed diagnosis of glioblastoma. Patients must have not responded to prior RT, with an interval of at least 12 weeks from RT completion to study entry. Tumor progression was confirmed by magnetic resonance imaging or computed tomography scan. Prior treatment with bevacizumab or vascular endothelial growth factor receptor44 inhibitors, PI3K, AKT, or mTOR inhibitors was not permitted. Patients had a Karnofsky performance status greater than or equal to 60, adequate organ and bone marrow function, fasting plasma glucose less than 120 mg/dL, hemoglobin A1C less than or equal to 8%, baseline left ventricular ejection fraction greater than or equal to 50%, and QTc less than 480 ms. Patients on enzyme-inducing anticonvulsants, warfarin, more than 4 mg/d dexamethasone, strong CYP3A inhibitors or inducers, or QT-prolonging medications were excluded, as were patients with a history of clinically significant cardiovascular events, intratumoral hemorrhage, or psychiatric disorders.
Histomolecular criteria for eligibility included PIK3CA or PIK3R1 mutation, loss of PTEN activity through PTEN mutation, homozygous deletion or negative PTEN expression (< 10% of tumor cells that stained positive), or positive phosphorylated AKTS473 (pAKTS473) by central immunohistochemistry (IHC) review. Cohort 1 surgical patients were required to have positive expression of pAKTS473 within the archival tumor.
Procedure
Buparlisib was supplied by Novartis (East Hanover, NJ). The dose of buparlisib was 100 mg administered orally daily.17,18
In cohort 1, participants received a pre-operative course of buparlisib for 7 to 13 days. The last dose of buparlisib was on the day of surgery. Whenever possible, tissue from both enhancing and nonenhancing tumor was collected. After recovery from surgery, participants resumed buparlisib in a manner consistent with cohort 2. In cohort 2, participants received buparlisib 100 mg daily for each 28-day cycle until disease progression or unacceptable toxicity.
Tumor assessments were performed with magnetic resonance imaging scans every 8 weeks using the Response Assessment in Neuro-Oncology criteria (Appendix Table A1, online only).19 PFS6 was defined as the proportion of participants alive and progression free at 6-months from cycle 1, day 1, of buparlisib. Only participants who had measurable disease at baseline and received at least one dose of therapy were evaluable for response, which was centrally reviewed for participants who achieved response or PFS6. Adverse events were evaluated using the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0). Additional analyses, including pharmacokinetics and IHC studies, tumor genomic profiling, reverse-phase protein array (RPPA), and matrix-assisted laser desorption/ionization-mass spectrometry imaging are described in the Appendix (online only).
Outcomes
The primary objectives in cohort 1 were to evaluate PI3K pathway modulation as a result of buparlisib in tumor tissue and to evaluate buparlisib concentration in enhancing and nonenhancing tumor tissue and plasma. Secondary end points included pharmacokinetics and safety of buparlisib in this population. Exploratory end points included correlation of 18F-fluorodeoxyglucose (FDG) positron emission tomography with response.
In cohort 2, the primary objective was to investigate the treatment efficacy of buparlisib in participants with recurrent glioblastoma as assessed by PFS6. Secondary end points were response rates and the median PFS, OS, and safety profile of buparlisib. Exploratory end points included correlation of outcomes with tumor genomics.
Sample Size Justification
The cohort 1 primary end point was the modulation of the PI3K pathway as assessed by IHC for pAKTS473 and phosphorylated S6 S235/236 (pS6S235/236). On the basis of historical data or mTOR complex 1 inhibitors in recurrent glioblastoma,6 a pharmacodynamic response rate less than 40% was considered to be low, a response rate of greater than or equal to 75% was considered to be high, and this portion of the trial was considered a success if nine (60%) of 15 participants showed a pathologic response. With a sample size of 15 patients, there was a 94% chance of this occurring if the true response rate was 75% and a 10% chance of this occurring if the true response rate was 40%.
The cohort 2 primary end point was PFS6. Historical comparison data suggested that ineffective therapies in recurrent glioblastoma have a PFS6 rate of approximately 9% to 16%.5,20,21 The trial was sized to differentiate between a 15% and a 32% PFS6. With a total sample size of 50 participants, this design yielded at least 90% power with a one-sided α less than .1 to detect a true PFS6 rate of at least 32%. If the number of successes was 12 or more, the therapy was to be considered worthy of additional study. More statistical analysis details are provided in the Appendix.
RESULTS
Patient Characteristics
Between May 9, 2011, and February 26, 2014, 136 patients were screened for eligibility from seven centers in the United States. Of these patients, 71 were excluded (Fig 1). Sixty-five patients were eligible and assigned to receive buparlisib (cohort 1, n = 15; cohort 2, n = 50; Fig 1).
FIG 1.

Study flow. PD, pharmacodynamics; PFS6, 6-month progression free survival; PK, pharmacokinetics.
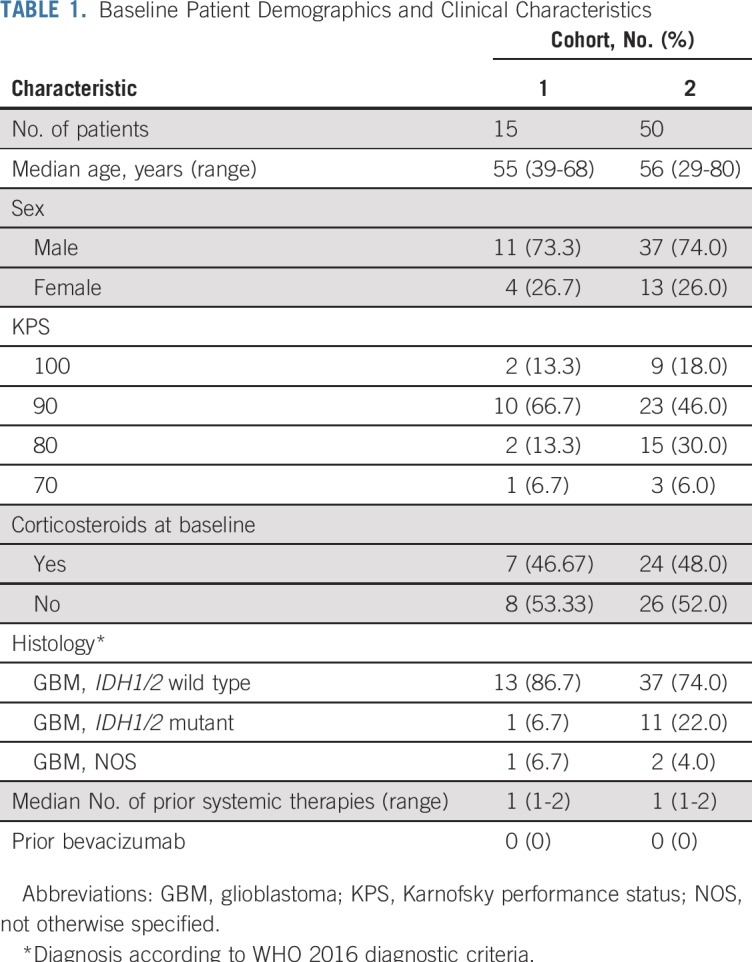
Baseline characteristics of patients are listed in Table 1. Sixty-four patients (98.5%) had received RT and temozolomide, and 31 (41.7%) and eight (12.3%) had received surgery or systemic therapy, respectively, for progressive disease. Demonstration of PI3K pathway activation in archival tumor tissue was based on IHC of PTEN and pAKTS473 status in 62 patients (95.4%) and genomic testing in three (4.6%). Overall, 31 enrolled patients (47.7%) had combined negative PTEN and positive pAKTS473 IHC; 24 (36.9%) had positive pAKTS473 IHC; seven (10.8%) had negative PTEN IHC; and three (4.6%) had genomic testing showing PTEN inactivation or PIK3CA mutation.
TABLE 1.
Baseline Patient Demographics and Clinical Characteristics
Pharmacokinetics and Pharmacodynamics of Buparlisib
Buparlisib was absorbed rapidly, achieving a maximum plasma concentration of 471 ± 147 ng/mL and 820 ± 192 ng/mL within a median of 1.5 hours postdose on days 1 and 8, respectively (Appendix Table A2, online only). Both maximum plasma concentration and exposure (0- to 5-hour area under the curve) increased between days 1 and 8, with an accumulation ratio of 1.88 ± 0.503 and 2.42 ± 0.726, respectively (Appendix Table A2). The accumulation of buparlisib was consistent with the reported half-life of approximately 40 hours reaching steady state by day 8.17,18
Resected tumor tissue was evaluable for pharmacokinetics and pharmacodynamics analyses in 14 patients. The average time between the last dose of buparlisib and the time of tumor sampling was 5 ± 1.61 hours. The average plasma concentration at the time of the tumor sampling was 585 ± 192 ng/mL. The geometric mean concentration of buparlisib in the tumor tissue was 612 ng/g (range, 86 to 6,947 ng/g), with a resulting tumor-to-plasma geometric mean ratio of 1.0 (range, 0.18 to 8.44). There was no significant difference between the non–contrast-enhancing (CE) and CE tumor tissue concentrations of buparlisib (mean, 404 ± 429 v 654 ± 363, respectively; P = 0.16; Fig 2A). Brain penetration of buparlisib also was confirmed by matrix-assisted laser desorption/ionization-mass spectrometry imaging performed on tumor specimens that showed the presence of drug in the tumor as well as in the infiltrated brain parenchyma (Fig 2B).
FIG 2.

Buparlisib pharmacokinetics and phosphatidylinositol 3-kinase pathway modulation as a result of buparlisib in tumor tissue in cohort 1. (A) Box plots of buparlisib concentration in non–contrast-enhancing (NCE) and contrast-enhancing (CE) tumor tissue assessed by liquid chromatography-tandem mass spectrometry. Difference between groups was calculated using the Mann-Whitney U test. (B) Histopathologic and matrix-assisted laser desorption/ionization-mass spectrometry imaging drug analysis on stereotactically registered specimens collected from a patient in cohort 1 treated with buparlisib. Image on the left demonstrates location of buparlisib (green) and vessels as measured by heme (red). Hematoxylin and eosin (HE) staining of the corresponding tissue. Outlined area delineates tumor and adjacent infiltrated brain parenchyma. (C) Representative microscopy images of the HE staining and PTEN, phosphorylated AKT (pAKT), and phosphorylated S6 (pS6) in tumor samples collected at baseline and on buparlisib treatment from a patient in cohort 1. (D) Quantification of pAKT immunohistochemistry (IHC) staining intensity in the surgical cohort. Data are mean ± SEM (n = 5 to 7 replicates for each sample). (E) Box plots of mean pAKT IHC staining intensity in the archival and resected tissues from the surgical cohort (n = 14). Difference between groups was calculated using the Wilcoxon test (See Appendix).
A decrease in pAKTS473 IHC score was achieved in six patients (42.9%) and was statistically significant in the overall cohort (Figs 2C to 2E), whereas the analysis of an independent set of pre- and post-treatment (RT + temozolomide) glioblastoma pairs did not show significant change in pAKTS473 or pS6S235/236 IHC (Appendix Fig A2, online only). A reduction in pAKTT308 only in post–buparlisib treatment tumor also was observed in RPPA analysis of 299 antibodies (Appendix Table A4, online only). Nevertheless, in seven patients (50%), no change in pAKTS473 IHC score was noted, and one patient (7.1%) had an increase in pAKTS473 IHC score, which suggested an incomplete blockade of PI3K signaling in approximately one half of patients. This finding was consistent with the absence of modulation of pS6S235/236 by IHC and lack of consistent changes in pS6S235/236, pS6S240/244, and p70 S6T389 kinases in the RPPA analysis (Appendix Fig A3, online only; Appendix Table A4). Moreover, RPPA analysis did not show a consistent change in members of the mitogen-activated protein kinase (MAPK) pathway when comparing buparlisib-treated versus control glioblastoma pairs (Appendix Table A4). Finally, there was no significant change in tumor cell proliferation between baseline and post-buparlisib treatment samples as assessed by the IHC proliferation marker Ki-67 (Appendix Fig A2).
Outcome
At final analysis (April 30, 2018), none of the 65 patients remained on treatment. The most frequent reason for treatment discontinuation in both groups was disease progression (59 [90.8%]; Fig 1). The median follow-up was 15.6 months (range, 3.6 to 36.6 months) in cohort 1 versus 9.8 months (range, 1.0 to 71.2 months) in cohort 2.
The study did not meet its primary end point for PFS6 with buparlisib in cohort 2 (n = 50); four patients (8%; 95% CI, 3% to 19%) reached PFS6, and the median PFS was 1.7 months (95% CI, 1.4 to 1.8 months; Table 2; Appendix Fig A4, online only). OS data were mature, with 58 deaths in the total population at the cutoff date (cohort 1, n = 13; cohort 2, n = 45). Two patients were still alive, and five were lost to follow-up. The median OS was 17.9 months (95% CI, 9.3 to 29.2 months) in cohort 1 and 9.8 months (95% CI, 8.4 to 12.1 months) in cohort 2 (Table 2; Appendix Fig A4).
TABLE 2.
Response to Treatment
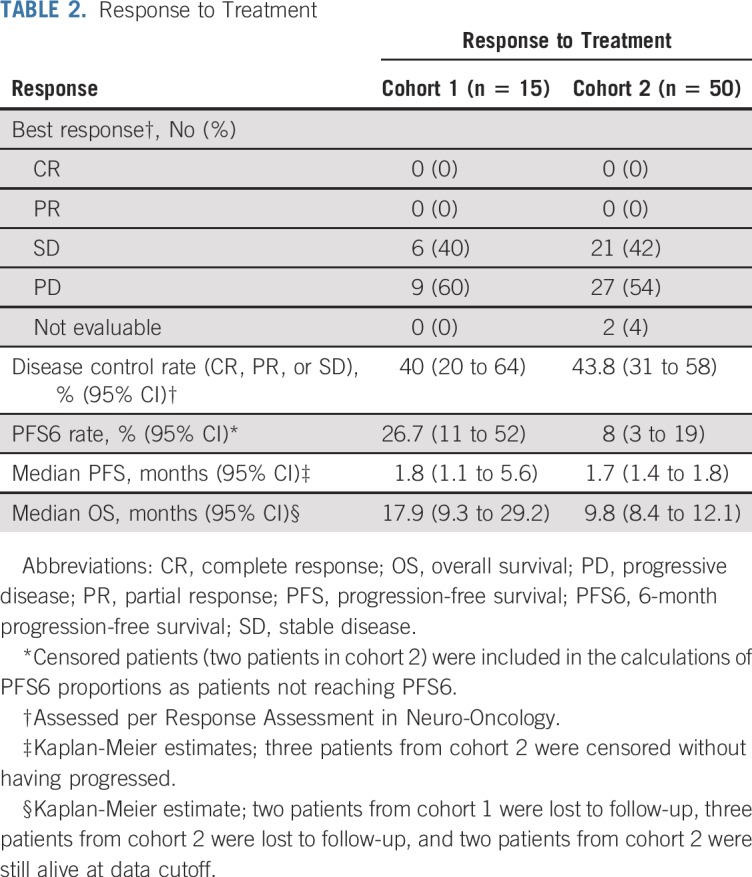
Best overall response was evaluable in 63 patients (96.9%). No patients achieved a radiographic response (Table 2). Six patients (40%; 95% CI, 20% to 64%) in cohort 1 and 21 (43.8%; 95% CI, 31% to 58%) in cohort 2 had disease stabilization as best response. In addition, 12 patients in cohort 1 had an FDG-positron emission tomography scan performed before and after treatment with buparlisib (mean delay, 11.4 days; range, 7 to 25 days). Six of the 12 patients had a modest decrease in tumor-to-background ratio in FDG uptake (−2.35% to −18.7%), but there was no correlation with outcome.
Correlation of Outcomes With Genotyping
Correlation between outcome and tumor genotyping was assessed in 46 patients (71.9%; eight [53.3%] in cohort 1 and 38 [76%] in cohort 2) for whom copy number array and/or tumor-targeted sequencing was available (Fig 3). Overall, pathogenic variants of PTEN, PIK3CA, or PIK3R1 were identified in 27 patients (56.3%; Fig 3).
FIG 3.

Relationship between mutations in phosphatidylinositol 3-kinase (PI3K) pathway members and response to buparlisib. Patient outcomes, including best overall response (BOR), median progression-free survival (PFS), and median overall survival (OS), are shown in the top rows; tumor genotyping and immunohistochemistry (IHC) results are shown in the middle rows; and integrative biomarker analysis for PI3K/PTEN signaling is shown in the bottom rows. Missense mutations are displayed in green, amplifications in orange, and deletions in blue. All mutations were reviewed by a molecular diagnostician to confirm that they were deemed pathogenic/hotspot mutations and were not commonly identified in normal databases as germline single nucleotide polymorphisms.
Candidate biomarkers, including PTEN, PIK3CA, PIK3R1, EGFR, PDGFRA, IDH1/2, and TP53 molecular alterations and IHC evidence for PTEN inactivation, pAKTS473, and pS6S235/236 activation, were evaluated for their association with outcome. Although no statistically significant association was found between PFS6 or OS and any of the candidate biomarkers, there was a shorter PFS in patients with IDH1/2-mutant versus IDH1/2 wild-type glioblastoma (median PFS, 0.9 months [interquartile range (IQR), 0.9-1.8 months] v 1.8 months [IQR, 1.1-3.6 months], respectively; log-rank P = .002; Appendix Fig A5, online only). No statistically significant association was found between PFS and PIK3CA/PIK3R1-mutant glioblastoma (median PFS, 2.2 months [IQR, 1.8-2.8 months] v 1.8 months [IQR, 0.9-2.8 months] in PIK3CA/PIK3R1 mutant v PIK3CA/PIK3R1 wild type, respectively; log-rank P = .67) or PTEN molecular alterations (median PFS, 1.8 months [IQR, 1.3-3.6 months] v 1.8 months [IQR, 0.9-2.8 months] in PTEN mutant v PTEN wild type, respectively; log-rank P = .57; Appendix Fig A5).
Toxicity
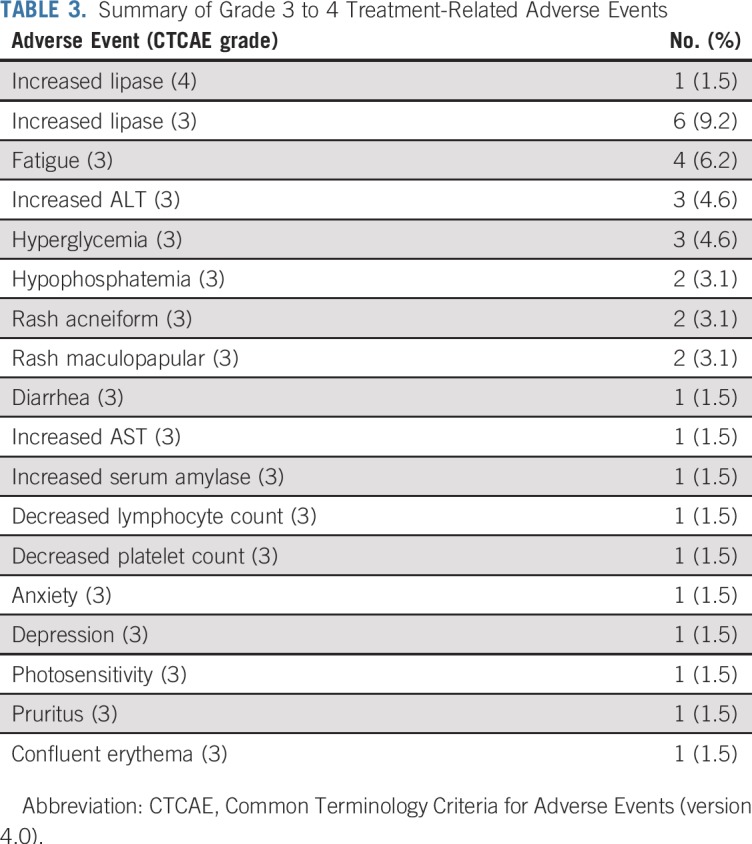
Table 3 lists the most common grade 3 and 4 adverse events. Overall, grade 3 to 4 treatment-related adverse events were reported in 40.0% (95% CI, 19.8% to 64.3%) of patients in cohort 1 and 32.0% (95% CI, 20.8% to 45.8%) of patients in cohort 2. Only one patient (1.5%) experienced a grade 4 toxicity that was at least possibly related to treatment, and consisted of an asymptomatic lipase increase. No suicidality was reported. No on-treatment deaths occurred. Buparlisib treatment was discontinued in two patients (3.1%) as a result of adverse events, one in each arm. The most common adverse events that led to dose reduction or discontinuation were increased lipase in six patients (9.2%) and increased ALT and hyperglycemia in five (7.7%).
TABLE 3.
Summary of Grade 3 to 4 Treatment-Related Adverse Events
DISCUSSION
This study shows that the brain-penetrant PI3K inhibitor buparlisib has minimal single-agent efficacy in patients with recurrent glioblastoma.5,20,21 Buparlisib did not meet the primary pharmacodynamic and efficacy end points of this study. These findings are consistent with previous results wherein PI3K/mTOR inhibitors alone or in combination with cytotoxic or targeted therapies in patients with glioblastoma unselected for PI3K pathway activation showed no clinical benefit.6,9-11,12,22-27 Although PI3K pathway activation was confirmed in all patients, post hoc analyses did not show a correlation between the mutation status of PTEN, PIK3CA, and PIK3R1 and outcome. Of note, a shorter PFS was observed in patients with IDH1/2-mutant tumors. However, because this was a post hoc analysis in a relatively small number of patients with IDH1/2-mutant tumors, definitive conclusions will require increased numbers.
To our knowledge, this study provides the first evidence that buparlisib can achieve adequate brain penetration in patients with glioblastoma. Buparlisib accumulation was seen in both CE and non-CE areas of tumor, with a trend to higher accumulation in CE areas. These findings are consistent with preclinical and early-phase studies of buparlisib14,16,28,29 as well as with the phase III trial BELLE-2 in human epidermal growth factor receptor 2–negative metastatic breast cancer.30 In this study, a higher rate of psychiatric adverse effects, including depression and anxiety, was observed with buparlisib, which were not reported with other PI3K inhibitors31-35 and were attributed to the high BBB penetration of buparlisib.
Our multidimensional analysis, including IHC, study of cell proliferation and signaling markers, RPPA, and tumor genomic profiling provide important insights for understanding mechanisms of resistance to single-agent PI3K inhibitors in glioblastoma. Although our analyses document inhibition of pAKTS473 in a subset of patients, blockade of the PI3K pathway activity was incomplete, as evidenced by the limited effects on downstream pS6S235/236. This was associated with minimal effects on tumor cell proliferation and outcome. The morphometric IHC and RPPA analyses suggest that persistent downstream signaling occured through incomplete blockade of PI3K pathway together with activity of complementary pathways, including MAPK signaling. Previous studies showed that buparlisib concentrations required to fully inhibit PI3K activity generate toxic off-target effects on cytoskeleton dynamics,36 which suggests that the therapeutic window of buparlisib might be too narrow in glioblastoma. The robust activity of PI3K signaling in glioblastoma may require more potent and selective inhibitors that would achieve greater pathway inhibition without causing dose-limiting adverse events.
Besides incomplete PI3K pathway inhibition, the persistent activity of p70 S6 kinases and the MAPK pathway observed in the RPPA analysis suggests activation of alternate prosurvival pathways that also may contribute to buparlisib resistance. This phenomenon might be overcome by a combination of buparlisib or other PI3K inhibitor with inhibiting complementary signaling or feedback molecules, such as sonic hedgehog, ribosomal S6 kinase, or insulin.37-40
A limitation of our study was the use of historical data as reference because of a lack of a control arm in the study design. Nevertheless, the absence of radiologic response observed in this study suggests that buparlisib has minimal single-agent efficacy in this population and is unlikely to demonstrate benefit against controls in future trials. Another limitation was that biomarker analyses for study entry were performed on archival tissues instead of use of an immediate pretreatment biopsy sample because the feasibility of performing sequential biopsies was limited by ethical considerations. However, recent studies that addressed clonal evolution of glioblastoma under therapy using whole-exome sequencing of pre- and post-treatment–paired tumors showed that molecular alterations of PTEN, PIK3CA, or PI3R1 are rarely lost in recurrent samples.41-43 This suggests that although a certain degree of clonal evolution occurs after treatment with DNA-damaging agents, alterations in the PI3K pathway are likely retained as targets in recurrent tumors in a majority of patients.
In conclusion, this study shows that buparlisib does not provide clinically meaningful benefit in patients with PI3K-activated recurrent glioblastoma. Our finding that pathway inhibition was incomplete suggests that additional studies are warranted to assess whether more potent and selective PI3K inhibitors may achieve greater pathway inhibition and clinical benefit. Careful assessment of tissue pharmacokinetics and pharmacodynamics through the surgical arm of the study was important to our understanding that poor pathway inhibition was likely the basis for the modest response seen in patients. This suggests that in glioblastoma, more routine use of studies specifically designed to confirm drug penetration and target engagement may be beneficial to perform before considering advancing a drug for more testing. In addition, investigation of how PI3K inhibitors can be combined with complementary therapeutics to provide clinical benefit in glioblastoma are still warranted and should include studies designed to understand the dependency and associations between biomarkers and response to PI3K inhibitors, including PTEN, PIK3CA, PI3R1, and IDH1/2 mutations.
ACKNOWLEDGMENT
We thank the patients who took part in the study and their families as well as the staff, research coordinators, and investigators at each participating institution. We acknowledge the support of the Ben and Catherine Ivy Foundation and Lisa Doherty, APRN, OCN; Debra Conrad LaFrankie, RN, OCN; Sandra French Ruland, RN, BSN, OCN; Jennifer Stefanik, NP, and Jann N. Sarkaria, MD, for sharing unpublished results. Finally, we acknowledge the Center for Cancer Genome Discovery; Paul Van Hummelen and Aaron Thorner; and the members of the Brigham and Women’s Hospital Center for Advanced Molecular Diagnostics, Clinical Cytogenetics Division.
Appendix
Pharmacokinetic Evaluation
The analytic method for the quantitative determination of buparlisib in human plasma and tumor was developed and validated by Novartis (Basel, Switzerland). The method consists of a solid phase extraction using a 96-well plate with Oasis HLB cartridge (10 mg, 30 μm; Waters, Milford, MA) and analysis by liquid chromatography-tandem mass spectrometry in multiple-reaction monitoring mode using electrospray ionization in the positive ion mode. The method is suitable for the determination of buparlisib in human plasma (EDTA) over the range of 1.00 ng/mL (lower limit of quantification) to 1,000 ng/mL (upper limit of quantification). No matrix effect was observed; mean recovery ranged from 59% to 61%. Buparlisib was stable in stock and diluted solutions, in matrix, and after multiple freeze-thaw cycles. The assay method exhibited sufficient specificity and selectivity, accuracy, precision, and sensitivity for the purposes and conclusions of the individual studies. For pharmacokinetic studies, blood samples were collected at the following time points on days 1 and 8 (± 1 day) before surgery: predose and at 0.5, 1.5, 3, and 5 hours postdose. Non–contrast-enhancing and contrast-enhancing brain tumor tissue and a concomitant blood sample were obtained at the time of surgery. Standard pharmacokinetic parameters were determined using noncompartmental methods. The tumor-to-plasma ratio was calculated by dividing the tumor geometric mean concentration by the plasma geometric mean concentration at the time of surgery.
Immunohistochemical Studies
Immunohistochemical (IHC) stainings for PTEN (#9559, 1:50 dilution, heated citrate retrieval; Cell Signaling Technologies, Danvers, MA), positive phosphorylated AKT (pAKT) S473 (#4060, 1:75 dilution, EDTA retrieval; Cell Signaling Technologies), and phosphorylated S6 (pS6) S235/236 (#4858, 1:75 dilution, EDTA retrieval; Cell Signaling Technologies) were performed on 5-μm formalin-fixed paraffin-embedded (FFPE) tissue sections.
In the surgical component of the trial, modulation of the phosphatidylinositol 3-kinase pathway response in tumor tissue was determined by pathologist-performed semiquantitative IHC scoring of pAKT and pS6 on the basis of previously established methods in preclinical models and clinical trials of glioblastoma.6,22 Sample staining was scored for intensity in tumor cells on a 0 to 2+ scale (0, no staining; 1+, weak positive staining; 2+, strong positive staining, with 1+ and 2+ being the average intensity of all positive cells in the cohort). Staining within nontumor elements (eg, macrophages, vessels) was not included in the scoring. Change in pAKT and pS6 IHC scores was the difference in score from baseline to surgery in each participant. Participants were classified into three groups; a reduction of staining score of one or more degrees qualified for response, whereas no change or an increase in score qualified for no response.
Matrix-Assisted Laser Desorption/Ionization-Mass Spectrometry Imaging Drug Analysis on Surgical Specimens
Surgical sections were flash frozen after surgery, stored at −80°C, and placed at −25°C 1 hour before use. Twelve-micrometer coronal tissue sections were prepared using a Microm HM550 cryostat (Thermo Fisher Scientific, Waltham, MA) and thaw mounted onto indium tinoxide–coated microscopic slides (Bruker, Billerica, MA) for matrix-assisted laser desorption/ionization-mass spectrometry imaging and onto optical slides for hematoxylin and eosin staining. Samples were dried for 15 minutes in a desiccator. 2,5-Dihydroxybenzoic acid (40 mg/mL solution in methanol OH/0.2% trifluoroacetic acid 70:30 volume-to-volume ratio) was deposited using the TM-Sprayer system (HTX Technologies, Chapel Hill, NC) as previously described (Sun Y et al: Neuro-oncol 19:774-785, 2017). Mass spectra were acquired using a 9.4-T solariX XR Fourier transform ion cyclotron resonance mass spectrometer (Bruker).
Tumor Genotyping
Targeted exome next-generation sequencing (OncoPanel) was performed at the Dana-Farber Cancer Institute Center for Cancer Genome Discovery on an approximately 50-mm thickness of FFPE tumor tissue using the OncoPanel version 2.0 custom targeted exome capture panel to examine the exons of 275 cancer-causing genes and their respective single nucleotide variants and indels. Data were annotated as previously described (Sholl LM, et al: JCI Insight 1:e87062, 2016; Ramkisson SH, et al: Neuro-oncol 19:986-996, 2017). Array comparative genomic hybridization copy number testing was performed using SurePrint G3 1M feature stock arrays (Agilent Technologies, Santa Clara, CA) from approximately 1 μg of total DNA extracted from FFPE tissue (approximately 200-μm thickness of tissue) using fragmentation simulation methodology (Craig JM, et al: PLoS One 7:e38881, 2012). Amplification was calculated as greater than 2.0 log-ratio, and single-copy losses generally were calculated as less than −0.3 log-ratio change compared with a pooled DNA normal. Results were analyzed using CytoGenomics and Genomic Workbench software (Agilent Technologies).
Reverse-Phase Protein Analysis
For comparison of signaling changes in buparlisib-treated patients, 11 paired untreated versus standard-of care–treated glioblastoma tumor sets obtained from The University of Texas M.D. Anderson Cancer Center were used as controls. Frozen tumor tissue approximately the size of a grain of rice was placed in 2-mL tubes with ceramic beads using a Precellys homogenizer (Bertin Instruments, Montigny-le-Bretonneux, France). Tissue was lysed using ice-cold lysis solution. Lysates from flash-frozen tissues were prepared and analyzed by reverse-phase protein analysis using 299 antibodies as described previously.45 Reverse phase protein microarrays were printed on nitrocellulose-coated glass FAST Slides (Schleicher & Schuell BioScience, Keene, NH) by a GeneTAC G3 arrayer (Genomic Solutions, Ann Arbor, MI). Antibody staining of each reverse phase protein microarray was done using an autostainer (BioGenex, Freemont, CA). The slide images were quantified using MicroVigene 4.0 (VigeneTech, Carlisle, MA). The spot-level raw data were processed using the R package SuperCurve (https://r-forge.r-project.org/R/?group_id=1899), which returns the estimated protein concentrations (raw concentration) and a quality control score for each slide. Raw concentration data were normalized by median centering of each sample across all proteins to correct loading bias.
Statistical Analyses
Data were summarized as frequencies and proportions for categorical variables and as medians and ranges for continuous variables. Intrapatient mean differences were evaluated with the paired Wilcoxon test. Intercohort mean differences were evaluated with the Mann-Whitney U test. Survival analyses (ie, progression-free survival [PFS], overall survival) were performed using the Kaplan-Meier method, and differences between groups were evaluated by the log-rank test. Survival for participants who were alive or lost to follow-up at the time of last contact on or before data cutoff was censored at the date of the last contact alive. Patients who were censored for PFS before 6-month PFS (PFS6) determination were included in the calculation of the PFS6 proportion as patients who did not reach PFS6. For biomarker evaluation, categorical groups were explored while considering variable distribution to evaluate the possible association with outcome using the Fisher’s exact test (PFS6) or the log-rank test (PFS, overall survival). Differentially expressed proteins in pre- and post-treatment samples were determined using Limma (Ritchie ME, et al: Nucleic Acids Res 43:e47, 2015), a software package used to perform differential expression analysis, and the R language (http://www.R-project.org). P = .05 was set for statistical significance for all evaluations.
FIG A1.

Treatment schema. CR, complete response; GBM, glioblastoma; IHC, immunohistochemistry; MRI, magnetic resonance imaging; pAKT, phosphorylated AKT; PD, progressive disease; PI3K, phosphatidylinositol 3-kinase; PR, partial response; SD, stable disease.
FIG A2.

Phosphatidylinositol 3-kinase pathway modulation as a result of buparlisib in tumor tissue in the control cohort. Quantification of (A and B) phosphorylated AKTS473 (pAKTS473) and (C and D) phosphorylated S6 S235/236 (pS6S235/236) immunohistochemistry staining in the surgical cohort. The control cohort consisted of seven patients treated with standard of care for whom surgical resection of tumor tissue was performed at initial diagnosis and recurrence. (B and D) Differences between groups were calculated using the paired Wilcoxon test.
FIG A3.

Phosphatidylinositol 3-kinase pathway modulation and changes in tumor cell proliferation as a result of buparlisib in tumor tissue in cohort 1. (A and B) Quantification of phosphorylated S6 S235/236 (pS6S235/236) immunohistochemistry staining and (C and D) tumor cell proliferation as assessed by the IHC proliferation marker Ki-67 in the surgical cohort. (B and D) Differences between groups were calculated using the paired Wilcoxon test.
FIG A4.

Kaplan-Meier curves of (A) overall survival (OS) and (B) progression-free survival (PFS) in cohorts 1 and 2. IQR, interquartile range.
FIG A5.

Kaplan-Meier curves of overall survival (OS) and progression-free survival (PFS) by (A and B) IDH1/2 status, (C and D) PIK3CA/PIK3R1 status, and (E and F) PFS by PTEN status. Differences between groups were evaluated using the log-rank test. IQR, interquartile range.
TABLE A1.
Summary of the Response Assessment in Neuro-Oncology Response Criteria

TABLE A2.
Buparlisib Pharmacokinetic Parameters

TABLE A3.
Buparlisib Pharmacokinetic Parameters in Cohort 1

TABLE A4.
Reverse-Phase Protein Array Analysis of Buparlisib-Treated Contrast-Enhancing Tumors Versus Unrelated SOC-Treated Control Tumors

Footnotes
Presented at the American Society of Clinical Oncology 2014 Annual Meeting, Chicago, IL, May 30-June 3, 2014.
Supported by the Ivy Foundation Early Phase Clinical Trials Consortium, DFHCC/MIT Bridge Project (R01CA188288, P50 CA165962), and by Novartis, which provided the drug and funding.
Clinical trial information: NCT01339052.
AUTHOR CONTRIBUTIONS
Conception and design: Patrick Y. Wen, Mehdi Touat, Shakti Ramkissoon, Howard Colman, Eudocia Q. Lee, Susan M. Chang, Michael D. Prados, W.K. Alfred Yung, Keith L. Ligon
Financial support: Keith L. Ligon
Administrative support: Christine S. McCluskey, Loreal E. Brown, W.K. Alfred Yung
Provision of study material or patients: Patrick Y. Wen, Ingo K. Mellinghoff, Howard Colman, Antonio M. Omuro, Lisa M. DeAngelis, Mark R. Gilbert, John F. de Groot, Timothy F. Cloughesy, Eudocia Q. Lee, Lakshmi Nayak, Tracy T. Batchelor, Susan M. Chang, Ian F. Dunn, Geoffrey S. Young, Michael D. Prados, W.K. Alfred Yung, Keith L. Ligon
Collection and assembly of data: Patrick Y. Wen, Mehdi Touat, Ingo K. Mellinghoff, Shakti Ramkissoon, Christine S. McCluskey, Kristine Pelton, Sankha S. Basu, Sarah C. Gaffey, Loreal E. Brown, Jungwoo Kim, Wei Wei, Mi-Ae Park, Jason T. Huse, Mikael L. Rinne, Howard Colman, Nathalie Y.R. Agar, Antonio M. Omuro, Lisa M. DeAngelis, Mark R. Gilbert, John F. de Groot, Andrew S. Chi, Eudocia Q. Lee, James R. Heath, Laura L. Horky, Tracy T. Batchelor, Rameen Beroukhim, Susan M. Chang, Ian F. Dunn, Dimpy Koul, Geoffrey S. Young, Michael D. Prados, David A. Reardon, Keith L. Ligon
Data analysis and interpretation: Patrick Y. Wen, Mehdi Touat, Brian M. Alexander, Ingo K. Mellinghoff, Shakti Ramkissoon, Kristine Pelton, Sam Haidar, Sankha S. Basu, Juan Emmanuel Martinez-Ledesma, Shaofang Wu, Wei Wei, Mi-Ae Park, John G. Kuhn, Nathalie Y.R. Agar, Antonio M. Omuro, Lisa M. DeAngelis, Mark R. Gilbert, John F. de Groot, Timothy F. Cloughesy, Thomas M. Roberts, Jean J. Zhao, Lakshmi Nayak, James R. Heath, Laura L. Horky, Susan M. Chang, Azra H. Ligon, Dimpy Koul, Michael D. Prados, Keith L. Ligon
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Buparlisib in Patients With Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Patrick Y. Wen
Honoraria: Merck
Consulting or Advisory Role: AbbVie, Agios Pharmaceuticals, AstraZeneca, Blue Earth Diagnostics, Eli Lilly, Genentech, Roche, Immunomic Therapeutics, Kadmon Corporation, KIYATEC, Puma Biotechnology, Vascular Biogenics, Taiho Pharmaceutical, Deciphera Pharmaceuticals, VBI Vaccines
Speakers’ Bureau: Merck, prIme Oncology
Research Funding: Agios Pharmaceuticals (Inst), AstraZeneca (Inst), BeiGene (Inst), Eli Lilly (Inst), Roche (Inst), Genentech (Inst), Karyopharm Therapeutics (Inst), Kazia Therapeutics (Inst), MediciNova (Inst), Novartis (Inst), Oncoceutics (Inst), Sanofi (Inst), Aventis (Inst), VBI Vaccines (Inst)
Travel, Accommodations, Expenses: Merck
Mehdi Touat
Consulting or Advisory Role: Agios Pharmaceuticals, Taiho Pharmaceutical
Travel, Accommodations, Expenses: Merck Sharp & Dohme
Brian M. Alexander
Employment: Foundation Medicine
Ingo K. Mellinghoff
Honoraria: Roche
Consulting or Advisory Role: Agios Pharmaceuticals, Puma Biotechnology, Debiopharm Group
Research Funding: General Electric, Amgen, Eli Lilly
Shakti Ramkissoon
Employment: Foundation Medicine
Stock and Other Ownership Interests: Foundation Medicine
Sam Haidar
Employment: Philips Healthcare
Stock and Other Ownership Interests: Johnson & Johnson
Wei Wei
Patents, Royalties, Other Intellectual Property: US patent application pending from a technology related to health or medicine (application No. 15/045241)
Jason T. Huse
Employment: Champions Oncology
Research Funding: Taiho Pharmaceutical (Inst)
John G. Kuhn
Consulting or Advisory Role: TG Therapeutics
Research Funding: Methodist Health System
Mikael L. Rinne
Employment: Novartis Institutes for BioMedical Research
Consulting or Advisory Role: N-of-One
Howard Colman
Honoraria: Merck Sharp & Dohme
Consulting or Advisory Role: Novocure, AbbVie, Foundation Medicine, Innocrin Pharmaceuticals, Tactical Therapeutics, Deciphera Pharmaceuticals, NewLink Genetics, Best Doctors, Merck
Research Funding: NewLink Genetics, Plexxikon, Kadmon Corporation, Orbus Therapeutics, Merck, DNAtrix, AbbVie, BeiGene, FORMA Therapeutics (Inst)
Nathalie Y.R. Agar
Consulting or Advisory Role: Bayesian Dx
Patents, Royalties, Other Intellectual Property: IP portfolio at Partners Healthcare for surgical margin delineation and intraoperative diagnosis largely based on mass spectrometry
Travel, Accommodations, Expenses: Bruker
Antonio M. Omuro
Consulting or Advisory Role: Stemline Therapeutics, Bristol-Myers Squibb, Alexion Pharmaceuticals, Novocure, Inovio Pharmaceuticals
Lisa M. DeAngelis
Consulting or Advisory Role: Roche, CarThera, BMJ Publishing Group, BTG, Tocagen, Sapience Therapeutics
John F. de Groot
Employment: Helsinn Therapeutics (I), Ziopharm Oncology (I)
Leadership: Ziopharm Oncology (I)
Stock and Other Ownership Interests: Gilead Sciences, Ziopharm Oncology (I)
Consulting or Advisory Role: Celldex, Deciphera Pharmaceuticals, Vascular Biogenics, Foundation Medicine, Genentech, Roche, Omniox, OXiGENE, AbbVie, Novogen, Kadmon Corporation, Merck, Five Prime Therapeutics, Insys Therapeutics, AstraZeneca, Boston Biomedical, GW Pharmaceuticals, CarThera
Research Funding: Deciphera Pharmaceuticals, Novartis, Eli Lilly, Sanofi, EMD Serono, Mundipharma Aventis, AstraZeneca
Timothy F. Cloughesy
Stock and Other Ownership Interests: Notable Labs
Consulting or Advisory Role: Roche, Genentech, Celgene, Tocagen, VBL Therapeutics, NewGen Therapeutics, Novartis, Agios Pharmaceuticals, Cortice Biosciences, Novocure, AbbVie, OXiGENE, Wellcome Trust, Pfizer, Notable Labs, Bristol-Myers Squibb, Merck, Insys Therapeutics, Human Longevity, Sunovion, Boston Biomedical, Novogen, Alexion Pharmaceuticals, GW Pharmaceuticals, Eli Lilly, Genocea Biosciences, Puma Biotechnology
Other Relationship: Global Coalition for Adaptive Research
Thomas M. Roberts
Leadership: iKang Healthcare Group, Geode Therapeutics, Crimson Pharmaceutical
Stock and Other Ownership Interests: iKang Healthcare Group, Crimson Pharmaceutical, Geode Therapeutics
Consulting or Advisory Role: Novartis Institutes for BioMedical Research
Patents, Royalties, Other Intellectual Property: Applied for patents combining PI3K inhibitors with immunotherapies
Jean J. Zhao
Leadership: Crimson Pharmaceutical, Geode Therapeutics
Stock and Other Ownership Interests: Crimson Pharmaceutical, Geode Therapeutics
Speakers’ Bureau: Ruijin Hospital
Research Funding: Eli Lilly, Puma Biotechnology
Patents, Royalties, Other Intellectual Property: Several intellectual properties at Dana-Faber Cancer Institute as a co-inventor
Travel, Accommodations, Expenses: Ruijin Hospital, Princess Margaret Cancer Centre
Eudocia Q. Lee
Honoraria: MedLink, UpToDate
Consulting or Advisory Role: Eli Lilly
Lakshmi Nayak
Honoraria: Bristol-Myers Squibb
Travel, Accommodations, Expenses: Bristol-Myers Squibb
James R. Heath
Leadership: Indi Molecular, PACT pharma, Sofie Biosciences, IsoPlexis, Nirmidas Biotech, Arivale
Stock and Other Ownership Interests: Indi Molecular, IsoPlexis, PACT pharma
Tracy T. Batchelor
Honoraria: Champions Oncology, UpToDate, Imedex, NXDC, Merck, GenomiCare Biotechnology
Consulting or Advisory Role: Merck, GenomiCare Biotechnology, NXDC, Amgen
Travel, Accommodations, Expenses: Merck, Roche, Genentech, GenomiCare Biotechnology
Rameen Beroukhim
Consulting or Advisory Role: Novartis, Merck (I), Gilead Sciences (I), ViiV Healthcare (I)
Research Funding: Novartis
Patents, Royalties, Other Intellectual Property: Prognostic Marker for Endometrial Carcinoma (US patent application 13/911456, filed June 6, 2013), SF3B1 Suppression as a Therapy for Tumors Harboring SF3B1 Copy Loss (international application No. WO/2017/177191, PCT/US2017/026693, filed July 4, 2017), Compositions and Methods for Screening Pediatric Gliomas and Methods of Treatment Thereof (international application No. WO/2017/132574, PCT/US2017/015448, filed 1/27/2017)
Susan M. Chang
Consulting or Advisory Role: Tocagen
Research Funding: Novartis (Inst), Agios Pharmaceuticals (Inst)
Azra H. Ligon
Leadership: Travera (I)
Stock and Other Ownership Interests: Travera (I)
Consulting or Advisory Role: Travera (I)
Geoffrey S. Young
Research Funding: Siemens Healthineers
Michael D. Prados
Stock and Other Ownership Interests: Quadriga BioSciences
Consulting or Advisory Role: Nativis, Tocagen
Research Funding: Genentech (Inst), Roche (Inst), Novartis (Inst), Nativis (Inst)
David A. Reardon
Honoraria: AbbVie, Cavion, Genentech, Roche, Merck, Midatech Pharma, Momenta Pharmaceuticals, Novartis, Novocure, Regeneron Pharmaceuticals, Stemline Therapeutics, Celldex, OXiGENE, Monteris Medical, Bristol-Myers Squibb, Juno Therapeutics, Inovio Pharmaceuticals, Oncorus, Agenus, EMD Serono, Merck, Merck KGaA, Taiho Pharmaceutical, Advantagene
Consulting or Advisory Role: Cavion, Genentech, Roche, Merck, Momenta Pharmaceuticals, Novartis, Novocure, Regeneron Pharmaceuticals, Stemline Therapeutics, Bristol-Myers Squibb, Inovio Pharmaceuticals, Juno Therapeutics, Celldex, OXiGENE, Monteris Medical, Midatech Pharma, Oncorus, AbbVie, Agenus, EMD Serono, Merck, Merck KGaA, Taiho Pharmaceutical
Research Funding: Celldex (Inst), Incyte (Inst), Midatech Pharma (Inst), Tragara Pharmaceuticals (Inst), Inovio Pharmaceuticals (Inst), Agenus (Inst), EMD Serono (Inst), Acerta Pharma (Inst), Omnivox
W.K. Alfred Yung
Stock and Other Ownership Interests: DNATrix
Honoraria: DNAtrix, Amgen
Consulting or Advisory Role: DNAtrix, Boehringer Ingelheim
Patents, Royalties, Other Intellectual Property: DNATrix
Keith L. Ligon
Stock and Other Ownership Interests: Travera
Consulting or Advisory Role: Midatech Pharma, Bristol-Myers Squibb
Research Funding: Plexxikon (Inst), Amgen (Inst), X4 Pharmaceuticals (Inst), Tragara Pharmaceuticals (Inst), Bristol-Myers Squibb (Inst), Eli Lilly (Inst)
Patents, Royalties, Other Intellectual Property: Molecular diagnostics assay patent
No other potential conflicts of interest were reported.
REFERENCES
- 1.Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro-oncol. 2017;19:v1–v88. doi: 10.1093/neuonc/nox158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Taillibert S, Kanner AA, et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: A randomized clinical trial. JAMA. 2015;314:2535–2543. doi: 10.1001/jama.2015.16669. [DOI] [PubMed] [Google Scholar]
- 5.Touat M, Idbaih A, Sanson M, et al. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann Oncol. 2017;28:1457–1472. doi: 10.1093/annonc/mdx106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cloughesy TF, Yoshimoto K, Nghiemphu P, et al. Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hegi ME, Diserens AC, Bady P, et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib--a phase II trial. Mol Cancer Ther. 2011;10:1102–1112. doi: 10.1158/1535-7163.MCT-11-0048. [DOI] [PubMed] [Google Scholar]
- 8. doi: 10.1016/j.cell.2013.09.034. Brennan CW, Verhaak RG, McKenna A, et al: The somatic genomic landscape of glioblastoma. Cell 155:462-477, 2013 [Erratum: Cell 157:753, 2014] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wen PY, Lee EQ, Reardon DA, et al. Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro-oncol. 2012;14:819–829. doi: 10.1093/neuonc/nos117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang SM, Wen P, Cloughesy T, et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357–361. doi: 10.1007/s10637-005-1444-0. [DOI] [PubMed] [Google Scholar]
- 11.Ma DJ, Galanis E, Anderson SK, et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro-oncol. 2015;17:1261–1269. doi: 10.1093/neuonc/nou328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pitz MW, Eisenhauer EA, MacNeil MV, et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro-oncol. 2015;17:1270–1274. doi: 10.1093/neuonc/nou365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burger MT, Pecchi S, Wagman A, et al. Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer. ACS Med Chem Lett. 2011;2:774–779. doi: 10.1021/ml200156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koul D, Fu J, Shen R, et al. Antitumor activity of NVP-BKM120--a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin Cancer Res. 2012;18:184–195. doi: 10.1158/1078-0432.CCR-11-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maire CL, Ramkissoon S, Hayashi M, et al. Pten loss in Olig2 expressing neural progenitor cells and oligodendrocytes leads to interneuron dysplasia and leukodystrophy. Stem Cells. 2014;32:313–326. doi: 10.1002/stem.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Netland IA, Førde HE, Sleire L, et al. Treatment with the PI3K inhibitor buparlisib (NVP-BKM120) suppresses the growth of established patient-derived GBM xenografts and prolongs survival in nude rats. J Neurooncol. 2016;129:57–66. doi: 10.1007/s11060-016-2158-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bendell JC, Rodon J, Burris HA, et al. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30:282–290. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 18.Rodon J, Braña I, Siu LL, et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2014;32:670–681. doi: 10.1007/s10637-014-0082-9. [DOI] [PubMed] [Google Scholar]
- 19.Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: Response assessment in Neuro-Oncology Working Group. J Clin Oncol. 2010;28:1963–1972. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 20.Wong ET, Hess KR, Gleason MJ, et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol. 1999;17:2572–2578. doi: 10.1200/JCO.1999.17.8.2572. [DOI] [PubMed] [Google Scholar]
- 21.Lamborn KR, Yung WK, Chang SM, et al. Progression-free survival: An important end point in evaluating therapy for recurrent high-grade gliomas. Neuro-oncol. 2008;10:162–170. doi: 10.1215/15228517-2007-062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wen PY, Chang SM, Lamborn KR, et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro-oncol. 2014;16:567–578. doi: 10.1093/neuonc/not247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reardon DA, Quinn JA, Vredenburgh JJ, et al. Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma. Clin Cancer Res. 2006;12:860–868. doi: 10.1158/1078-0432.CCR-05-2215. [DOI] [PubMed] [Google Scholar]
- 24.Doherty L, Gigas DC, Kesari S, et al. Pilot study of the combination of EGFR and mTOR inhibitors in recurrent malignant gliomas. Neurology. 2006;67:156–158. doi: 10.1212/01.wnl.0000223844.77636.29. [DOI] [PubMed] [Google Scholar]
- 25.Kreisl TN, Lassman AB, Mischel PS, et al. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM) J Neurooncol. 2009;92:99–105. doi: 10.1007/s11060-008-9741-z. [DOI] [PubMed] [Google Scholar]
- 26.Reardon DA, Desjardins A, Vredenburgh JJ, et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol. 2010;96:219–230. doi: 10.1007/s11060-009-9950-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lassen U, Sorensen M, Gaziel TB, et al. Phase II study of bevacizumab and temsirolimus combination therapy for recurrent glioblastoma multiforme. Anticancer Res. 2013;33:1657–1660. [PubMed] [Google Scholar]
- 28.Nanni P, Nicoletti G, Palladini A, et al. Multiorgan metastasis of human HER-2+ breast cancer in Rag2-/-;Il2rg-/- mice and treatment with PI3K inhibitor. PLoS One. 2012;7:e39626. doi: 10.1371/journal.pone.0039626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maira M, Schnell C, Lollini P, et al: Preclinical and preliminary clinical activity of NVP-BKM120, an oral pan-class I PI3K inhibitor, in the brain. Ann Oncol 23, 2012 (suppl 9; abstr 1675P) [Google Scholar]
- 30.Baselga J, Im SA, Iwata H, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:904–916. doi: 10.1016/S1470-2045(17)30376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gopal AK, Kahl BS, de Vos S, et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370:1008–1018. doi: 10.1056/NEJMoa1314583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saura C, Sachdev J, Patel MR, et al: Ph1b study of the PI3k inhibitor taselisib (GDC-0032) in combination with letrozole in patients with hormone receptor-positive advanced breast cancer. Cancer Res 75, 2015 (suppl 9; abstr PD5-2)
- 33.Sarker D, Ang JE, Baird R, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21:77–86. doi: 10.1158/1078-0432.CCR-14-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayer IA, Abramson VG, Formisano L, et al. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2- metastatic breast cancer. Clin Cancer Res. 2017;23:26–34. doi: 10.1158/1078-0432.CCR-16-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dreyling M, Santoro A, Mollica L, et al. Copanlisib in patients with relapsed or refractory indolent B-cell lymphoma (Chronos-1) Hematol Oncol. 2017;35:119–120. [Google Scholar]
- 36.Brachmann SM, Kleylein-Sohn J, Gaulis S, et al. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Mol Cancer Ther. 2012;11:1747–1757. doi: 10.1158/1535-7163.MCT-11-1021. [DOI] [PubMed] [Google Scholar]
- 37.Filbin MG, Dabral SK, Pazyra-Murphy MF, et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: New therapeutic opportunities. Nat Med. 2013;19:1518–1523. doi: 10.1038/nm.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serra V, Eichhorn PJ, García-García C, et al. RSK3/4 mediate resistance to PI3K pathway inhibitors in breast cancer. J Clin Invest. 2013;123:2551–2563. doi: 10.1172/JCI66343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. doi: 10.1038/s41586-018-0343-4. Hopkins B, Pauli C, Du X, et al: Suppression of insulin feedback enhances the efficacy of PI3k inhibitors. Nature 560:499-503, 2018 [Erratum: Nature 563;E24, 2018] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ni J, Ramkissoon SH, Xie S, et al. Combination inhibition of PI3K and mTORC1 yields durable remissions in mice bearing orthotopic patient-derived xenografts of HER2-positive breast cancer brain metastases. Nat Med. 2016;22:723–726. doi: 10.1038/nm.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim J, Lee IH, Cho HJ, et al. Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell. 2015;28:318–328. doi: 10.1016/j.ccell.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 42.Kim H, Zheng S, Amini SS, et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015;25:316–327. doi: 10.1101/gr.180612.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J, Cazzato E, Ladewig E, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48:768–776. doi: 10.1038/ng.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Louis DN, Ohgaki H, Wiestler OD, et al. World Health Organization Histological Classification of Tumours of the Central Nervous System (ed 2). Lyon, France, International Agency for Research on Cancer, 2016.
- 45.Akbani R, Ng KS, Werner HMJ, et al. doi: 10.1038/ncomms4887. A pan-cancer proteomic perspective on the Cancer Genome Atlas Nature Comms: 5:3887, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]