

Abstract
Purpose
DICER1 syndrome is an autosomal-dominant, pleiotropic tumor-predisposition disorder caused by pathogenic germline variants in DICER1. We sought to quantify risk, hazard rates, and the probability of neoplasm incidence accounting for competing risks (“cumulative incidence”) of neoplasms (benign and malignant) and standardized incidence ratios for malignant tumors in individuals with DICER1 pathogenic variation.
Patients and Methods
We combined data from three large cohorts of patients who carry germline pathogenic variation in DICER1. To reduce ascertainment bias, we distinguished probands from nonprobands. Neoplasm diagnoses were confirmed by review of pathology reports and/or central review of surgical pathology materials. Standardized cancer incidence ratios were determined relative to the SEER program, which does not capture all DICER1-associated neoplasms. For all malignancies and benign tumors (“neoplasms,” excluding type Ir pleuropulmonary blastoma and thyroid nodules), we used the Kaplan-Meier method and nonparametric cumulative incidence curves to estimate neoplasm-free survival.
Results
We calculated the age at first neoplasm diagnosis (systematically ascertained cancers plus DICER1-associated neoplasms pleuropulmonary blastoma, cystic nephroma, and nasal chondromesenchymal hamartoma) in 102 female and male nonproband DICER1 carriers. By age 10 years, 5.3% (95% CI, 0.6% to 9.7%) of nonproband DICER1 carriers had developed a neoplasm (females, 4.0%; males, 6.6%). By age 50 years, 19.3% (95% CI, 8.4% to 29.0%) of nonprobands had developed a neoplasm (females, 26.5%; males, 10.2%). After age 10 years, female risk was elevated compared with male risk. Standardized cancer incidence ratio analysis of 102 nonproband DICER1 carriers, which represented 3,344 person-years of observation, showed significant cancer excesses overall, particularly of gynecologic and thyroid cancers.
Conclusion
This work provides the first quantitative analysis of site-specific neoplasm risk and excess malignancy risk in 102 systematically characterized nonproband DICER1 carriers. Our findings inform DICER1 syndrome phenotype, natural history, and genetic counseling.
INTRODUCTION
DICER1 syndrome is an autosomal-dominant, familial pleiotropic tumor-predisposition disorder1 caused by pathogenic germline variants in DICER1, an essential component of the microRNA processing pathway.2 The hallmark neoplasm of DICER1 syndrome is pleuropulmonary blastoma (PPB), a pediatric dysembryonic sarcoma of the lung and pleura.3 Several other distinctive neoplasms, which often present in childhood, have been reported among DICER1 pathogenic variant carriers, including cystic nephroma (CN), anaplastic renal sarcoma, Wilms tumor, Sertoli-Leydig cell tumor (SLCT) and gynandroblastoma, differentiated thyroid carcinoma, nasal chondromesenchymal hamartoma (NCMH), ciliary body medulloepithelioma (CBME), embryonal rhabdomyosarcoma (RMS), and primary brain tumors, including intracranial sarcoma, pituitary blastoma, and pineoblastoma.4 DICER1-associated neoplasms are believed to evolve via an unusual form of Knudson’s two-hit hypothesis. Instead of a second DICER1 somatic loss-of-function mutation, they arise after acquisition of a trans-somatic missense DICER1 mutation in an individual who harbors a pathogenic germline DICER1 variant.5-7 Critically, the somatic missense mutations in DICER1-associated neoplasms target one of five hotspot codons and result in hypomorphic DICER1 RNase IIIb function.8-10 Incomplete penetrance, especially for cancer phenotypes, and variable expressivity were recognized from early analyses of familial PPB pedigrees. The apparent requirement for somatic mutation in one of five specific codons may explain, in part, the incomplete penetrance noted in families.
The International PPB/DICER1 Registry was established in 1988, shortly after the first clinicopathologic report of PPB to establish this neoplasm as a specific entity.3 Although the registry is not population based, it nevertheless captures 68% to 84% of PPB in SEER-18 catchment areas,11 increasing confidence in the generalizability of registry findings. Before 2016, all enrolled individuals had a documented DICER1-related condition, such as PPB. Since December 2016, DICER1 carriers without a tumor history have also been enrolled. In 2011, the International Ovarian and Testicular Stromal Tumor Registry was founded as a parallel registry and applies similar strategies to study these rare ovarian and testicular neoplasms, including the link between DICER1 and sex cord-stromal tumors. The DICER1 study at the National Cancer Institute (NCI) also opened in 2011 with a goal to comprehensively study the phenotype of proband and nonproband DICER1 carriers. Together, these three studies have contributed to the delineation of the phenotypic spectrum and natural history of DICER1-associated tumors and have elucidated treatment-related outcomes in PPB.11
To date, no published analyses have quantified risk, hazard rates, and probability of neoplasm incidence accounting for competing risks (hereafter, “cumulative incidence”) of neoplasms (benign and malignant) in individuals with germline DICER1 pathogenic variation (hereafter, “DICER1 carriers”). This is especially important as germline pathogenic DICER1 variation is estimated to occur in the general population in approximately one in 10,600 people.12 To address these knowledge gaps, we determined the cumulative neoplasm incidence in probands and nonproband (proband relatives) DICER1 carriers. We quantified the standardized incidence ratios of malignancy for SEER Program–tracked cancers in nonprobands and probands. Lastly, in DICER1 carriers with one or more neoplasm, we determined pathologic type of neoplasm, age at onset, and frequency of multiple primary neoplasms.
PATIENTS AND METHODS
Study Participants
Individuals were enrolled in the International PPB/DICER1 Registry (ClinicalTrials.gov identifier: NCT01464606), the NCI Natural History of DICER1 Syndrome study (ClinicalTrials.gov identifier: NCT01247597), and/or the International Ovarian and Testicular Stromal Tumor Registry (ClinicalTrials.gov identifier: NCT01970696). Eligibility criteria included a history of DICER1-associated neoplasm or documentation of a pathogenic germline DICER1 variant. In the NCI Natural History of DICER1 Syndrome study (hereafter, the NCI DICER1 study), the proband—the enrolled individual through whom each family was ascertained—and his or her biologic relatives were recruited and tested for DICER1 pathogenic variation. Nonproband DICER1 carriers are biologic relatives of an enrolled proband harboring the familial DICER1 variant. Medical and family history, relationship to proband, germline DICER1 testing results, and vital status were recorded for all participants. Medical records and questionnaires from participants (probands and nonprobands) in the three studies were systematically reviewed for neoplasm (benign and malignant) diagnoses. We conducted follow-ups at regular intervals with participants to document new diagnoses. Whenever possible, neoplasm diagnoses were confirmed by review of surgical pathology reports and/or central review of pathology materials. A subset of families was invited to a comprehensive, 3-day NIH Clinical Center evaluation, including imaging and physical examination. We considered individuals with a DICER1-associated neoplasm and no pathogenic germline DICER1 variant (mosaic/sporadics) separately. As enrollment in more than one of these studies was common, we removed duplicate entries. All protocols were approved by the relevant institutional human subjects’ committees, and informed written consent/assent was obtained for each participant.
Standardized incidence ratios (SIRs) for malignancy (collectively, cancers, which excluded nonmelanoma skin cancers) were determined assuming a Poisson distribution for the observed case counts. We compared the observed number of cancers in DICER1 carriers with expected frequencies from the SEER program13 on the basis of age-, sex-, race-, and birth cohort–specific cancer incidence data. We calculated the ratio of observed-to-expected cancers for the cohort. We described continuous variables using medians and ranges, testing for differences using the Wilcoxon rank-sum test. We assessed differences in proportions using the χ2 test or an exact test when limited by infrequent observations (n < 5). Analyses were stratified by gender and/or adulthood (age 18 years and older). SEER analyses used SAS (SAS/STAT User’s Guide, Version 9.3; SAS Institute, Cary, NC) and Surveillance Research Program, NCI SEER*Stat software (version 8.3.4; http://www.seer.cancer.gov/seerstat).
Analyses of survival, probability of neoplasm incidence accounting for competing risks (cumulative incidence), age at onset, and counts of multiple primaries included all malignancies except nonmelanoma skin cancer, plus NCMH and CN (collectively, “neoplasms”). We excluded type Ir PPB (lung cysts without a primitive malignant component) and thyroid nodules, previously reported,14 from primary analyses. We excluded neoplasm recurrence and metastasis end points in our analysis, but included metachronous and synchronous neoplasms when this distinction could be determined confidently. The Kaplan-Meier method provided neoplasm-free survival estimates, with spline functions generating annual hazard of neoplasm diagnosis smoothed estimates15,16 for NCI cohort male and female nonproband carriers. We estimated nonparametric cumulative incidence curves for competing first neoplasm diagnoses (types I to III PPB, CN, SLCT, and all other neoplasm types) using all NCI cohort participants.17 Onset dates for first observed neoplasms within all three cohorts were plotted by site. Neoplasms in individuals with multiple neoplastic diagnoses were tabulated, and pairwise counts from the most common neoplasm diagnoses were plotted in an undirected graph. We estimated neoplasm-free survival and hazard of neoplasm using MATLAB (version R2017a; MathWorks, Natick, MA). All other analyses were conducted using R (version 3.3.1)18 with survival package (version 2.38).19,20
RESULTS
Table 1 lists the clinical data pooled from the registries and the NCI DICER1 study (all participants; N = 207), the NCI DICER1 study only (n = 148), and the NCI nonproband DICER1 carriers (n = 102). The majority of participants were non-Hispanic whites. To evaluate survival bias, we considered the relationships of the nonprobands to the probands (Appendix Table A1, online only). Of note, of the 17 nonproband participants who were diagnosed with a neoplasm, six (19%) of the 32 parents and one (10%) of the 10 grandparents had an observed neoplasm versus four (14%) of 28 siblings and one (20%) of five offspring. Appendix Table A2 (online only) lists the number and neoplasm types observed in all DICER1 carriers and in only nonprobands from the NCI DICER1 study. Appendix Table A3 (online only) describes the 26 participants without a detectable pathogenic germline DICER1 variant but with a history of at least one DICER1-associated neoplasm. These were either sporadic neoplasms or neoplasms arising in participants with suspected mosaicism. Appendix Table A4 (online only) lists the 31 neoplasms in the 26 mosaic/sporadic participants.
TABLE 1.
Demographics, Neoplasm Burden, and Mortality in DICER1 Carriers From the NCI DICER1 Study, International Pleuropulmonary Blastoma/DICER1 Registry, and Ovarian and Testicular Stromal Tumor Registry

To better understand the full spectrum of malignancy in DICER1 carriers, we conducted SIR analyses of 207 DICER1 carriers (probands and nonprobands) in the entire cohort (Appendix Table A5, online only). Given that many study participants were ascertained because they had a DICER1-associated neoplasm, there were greatly increased, significant risks for known syndrome-associated neoplasms, including gynandroblastoma, PPB, RMS, SLCT, and thyroid carcinoma. We observed one neuroblastoma. SIR analysis of 148 DICER1 carriers—probands and nonprobands (Appendix Table A6, online only)—from the NCI DICER1 study cohort demonstrated a neoplasm profile that was similar to the DICER1 carriers in the entire cohort.
To quantify the excess risk of cancer in the cohort while minimizing potential bias, we performed SIR analysis on 102 nonproband DICER1 carriers from the NCI DICER1 study, which represented 3,344 person-years of observation (Table 2). We observed evidence for elevated malignancy risk on the basis of SIR calculations. In addition to the malignancies listed in Table 2, we also observed one type I PPB, two CBMEs, two CNs, and one NCMH in the 102 nonproband DICER1 carriers; however, as CBME, CN, NCMH, and type I PPB are not tracked by SEER, risk estimates could not be calculated.
TABLE 2.
Standardized Incidence Ratios of 11 Cancers Observed in a Cohort of 102 Nonproband Carriers (3,344 person-years of risk) With a Germline DICER1 Pathogenic Variant From the National Cancer Institute DICER1 Study Only

To better understand the natural history of PPB, we quantified the number of type Ir PPB (regressed or nonprogressed type I PPB) in the 102 nonproband DICER1 carriers. There were 28 type Ir PPBs in 28 participants (27%; female, n = 15; male, n = 13; P > .05). Of these, five were resected and pathologically confirmed and 23 were detected using imaging in individuals above the typical age of progression.
Figure 1 compares the age at first neoplasm diagnosis (systematically ascertained cancers plus DICER1-associated neoplasms type I PPB, CN, and NCMH) in the NCI study in all female and male nonproband DICER1 carriers to age 20 (Fig 1A) and 60 years (Fig 1B), respectively. By age 10 years, 5.3% (95% CI, 0.6% to 9.7%) of nonproband DICER1 carriers had developed a neoplasm (females, 4.0%; males, 6.6%). Penetrance by decade for nonproband DICER1 carriers age 10 to 60 years is shown in Appendix Table A7 (online only). Care should be taken in the interpretation of cumulative incidence in the older age ranges as the small number of individuals observed at those ages leads to reduced precision in estimation. The hazards of a neoplasm diagnosis (percent-per-year risk for cancers plus DICER1-associated neoplasms CN and NCMH) in nonproband DICER1 carriers to age 20 years and age 60 years are shown in Figures 1C and 1D, respectively.
FIG 1.

Estimated neoplasm-free survival and hazard of first neoplasm—cancers plus DICER1-associated neoplasms cystic nephroma and nasal chondromesenchymal hamartoma—incidence among nonproband DICER1 carriers of the National Cancer Institute DICER1 study. (A and B) Estimated neoplasm-free survival among these individuals. See Appendix Table A7 (online only) for penetrance of neoplasm diagnosis by decade. (C and D) Estimated hazard of first neoplasm diagnosis. Female participants are denoted in red and male in blue. Dashed lines indicate nonparametric estimates (Kaplan-Meier curves). Solid lines indicate spline-estimated cumulative incidence and hazard. Left panels display curves to age 20 years and right panels to age 60 years. Common vertical axis markers of 0.5% event-free survival and 0.025 hazard are indicated by horizontal dashed lines and bold axis markers, and age 20 years is similarly marked on the horizontal axis.
We systematically ascertained neoplasms in the 148 probands and nonprobands in the NCI DICER1 study. Figure 2 shows the early childhood and lifetime cumulative incidence of types I, II, and III PPB, all PPB, CN, SLCT, other neoplasms, and all neoplasms combined, as a first malignancy event, in this cohort. Cumulative incidence is calculated with each PPB type, CN, SLCT, and other neoplasms as competing first malignancy events. As a result of the small numbers, deaths were censored rather than considered as a competing risk.
FIG 2.

Estimated probability of neoplasm development accounting for competing risks—cumulative incidence—of specific DICER1-associated neoplasms among probands and nonprobands from the National Cancer Institute DICER1 study: type I pleuropulmonary blastoma (PPB; red), type II PPB (green), type III PPB (orange), cystic nephroma (purple), and Sertoli-Leydig cell tumor (SLCT; gray). Hatched multicolor lines denote overlap of multiple neoplasm cumulative risks. Cumulative incidence to (A) age 6 years and (B) age 60 years. Tick marks denote censoring times.
Finally, we plotted the age at neoplasm diagnosis and neoplasm type for the pooled 116 DICER1 carriers with one or more neoplasms (proband and nonproband) in early childhood (Fig 3A) and to age 60 years (Fig 3B). Three individuals were diagnosed with CN in adolescence, older than the typically cited age risk of 4 years or younger. The wide age-at-onset range for SLCT was notable. Risk for thyroid carcinoma begins at younger than age 10 years and continues into adulthood, as we have previous reported.14 NCMH was observed in children and young adults.21 Of 116 DICER1 carriers with a neoplasm, 26 (22%) developed multiple neoplasms over time. The most frequent co-occurrences involved known DICER1-associated neoplasms (eg, PPB, CN, NCMH, SLCT, or thyroid carcinoma; Appendix Fig. A1, online only). Among females with a DICER1-related ovarian neoplasm, we observed three (13%) of 24 with a contralateral metachronous SLCT and one (17%) of six with multiple primary RMS (Appendix Fig. A1).
FIG 3.

Age of neoplasm diagnosis and neoplasm type for the 116 DICER1 carriers (151 neoplasms; proband and nonproband) from all three studies. Specific DICER1-associated neoplasms are highlighted in color: type I pleuropulmonary blastoma (PPB; red), type II PPB (green), type III PPB (orange), Sertoli-Leydig cell tumor (SLCT; purple), and cystic nephroma (gray). Diagnoses to (A) age 10 years and (B) age 60 years. Gray boxes span from the first to the third quartile of observed diagnoses, and vertical bars in the boxes indicate median diagnosis age. NCMH, nasal chondromesenchymal hamartoma.
DISCUSSION
This report provides the first quantitative analysis of site-specific neoplasm risk and excess malignancy risk in a large cohort of systematically characterized nonproband DICER1 carriers. Follow-up of participants allowed for the prospective documentation of additional incident neoplasms over time. In nonprobands, we observed the spectrum of DICER1-associated neoplasms (thyroid carcinoma, type I PPB, CN, CBME, and gynecologic tumors) and, for the first time, quantified neoplasm penetrance and demonstrated significantly increased risks for all cancers combined. Of note, there were no deaths observed in nonproband DICER1 carriers. Our findings inform DICER1 syndrome phenotype, natural history, and genetic counseling.
Among all DICER1 carriers, the most common neoplasms were PPB; gynecologic tumors, especially SLCT and RMS; and CN. Thyroid carcinoma was the most common neoplasm in nonproband DICER1 carriers. Other less common but established DICER1-associated cancers, such as pineoblastoma, pituitary blastoma, and Wilms tumor, were not observed in this study. The six RMSs arose exclusively from the gynecologic tract. Case reports describe neuroblastoma in children with DICER1 syndrome,4 but we observed no significant neuroblastoma excess and no participant developed testicular cancer, which is consistent with previous reports.22,23 Among all participants, we observed individual examples of rare cancers not previously reported in DICER1 carriers (thymoma, spindle cell carcinoma of the kidney, and teratoma). The significantly elevated SIRs were driven by the exceedingly small (essentially zero) expected case numbers. The significant excess of malignant peripheral nerve sheath tumors in our SIR analysis is likely explained by the coexistence of neurofibromatosis type I in the two affected children. There were nonsignificant excesses of common adult tumors (melanoma, prostate, and breast cancer).
Most individuals with pathogenic germline DICER1 variation live generally healthy lives. Although risks of malignancy are elevated, we note that many DICER1-related cancers, including PPB, SLCT, and gynandroblastoma, are most curable when found at an early stage. Neoplastic risk increases with age, especially in girls and women, predominantly because of gynecologic cancers. Ultrasound represents a feasible surveillance strategy for these tumors, which are largely curable with surgery alone when found before rupture or capsular invasion. With CN, a histologically benign condition, early diagnosis may be nephron sparing and resection may prevent progression to renal sarcoma.
With that in mind, we recommend familial cascade DICER1 testing to identify additional family members who are at risk for DICER1-related neoplasms. We emphasize that, given the moderate penetrance in the DICER1 syndrome, a negative family history should not dissuade an individual from genetic testing when an individual presents with a condition known to be associated with DICER1, including PPB, CN, ovarian SLCT, or gynecologic sarcoma.
Surveillance strategies to manage risk in DICER1 carriers have been recently published.24,25 We recommend a surveillance chest computed tomography scan in the first year of life and, if normal, again at age 2 years, shortly before the peak incidence of types II and III PPB. Intermittent chest radiography during childhood is also recommended. The risk of sedation, which may be needed for cross-sectional imaging, and radiation exposure with computed tomography scan or chest radiographs must be balanced with the dramatic increase in survival and the decreased need for intensive therapies when PPB is found as the purely cystic type I (5-year overall survival of 91% compared with that of type II or type III [5-year overall survival, 74% and 53%, respectively]).11 Given the markedly increased risk for ovarian sex cord-stromal tumors associated with a favorable prognosis if found at stage IA, we also recommend surveillance pelvic ultrasounds beginning in childhood and extending through adulthood. If an ovarian tumor is found, surgical resection should include appropriate staging. Thyroid evaluation is recommended every 3 years unless nodules are detected. When nodules are found, age-appropriate guidelines for the evaluation of thyroid nodules from established endocrine societies should be followed. To place these results and surveillance recommendations in context, the cumulative risk of breast cancer in BRCA1 or BRCA2 carriers is approximately 4% between the age of 21 and 30 years, increasing thereafter.26 For this risk, annual surveillance breast magnetic resonance imaging is recommended from age 25 to 29 years.27
In DICER1 carriers, penetrance for less serious conditions, such as thyroid nodules, is substantially higher than that for malignant conditions, such as types II or III PPB or SLCT. Although penetrance for benign thyroid nodules is high, only a small percentage progress to thyroid carcinoma.14 We observed a similar natural history in PPB in this study. In the 102 nonprobands, we observed one type I PPB and 28 type Ir PPB. The latter are type I PPB that either experienced regression or, more likely, never experienced disease progression beyond a simple lung cyst and harbor little-to-no malignant potential. The etiology and mechanism of this regression/nonprogression is unknown. Thus, in the 102 nonproband DICER1 carriers, there was radiographic or histologic evidence of type I or Ir PPB in 29 individuals (28%). Our findings suggest that cystic PPB is common in DICER1 carriers and that only a minority experience progression to type II or III PPB, given the rarity of these tumors. Factors that contribute to this transformation merit investigation, and additional studies are underway to determine what methods will best predict progression and/or detect progression at the earliest possible time point.
Strengths of this analysis include the large cohort of DICER1 carriers that was pooled from multiple studies and detailed follow-up information from study participants. More heavily affected, multigenerational families may have a greater likelihood of undergoing DICER1 genetic testing and may therefore inflate the risk of neoplasm estimates. SIR results are descriptive of those included in the NCI cohort and must be interpreted by accounting for the operative convenience sampling associated with study participation. SIR analysis is likely an underestimate as it is constrained to consider only cancers ascertained or precisely identified by the SEER system, which does not capture type I PPB, CBME, CN, or NCMH, all clinically relevant neoplasms observed in these participants. In families in which the original proband died before enrollment or did not enroll in the NCI study, the first ascertained enrolled individual was considered the proband for the purpose of proband/nonproband analysis. If deceased individuals who were enrolled only in a registry study had been included in the proband/nonproband analyses, at least one additional case of type I PPB in a nonproband would have been noted in this analysis. Survival bias is also reflected in higher neoplasm counts among younger relatives (ie, siblings and offspring) compared with older relatives (ie, parents and grandparents). Our study population was overwhelmingly non-Hispanic white. There are no racial groups with a known increased risk for DICER1-associated neoplasms, although this merits additional study. We acknowledge the limited ethnic and racial heterogeneity of the three studies and believe it reflects known, long-standing biases in biomedical research recruitment. Although this analysis represents the largest available cohort, the sample size confers relatively wide CIs.
We are now refining DICER1 syndrome prevalence and penetrance estimates from large-scale exome sequencing data. Additional clinical strategies are needed to identify the subset of DICER1 carriers who are at highest risk of the most aggressive disease phenotypes, particularly type II and III PPB and gynecologic tumors.
Appendix
FIG A1.

Diagram of counts of multiple primary DICER1-associated neoplasms: Pleuropulmonary blastoma (PPB, all types, except type Ir), malignant peripheral nerve sheath tumor (MPNST), nasal chondromesenchymal hamartoma (NCMH), Sertoli-Leydig cell tumor (SLCT), rhabdomyosarcoma, cystic nephroma, and thyroid cancer. Each neoplasm type is displayed as a vertex; for each pair of neoplasm, a line between their corresponding vertices indicates the number of individuals with at least one primary neoplasm of each type. The weight and color of the line, along with the number printed in a bubble on each line, indicate the number of individuals. Vertices with no shared line (eg, cystic nephroma and MPNST) indicate that no participants had a primary neoplasm of each type. One participant had multiple primary rhabdomyosarcomas, and three participants had multiple primary SLCTs. Data are also presented in Appendix Table A8 (online only).
FIG A2.

Study flow diagram detailing participant, neoplasm, and malignancy counts in the study (excluding type Ir PPB and thyroid nodules). NCI, National Cancer Institute; PPB, pleuropulmonary blastoma.
TABLE A1.
Non-Proband NCI DICER1 Study Participants, by Relation to Proband, With Corresponding Numbers of Neoplasms in Participants With One or More Neoplasms
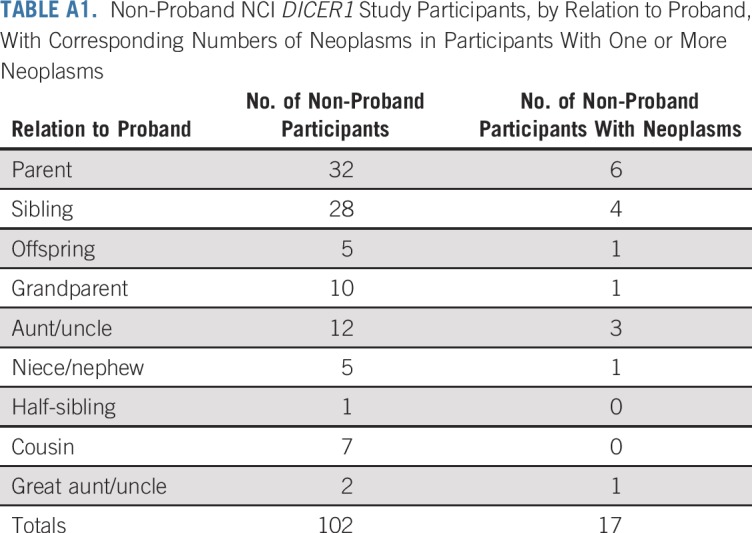
TABLE A2.
Counts of Observed Neoplasms by Type, Sex, and Participant Type in All DICER1-Carriers Participants and in Non-Proband DICER1 Carriers From the NCI DICER1 Study

TABLE A3.
Demographics, Neoplasm Burden, and Mortality in DICER1-Mosaic/Sporadic Participants From the NCI DICER1 Study, International Pleuropulmonary Blastoma Registry, and Ovarian and Testicular Stromal Tumor Registry
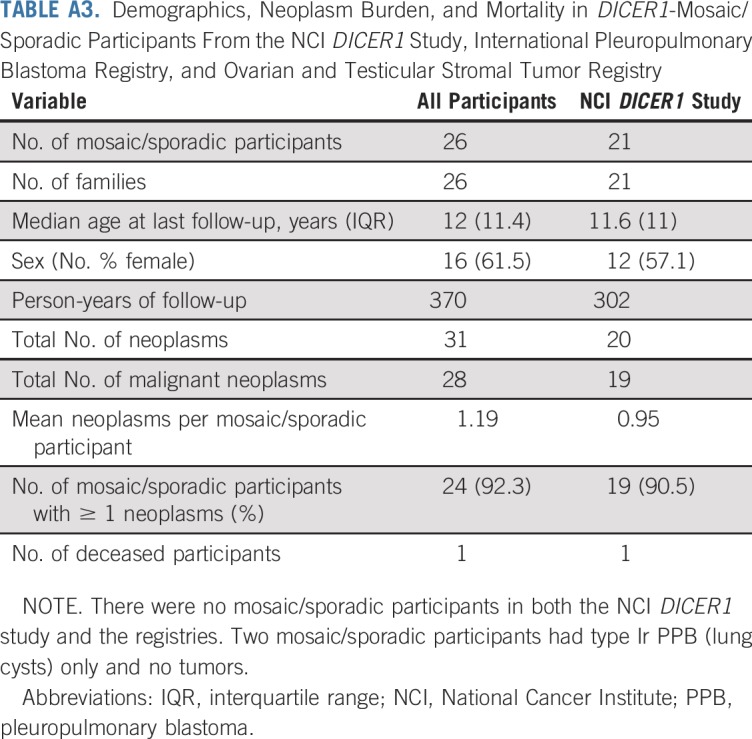
TABLE A4.
Neoplasm Counts Among DICER1-Mosaic/Sporadic Participants
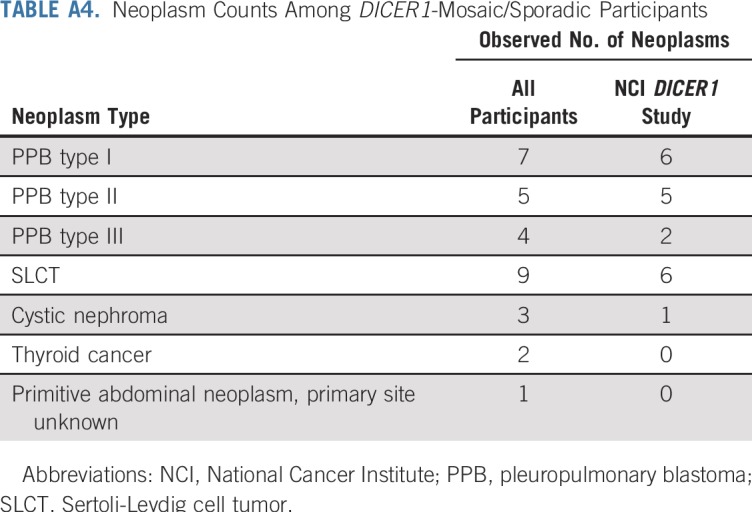
TABLE A5.
SIR of 102 Cancers Observed in Entire Cohort of 207 DICER1 Carriers (probands and non-probands; 4,747 person-years of risk)

TABLE A6.
SIR of 45 Cancers Observed in Cohort of 148 DICER1 Carriers (probands and non-probands; 3,951 person-years of risk) From the NCI DICER1 Study Only

TABLE A7.
Non-Proband DICER1-Carrier Cumulative Age-Specific Neoplasm Risk (cancers plus DICER1-associated neoplasms CN and NCMH), Per Decade

TABLE A8.
Table of DICER1-Carriers With More Than One Reported Neoplasm Diagnosis

Footnotes
Presented at the Annual Meeting of the American College of Medical Genetics and Genomics, Charlotte, NC, April 10-14, 2018.
Supported by the National Cancer Institute Intramural Research Program, Division of Cancer Epidemiology and Genetics, Grant No. R01-CA143167 and The Parson’s Foundation (D.A.H.). The International PPB Registry is supported by the Pine Tree Apple Classic Fund, the Pediatric Cancer Research Foundation, and the Rein in Sarcoma Foundation. The International Ovarian and Testicular Stromal Tumor Registry is supported by St. Baldrick’s Foundation, Pine Tree Apple Classic Fund, Hyundai Hope on Wheels, and the Randy Shaver Cancer Research and Community Fund.
AUTHOR CONTRIBUTIONS
Conception and design: Douglas R. Stewart, Ana F. Best, Louis P. Dehner, Yoav H. Messinger, Philip S. Rosenberg, D. Ashley Hill, Kris Ann P. Schultz
Financial support: Douglas R. Stewart, Kris Ann P. Schultz, D. Ashley Hill
Administrative support: Gretchen M. Williams, Laura A. Harney, Ann G. Carr, Anne K. Harris
Provision of study materials or patients: Douglas R. Stewart, Louis P. Dehner, Yoav H. Messinger, D. Ashley Hill, Kris Ann P. Schultz
Collection and assembly of data: Douglas R. Stewart, Ana F. Best, Gretchen M. Williams, Laura A. Harney, Ann G. Carr, Anne K. Harris, Christian P. Kratz, Louis P. Dehner, Yoav H. Messinger, D. Ashley Hill, Kris Ann P. Schultz
Data analysis and interpretation: Douglas R. Stewart, Ana F. Best, Laura A. Harney, Ann G. Carr, Louis P. Dehner, Yoav H. Messinger, Philip S. Rosenberg, D. Ashley Hill, Kris Ann P. Schultz
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Neoplasm Risk Among Individuals With a Pathogenic Germline Variant in DICER1
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Douglas R. Stewart
Consulting or Advisory Role: Genome Medical
Ana F. Best
Travel, Accommodations, Expenses: Foundation Medicine
Yoav H. Messinger
Honoraria: Sanofi
D. Ashley Hill
Stock or Other Ownership: ResourcePath
Travel, Accommodations, Expenses: Driver
No other potential conflicts of interest were reported
REFERENCES
- 1.Priest JR, Watterson J, Strong L, et al. : Pleuropulmonary blastoma: A marker for familial disease. J Pediatr 128:220-224, 1996 [DOI] [PubMed] [Google Scholar]
- 2.Hill DA, Ivanovich J, Priest JR, et al. : DICER1 mutations in familial pleuropulmonary blastoma. Science 325:965, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manivel JC, Priest JR, Watterson J, et al. : Pleuropulmonary blastoma. The so-called pulmonary blastoma of childhood. Cancer 62:1516-1526, 1988 [DOI] [PubMed] [Google Scholar]
- 4.Pagon RA: DICER1-related disorders, in GeneReviews. Seattle, WA, University of Washington, 2014. https://www.ncbi.nlm.nih.gov/books/NBK196157/ [Google Scholar]
- 5.Seki M, Yoshida K, Shiraishi Y, et al. : Biallelic DICER1 mutations in sporadic pleuropulmonary blastoma. Cancer Res 74:2742-2749, 2014 [DOI] [PubMed] [Google Scholar]
- 6.Pugh TJ, Yu W, Yang J, et al. : Exome sequencing of pleuropulmonary blastoma reveals frequent biallelic loss of TP53 and two hits in DICER1 resulting in retention of 5p-derived miRNA hairpin loop sequences. Oncogene 33:5295-5302, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brenneman M, Field A, Yang J, et al. : Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in pleuropulmonary blastoma / DICER1 syndrome: A unique variant of the two-hit tumor suppression model. F1000 Res 4:214, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heravi-Moussavi A, Anglesio MS, Cheng SW, et al. : Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med 366:234-242, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Gurtan AM, Lu V, Bhutkar A, et al. : In vivo structure-function analysis of human Dicer reveals directional processing of precursor miRNAs. RNA 18:1116-1122, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anglesio MS, Wang Y, Yang W, et al. : Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. J Pathol 229:400-409, 2013 [DOI] [PubMed] [Google Scholar]
- 11.Messinger YH, Stewart DR, Priest JR, et al. : Pleuropulmonary blastoma: A report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the International Pleuropulmonary Blastoma Registry. Cancer 121:276-285, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Field A, Schultz KAP, et al. : The prevalence of DICER1 pathogenic variation in population databases. Int J Cancer 141:2030-2036, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.National Cancer Institute : SEER*Stat databases: November 2014 submission. https://seer.cancer.gov/data-software/documentation/seerstat/nov2014/ [Google Scholar]
- 14.Khan NE, Bauer AJ, Schultz KAP, et al. : Quantification of thyroid cancer and multinodular goiter risk in the DICER1 syndrome: A family-based cohort study. J Clin Endocrinol Metab 102:1614-1622, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenberg PS, Greene MH, Alter BP: Cancer incidence in persons with Fanconi anemia. Blood 101:822-826, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Rosenberg PS: Hazard function estimation using B-splines. Biometrics 51:874-887, 1995 [PubMed] [Google Scholar]
- 17.Putter H, Fiocco M, Geskus RB: Tutorial in biostatistics: Competing risks and multi-state models. Stat Med 26:2389-2430, 2007 [DOI] [PubMed] [Google Scholar]
- 18.R Core Team : R: A Language and Environment for Statistical Computing. Vienna, Austria, R Foundation for Statistical Computing, 2016 [Google Scholar]
- 19.Therneau TM: A package for survival analysis in S (version 2.38). https://cran.r-project.org/web/packages/survival/index.html
- 20.Therneau TM, Grambsch PM: Modeling Survival Data: Extending the Cox Model. New York, NY, Springer, 2000 [Google Scholar]
- 21.Stewart DR, Messinger Y, Williams GM, et al. : Nasal chondromesenchymal hamartomas arise secondary to germline and somatic mutations of DICER1 in the pleuropulmonary blastoma tumor predisposition disorder. Hum Genet 133:1443-1450, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Boer CM, Eini R, Gillis AM, et al. : DICER1 RNase IIIb domain mutations are infrequent in testicular germ cell tumours. BMC Res Notes 5:569, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabbaghian N, Bahubeshi A, Shuen AY, et al. : Germ-line DICER1 mutations do not make a major contribution to the etiology of familial testicular germ cell tumours. BMC Res Notes 6:127, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schultz KAP, Rednam SP, Kamihara J, et al. : PTEN, DICER1, FH, and their associated tumor susceptibility syndromes: Clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res 23:e76-e82, 2017 [DOI] [PubMed] [Google Scholar]
- 25.Schultz KAP, Williams GM, Kamihara J, et al. : DICER1 and associated conditions: Identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res 24:2251-2261, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuchenbaecker KB, Hopper JL, Barnes DR, et al. : Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 317:2402-2416, 2017 [DOI] [PubMed] [Google Scholar]
- 27.National Comprehensive Cancer Network : NCCN guides for genetic/familial high-risk assessment: Breast and ovarian (version 1.2018). https://www.nccn.org/Common/FileManager.ashx?fileManagerId=365c45f8-a09b-4d0a-844b-a7c0bd140dc2