

The mitochondrial proteins processing system has been extensively studied with proteomics, but in a rather static way. In this work, human cells were treated by two compounds known to impact mitochondria, rapamycin and zinc. A combined N-terminomics and shotgun proteomics strategy allowed studying the impact of these compounds on human mitochondria. Moderate but significant changes were detected at the proteome level, impacting core mitochondrial functions. The mitochondrial processing appeared robust under subtoxic conditions but perturbed in its efficiency.
Keywords: Mitochondria function or biology, Cell biology*, Mass Spectrometry, Cellular organelles*, Enzymes*, N-terminomics, rapamycin, zinc
Graphical Abstract
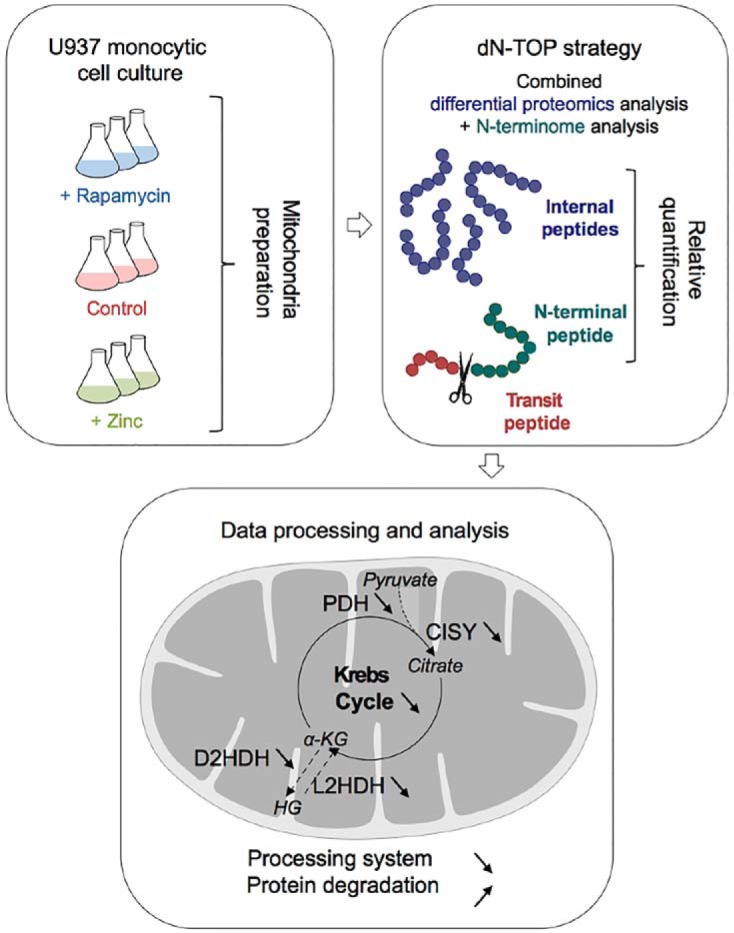
Highlights
Rapamycin and zinc induce moderate but significant mitochondrial proteome changes.
The mitochondrial proteins processing system is robust under subtoxic conditions.
Rapamycin and zinc perturb the mitochondrial proteins processing system.
Rapamycin and zinc perturb the mitochondrial proteins homeostasis.
Abstract
All but thirteen mammalian mitochondrial proteins are encoded by the nuclear genome, translated in the cytosol and then imported into the mitochondria. For a significant proportion of the mitochondrial proteins, import is coupled with the cleavage of a presequence called the transit peptide, and the formation of a new N-terminus. Determination of the neo N-termini has been investigated by proteomic approaches in several systems, but generally in a static way to compile as many N-termini as possible. In the present study, we have investigated how the mitochondrial proteome and N-terminome react to chemical stimuli that alter mitochondrial metabolism, namely zinc ions and rapamycin. To this end, we have used a strategy that analyzes both internal and N-terminal peptides in a single run, the dN-TOP approach. We used these two very different stressors to sort out what could be a generic response to stress and what is specific to each of these stressors. Rapamycin and zinc induced different changes in the mitochondrial proteome. However, convergent changes to key mitochondrial enzymatic activities such as pyruvate dehydrogenase, succinate dehydrogenase and citrate synthase were observed for both treatments. Other convergent changes were seen in components of the N-terminal processing system and mitochondrial proteases. Investigations into the generation of neo-N-termini in mitochondria showed that the processing system is robust, as indicated by the lack of change in neo N-termini under the conditions tested. Detailed analysis of the data revealed that zinc caused a slight reduction in the efficiency of the N-terminal trimming system and that both treatments increased the degradation of mitochondrial proteins. In conclusion, the use of this combined strategy allowed a detailed analysis of the dynamics of the mitochondrial N-terminome in response to treatments which impact the mitochondria.
Organellar proteomics and especially mitochondrial proteomics are almost as old as proteomics itself; the first primitive draft of a human mitochondrial proteome was published only a few years after the word “proteomics” was coined (1). Mitochondrial proteomics has been used over the years for numerous applications (reviewed in (2)), such as the elucidation of the repertoire of mitochondrial proteins (e.g. in (3–6)). The approach has been refined to investigate the events that occur during protein import into the mitochondria. Almost all mitochondrial proteins are encoded in the nuclear DNA and imported into the mitochondria by a complex system (reviewed in (7–9)). In the case of at least half of the mitochondrial proteins, a transit peptide is cleaved during import, generating a new N-terminus. Therefore, N-terminomics approaches have been used to characterize the new N-termini in various organisms, from yeast to mammals (10–12). These studies have shown that besides the major mitochondrial processing peptidase that cleaves the transit peptide, the system includes aminopeptidases that trim the N-terminal end to create ragged termini and stabilize the proteins (10, 13).
Mitochondrial proteomics has also been used to investigate the modulations of the mitochondrial proteome in response to various biological situations, ranging from alterations of mitochondrial DNA (14, 15) to various physiopathological situations such as aging (16–18), exposure to ionizing radiations (19), metal toxicity (20) and various metabolic (21–25) and iatrogenic (21, 26–29) perturbations.
Although determination of the neo-N-termini of mitochondrial proteins is still an active research field (10–12), nothing is yet known about the robustness of the mitochondrial protein processing system under conditions of mitochondrial stress, particularly nonlethal stress. It is not known, for example, whether errors occur in the precursor cleavage during stress, or how the other components of the mitochondrial protein processing system behave in such conditions.
In this context, the aim of this study was to investigate the impact of sublethal doses of two known mitochondrial stressors, namely rapamycin and zinc, on human mitochondria. These two stressors were chosen as they act via different mechanisms and therefore allow determining whether the effects on the mitochondrial proteome and/or the processing system could be stress-generic or agent specific.
On the one side, zinc is both a trace element essential for the proper functioning of the immune system (30, 31)) and a toxic element at high doses, causing for example the metal fume fever (32, 33). The zinc ion has a strong affinity to sulfur and binds to glutathione (34) and to cysteine residues in protein active sites (35, 36). This can result in the inhibition of key metabolic enzymes, ranging from glyceraldehyde phosphate dehydrogenase to mitochondrial enzymes (37–39). Interestingly, zinc toxicity is at least partly reversed by supplementation with metabolic end-products such as pyruvate and/or oxaloacetate (40–45). Thus, zinc toxicity is linked to metabolic dysfunction and with a clear involvement of the mitochondria.
On the other side, the drug rapamycin has a known strong impact on mitochondrial function. Its effects on the organelle have been described from the very beginning of its description (46) and refined over time (47, 48). Proteomics has contributed to the understanding of its effects (49).
To probe the effects of these two mitochondrial stressors on the mitochondrial proteome, as well as their impact on the mitochondrial protein processing system, the previously-described combined shotgun proteomics and N-terminomics approach afforded by the doublet N-terminal oriented (dN-TOP) strategy (50) was used in the present study.
MATERIALS AND METHODS
Cell Culture
The U937 cells were grown in suspension in RPMI1640 medium supplemented with 10% fetal bovine serum and 10 mm Hepes pH 7.5 buffer. Small-scale cultures were carried out in 75 cm2 or 175 cm2 flasks (culture volumes were 25 ml or 60 ml respectively) and used for targeted assays such as mitochondrial potential or enzyme activities. Large-scale cultures for mitochondrial preparations were carried out in 1-liter spinner bottles. Cells were grown to a density of 500,000 cells/ml and then treated with either 10 nm rapamycin or 100 μm zinc acetate. Both treatments induced <20% cell death, as determined by dye exclusion. All experiments were carried out on three independent cultures.
Mitochondrial Transmembrane Potential Measurement
The mitochondrial transmembrane potential was assessed by Rhodamine 123 uptake. Cells were incubated with 80 nm Rhodamine 123 for 30 min at 37 °C in the incubator, then rinsed twice in cold glucose (1 mg/ml) in PBS and harvested in cold glucose (1 mg/ml) - PBS with propidium iodide (1 μg/ml). The mitochondrial potential of cells was analyzed by flow cytometry on a FACScalibur instrument (Beckton Dickinson, Franklin Lakes, NJ). Dead cells (propidium positive) were excluded from analysis. A low rhodamine concentration (80 nm) was used to avoid intramitochondrial fluorescence quenching, which would result in a poor estimation of the mitochondrial potential (51).
Isolation of Mitochondria
Mitochondria were isolated as described in (52). Briefly, cells were harvested, rinsed three times in PBS and resuspended in a 10-fold pellet volume of hypotonic buffer (10 mm Hepes-NaOH pH 7.5, 2 mm MgCl2, 1 mm EGTA, 50 mm KCl). After 10 min on ice, cells were lysed with ten strokes of a Dounce homogenizer (tight pestle). A volume of 2 m sucrose equivalent to one-tenth that of buffer was added, and the mixture homogenized with two additional strokes. The suspension was centrifuged at 1000 × g for 5 min to pellet the nuclei, unbroken cells and debris. The supernatant was collected then centrifuged at 7000 × g for 10 min to pellet the mitochondria. The mitochondrial pellet was resuspended in washing buffer (10 mm Hepes-NaOH pH 7.5, 2 mm MgCl2, 250 mm sucrose) and centrifuged at 7000 × g for 10 min. The protein concentration of the mitochondrial suspension was determined by a dye-binding assay (53) and the extracts were aliquoted and stored at −80 °C until use. This protocol afforded the best compromise between yield and purity (11).
Experimental Design and Statistical Rationale
Each rapamycin- and zinc acetate-treated cell culture was performed in biological triplicate (n = 3). In parallel, three independent cell cultures underwent no treatment to generate control samples. Each sample was processed as described below and measured three times by mass spectrometry (MS), resulting in a total of 27 analyses.
Proteomics Analysis and dN-TOP Strategy
Sample Preparation
The protocol used in the present study was adapted from previous work (11) and (52). Briefly, aliquots containing 400 μg of mitochondria-enriched protein extract was taken for each sample. Aliquots were centrifuged at 10,000 × g for 20 min and at 20,000 × g for 5 min at 4 °C. The mitochondria were resuspended in labeling buffer (50 mm Tris-HCl, 8 m urea, 2 m thiourea, 1% SDS, pH 8.2), reduced by incubating with 5 mm tributylphosphine (TBP) for 1 h and alkylated by incubating with 50 mm iodoacetamide for 1 h at room temperature. An equimolar solution of 100 mm light and heavy N-succinimidyloxycarbonylmethyl tris(2,4,6-trimethoxyphenyl)phosphonium bromide (TMPP) 80% acetonitrile (ACN), 200:1 reagent/protein molar ratio was added, and the resulting solution incubated for 1 h with shaking. Any residual derivatizing reagent was quenched by incubating with a solution of 0.1 m hydroxylamine at room temperature for 1 h. Glycerol (10%) and bromphenol blue (1%) were added to the samples and stacking gel bands prepared for 50 μg of each sample replicate. Stacking gel bands were excised, cut, in-gel reduced (using 10 mm dithiothreitol in 25 mm NH4HCO3), alkylated (using 55 mm iodoacetamide in 25 mm NH4HCO3), and the proteins digested overnight using modified porcine trypsin (Promega, Madison, WI) at 37 °C. Tryptic peptides were extracted using 60% ACN in 0.1% formic acid. After vacuum centrifugation, tryptic peptides were resuspended in 190 μl of 0.1% formic acid (FA) in water.
Nano-LC-MS/MS Analysis
Nano-LC-MS/MS analysis was performed on a nanoAcquity UPLC device (Waters, Milford, MA) coupled to a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). The solvents consisted of 0.1% FA in H2O (solvent A) and 0.1% FA in ACN (solvent B). Sample volumes of 2 μl were loaded onto a Symmetry C18 precolumn (20 mm × 180 μm, 5 μm diameter particles; Waters) over 3 min at 5 μl/min with 1% solvent B. Peptides were eluted on an Acquity UPLC BEH130 C18 column (250 mm × 75 μm, 1.7 μm particles; Waters) at 450 nl/min with the following gradients of solvent B: linear from 1% to 20% in 48 min, linear from 20% to 40% in 37 min, up to 90% in 1 min, isocratic at 90% for 8 min, down to 1% in 1 min, isocratic at 1% for 25 min.
The Q-Exactive Plus was operated in data-dependent acquisition mode by automatically switching between full MS and consecutive MS/MS acquisitions. Full-scan MS spectra were collected from 300–1,800 m/z at a resolution of 70,000 at 200 m/z with an automatic gain control target fixed at 3 × 106 ions and a maximum injection time of 50 ms. The top 10 precursor ions with an intensity exceeding 2 × 105 ions and charge states ≥ 2 were selected from each MS spectrum for fragmentation by higher-energy collisional dissociation. Spectra were collected at a resolution of 17,500 at 200 m/z with a fixed first mass of 100 m/z, an automatic gain control target fixed at 1 × 105 ions and a maximum injection time of 100 ms. Dynamic exclusion time was set to 60 s.
The Data Sets Were Used for Shotgun and TMPP Data Analyses
Shotgun Data Analysis
Shotgun analysis was carried out using MaxQuant (version 1.6.0.16) (54). Raw data-derived peak lists were searched using the Andromeda search engine against an in-house database containing all Homo sapiens entries extracted from UniProtKB-SwissProt (release of 01/2016, 20,175 entries), common contaminants (119 entries) and the corresponding 20,294 reverse entries. The database was generated using the database toolbox from Mass Spectrometry Data Analysis (MSDA1, publicly available from https://msda.unistra.fr) (55). The precursor mass tolerance was set to 20 ppm for the first search and 4.5 ppm for the main search. The fragment ion mass tolerance was set to 20 ppm. Trypsin was chosen as the digestion enzyme and one missed cleavage was tolerated. Carbamidomethylation of cysteine residues was set as a fixed modification, and the oxidation of methionine residues and acetylation of protein N-termini as variable modifications. The minimum peptide length was set to seven amino acids. A maximum false discovery rate (FDR) of 1% was set at both the peptide and protein levels. Quantification, normalization and protein abundance estimation were performed using the MaxLFQ method implemented in MaxQuant (56). The option “match between runs” was enabled. Both modified and unmodified peptides (oxidation of methionine residues and acetylation of protein N-termini) were excluded from protein quantification. All other parameters were set as default. Proteins identified as “reversed”, “contaminants” or “only identified by site” and proteins identified with no unique peptide were discarded from the list of identified proteins. Protein identification was performed using internal peptides only and not the N-terminal peptides, because they carried a TMPP label that could not be identified through this analysis. Mitochondrial proteins were extracted based on neXtProt gold and silver annotations for the GO term (GO:0005739) and the UniProt term (SL-0173) for “Mitochondrion” (https://snorql.nextprot.org/).
Statistical Analysis of Shotgun Proteomics Data
Proteins with two or more missing values per injection triplicate were discarded. In each analysis, protein abundance was normalized to the sum of all protein abundances. Finally, the median of the injection triplicate data was calculated for each protein, and this value was used as the protein abundance for this biological point. This led to a data set with one value per protein and biological point, so that the final data set contained three values (biological replicates) per biological condition (control, rapamycin-treated, zinc-treated). Pairwise comparisons of the mitochondrial protein abundances in the treated samples and the control samples were carried out using the Perseus package (version 1.5.5.3). The log2-transformed quantities were checked for normality distribution (Shapiro-Wilk test) and homoscedasticity (Bartlett test) in RStudio (version 1.0.136), then submitted to a Student's T test. Proteins with p ≤ 0.05 were considered as variants. To account for the multiple testing issue, we used several approaches such as the Benjamini-Hochberg FDR (57), the sequential Fisher approach (58) and the sequential goodness of fit approach (59). Global analysis of the shotgun data was performed using the PAST software (60).
TMPP Data Analysis
Raw data were converted into mgf files using the MSConvert tool from ProteomeWizard (v3.0.6090; http://proteowizard.sourceforge.net/). The peak lists were searched using Mascot (version 2.5.1; Matrix Science, London, UK) against the previously described in-house database. The following parameters were applied: semiTrypsin as the digestion enzyme, one permitted missed cleavage per peptide, a mass tolerance of 5 ppm for the precursor ions and 0.07 Da for the peptide fragments, carbamidomethylation of cysteine residues as a fixed modification, and oxidation of methionine residues and light (+572.18 Da) and heavy (+581.21 Da) TMPP derivatization on any N-terminal amino acid or side chain of tyrosine and lysine residues as variable modifications. Mascot result files were loaded into Proline software (http://proline.profiproteomics.fr (61)) and proteins were validated on pretty rank equal to one, and a Mascot ion score ≥ 13. The N-terminal peptides were validated using the “dN-TOP Validation Tool” available at https://msda.unistra.fr (55), by confirming the identification of both the light and heavy labeled peptide forms and their chromatographic co-elution (11).
Statistical Analysis of TMPP Data
For each analysis, light and heavy TMPP-labeled peptide abundances were summed and normalized to the sum of all measured abundances of TMPP-labeled peptides. The median of the injection triplicates measurements was calculated for each peptide. Then peptides were classified as transit peptides or degradation peptides. When a mitochondrial protein bears a transit peptide, it is first cleaved by the intermediate peptidase. Then aminopeptidases may act on the new N-terminus and remove a few amino acids (10), leading to the mature form(s) of the protein. Further protein cleavage is indicative of protein degradation. Transit peptides were therefore defined as the most upstream position experimentally observed, either from the present study or from the literature (11, 12, 62). Positions up to 10 aa downstream of this most upstream position were also classified as transit peptides, to take into account the aminopeptidase processing. More downstream positions observed were classified as degradation peptides. The following ratio was calculated (Equation 1) for each condition:
| (1) |
where “i” represents a TMPP-labeled peptide, “j” the corresponding protein, and “all internal peptides” refers to all peptides belonging to the protein quantified with the MaxQuant workflow. Finally, only TMPP-labeled peptides that showed no missing value for their corresponding internal peptide abundances and that showed a non-null abundance value in the control condition were retained. This restriction was relevant as we should detect the N-terminus in the control condition.
Statistical analyses were carried out using a Wilcoxon rank-sum test or a Wilcoxon signed-rank test in RStudio (version 1.0.136) to evaluate the influence of rapamycin or zinc treatment on the N-terminal processing.
For the assessment of N-terminal trimming, a ragging index was calculated and defined using Equation 2:
| (2) |
This index was tested using a Wilcoxon signed-rank test for modulation by the treatments. For distal peptides, where null values can be expected, at least in the control samples, the test variable was defined using Equation 3
| (3) |
where Ri,j is defined in Eq. 1. This value was tested by a Wilcoxon signed-rank test for modulation by the rapamycin or zinc treatment.
For the statistical analysis of the distal peptides, where null values can be expected, at least in the control samples, the test variable was defined as (Equation 4):
| (4) |
where Ri,j is defined in Eq. 1.
This value was tested by a Wilcoxon signed-rank test for modulation by the rapamycin or zinc treatment. Complete LC-MS/MS data sets were deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD010220 for shotgun data and PXD010572 for TMPP data (63).
Enzyme Assays
To compensate for different mitochondrial purity and/or yield issues that could have biased the proteomic results, enzyme assays were carried out on complete cell extracts. Briefly, cells were harvested, rinsed twice with PBS and resuspended in a 10-fold pellet volume of lysis buffer (10 mm Hepes-NaOH pH 7.5, 2 mm MgCl2, 1 mm EGTA, 50 mm KCl, 0.1% (w/v) tetradecyldimethylammonio propane sulfonate [SB 3–14]). Cells were lysed by incubating on ice for 20 min with occasional vortexing. The suspension was then centrifuged at 1000 × g for 5 min, the supernatant collected, protein concentration determined by a dye-binding assay (53). Extracts were aliquoted and stored at −80 °C until use.
Citrate synthase (CISY) activity was measured as described in (64), using liberation of coenzyme A and 5–5′-thio-bis-(2nitrobenzoic acid) (DTNB) reduction at 412 nm. Biphenyl hydrolase-like protein (BPHL) activity was measured through the homocysteine thiolactonase activity (65), using DTNB reduction.
Succinate dehydrogenase (SDH) (66), d- and l-hydroxybutyrate dehydrogenase (D2HDH and L2HDH) (67), NAD-dependent isocitrate dehydrogenase (IDH) (68) and pyruvate dehydrogenase (PDH) (69) activities were all measured by the PMS-coupled iodonitrotetrazolium reduction method. Glutaminase (GLSK) activity was measured with a glutamate dehydrogenase-coupled assay (70). All results were converted into nmol substrate converted per min and mg protein, using the extinction coefficient of the final product.
RESULTS
Modulation of the Mitochondrial Proteome by Rapamycin and Zinc
A classical differential analysis using internal peptides was performed to investigate the effects of rapamycin and zinc on the mitochondrial proteome after a 24 h treatment (supplemental Tables S1 and S2). Clustering analysis on all the experimental replicates (Fig. 1) indicated that technical replicates are very closely related, validating our data reduction approach from the technical replicates. At a higher level, it appeared that treatment with rapamycin induced the strongest effects, whereas the effects of the treatment with zinc were less pronounced.
Fig. 1.

Global analysis of the proteomic experiment by hierarchical clustering. The clustering was calculated by the PAST software using a Paired group algorithm (UGPM) and an Euclidean similarity index. Ctrl, Control; Rapa, Rapamycin; Zn, Zinc.
Over 100 mitochondrially annotated proteins were modulated by at least one of the treatments. A selection of these proteins is shown in Table I (the complete list of differentially expressed proteins being given in supplemental Table S3). The expected false discovery rates for these proteins are provided for information in supplemental Table S4.
Table I. Selected mitochondrial proteins after the proteomic shotgun analysis.
| Fasta headers | UniProt ID | Rapa/control | T-test rapa | Zn/control | T-test Zn |
|---|---|---|---|---|---|
| Complex II subunits | |||||
| Succ. Dehydrogenase sub.Ac | P31040 | 1.29 | 0.048 | 1.19 | 0.057 |
| Succ. Dehydrogenase sub.B | P21912 | 1.20 | 0.0047 | 1.132 | 0.19 |
| Complex IIIa subunits | |||||
| Complex III subunit 1 | P31930 | 1.10 | 0.16 | 1.22 | 0.015 |
| Complex III subunit 2 | P22695 | 1.08 | 0.17 | 1.23 | 0.012 |
| Complex III subunit 6 | P07919 | 1.05 | 0.98 | 1.08 | 0.88 |
| Complex III subunit 7 | P14927 | 1.02 | 0.88 | 1.18 | 0.54 |
| Complex III subunit 8 | O14949 | 0.91 | 0.65 | 1.04 | 0.93 |
| Complex III subunit 9 | Q9UDW1 | 0.87 | 0.37 | 0.78 | 0.21 |
| Complex V subunits | |||||
| ATP synthase subunit alpha | P25705 | 1.01 | 0.88 | 1.12 | 0.34 |
| ATP synthase subunit beta | P06576 | 1.93 | 0.050 | 1.61 | 0.13 |
| ATP synthase subunit gamma | P36542 | 0.94 | 0.84 | 1.03 | 0.82 |
| ATP synthase subunit delta | P30049 | 2.25 | 0.0050 | 1.63 | 0.022 |
| ATP synthase subunit d | O75947 | 1.10 | 0.49 | 1.18 | 0.36 |
| ATP synthase subunit e | P56385 | 0.828 | 0.29 | 0.76 | 0.29 |
| ATP synthase subunit f | P56134 | 0.90 | 0.51 | 1.14 | 0.50 |
| ATP synthase subunit g | O75964 | 0.87 | 0.36 | 0.99 | 0.90 |
| ATP synthase subunit O | P48047 | 0.96 | 0.85 | 1.14 | 0.59 |
| ATP synthase subunit s | Q99766 | 1.01 | 0.99 | 1.03 | 0.98 |
| ATP synthase subunit 8 | P03928 | 1.14 | 0.47 | 1.06 | 0.75 |
| ATP synthase coupling fact. 6 | P18859 | 1.78 | 0.14 | 1.82 | 0.022 |
| ATP synth. assembly fact. 2 | Q8N5M1 | 0.53 | 0.026 | 0.44 | 0.14 |
| Enzymesb | |||||
| Valacyclovir hydrolase | Q86WA6 | 0.47 | 0.0033 | 0.60 | 0.028 |
| d-2 hydroxyglutarate DH | Q8N465 | 0.51 | 0.029 | 1.01 | 0.89 |
| l-2 hydroxyglutarate DH | Q9H9P8 | 0.82 | 0.040 | 0.95 | 0.62 |
| Pyruvate DH phosphatase 1 | Q9P0J1 | 0.73 | 0.029 | 0.70 | 0.079 |
| Pyruvate DH phosphatase reg | Q8NCN5 | 0.72 | 0.031 | 0.64 | 0.061 |
| Pyruvate DH kinase 1 | Q15118 | 1.43 | 0.048 | 1.28 | 0.35 |
| Pyruvate DH kinase 3 | Q15120 | 1.07 | 0.38 | 1.38 | 0.0061 |
| Isocitrate DH (NAD) sub. beta | O43837 | 0.76 | 0.059 | 0.82 | 0.049 |
| Isocitrate DH (NAD) sub. alpha | P50213 | 0.97 | 0.91 | 0.85 | 0.49 |
| Isocitrate DH sub. gamma | P51553 | 0.72 | 0.034 | 0.86 | 0.21 |
| Glutaminase | O94925 | 0.48 | 0.0038 | 0.81 | 0.096 |
| Citrate synthase | O75390 | 0.72 | 0.09 | 0.70 | 0.017 |
| Proteases | |||||
| Lon protease homolog | P36776 | 0.71 | 0.18 | 0.56 | 0.051 |
| Clp Protease catal. subunit | Q16740 | 0.51 | 0.04 | 0.61 | 0.031 |
| Mito. Intermediate peptidasec | Q99797 | 0.553 | 0.0017 | 0.65 | 0.011 |
| Mito. processing protease betac | O75439 | 0.64 | 0.079 | 0.69 | 0.045 |
| Mito. processing protease alpha | Q10713 | 0.80 | 0.563 | 0.85 | 0.691 |
a complex III: ubiquinol-cytochrome c reductase complex.
b DH = dehydrogenase.
c normalized peptide intensities for these proteins are provided in Supplementary Table V.
Rapa, Rapamycin; Zn, Zinc.
To gain more detailed insights into the physiological translation of the modulations found through this global proteomic screen, enzyme activity assays were performed. To alleviate possible artifacts because of variations in mitochondrial preparations purity, the enzymatic assays were carried out on total cell extracts. This prevented us from testing enzymes that have multiple locations (e.g. acyl coenzyme A ligases, nucleotide-modifying enzymes) or activities for which cytosolic and mitochondrial isoforms coexist (e.g. NADP-dependent isocitrate dehydrogenase or adenylate kinase). Although this severely limited the number of enzymes that could be tested, clearcut results were obtained for glutaminase, d- and l-hydroxybutyrate dehydrogenase, NAD-dependent isocitrate dehydrogenase, succinate dehydrogenase, citrate synthase and BPHL as summarized in Table II. Good agreement between the enzymatic assays and the proteomic results was observed for citrate synthase (CISY), succinate dehydrogenase (SDHA and SDHB) and l-hydroxybutyrate dehydrogenase (L2HDH), moderate agreement was observed for d-hydroxybutyrate dehydrogenase (D2HDH) and glutaminase (GLSK), and poor agreement was seen for BPHL and NAD-dependent isocitrate dehydrogenase (IDH). In the case oh IDH, it should be noted that the amount of only one subunit (IDH3G) appeared to be modulated. As the complete active enzyme is a tetramer (2xIDH3A, IDH3B and IDH3G) (71), it is unsurprising that the enzymatic activity does not follow the trend given by only one of the subunits.
Table II. Mitochondrial enzyme activities.
| Enzyme | Control | Rapamycin | Zinc | Ratio rapa/ctrl | Ratio zinc/ctrl | Shotgun ratio rapa/ctrl | Shotgun ratio zinc/ctrl |
|---|---|---|---|---|---|---|---|
| Glutaminase | 196 ± 35 | 152 ± 16 | 121 ± 8 | 0.77 | 0.62* | 0.48 | 0.81 |
| l-hydroxyglutarate dehydrogenase | 164 ± 30 | 122 ± 10 | 114 ± 22 | 0.74 | 0.70* | 0.82 | 0.95 |
| d-hydroxyglutarate dehydrogenase | 137 ± 23 | 109 ± 11 | 102 ± 13 | 0.80 | 0.75 | 0.51 | 1.01 |
| NAD-isocitrate dehydrogenase | 276 ± 11 | 270 ± 27 | 219 ± 40 | 0.98 | 0.79 | ND | ND |
| Succinate dehydrogenase | 82 ± 6 | 114 ± 8 | 112 ± 8 | 1.40** | 1.37** | >1.20 | >1.13 |
| BPHL (Valacyclovir hydrolase) | 842 ± 81 | 677 ± 72 | 790 ± 56 | 0.80* | 0.94 | 0.47 | 0.60 |
| Citrate synthase | 900 ± 97 | 573 ± 69 | 614 ± 72 | 0.64** | 0.68** | 0.72 | 0.70 |
| Pyruvate dehydrogenase | 188 ± 20 | 155 ± 14 | 141 ± 21 | 0.83* | 0.75* | NR | NR |
All the activities are expressed in nmole substrate converted/min/mg total protein (mean ± standard deviation). The experiments were carried out on total cell extracts (n = 3, biological replicates). Statistical significance of the results in the heteroscedastic Student T test: * p < 0.05; ** p < 0.01.
ND: not determined, as the enzyme is composed of several subunits.
NR: not relevant, as the pyruvate dehydrogenase activity is controlled by phosphorylation.
ctrl, control; rapa, rapamycin.
In addition to these enzymes, we also tested the pyruvate dehydrogenase (PDH) activity. The PDH subunits did not appear in the list of modulated proteins, but PDH activity is regulated by phosphorylation (72) and both the kinase and the phosphatase subunits appeared in the list of modulated proteins, with opposite trends. These changes in the PDH phosphatases (PDP1 and PDPR) and PDH kinases (PDK1 and PDK3) amounts suggest a decrease in the PDH activity upon treatment by rapamycin or zinc, which was confirmed by the enzymatic assay (Table II).
Altogether, these results pointed to a decrease in the mitochondrial metabolism upon treatment by zinc or rapamycin. To investigate possible consequences of such a decrease on the mitochondrial function, we checked the mitochondrial transmembrane potential via a fluorescent dye accumulation assay. The fluorescence values (mean ± standard deviation, normalized fluorescence units on independent biological triplicates) were 100 ± 4 in the control samples, 98 ± 9 in the rapamycin-treated samples, and 96 ± 6 in the zinc-treated samples. None of these changes were statistically significant. As the fluorescence intensity depends on both the intrinsic activity of each mitochondrion and on the number of mitochondria, it can be reasonably inferred that neither treatment induced a major change in the number of mitochondria.
Analysis of shotgun proteomic data indicated that both treatments induced alterations of the mitochondrial proteome, but no gross cellular dysfunction nor massive cell death was observed. Thus, the dynamics of the mitochondrial protein processing system under moderate stress could be assessed.
Indeed, two of the proteins identified as strongly decreased was the mitochondrial intermediate peptidase (MIPEP) and the beta (catalytic) subunit of the mitochondrial processing protease (MPPB), pointing to possible defects in the processing of mitochondrial proteins upon treatment with rapamycin or zinc. To investigate such effects, the shotgun proteomic study was combined with a N-terminomics investigation using the dN-TOP approach (11), which allows simultaneous and parallel analysis of internal and N-terminal peptides (supplemental Table S8). We sorted the N-terminal peptides according to their positions as described in the methods section. Upstream peptides were classified as possible transit/signal peptides (supplemental Table S7). More downstream peptides were classified as peptides indicative of protein degradation (supplemental Table S8).
First and foremost, new, alternate N-termini were not detected following treatment with rapamycin or zinc. Despite the decrease in MIPEP and MPPB, the N-termini detected in the rapamycin- or zinc-treated samples were always like those detected in the control samples, indicating that the mitochondrial processing system is robust, at least under those conditions. Besides MIPEP, the processing system also includes aminopeptidases, which sometimes results into ragged ends. Evaluation of the quantitative ratio between the various N-terminal positions by a Wilcoxon signed-rank test indicated that the trimming process was not significantly altered by the rapamycin treatment, although only twenty-two proteins with ragged ends at positions compatible with a transit peptide (i.e. between positions 10 and 100) could be analyzed. Zinc, however, caused a small but consistent and statistically significant decrease in N-terminal trimming (Fig. 2A–2B).
Fig. 2.

Wilcoxon signed-rank test results (A to F) and Wilcoxon rank-sum test results (G, H). Box plots represent the ranking of the compared groups, while evaluating the influence of the treatments on the ragging process (A, rapamycin treatment; B, zinc treatment), on the mitochondrial processing system (C, rapamycin treatment; D, zinc treatment), on mitochondrial protein degradation (E, rapamycin treatment; F, zinc treatment) and on protein processing systems (G, rapamycin treatment; H, zinc treatment). Ratio values are plotted on the y-axes. For each group, dots represent the evaluated ratio value for N-terminal peptides. Peptides of proteins contained in Table I, if present in the compared groups, are highlighted using colors. Tests are performed at a predefined significance level of p = 0.05.
We then used the quantitative data to check whether treatment with rapamycin or zinc induced a quantitative alteration in the N-terminal processing. As the mitochondrial stressors used in this study did induce changes in the mitochondrial proteome as shown by the shotgun proteomics part of the study, the intensity of the N-terminal peptides signals alone were not enough to study the changes occurring in the processing system, and we had to compensate for the changes brought to protein amounts. To this purpose, we calculated for each protein and each point the ratio between the intensity of the TMPP-labeled peptide and the sum of all identified internal peptides belonging to the target protein. This ratio is an index of the properly processed protein form.
So for each protein, if there is only a change in the protein amount without any difference in the efficiency of the N-terminal processing, the signals for the N-terminal peptides and for the internal peptides should change in parallel, leading to a theoretically constant N-terminal/internal peptides ratio.
If there is an altered N-terminal processing, this may result into minor N-terminal peptides different from the canonical one, which may go undetected if they are too minor in abundance. This would however result in a decrease of the signal of the canonical peptide, and thus in a decrease of the TMPP/internal peptides ratio. To detect a general trend in the accurate processing of mitochondrial proteins without being biased by quantitative outliers, we performed a rank statistical analysis of this ratio by a Wilcoxon signed-rank test. This test revealed that rapamycin significantly affected the mitochondrial processing system (p < 0.00001), whereas zinc did not (p = 0.29) (Fig. 2C–2D). The same analysis performed on distal peptides, indicative of protein degradation, revealed that the TMPP/internal peptides ratios were significantly higher for distal peptides (p < 0.01 for both rapamycin and zinc treatments), showing that these treatments promote protein degradation (Fig. 2E–2F).
In these statistical tests, the test variable is compared with a random distribution to determine whether it differs from random or not. Such analyses cannot compare the measured effects of treatment with other induced cellular effects. To address this question, we used the fact that both mitochondrial and nonmitochondrial proteins are present in our analyses and performed a Wilcoxon rank-sum test using all proteins (mitochondrial and nonmitochondrial) with a labeled N-terminal peptide at a position between 1 and 100. The TMPP/internal peptides ratios were significantly lower for mitochondrial proteins compared with nonmitochondrial ones following rapamycin treatment (p < 0.001), showing at least that the mitochondrial processing system is more sensitive to rapamycin than other cellular processing systems (e.g. the signal peptidase). Zinc treatment caused a much less dramatic effect (p ≈ 0.08) (Fig. 2G–2H).
DISCUSSION
Most current N-terminomics approaches focus on the analysis of terminal peptides (10, 73, 74), whereas the dN-TOP approach singles out by its ability to analyze within a single run both the N-terminal and internal peptides, at the expense of a requirement for free N-termini. This makes the method suitable for degradome and processome studies. In the present study, it enabled investigation of the changes in the N-terminal processing system following various treatments. Neither zinc nor rapamycin exposure caused massive, detectable alterations of the mitochondrial proteins, such as new N-terminal positions, which is to be expected given the critical role of mitochondria in eukaryotic life. The mitochondrial transmembrane potential, and hence, mitochondrial function, were not affected by the treatments. From the results of fluorescence and shotgun analyses, we can conclude that the intrinsic activity of each mitochondrion, the number of mitochondria and the protein-processing system were unaffected by the treatments. However, the efficiency of protein processing may be altered in such sublethal stress, leading to minor errors which were not directly detected in the proteomic analysis.
These minor alterations can be globally investigated using the multiplicity of the N-terminomics data combined with statistical analyses. If the applied stress causes no biological effects on the protein-processing system, observed variations should follow a random distribution. Our statistical analysis of the quantitative data showed that this was not the case, suggesting an overall reduction in the efficiency of the processing system.
Interestingly, the N-terminal trimming process was slightly but significantly inhibited by zinc. The three mitochondrial aminopeptidases known to date (MAP12, MAP2, and XPP3) are all metalloproteases containing respectively cobalt for MAP12 (75) and manganese for MAP2 and XPP3 (76, 77) as metal cofactors, and are thus very likely to be directly inhibited by excess zinc, as previously described for other metalloproteases (78, 79). This mechanism also probably applies to the mitochondrial processing protease, which is known to be a zinc metalloprotease (80), and has been described to be inhibited by excess zinc (81).
A slight but significant change was also detected when distal N-termini were analyzed. In this case, an increase of the distal N-termini was detected after exposure to either zinc or rapamycin. However, the detection of cleaved proteins forms indicate that they were stable enough to accumulate and were immediately destroyed by the intramitochondrial proteases. Thus, our results can be correlated with the observed decrease in the amounts of the ClpP and Lon proteases upon zinc and rapamycin treatments (Table I). Interestingly, several mitochondrial metalloproteases classified as quality control proteases (13) were detected in our proteomic screen but did not exhibit significant quantitative changes upon zinc treatment, as shown in supplemental Table S3 for the PREP, OMA1, SPG7, and AFG32 proteases. However, the zinc inhibition mechanism described above may apply to these proteases too and explain the increase in distal termini observed upon zinc treatment.
Our Data Highlighted New Players in the Mitochondrial Response to Zinc or Rapamycin Exposure
Regarding zinc, citrate synthase appears as an important enzyme, in addition to ketoglutarate dehydrogenase (38). Citrate synthase active site contains two histidines (82, 83), which are known to bind zinc with a high affinity (84) and lead sometimes to protein activation, as described for example for kallikrein 5 (85). This hypothesis is further substantiated by the binding of citrate synthase to zinc (86). This may explain the observed decreased activity, and, through a zinc-induced destabilization mechanism, the observed decrease in amount of this protein.
The observed concomitant decrease in pyruvate dehydrogenase activity may explain the protective roles of pyruvate and oxaloacetate (41). Increased concentrations of these two substrates of the citrate synthase and pyruvate dehydrogenase may overcome the moderate decrease in the enzymes' activities, restoring normal mitochondrial function (41).
In the same trend, the decrease in the two hydroxyglutarate dehydrogenase activities upon zinc treatment is also of interest, as these enzymes appear to be “metabolic repair” enzymes which degrade byproducts of isocitrate dehydrogenase (d-hydroxyglutarate (87)) and malate dehydrogenase (l-hydroxyglutarate (88)) activities. The observed decrease in these activities following zinc exposure may contribute to the toxicity of zinc toward mitochondria.
The effects of rapamycin on mitochondria were expected to be indirect, and our results support previously published data (47, 49). Decrease in mitochondrial metabolism was indicated by the decrease in the activity of the citrate cycle (revealed by the decreased activities of citrate synthase and pyruvate dehydrogenase). However, in contrast to results previously published on T lymphocytes (49), the activity changes that we observed in our macrophage system did not result into any gross alterations of the mitochondria such as a decreased transmembrane potential. Our study demonstrates that proteomics can detect moderate changes in cell and organelle systems before large physiological consequences occur.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD010220 for shotgun data and PXD010572 for TMPP data.
Supplementary Material
Footnotes
* This work was financially supported by the “Agence Nationale de la Recherche” (ANR eNergiome, ANR-13-BSV6-0004) and the French Proteomic Infrastructure (ProFI; ANR-10-INBS-08-03).
 This article contains supplemental Tables.
This article contains supplemental Tables.
1 The abbreviations used are:
- MSDA
- mass spectrometry data analysis
- FDR
- false discovery rate
- IDH
- isocitrate dehydrogenase
- PDH
- pyruvate dehydrogenase
- MAP
- mitochondrial aminopeptidase.
REFERENCES
- 1. Rabilloud T., Kieffer S., Procaccio V., Louwagie M., Courchesne P. L., Patterson S. D., Martinez P., Garin J., and Lunardi J. (1998) Two-dimensional electrophoresis of human placental mitochondria and protein identification by mass spectrometry: Toward a human mitochondrial proteome. Electrophoresis 19, 1006–1014 [DOI] [PubMed] [Google Scholar]
- 2. Chen X. L., Li J., Hou J. J., Xie Z. S., and Yang F. Q. (2010) Mammalian mitochondrial proteomics: insights into mitochondrial functions and mitochondria-related diseases. Expert Rev. Proteomics 7, 333–345 [DOI] [PubMed] [Google Scholar]
- 3. Lescuyer P., Strub J. M., Luche S., Diemer H., Martinez P., Van Dorsselaer A., Lunardi J., and Rabilloud T. (2003) Progress in the definition of a reference human mitochondrial proteome. Proteomics 3, 157–167 [DOI] [PubMed] [Google Scholar]
- 4. Gaucher S. P., Taylor S. W., Fahy E., Zhang B., Warnock D. E., Ghosh S. S., and Gibson B. W. (2004) Expanded coverage of the human heart mitochondrial proteome using multidimensional liquid chromatography coupled with tandem mass spectrometry. J. Proteome Res. 3, 495–505 [DOI] [PubMed] [Google Scholar]
- 5. Pagliarini D. J., Calvo S. E., Chang B., Sheth S. A., Vafai S. B., Ong S. E., Walford G. A., Sugiana C., Boneh A., Chen W. K., Hill D. E., Vidal M., Evans J. G., Thorburn D. R., Carr S. A., and Mootha V. K. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Taylor S. W., Fahy E., Zhang B., Glenn G. M., Warnock D. E., Wiley S., Murphy A. N., Gaucher S. P., Capaldi R. A., Gibson B. W., and Ghosh S. S. (2003) Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 21, 281–286 [DOI] [PubMed] [Google Scholar]
- 7. Schatz G. (1996) The protein import system of mitochondria. J. Biol. Chem. 271, 31763–31766 [DOI] [PubMed] [Google Scholar]
- 8. Schmidt O., Pfanner N., and Meisinger C. (2010) Mitochondrial protein import: from proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 11, 655–667 [DOI] [PubMed] [Google Scholar]
- 9. Wiedemann N., and Pfanner N. (2017) Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 86, 685–714 [DOI] [PubMed] [Google Scholar]
- 10. Vogtle F. N., Wortelkamp S., Zahedi R. P., Becker D., Leidhold C., Gevaert K., Kellermann J., Voos W., Sickmann A., Pfanner N., and Meisinger C. (2009) Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell 139, 428–439 [DOI] [PubMed] [Google Scholar]
- 11. Vaca Jacome A. S., Rabilloud T., Schaeffer-Reiss C., Rompais M., Ayoub D., Lane L., Bairoch A., Van Dorsselaer A., and Carapito C. (2015) N-terminome analysis of the human mitochondrial proteome. Proteomics 15, 2519–2524 [DOI] [PubMed] [Google Scholar]
- 12. Calvo S. E., Julien O., Clauser K. R., Shen H., Kamer K. J., Wells J. A., and Mootha V. K. (2017) Comparative analysis of mitochondrial N-termini from mouse, human, and yeast. Mol. Cell. Proteomics 16, 512–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lebeau J., Rainbolt T. K., and Wiseman R. L. (2018) Coordinating mitochondrial biology through the stress-responsive regulation of mitochondrial proteases. Int. Rev. Cell Mol. Biol. 340, 79–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chevallet M., Lescuyer P., Diemer H., van Dorsselaer A., Leize-Wagner E., and Rabilloud T. (2006) Alterations of the mitochondrial proteome caused by the absence of mitochondrial DNA: A proteomic view. Electrophoresis 27, 1574–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tun A. W., Chaiyarit S., Kaewsutthi S., Katanyoo W., Chuenkongkaew W., Kuwano M., Tomonaga T., Peerapittayamongkol C., Thongboonkerd V., and Lertrit P. (2014) Profiling the mitochondrial proteome of Leber's hereditary optic neuropathy (LHON) in Thailand: down-regulation of bioenergetics and mitochondrial protein quality control pathways in fibroblasts with the 11778G > a mutation. Plos One 9, e106779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stauch K. L., Purnell P. R., Villeneuve L. M., and Fox H. S. (2015) Proteomic analysis and functional characterization of mouse brain mitochondria during aging reveal alterations in energy metabolism. Proteomics 15, 1574–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chang J., Cornell J. E., Van Remmen H., Hakala K., Ward W. F., and Richardson A. (2007) Effect of aging and caloric restriction on the mitochondrial proteome. J. Gerontol. 62, 223–234 [DOI] [PubMed] [Google Scholar]
- 18. Musicco C., Capelli V., Pesce V., Timperio A. M., Calvani M., Mosconi L., Cantatore P., and Gadaleta M. N. (2011) Rat liver mitochondrial proteome: Changes associated with aging and acetyl-L-carnitine treatment. J. Proteomics 74, 2536–2547 [DOI] [PubMed] [Google Scholar]
- 19. Barjaktarovic Z., Schmaltz D., Shyla A., Azimzadeh O., Schulz S., Haagen J., Dorr W., Sarioglu H., Schafer A., Atkinson M. J., Zischka H., and Tapio S. (2011) Radiation-induced signaling results in mitochondrial impairment in mouse heart at 4 weeks after exposure to X-rays. Plos One 6, e27811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang S. R., Fu J. L., and Zhou Z. C. (2005) Changes in the brain mitochondrial proteome of male Sprague-Dawley rats treated with manganese chloride. Toxicol. Appl. Pharmacol. 202, 13–17 [DOI] [PubMed] [Google Scholar]
- 21. Strong R., Nakanishi T., Ross D., and Fenselau C. (2006) Alterations in the mitochondrial Proteome of adriamycin resistant MCF-7 breast cancer cells. J. Proteome Res. 5, 2389–2395 [DOI] [PubMed] [Google Scholar]
- 22. Oshima R., Nakano H., Katayama M., Sakurai J., Wu W., Koizumi S., Asano T., Watanabe T., Asakura T., Ohta T., and Otsubo T. (2008) Modification of the hepatic mitochondrial proteome in response to ischemic preconditioning following ischemia-reperfusion injury of the rat liver. Eur. Surgical Res. 40, 247–255 [DOI] [PubMed] [Google Scholar]
- 23. Shi L., Wang Y., Tu S. Y., Li X. L., Sun M. M., Srivastava S., Xu N. Z., Bhatnagar A., and Liu S. Q. (2008) The responses of mitochondrial proteome in rat liver to the consumption of moderate ethanol: The possible roles of aldo-keto reductases. J. Proteome Res. 7, 3137–3145 [DOI] [PubMed] [Google Scholar]
- 24. Eccleston H. B., Andringa K. K., Betancourt A. M., King A. L., Mantena S. K., Swain T. M., Tinsley H. N., Nolte R. N., Nagy T. R., Abrams G. A., and Bailey S. M. (2011) Chronic exposure to a high-fat diet induces hepatic steatosis, impairs nitric oxide bioavailability, and modifies the mitochondrial proteome in mice. Antioxidants Redox Signaling 15, 447–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen X. L., Cui Z. Y., Wei S. S., Hou J. J., Xie Z. S., Peng X., Li J., Cai T. X., Hang H. Y., and Yang F. Q. (2013) Chronic high glucose induced INS-1 beta cell mitochondrial dysfunction: A comparative mitochondrial proteome with SILAC. Proteomics 13, 3030–3039 [DOI] [PubMed] [Google Scholar]
- 26. Burte F., De Girolamo L. A., Hargreaves A. J., and Billett E. E. (2011) Alterations in the mitochondrial proteome of neuroblastoma cells in response to complex 1 inhibition. J. Proteome Res. 10, 1974–1986 [DOI] [PubMed] [Google Scholar]
- 27. Pienaar I. S., Schallert T., Hattingh S., and Daniels W. M. U. (2009) Behavioral and quantitative mitochondrial proteome analyses of the effects of simvastatin: implications for models of neural degeneration. J. Neural Transmission 116, 791–806 [DOI] [PubMed] [Google Scholar]
- 28. Lee Y. H., Chung M. C. M., Lin Q. S., and Boelsterli U. A. (2008) Troglitazone-induced hepatic mitochondrial proteome expression dynamics in heterozygous Sod2(+/-) mice: Two-stage oxidative injury. Toxicol. Appl. Pharmacol. 231, 43–51 [DOI] [PubMed] [Google Scholar]
- 29. Hwang H. J., Dornbos P., Steidemann M., Dunivin T. K., Rizzo M., and LaPres J. J. (2016) Mitochondrial-targeted aryl hydrocarbon receptor and the impact of 2,3,7,8-tetrachlorodibenzo-p-dioxin on cellular respiration and the mitochondrial proteome. Toxicol. Appl. Pharmacol. 304, 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brieger A., Rink L., and Haase H. (2013) Differential regulation of TLR-dependent MyD88 and TRIF signaling pathways by free zinc ions. J. Immunol. 191, 1808–1817 [DOI] [PubMed] [Google Scholar]
- 31. Haase H., and Rink L. (2014) Multiple impacts of zinc on immune function. Metallomics 6, 1175–1180 [DOI] [PubMed] [Google Scholar]
- 32. Drinker P., Thomson R. M., and Finn J. L. (1927) Metal Fume Fever: IV. Threshold doses of zinc oxide, preventive measures, and the chronic effects of repeated exposures. J. Industrial Hygiene 9, 331–345 [Google Scholar]
- 33. Fine J. M., Gordon T., Chen L. C., Kinney P., Falcone G., and Beckett W. S. (1997) Metal fume fever: characterization of clinical and plasma IL-6 responses in controlled human exposures to zinc oxide fume at and below the threshold limit value. J. Occup. Environ. Med. 39, 722–726 [DOI] [PubMed] [Google Scholar]
- 34. Chen C. J., and Liao S. L. (2003) Zinc toxicity on neonatal cortical neurons: involvement of glutathione chelation. J. Neurochem. 85, 443–453 [DOI] [PubMed] [Google Scholar]
- 35. Gazaryan I. G., Krasnikov B. F., Ashby G. A., Thorneley R. N., Kristal B. S., and Brown A. M. (2002) Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. J. Biol. Chem. 277, 10064–10072 [DOI] [PubMed] [Google Scholar]
- 36. Krotkiewska B., and Banas T. (1992) Interaction of Zn2+ and Cu2+ ions with glyceraldehyde-3-phosphate dehydrogenase from bovine heart and rabbit muscle. Int. J. Biochem. 24, 1501–1505 [DOI] [PubMed] [Google Scholar]
- 37. Aude-Garcia C., Dalzon B., Ravanat J. L., Collin-Faure V., Diemer H., Strub J. M., Cianferani S., Van Dorsselaer A., Carriere M., and Rabilloud T. (2016) A combined proteomic and targeted analysis unravels new toxic mechanisms for zinc oxide nanoparticles in macrophages. J. Proteomics 134, 174–185 [DOI] [PubMed] [Google Scholar]
- 38. Brown A. M., Kristal B. S., Effron M. S., Shestopalov A. I., Ullucci P. A., Sheu K. F., Blass J. P., and Cooper A. J. (2000) Zn2+ inhibits alpha-ketoglutarate-stimulated mitochondrial respiration and the isolated alpha-ketoglutarate dehydrogenase complex. J. Biol. Chem. 275, 13441–13447 [DOI] [PubMed] [Google Scholar]
- 39. Dineley K. E., Votyakova T. V., and Reynolds I. J. (2003) Zinc inhibition of cellular energy production: implications for mitochondria and neurodegeneration. J. Neurochem. 85, 563–570 [DOI] [PubMed] [Google Scholar]
- 40. Kim H. J., Kim J. M., Park J. H., Sung J. J., Kim M., and Lee K. W. (2005) Pyruvate protects motor neurons expressing mutant superoxide dismutase I against copper toxicity. Neuroreport 16, 585–589 [DOI] [PubMed] [Google Scholar]
- 41. Berry E. V., and Toms N. J. (2006) Pyruvate and oxaloacetate limit zinc-induced oxidative HT-22 neuronal cell injury. Neurotoxicology 27, 1043–1051 [DOI] [PubMed] [Google Scholar]
- 42. Wang X., Perez E., Liu R., Yan L. J., Mallet R. T., and Yang S. H. (2007) Pyruvate protects mitochondria from oxidative stress in human neuroblastoma SK-N-SH cells. Brain Res. 1132, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sheline C. T., Behrens M. M., and Choi D. W. (2000) Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J. Neurosci. 20, 3139–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kawahara M., Kato-Negishi M., and Kuroda Y. (2002) Pyruvate blocks zinc-induced neurotoxicity in immortalized hypothalamic neurons. Cell. Mol. Neurobiol. 22, 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yoo M. H., Lee J. Y., Lee S. E., Koh J. Y., and Yoon Y. H. (2004) Protection by pyruvate of rat retinal cells against zinc toxicity in vitro, and pressure-induced ischemia in vivo. Investigative Ophthalmol. Visual Sci. 45, 1523–1530 [DOI] [PubMed] [Google Scholar]
- 46. Singh K., Sun S., and Vezina C. (1979) Rapamycin (AY-22,989), a new antifungal antibiotic. IV. Mechanism of action. J. Antibiot. 32, 630–645 [DOI] [PubMed] [Google Scholar]
- 47. Simon N., Morin C., Urien S., Tillement J. P., and Bruguerolle B. (2003) Tacrolimus and sirolimus decrease oxidative phosphorylation of isolated rat kidney mitochondria. Br. J. Pharmacol. 138, 369–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Paglin S., Lee N. Y., Nakar C., Fitzgerald M., Plotkin J., Deuel B., Hackett N., McMahill M., Sphicas E., Lampen N., and Yahalom J. (2005) Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 65, 11061–11070 [DOI] [PubMed] [Google Scholar]
- 49. Schieke S. M., Phillips D., McCoy J. P. Jr, Aponte A. M., Shen R. F., Balaban R. S., and Finkel T. (2006) The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J. Biol. Chem. 281, 27643–27652 [DOI] [PubMed] [Google Scholar]
- 50. Bertaccini D., Vaca S., Carapito C., Arsene-Ploetze F., Van Dorsselaer A., and Schaeffer-Reiss C. (2013) An improved stable isotope N-terminal labeling approach with light/heavy TMPP to automate proteogenomics data validation: dN-TOP. J. Proteome Res. 12, 3063–3070 [DOI] [PubMed] [Google Scholar]
- 51. Perry S. W., Norman J. P., Barbieri J., Brown E. B., and Gelbard H. A. (2011) Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. BioTechniques 50, 98–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carapito C., Kuhn L., Karim L., Rompais M., Rabilloud T., Schwenzer H., and Sissler M. (2017) Two proteomic methodologies for defining N-termini of mature human mitochondrial aminoacyl-tRNA synthetases. Methods 113, 111–119 [DOI] [PubMed] [Google Scholar]
- 53. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 54. Tyanova S., Temu T., and Cox J. (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 [DOI] [PubMed] [Google Scholar]
- 55. Carapito C., Burel A., Guterl P., Walter A., Varrier F., Bertile F., and Van Dorsselaer A. (2014) MSDA, a proteomics software suite for in-depth Mass Spectrometry Data Analysis using grid computing. Proteomics 14, 1014–1019 [DOI] [PubMed] [Google Scholar]
- 56. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yekutieli D., and Benjamini Y. (1999) Resampling-based false discovery rate controlling multiple test procedures for correlated test statistics. J. Statistical Planning Inference 82, 171–196 [Google Scholar]
- 58. Diz A. P., Carvajal-Rodriguez A., and Skibinski D. O. (2011) Multiple hypothesis testing in proteomics: a strategy for experimental work. Mol. Cell. Proteomics 10, M110 004374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Carvajal-Rodriguez A., and de Una-Alvarez J. (2011) Assessing significance in high-throughput experiments by sequential goodness of fit and q-value estimation. PLoS ONE 6, e24700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hammer O., Harper D. A. T., and Ryan P. D. (2001) Paleontological statistics software package for education and data analysis. Palaeontol. Electronica 4, 9 p [Google Scholar]
- 61. Carapito C., Lane L., Benama M., Opsomer A., Mouton-Barbosa E., Garrigues L., Gonzalez de Peredo A., Burel A., Bruley C., Gateau A., Bouyssie D., Jaquinod M., Cianferani S., Burlet-Schiltz O., Van Dorsselaer A., Garin J., and Vandenbrouck Y. (2015) Computational and mass-spectrometry-based workflow for the discovery and validation of missing human proteins: application to chromosomes 2 and 14. J. Proteome Res. 14, 3621–3634 [DOI] [PubMed] [Google Scholar]
- 62. Gevaert K., Goethals M., Martens L., Van Damme J., Staes A., Thomas G. R., and Vandekerckhove J. (2003) Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides. Nat. Biotechnol. 21, 566–569 [DOI] [PubMed] [Google Scholar]
- 63. Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Rios D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolome S., Apweiler R., Omenn G. S., Martens L., Jones A. R., and Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alp P. R., Newsholme E. A., and Zammit V. A. (1976) Activities of citrate synthase and NAD+-linked and NADP+-linked isocitrate dehydrogenase in muscle from vertebrates and invertebrates. Biochem. J. 154, 689–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Marsillach J., Suzuki S. M., Richter R. J., McDonald M. G., Rademacher P. M., MacCoss M. J., Hsieh E. J., Rettie A. E., and Furlong C. E. (2014) Human valacyclovir hydrolase/biphenyl hydrolase-like protein is a highly efficient homocysteine thiolactonase. PLoS ONE 9, e110054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Munujos P., Coll-Canti J., Gonzalez-Sastre F., and Gella F. J. (1993) Assay of succinate dehydrogenase activity by a colorimetric-continuous method using iodonitrotetrazolium chloride as electron acceptor. Anal. Biochem. 212, 506–509 [DOI] [PubMed] [Google Scholar]
- 67. Rzem R., Veiga-da-Cunha M., Noel G., Goffette S., Nassogne M. C., Tabarki B., Scholler C., Marquardt T., Vikkula M., and Van Schaftingen E. (2004) A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. U.S.A. 101, 16849–16854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Niehaus W. G., Richardson S. B., and Wolz R. L. (1996) Slow-binding inhibition of 6-phosphogluconate dehydrogenase by zinc ion. Arch. Biochem. Biophys. 333, 333–337 [DOI] [PubMed] [Google Scholar]
- 69. Schwab M. A., Kolker S., van den Heuvel L. P., Sauer S., Wolf N. I., Rating D., Hoffmann G. F., Smeitink J. A., and Okun J. G. (2005) Optimized spectrophotometric assay for the completely activated pyruvate dehydrogenase complex in fibroblasts. Clin. Chem. 51, 151–160 [DOI] [PubMed] [Google Scholar]
- 70. Kvamme E., Torgner I. A., and Svenneby G. (1985) Glutaminase from mammalian tissues. Methods Enzymol. 113, 241–256 [DOI] [PubMed] [Google Scholar]
- 71. Ma T., Peng Y., Huang W., Liu Y., and Ding J. (2017) The beta and gamma subunits play distinct functional roles in the alpha2betagamma heterotetramer of human NAD-dependent isocitrate dehydrogenase. Sci. Rep. 7, 41882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Denton R. M., McCormack J. G., Rutter G. A., Burnett P., Edgell N. J., Moule S. K., and Diggle T. A. (1996) The hormonal regulation of pyruvate dehydrogenase complex. Adv. Enzyme Regul. 36, 183–198 [DOI] [PubMed] [Google Scholar]
- 73. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., and Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kleifeld O., Doucet A., auf dem Keller U., Prudova A., Schilling O., Kainthan R. K., Starr A. E., Foster L. J., Kizhakkedathu J. N., and Overall C. M. (2010) Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 28, 281–288 [DOI] [PubMed] [Google Scholar]
- 75. Hu X. V., Chen X., Han K. C., Mildvan A. S., and Liu J. O. (2007) Kinetic and mutational studies of the number of interacting divalent cations required by bacterial and human methionine aminopeptidases. Biochemistry 46, 12833–12843 [DOI] [PubMed] [Google Scholar]
- 76. Wang J., Sheppard G. S., Lou P., Kawai M., Park C., Egan D. A., Schneider A., Bouska J., Lesniewski R., and Henkin J. (2003) Physiologically relevant metal cofactor for methionine aminopeptidase-2 is manganese. Biochemistry 42, 5035–5042 [DOI] [PubMed] [Google Scholar]
- 77. Singh R., Jamdar S. N., Goyal V. D., Kumar A., Ghosh B., and Makde R. D. (2017) Structure of the human aminopeptidase XPNPEP3 and comparison of its in vitro activity with Icp55 orthologs: Insights into diverse cellular processes. J. Biol. Chem. 292, 10035–10047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gomez-Ortiz M., Gomis-Ruth F. X., Huber R., and Aviles F. X. (1997) Inhibition of carboxypeptidase A by excess zinc: analysis of the structural determinants by X-ray crystallography. FEBS Lett. 400, 336–340 [DOI] [PubMed] [Google Scholar]
- 79. Hwang S. R., and Hook V. (2008) Zinc regulation of aminopeptidase B involved in neuropeptide production. Febs Lett. 582, 2527–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Taylor A. B., Smith B. S., Kitada S., Kojima K., Miyaura H., Otwinowski Z., Ito A., and Deisenhofer J. (2001) Crystal structures of mitochondrial processing peptidase reveal the mode for specific cleavage of import signal sequences. Structure 9, 615–625 [DOI] [PubMed] [Google Scholar]
- 81. Luciano P., Tokatlidis K., Chambre I., Germanique J. C., and Geli V. (1998) The mitochondrial processing peptidase behaves as a zinc-metallopeptidase. J. Mol. Biol. 280, 193–199 [DOI] [PubMed] [Google Scholar]
- 82. Wiegand G., and Remington S. J. (1986) Citrate synthase: structure, control, and mechanism. Annu. Rev. Biophys. Biophys. Chem. 15, 97–117 [DOI] [PubMed] [Google Scholar]
- 83. Usher K. C., Remington S. J., Martin D. P., and Drueckhammer D. G. (1994) A very short hydrogen bond provides only moderate stabilization of an enzyme-inhibitor complex of citrate synthase. Biochemistry 33, 7753–7759 [DOI] [PubMed] [Google Scholar]
- 84. Maret W. (2013) Inhibitory zinc sites in enzymes. Biometals 26, 197–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Debela M., Goettig P., Magdolen V., Huber R., Schechter N. M., and Bode W. (2007) Structural basis of the zinc inhibition of human tissue kallikrein 5. J. Mol. Biol. 373, 1017–1031 [DOI] [PubMed] [Google Scholar]
- 86. Tan Y. F., O'Toole N., Taylor N. L., and Millar A. H. (2010) Divalent metal ions in plant mitochondria and their role in interactions with proteins and oxidative stress-induced damage to respiratory function. Plant Physiol. 152, 747–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ward P. S., Patel J., Wise D. R., Abdel-Wahab O., Bennett B. D., Coller H. A., Cross J. R., Fantin V. R., Hedvat C. V., Perl A. E., Rabinowitz J. D., Carroll M., Su S. M., Sharp K. A., Levine R. L., and Thompson C. B. (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nadtochiy S. M., Schafer X., Fu D., Nehrke K., Munger J., and Brookes P. S. (2016) Acidic pH Is a Metabolic Switch for 2-Hydroxyglutarate Generation and Signaling. J. Biol. Chem. 291, 20188–20197 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD010220 for shotgun data and PXD010572 for TMPP data.