

Supplemental Digital Content is Available in the Text.
Our work reveals that the platelet-derived growth factor-BB, by inhibiting nociceptive M-type potassium channels, acts as a pain-inducing proinflammatory factor that significantly contributes to inflammatory pain.
Keywords: Inflammatory pain, Nociceptor excitability, Kv7 channels, M-current, PDGF
Abstract
Endogenous inflammatory mediators contribute to the pathogenesis of pain by acting on nociceptors, specialized sensory neurons that detect noxious stimuli. Here, we describe a new factor mediating inflammatory pain. We show that platelet-derived growth factor (PDGF)-BB applied in vitro causes repetitive firing of dissociated nociceptor-like rat dorsal root ganglion neurons and decreased their threshold for action potential generation. Injection of PDGF-BB into the paw produced nocifensive behavior in rats and led to thermal and mechanical pain hypersensitivity. We further detailed the biophysical mechanisms of these PDGF-BB effects and show that PDGF receptor–induced inhibition of nociceptive M-current underlies PDGF-BB–mediated nociceptive hyperexcitability. Moreover, in vivo sequestration of PDGF or inhibition of the PDGF receptor attenuates acute formalin-induced inflammatory pain. Our discovery of a new pain-facilitating proinflammatory mediator, which by inhibiting M-current activates nociceptive neurons and thus contributes to inflammatory pain, improves our understanding of inflammatory pain pathophysiology and may have important clinical implications for pain treatment.
1. Introduction
Tissue injury initiates an innate inflammatory response promoted by the secretion of multiple factors from the injured tissue and from resident and recruited immune cells.9,20,37 The activity of this wide array of inflammatory mediators leads to dramatic alterations in somatosensory function.32,56 Some factors increase the sensitivity of peripheral nociceptive neurons to stimulation (sensitization), whereas others directly cause neuronal activation.10,12,28,32 The identification of specific inflammatory mediators and characterization of their mechanism of action is of utmost importance for the development of selective and effective therapeutic approaches to inflammatory pain. Here, we describe a previously unknown role of a well-known factor, platelet-derived growth factor (PDGF), in activating peripheral nociceptive neurons and in mediating inflammatory pain.
Platelet-derived growth factor comprises a family of homodimeric and heterodimeric growth factors, which exerts their action through tyrosine kinase receptors—PDGF receptors (PDGFR)-α and PDGFR-β.3 It has been demonstrated in vivo that PDGF isoforms, PDGF-AA and PDGF-CC, act mainly through PDGFR-α, whereas platelet-derived growth factor-BB (PDGF-BB) activates PDGFR-β.3,68 Platelet-derived growth factor was initially shown to be essential for the development and migration of fibroblasts, muscle cells, and glial cells, and also functioned as a mitogen.3,21,50,52 Platelet-derived growth factor has also been shown to play a modulatory role in the inflammatory process.3,68 Platelet-derived growth factor was found in peripheral tissues on the first day after injury, whereas PDGFR-β upregulation was shown to occur minutes after injury.5,6,43,53 These data implicate PDGF as one of the factors secreted during inflammation. These facts, together with the data showing that PDGFR are expressed in dorsal root ganglion (DRG) neurons and the sciatic nerve,26 and that PDGF and its receptors have been previously implicated in pain hypersensitivity45,48 led us to hypothesize that PDGF may modulate the activity of peripheral sensory neurons, thus contributing to inflammatory pain.
Using in vivo behavioral approaches, as well as electrophysiological, immunohistochemical, and pharmacological techniques, we show that PDGF-BB–mediated signaling inhibits the KV7/M class of potassium (K+) channels. These channels underlie the low voltage-activating, noninactivating potassium (K+) M-current, or IM.14,15 Kv7/M channels have been established as a major regulator of nociceptive excitability.8,15,24,25 The inhibition of Kv7/M-currents has been shown to produce nociceptive firing, decrease in action potential threshold, and increase in firing gain,8,23,51,58,65 as well as to lead to pain hypersensitivity in behavioral models.39,40 Our findings suggest that PDGF elicits action potential firing in nociceptors and produces sensitization to painful stimuli in vivo, by PI3K-mediated suppression of Kv7/M-currents. Moreover, injection of PDGF is sufficient to induce nocifensive behavior. We show that PDGF-BB–mediated inhibition of IM is a primary mechanism underlying the PDGF-BB–induced increase in nociceptive excitability. Finally, we show that sequestration of PDGF or inhibition of the PDGFR during tissue inflammation reduces inflammatory pain.
2. Materials and methods
2.1. Animals
All procedures were conducted in accordance with the guidelines of the Animal Ethics Committee of the Hebrew University of Jerusalem and the University of Pittsburgh, and were approved by the ethics committees of the Hebrew University and the University of Pittsburgh. Five- to 7-week-old Sprague-Dawley (SD) male rats (150-225 gr) were housed under controlled temperature (23 ± 2°C) and environment, with ad libitum access to food and water, and kept in a 12-hour light/dark cycle.
2.2. Immunohistochemistry procedures
2.2.1. Perfusions and tissue extraction
Naive rats were anesthetized with beuthanasia and perfused with saline (0.9%). The glabrous skin from hind paws was dissected and postfixed in 2% paraformaldehyde (PFA) in phosphate buffered saline (PBS) for 48 hours at 4°C. The rat was then perfused with 4% PFA in PBS. Lumbar DRGs were dissected and postfixed overnight in 4% PFA.
2.2.2. Processing dorsal root ganglion samples
Dorsal root ganglion samples were transferred to 20% and 30% sucrose diluted in 0.1 M PBS for cryoprotection. After equilibrating tissue in embedding matrix for 20 minutes (OCT, TissueTek), specimens were snap frozen in cooled isopentane (−55°C) for 15 seconds and stored at −80°C. Frozen tissue samples were sectioned using a cryostat set at −18°C. Dorsal root ganglion sections (10 µm) were mounted on “super frost + slides” (Fisher) and dried overnight under a hood, at room temperature (RT). They were then stored at −80°C until used. Tissue was rinsed 2 × 5 minutes with PBS. To block nonspecific antibody binding, tissue sections were blocked in 10% normal goat serum (NGS) containing 0.2% Triton X-100 for tissue permeabilization in PBS, for 1 hour at RT. Tissue slides were then incubated with primary antibodies (anti-PDGFR-β, rabbit polyclonal, 1:400, Santa Cruz Biotechnology; anti-CGRP, mouse monoclonal, 1:500, Abcam; anti-NF200, mouse monoclonal, 1:2000, Sigma Aldrich; Isolectin-B4 Alexa Fluor 568 conjugated, 1:1000, Life Technologies) in a buffer containing 2% NGS and 0.2% Triton X-100, in 0.1 M PBS, overnight at 4°C and rinsed 3 × 5 minutes with PBS. They were then incubated with Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 568 goat anti-mouse (1:2000; Life technologies, Carlsbad, CA) secondary antibodies diluted in 2% NGS in PBS in the dark at room temperature for 1 hour. After rinsing 3 × 5 minutes with PBS, they were incubated with 4′,6-diamidino-2-phenylindole (50 ng/mL; Cell Signaling Technologies, Danvers, MA) in PBS for 5 minutes at RT and rinsed 3 × 5 minutes. Finally, sections were allowed to air dry at RT, coverslipped using ProLong Gold antifade mounting media (Invitrogen Molecular Probes, Eugene, OR), and stored at 4°C until imaging. All the experiments were controlled by omission of primary antibody.
Quantification was performed using Nikon NIS-Elements software (Nikon Instruments Inc, Melville, NY). Colocalization of PDGFR-β with the cell markers was determined after applying uniformly set intensity thresholds based on the background signal from negative controls.
2.2.3. Processing of skin samples
Skin samples were transferred to successive baths (5 minutes each) of 70%, 85%, 95%, and 100% ethanol and embedded in paraffin. Paraffin-embedded tissue samples were sectioned using a vibratome, directly mounted onto poly-L-lysine coated slides and stored at RT until used. Before starting the immunohistochemistry procedure, slides were baked at 60°C for a minimum of 2 hours. The tissue was then rehydrated with successive immersions (5 minutes each) in xylenes 100%, xylenes 1:1 ethanol, ethanol 100%, 95%, 85%, 70%, and then running MilliQ water. An antigen retrieval procedure was performed by immersion of the samples in a sodium citrate buffer (10 mM), 0.5% Tween 20, at pH = 6, and heated in a steamer for 25 minutes. Samples were then allowed to cool down in buffer for 30 minutes and transferred to 0.2% TX100 in PBS for 10 minutes. The blocking was performed with 5% NGS in PBS for 1 hour at RT. Primary antibody incubation was done overnight at 4°C in 1% NGS (antibodies: anti-PDGFR-β, rabbit polyclonal, Novus biologicals; anti-CGRP, mouse monoclonal, 1:250, Abcam; anti-NF200, mouse monoclonal, 1:250, Sigma; Isolectin-B4 Alexa Fluor 568 conjugated, 1:1000, Life Technologies).
Tissue samples were imaged using a confocal microscope, Nikon A1.
2.3. Rat lumbar dorsal root ganglion cell culture preparation
Primary DRG neurons were isolated from ∼150 g Sprague-Dawley male rat lumbar DRGs, as previously described.8,12,28 Briefly, lumbar DRG neurons (L1-L6) were removed and placed into Dulbecco's modified eagle medium with 1% penicillin–streptomycin, then digested in 5 mg/mL collagenase, 1 mg/mL Dispase II (Roche), and 0.25% trypsin, followed by addition of 0.25% trypsin inhibitor. Cells were triturated with Pasteur pipettes in the presence of DNase I (250 U), and centrifuged through 15% BSA. The cell pellet was resuspended in 1 mL of Neurobasal media, containing B27 supplement (Invitrogen, Carlsbad, CA), penicillin and streptomycin, 10 mM AraC, 2.5S NGF (100 ng/mL, Promega), and glial cell-derived neurotrophic factor (2 ng/mL). Cells were plated onto poly-D-lysine (100 g/mL) and laminin (1 mg/mL)-coated 35-mm tissue culture dishes. Cells were kept in the incubator at 37°C with 5% carbon dioxide.
2.4. General electrophysiology
Recordings were performed from small (∼25 μm) dissociated rat DRG neurons, up to 48 hours after plating. These neurons have been described in the literature to be nociceptive.17 Indeed, in some of the experiments, 1 µM of capsaicin was added to the bath solution at the end of the experiment and the resulting inward current was examined. All the examined neurons (n = 9) showed capsaicin-induced robust inward current (see Results). Cell diameter was measured online using Nikon Elements AR software (Nikon), from images acquired using a CCD camera (Q-Imaging). Whole-cell membrane currents and voltages were recorded using a nystatin-based perforated patch technique31 in voltage-clamp and fast current-clamp modes, respectively, using a MultiClamp 700B amplifier (Molecular Devices), at RT (24 ± 2°C). Data were sampled at 20 kHz and were low-pass filtered at 500 Hz (−3 dB, 8 pole Bessel filter). Patch pipettes (2-5 MΩ) were pulled from borosilicate glass capillaries (1.5/1.1 mm OD/ID; Sutter Instrument Co, Novato, CA) on a P-1000 puller (Sutter Instrument Co., Novato, CA) and fire-polished (LW Scientific, Lawrenceville, GA). Command voltage and current protocols were generated with a Digidata 1440A A/D interface (Molecular Devices, San Jose, CA). Data were digitized using pCLAMP 10.3 (Molecular Devices). Data averaging and peak detection were performed using Clampfit 10.3 software. Data were fitted and analyzed using Origin 9 (OriginLab) and Matlab.
Nystatin-based pipette solution (290 mOsm) for perforated patch recordings was freshly prepared in the dark every 2 to 3 hours and contained (in mM): 140 KCl, 1.6 MgCl2, 2 EGTA, 10 HEPES, 2.5 Mg-ATP, 0.5 Na-GTP, pH 7.2 adjusted with KOH. Nystatin (Sigma-Aldrich, St. Louis, MO) was dissolved in dimethyl sulfoxide (DMSO) (Sigma-Aldrich) to a 50 mg/mL stock solution, ultrasonicated for 1 minute, and then diluted to a working concentration of 125 μg/mL, in the pipette solution.
The extracellular solution contained (in mM): 145 NaCl, 5 KCl, 1 MgCl2, 10 HEPES, and 10 D-glucose, pH was adjusted to 7.4.
Pipette potential was zeroed before seal formation, and analyzed membrane potential was corrected for a −4.5-mV liquid junction potential. For voltage-clamp recordings, capacitive currents were minimized.
2.4.1. Current-clamp recordings
Only cells with stable resting potential and stable action potential (AP) threshold during a 5-minute application of vehicle (extracellular solution) were analyzed. To rule out possible time-dependent changes in neuronal excitability, we applied vehicle solution for ∼15 minutes. No changes in neuronal excitability were observed during the period of vehicle application (n = 5 neurons). The action potential thresholds and neuronal firing properties were assessed by analyzing the neuronal responses to a series of depolarizing steps (1-2 nA in increments of 0.05 nA each). Action potential threshold was calculated from phase plot (dV/dt) analysis of a single AP with a 10-ms step when plotted vs time or vs membrane voltage, as previously described.28 The voltage of the threshold was measured using “first local minimum” of the function before the peak. The “first local minimum” was determined as the first minimal value of dV/dt after the peak, followed by an additional increase in dV/dt, while analyzing the function from its positive peak to time “0.” The time for the first local minimum was defined as the time of threshold, and its voltage was then detected from the original trace. The properties of a single AP were compared with the same above rheobase AP, after the treatment. Input resistance (Rin) was calculated by analyzing the peak change in membrane voltage for 500 ms hyperpolarizing steps of −0.1 nA. Measurements of changes in action potential threshold and mean evoked firing (f-I curve) after application of PDGF-BB were performed at the original (control) resting membrane potential (Vm) obtained for each cell (average −56.8 ± 0.9 mV, n = 40 neurons), maintained by injecting steady DC current. Changes in resting membrane potentials ΔV were measured as the difference in Vm at different posttreatment times (Vt) subtracted from the resting Vm values measured 3 minutes before application of either PDGF-BB or vehicle (V0; Vt − V0). The changes were calculated for each individual cell and averaged for a “time-point-mean change.” In the “PDGF-BB” group, 12 of 18 examined cells fired action potentials after PDGF-BB application. These neurons were omitted from the analysis and only neurons that showed depolarization without firing (6 neurons) were used for the analysis of the changes in resting membrane potential.
For the negative control experiments, vehicle (5 μΜ of HCl in the extracellular solution) was focally and continuously applied to the cells, and excitability parameters were measured at the same time points as PDGF-BB.
In some experiments, cells were incubated with 10 μM of imatinib for at least 30 minutes before PDGF-BB or XE991 was applied. In these experiments, 10 μM of imatinib was also included in the extracellular solution.
2.4.2. Voltage-clamp recordings
To characterize IM, a deactivation protocol29,59,65 was used as shown in the study by Barkai et al.8 Briefly, from a holding potential of −20 mV, 1-second hyperpolarizing voltage steps were applied in increments of −5 mV. These steps induced slow current “relaxations” after the instantaneous inward current drops, which represented slow IM deactivation. Current relaxations were fitted to biexponential curves (starting after the capacitance artifact) and were extrapolated back to the beginning of the hyperpolarizing command pulses. IM amplitudes were calculated as the differences between the instantaneous peak currents at command onset and the steady-state currents just before command offset.
To examine whether PDGF-BB caused an inward current, long-lasting (more than 15 minutes) free-run (gap-free) experiments were performed. In these cases, cells were held at −60 mV during the recordings.
For the negative control experiments, vehicle (5 μΜ of HCl in the extracellular solution) was focally and continuously applied on the cells, and IM amplitudes were measured at the same time points as for the PDGF-BB.
In some experiments, cells were incubated with either 10 μM of imatinib for at least 30 minutes or 20 nM of wortmannin for at least 5 minutes before PDGF-BB or XE991 was applied. In these experiments, 10 μM of imatinib or 20 nM of wortmannin were also included in the extracellular solution. Treatment with either imatinib or wortmannin did not affect IM amplitudes (IM, SES = −57.2 ± 8.3 pA; IM, imatinib = −63.8 ± 9.4 pA, P = 0.6, unpaired Student t-test, n = 7 cells; IM, SES = −56.5 ± 10.6 pA; IM, wortmannin = −53.7 ± 12.6 pA, P = 0.88, unpaired Student t-test, n = 6 cells).
2.4.3. Single-cell calcium (Ca2+) imaging
Fluorescent Ca2+ imaging of cultured DRG neurons was performed using an inverted microscope (Nikon Eclipse Ti), equipped with an Epi-Fl attachment, perfect focus system (Nikon, Tokyo, Japan), and Exi Aqua monochromator (QImaging). Acutely dissociated (see above) DRG neurons were loaded with fura-2 acetoxymethyl (fura-2 AM, stock in DMSO), for 45 to 60 minutes in a standard extracellular bath solution (SES) composed of (in mM): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES. They were then rinsed for 45 to 60 minutes for deesterification of intracellular AM esters. Intracellular [Ca2+]i was measured fluoroimetrically as ratiometric excitation at 340 nm and 380 nm (F = ΔF340/380). Emission was collected at 510 nm. At the beginning of the experiment, cells were bathed in SES. After ∼5 minutes, cells were perfused with nominally Ca2+-free SES composed of (in mM): 145 NaCl, 5 KCl, 1 MgCl2, 10 glucose, and 10 HEPES. Under these conditions, PDGF-BB was focally and continuously applied (see below) onto individual small (∼25 μm) dissociated rat DRG neurons. Five to 7 minutes after cessation of PDGF-BB application, cells were perfused with nominally Ca2+-free SES containing 1 μM of thapsigargin. For all experiments, images were taken every 1 second, monitored online, and analyzed offline using Nikon Elements AR Software (Nikon). For quantification of changes in fluorescent intensity, values of fluorescence at the specific time points (F) were normalized to baseline signal (F − F0). F0 was measured 5 minutes before application of PDGF-BB.
2.4.4. Solutions and drugs for in vitro experiments
Recombinant rat carrier-free (CF) PDGF-BB (520-BB/CF, R&D Systems, Minneapolis, MN) was reconstituted to 100 µg/mL in 4 mM of HCl stock solution. Then, PDGF-BB was diluted to 125 ng/mL in extracellular solution. When indicated, PDGF-BB or its vehicle was focally and continuously applied onto plated DRG neurons using pressure puffs supplied by a pneumatic PicoPump PV820 (World Precision Instruments, Sarasota, FL), connected to a glass pipette (2-5 MΩ). The pipette was placed 25 to 50 μm away from the recorded neuron.
The vehicle solution for control experiments was 5 μΜ of HCl in the extracellular solution.
Imatinib (I-5577, LC laboratories, Woburn, MA) was dissolved in DMSO in a 10-mM stock solution. On the day of experiments, it was diluted in extracellular solution to a working concentration of 10 μM.
Wortmannin (W1628; Sigma) was dissolved with DMSO into stock aliquots. On experiment day, the stock was diluted to a working concentration of 20 nM.13,61
XE991 (2000, Tocris Bioscience, Bristol, United Kingdom) was dissolved in water in 10-mM stock aliquots. On experiment days, the stock was diluted to a working concentration of 10 µM and applied by bath perfusion.
Capsaicin (M2028, Sigma) was applied in the bath solution at a concentration of 1 μM.
2.5. Behavioral experiments
In this study, ∼200 g (6 weeks) SD male rats (purchased from Harlan Laboratories, Jerusalem, Israel) were used. The rats were habituated to handling and experimental procedures for 1 week before injections. The behavioral baseline was obtained by 2 preliminary measurements on the day before and the first day of experiment. The rats were habituated to the test environment for 24 hours in Plexiglas chambers and the experimental surfaces before the behavioral tests. The same investigator performed the scoring in all the behavior tests and was blind to the administered treatments. All injected solutions were freshly prepared on the day of experiment.
2.5.1. Measurements of thermal and mechanical sensitivity
The behavioral experiment contained the following groups: “vehicle,” “PDGF-BB,” “imatinib,” and “PDGF-BB + imatinib”. Single intraplantar injections of 40 μL of vehicle, PDGF-BB, imatinib, or a combination of PDGF-BB and imatinib into the rat hind paw were performed. Thermal sensitivity was determined using paw withdrawal latency (PWL) determined with a Hargreaves device (Ugo Basile, Varese, Italy). To avoid tissue damage, a 20-seconds cutoff time was used. Mechanical sensitivity was measured using an in-house custom-built electronic von Frey apparatus. A tip was mounted on the pressure sensor (Ugo Basile) and manually applied in the vertical direction with gradual increase of pressure intensity until the paw withdrew from the tip. The force at which the animal withdrew its paw was displayed and recorded.
2.5.2. Formalin test
Forty microliter of formalin (2% in saline; F1635, Sigma) with PBS (“Formalin + vehicle” group), formalin with PDGFR-β-Fc (“Formalin + PDGFR-β-Fc” group), or PDGFR-β-Fc alone (in saline; “PDGFR-β-Fc alone” group) were injected into the left hind paw. In another experiment, 40 μL of formalin (2% in saline) with either saline (“Formalin alone” group) or imatinib (60 ng/g; (“Formalin + imatinib” group) were injected. For the positive control experiment, either 1.5 or 5 mg/kg of morphine were injected into the loose skin under the neck, 30 minutes before injection of 40 μL of formalin (2% in saline) to the rat's hind paw (“Formalin + morphine” group). Spontaneous pain behavior was video-recorded for 60 minutes and analyzed post hoc. Time spent licking and biting was calculated in 5-minute intervals, starting immediately after injection. The total time spent licking and biting in 60 minutes was also analyzed.
2.5.3. Measurement of spontaneous nocifensive behavior
Twelve or 50 μg/mL of PDGF-BB prepared as described above were injected (40 µL) into the rat's hind paw. The time spent licking, biting, flinching, and guarding of the injected paw was collected over a period of 45 minutes after injection of PDGF-BB and vehicle.
2.5.4. Complete Freund's adjuvant—induced inflammation
Baseline thermal sensitivity was measured using PWL using a Hargreaves device (Ugo Basile). Mechanical sensitivity was measured using von Frey apparatus filaments. Animals then received a 100-μL injection of complete Freund's adjuvant (CFA; Calbiochem) in the left hind paw under anesthesia. Three days after CFA injection, animals received intraplantar injections of either imatinib (50 μg in 40 μL) or saline (40 μL) both ipsilateral and contralateral to the CFA injection site, and then PWL and mechanical threshold were tested in both hind paws. These injections and behavioral tests were repeated on days 4 and 5.
2.5.5. Solutions for behavioral experiments
Recombinant rat PDGF-BB protein was taken out from the frozen stock and prepared freshly in saline (0.9% NaCl, B. Braun, Melsungen) to reach a working concentration of either 12 or 50 μg/mL.
Imatinib methanesulfonate salt (I-5508) was dissolved in saline to a final concentration of 12 or 60 ng/g as indicated in the Results section.
Formalin (2%, Sigma) was prepared in saline solution.
Recombinant human CF PDGFR-β-Fc (385-PR/CF; R&D Systems) stock was prepared to a concentration of 500 μg/mL in sterile PBS. On the day of the experiment, PDGFR-β-Fc was prepared freshly out of frozen stock to make a solution of 500 ng per injection (40 μL) in formalin 2% or in saline solution.
Morphine (obtained from Hadassah Medical Center pharmacy) was diluted in saline on the day of experiment and administrated subcutaneously (120 μL) into the loose skin under the neck, at 1.5 and 5 mg/kg, 30 minutes before injection of formalin 2% in the hind paw.
For all injected solutions, the corresponding vehicle used to dissolve the drug served as a negative control.
2.6. Western blot
Paws were injected with saline or 2% formalin as described above (see Formalin Test). An additional noninjected animal was used as a negative control. Rats were killed 35 minutes after injection, and paw skin tissue samples were removed and homogenized in radioimmunoprecipitation assay lysis buffer containing protease inhibitors. After removing precipitated cell debris by high-speed centrifugation (12,000 rpm for 10 minutes), loading buffer was added to the supernatants and they were then denatured at 95°C for 5 minutes. Fifty μg of each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on a 4% to 20% polyacrylamide gel. A Western blotting system (Bio-Rad, Hercules, CA) was used to transfer the proteins to the nitrocellulose membrane, which was blocked with 5% (wt/vol) fat-free milk for 1 hour at room temperature and then incubated with an appropriate primary antibody overnight at 4°C. The membrane was then washed in Tris-buffered saline containing 0.1% (vol/vol) Tris-buffered saline Tween 20 (TBST) 3 times and incubated with an appropriate secondary antibody conjugated with horseradish peroxidase for 1 hour. Finally, the membrane was repeatedly washed in TBST, and the bound antibodies were visualized using an enhanced chemiluminescence system (Western Bright ECL, San Jose, CA). Immunoblot densitometry and normalization was performed using BioRad Image Lab 4.1 software.
The following antibodies were used: rabbit polyclonal Ab against platelet-derived growth factor (PDGF)-B (#ab178409; Abcam, Cambridge, United Kingdom) at a dilution of 1:500, rabbit polyclonal Ab against GAPDH (#ab37168; Abcam) at a dilution of 1:500, and a horseradish peroxidase–conjugated goat anti-rabbit antibody IgG (#ab97080; Abcam) at a dilution of 1:10,000.
2.7. Experimental design and statistical analysis
Data are shown as mean ± SEM. Differences between groups were analyzed using a 2-tailed Student t-test or one-/two-way analysis of variance followed by Bonferroni post hoc tests, when appropriate. The criterion for statistical significance was P < 0.05.
2.7.1. Sample size calculation
We did not conduct a power analysis because we were studying the effect of a new mediator and had no way to estimate the effect size. For in vivo experiments, we collected and analyzed all the data using a minimum of 6 animals per group, and for in vitro experiments, we always compared the same cells (at least 5) before and after treatment.
The number of replicates (n) for each experiment is given either in the Figure legends or in the Results. If a representative example is shown, we always explain how representative it is, that is, how many cells/animals showed a similar effect. The inclusion criteria for each experiment are described in the Materials and Methods section above.
All figures and tables are accompanied with an extended data file that contains exact P values as well as other statistical analysis parameters.
3. Results
3.1. Platelet-derived growth factor-BB activates nociceptor-like cultured dorsal root ganglion neurons and causes nocifensive behavior and pain hypersensitivity when injected intraplantarly in vivo
Platelet-derived growth factor has been shown to participate in the inflammatory process, and PDGFR is expressed in DRG neurons.26 Therefore, we thought to first determine whether PDGF-BB, one of the PDGF isoforms, modulated the activity of dissociated nociceptive (∼25-μm diameter) neurons in vitro using the perforated patch approach (see Methods). At resting potential (−56.8 ± 0.9 mV), all neurons were quiescent (n = 40). Focal continuous application of PDGF-BB (125 ng/mL) caused a slow depolarization of about 20 mV in all recorded neurons within 5 to 10 minutes (n = 18 cells, Figs. 1A and B). In 67% of the neurons (12 of 18 neurons), application of PDGF-BB led to repetitive firing at a frequency of about 1 Hz (1.4 ± 0.4 Hz; Fig. 1A). Under control conditions, when vehicle solution was focally applied on the neurons, neither depolarization (Fig. 1B) nor increase in neuronal excitability were observed during sustained recordings (up to 15 minutes, data not shown). The excitatory effects of PDGF-BB persisted throughout drug application. In some neurons, we examined the reversibility of the PDGF-BB effects and found that the PDGF-BB–mediated increase in neuronal excitability were sustained during washout in 3 of 6 neurons, whereas PDGF-BB–mediated depolarization was sustained during washout in all neurons (n = 6). These results show that application of PDGF-BB activates nociceptor-like DRG neurons and suggest that this activation could potentially cause nociception when injected in vivo.
Figure 1.
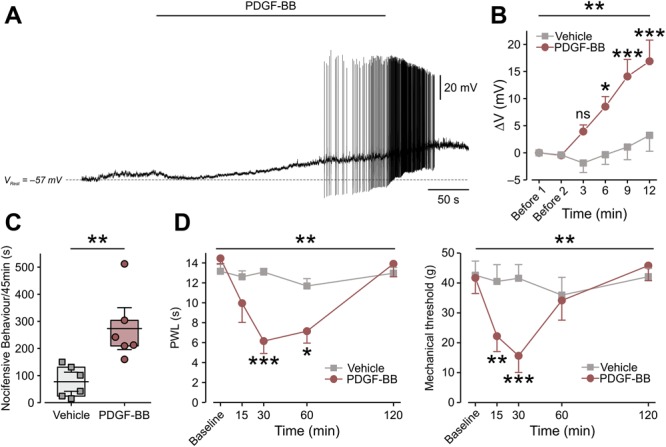
Platelet-derived growth factor-BB activates nociceptor-like cultured DRG neurons and induces nocifensive behavior and pain hypersensitivity. (A) Representative trace (12 of 18 neurons) of current-clamp perforated patch recordings from nociceptor-like cultured DRG neurons showing membrane voltage response to continuous focal application (marked by horizontal bar) of 125 ng/mL of PDGF-BB. Dashed lines indicate resting potentials before drug application (−57 mV). (B) Mean ± SEM of changes in resting membrane potential ΔV (Vt − V0) after focal application of PDGF-BB alone (red) or vehicle (5 μM HCl, light gray). Time point “Before 1” indicates the time where V0 values were measured (3 minutes before application of either PDGF-BB or vehicle). Time point “Before 2” indicates the time just before application of either PDGF-BB or vehicle. ns, not significant, *P < 0.05, **P < 0.01; ***P < 0.001; Repeated-measures (RM) 2-way ANOVA with post hoc Bonferroni. In the “PDGF-BB,” only neurons that showed depolarization without firing (n = 6 neurons) were used for the analysis; n = 5 cells for the “Vehicle” group. (C) Graph comparing box plots and individual values of the total time (in seconds) spent by rats licking, biting, flinching, and guarding the hind paw (nocifensive behavior) during 45 minutes after intraplantar injection of 50 μg/mL of PDGF-BB or its vehicle (2 mM of HCl). **P < 0.01; Student t-test; n = 6 rats per group. Box plots depict mean, 25th, 75th percentile, and SD. (D) The decrease in thermal (radiant heat) paw withdrawal latency (PWL, left) and mechanical threshold (electronic von Frey, right) after intraplantar injection of 12 μg/mL of PDGF-BB as compared to injection of vehicle (500 μM HCl, n = 6 rats per group, ***P < 0.001; **P < 0.01; RM 2-way ANOVA with post hoc Bonferroni). ANOVA, analysis of variance; DRG, dorsal root ganglion; PDGF, platelet-derived growth factor.
To examine this notion more directly, we injected PDGF-BB subcutaneously into the left hind paw of naive rats. Platelet-derived growth factor-BB at 12 μg/mL did not produce spontaneous nocifensive behavior (time spent licking, biting, flinching, and guarding; supplementary Fig. 1A, available at http://links.lww.com/PAIN/A758). We next increased the dose and showed that intraplantar injection of 50 μg/mL PDGF-BB caused ongoing pain, as reflected by a significant increase in nocifensive behavior, after the injection of PDGF-BB but not its vehicle (Fig. 1C). This increased spontaneous behavior started 25 minutes after PDGF-BB injection and lasted for about 15 minutes (supplementary Fig. 1B, available at http://links.lww.com/PAIN/A758). Importantly, intraplantar injection of a lower PDGF-BB dose (12 μg/mL), although not effective in producing spontaneous nocifensive behavior, was sufficient to significantly decrease mechanical paw withdrawal thresholds and paw withdrawal latencies to noxious heat (Fig. 1D). The effect of 12 μg/mL of PDGF-BB on mechanical threshold started 15 minutes after PDGF-BB injection, peaked at 30 minutes, and ended 1 hour after injection (Fig. 1D, right). The PDGF-BB–mediated decease in thermal latency started 30 minutes after PDGF-BB injection and wore off within 2 hours (Fig. 1D, left). These results suggest that PDGF-BB injected into the intact paw activates primary sensory neurons, thereby increasing their responsiveness to noxious stimuli and producing nociception.
3.2. Platelet-derived growth factor-BB increases gain and decreases threshold in nociceptor-like dorsal root ganglion neurons
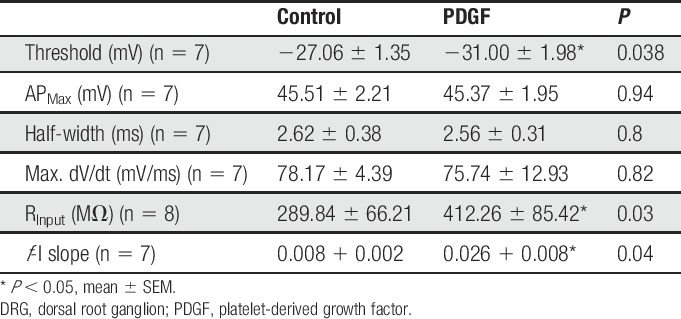
To elucidate the biophysical mechanism of PDGF-BB–mediated hyperexcitability, we first determined whether PDGF-BB affects passive and active membrane properties of nociceptor-like DRG neurons. To maintain a steady membrane potential (Vm) at the original resting membrane potential for each cell despite PDGF-BB–mediated membrane depolarization, a negative DC current was applied. This enabled us to accurately measure changes in neuronal excitability in the presence of PDGF-BB. Treatment with PDGF-BB, but not vehicle (supplementary Table 1, available at http://links.lww.com/PAIN/A758), increased the frequency of spikes evoked by depolarizing current steps (Fig. 2A), causing a significant increase in neuronal gain (ie, slope of the f-I curve, m; Fig. 2B). In addition, treatment with PDGF-BB significantly decreased the action potential threshold (Figs. 2C–E and Table 1) and increased the input resistance (Rin; Fig. 2F, also see Table 1) in 6 of 8 nociceptor-like DRG neurons. The action potential amplitude and width (Table 1) or the maximal rate of change of the membrane voltage (dV/dt)max of the action potential was not affected (Table 1). Application of vehicle did not alter the excitable properties of neurons (supplementary Table 1, available at http://links.lww.com/PAIN/A758).
Figure 2.
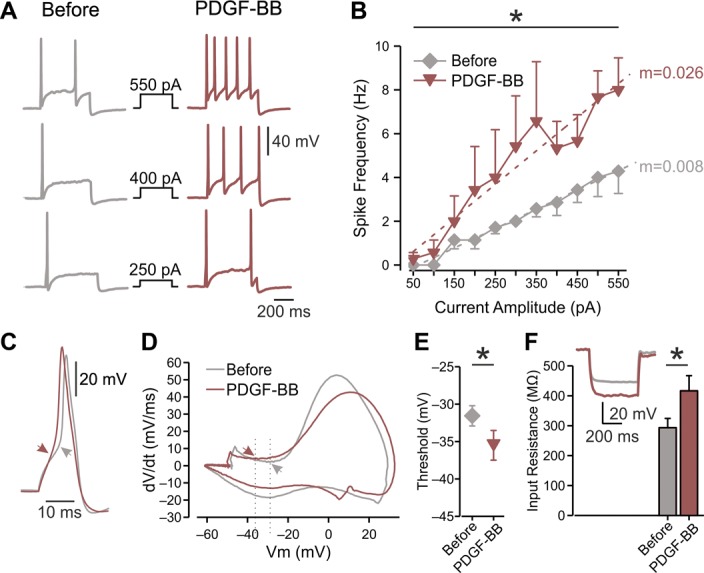
Platelet-derived growth factor-BB increases the excitability of nociceptor-like DRG neurons and increases the Rin. (A) Representative traces of typical voltage responses to 500 ms current steps (shown in middle) recorded from the same neuron before (left) and after ∼15 minutes of focal continuous application of 125 ng/mL of PDGF-BB (right, representative of n = 7). (B) Mean frequency–intensity (f-I) curves of DRG neurons recorded before (gray diamonds) and ∼15 minutes after (red inverted triangles) application of PDGF-BB. Note that PDGF-BB induced a significant increase in gain (m, dotted lines; *P < 0.05, paired t test; n = 7, see also Table 1). (C) Superimposed representative traces of a single action potential recorded from the same neuron before (gray) and after (red) the application of PDGF-BB. Arrows indicate the AP threshold, derived from the phase plots shown in (D). (D) Representative phase plots of rate of change of the membrane potential (dV/dt) vs membrane potential (Vm) during an action potential recorded from the same neuron before (gray) and 10 to 20 minutes after the application of PDGF-BB (red). Arrows and dashed lines indicate shift in threshold voltage and quantified in (E). (E) Thresholds for generation of action potentials calculated from the same neurons before and ∼15 minutes after application of PDGF-BB (mean ± SEM, *P < 0.05, paired t test, n = 7). (F) Bar graph depicting Rin before (gray) and after ∼15 minutes exposure to PDGF-BB (red); *P < 0.05, paired t test, n = 8. Inset: representative typical voltage responses to 500 ms, −100 pA current before (gray) and after (red) the treatment with PDGF-BB. All measurements described in this figure were performed at the native resting potential of each cell, adjusted after PDGF-BB application by injecting the appropriate repolarizing DC currents. AP, action potential; DRG, dorsal root ganglion; PDGF, platelet-derived growth factor.
Table 1.
Parameters of excitability of nociceptor-like DRG neurons before and after the application of PDGF (125 ng/mL).
3.3. Platelet-derived growth factor-BB inhibits IM in nociceptor-like dorsal root ganglion neurons
The changes induced by PDGF-BB application, including mild depolarization followed by action potential firing, increased spike threshold without concomitant changes in spike amplitude or width, and the increase in Rin (Table 1), have all been previously associated with the inhibition of IM.8,24,27,65 Therefore, we tested whether the PDGF-BB–induced increase in neuronal excitability was due to IM inhibition. We used a well-established voltage-clamp protocol in perforated patch configuration41 to isolate IM from other currents (Fig. 3A). Focal application of PDGF-BB inhibited the IM in all (16 of 16) recorded neurons (Fig. 3A, middle, Figs 3B and C, left, see also Fig. 7D). Subsequent bath application of 10 μM of XE991, a specific IM blocker,66 did not further inhibit IM in all recorded neurons (Fig. 3A, right, Fig. 3C, left, see also Fig. 7D). Similar to the effect of PDGF-BB on excitability (Figs. 1A and B), the PDGF-BB–mediated inhibition of IM started 6 minutes after PDGF-BB application, and was sustained at 9 minutes (Fig. 3D, see also Fig. 7D). In 6 of 9 cells, this PDGF-BB–mediated inhibition was reversed after 20 minutes of PDGF-BB washout (Fig. 3D). Focal application of vehicle did not affect the amplitude of IM (Fig. 3C, right, see also Fig. 7D). These results show that PDGF-BB leads to inhibition of IM in nociceptor-like DRG neurons.
Figure 3.
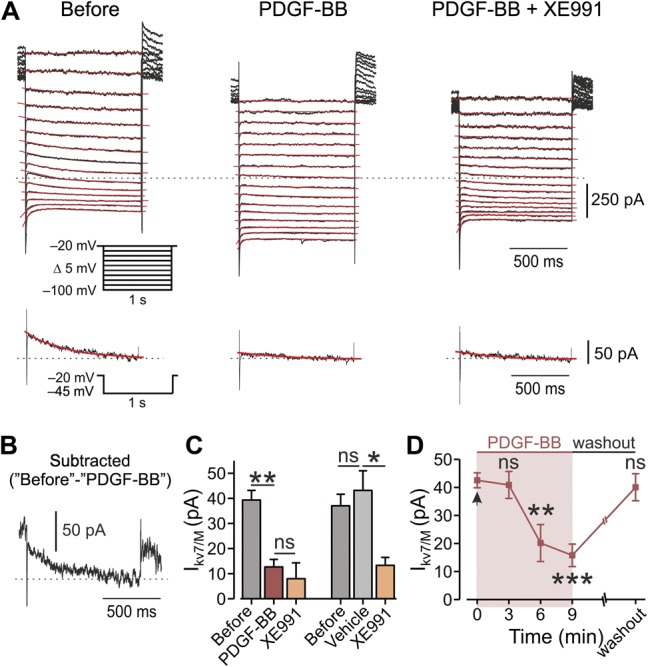
Application of PDGF-BB leads to inhibition of IM in DRG neurons. (A) Voltage-clamp perforated patch recordings from DRG neurons showing families of IM evoked by a series of 1-second, 5-mV hyperpolarizing voltage steps from a holding potential of −20 mV (shown in inset). The 3 subpanels show the current responses before (left), after focal application of 125 ng/mL of PDGF-BB (middle), and after bath application of 10 μM of XE991 on top of focal application of PDGF-BB (right). The dotted line indicates zero current level. The current response obtained by stepping to −45 mV is shown at the bottom of each subpanel. The IM relaxation was fitted with a biexponential line (red), which was extrapolated to the beginning of the voltage step. (B) Subtracted trace of IM evoked by stepping to −45 mV before the application of PDGF-BB minus IM trace evoked by stepping to −45 mV after PDGF. (C) Left, bar graph depicting mean ± SEM of peak IM amplitude (measured by stepping to −45 mV), before (gray), 10 minutes after the application of PDGF-BB (red), and 10 minutes after the treatment with XE991 on top of PDGF-BB (orange). **P < 0.01, ns, not significant, RM one-way ANOVA with post hoc Bonferroni test, n = 7 neurons. Right, same as left but showing that a 10-minute application of vehicle (light gray) has no effect on IM amplitude, ns, not significant, *P < 0.05, RM one-way ANOVA with post hoc Bonferroni test n = 5 neurons. (D) Peak IM amplitude (measured by stepping to −45 mV, mean ± SEM), plotted vs time of application of PDGF-BB. Platelet-derived growth factor-BB application is indicated by the arrow (time “0”). One-way ANOVA with post hoc Bonferroni test, n = 13 neurons for 6 minutes, n = 11 neurons for 9 minutes. In 7 out of 9 neurons, ∼15 minutes washout of PDGF-BB led to full recovery of IM. ns, not significant; **P < 0.01. ANOVA, analysis of variance; DRG, dorsal root ganglion; PDGF, platelet-derived growth factor; RM, repeated-measures.
Figure 7.
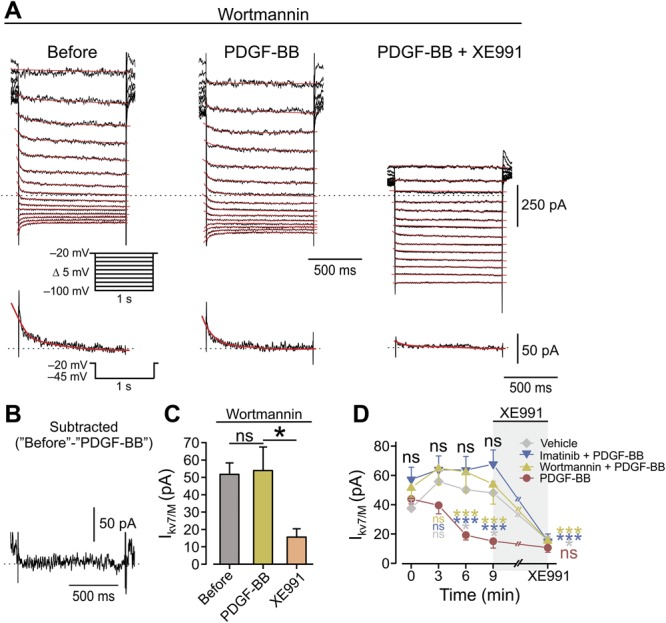
Inhibition of PI3K pathway prevents PDGF-induced inhibition of IM. (A) Voltage-clamp perforated patch recordings from DRG neurons pretreated before the experiment and perfused during the experiment with extracellular solution containing 20 nM of wortmannin. Families of IM evoked by a series of 1-second, 5-mV hyperpolarizing voltage steps from a holding potential of −20 mV (shown in inset). The 3 subpanels show the current responses in cells treated with wortmannin, before (left), after the focal application of 125 ng/mL of PDGF-BB (middle), and after the bath application of 10 μM of XE991 on top of focal application of PDGF-BB (right). The dotted line indicates zero current level. The current response obtained by stepping to −45 mV is shown at the bottom of each subpanel. (B) Subtracted trace of IM trace evoked by stepping to −45 mV before application of PDGF-BB minus IM trace evoked by stepping to −45 mV after PDGF-BB. (C) Bar graph depicting mean ± SEM of peak IM amplitude in cells pretreated with 20 nM of wortmannin and measured by stepping to −45 mV, before (gray), 10 minutes after the application of PDGF-BB (yellow), and 10 minutes after the bath application of XE991 on top of PDGF-BB (orange). ns, not significant, *P < 0.05, RM one-way ANOVA with post hoc Bonferroni test, n = 7. (D) Comparison of changes in peak IM amplitude (measured by stepping to −45 mV, mean ± SEM) with time, after focal application of PDGF-BB (red circles), vehicle (light gray diamonds), PDGF-BB onto cells treated with imatinib (blue inverted triangles), and PDGF-BB onto cells treated with wortmannin (yellow triangles). Black letters “ns”, comparison by the time points between the “Vehicle,” “Imatinib + PDGF-BB,” and “Wortmannin + PDGF-BB” groups; light gray letters and asterisks—comparison by the time points between the “PDGF-BB” and the “Vehicle” groups; blue letters and asterisks—comparison by the time points between the “PDGF-BB” and the “Imatinib + PDGF-BB” groups; orange letters and asterisks—comparison by the time points between the “PDGF-BB” and the “Wortmannin + PDGF-BB” groups. RM 2-way ANOVA with post hoc Bonferroni test, n = 9 for the “PDGF-BB” group; n = 7 neurons for the “Imatinib + PDGF-BB” and the “Wortmannin + PDGF-BB” groups, n = 4 for the “Vehicle” group. ns, not significant; *P < 0.05; ***P < 0.001. In each experiment, 10 minutes after the application of either PDGF-BB or vehicle, XE991 was added for 7 minutes (light gray shading). At time point “XE991,” the statistical comparison shown is between the peak IM amplitude values at the “9 minute” time point and the values after ∼7 minutes of XE991. ns, not significant; *P < 0.05; ***P < 0.001. ANOVA, analysis of variance; DRG, dorsal root ganglion; PDGF, platelet-derived growth factor; RM, repeated-measures.
Importantly, in cells pretreated with 10 μM of XE991 to completely block IM,8 PDGF-BB did not further affect membrane properties (thresholdbefore = −35.1 ± 1.9 mV, thresholdXE991 = −40.3 ± 1.5 mV, thresholdXE991+PDGF = −38.9 ± 2.3 mV: for “before” vs“XE991” groups—P = 0.004, for “XE991” vs “XE991 + PDGF” groups—P = 0.14; n = 7, Student t-test) and excitability (mBefore = 0.009 + 0.003, mXE991 = 0.024 + 0.01, mXE991+PDGF = 0.024 + 0.02: for “before” vs “XE991” groups—P = 0.03, for “XE991” vs “XE991 + PDGF” groups—P = 0.42; n = 6, Student t-test).
Finally, we examined whether in addition to IM inhibition, PDGF-BB may also induce depolarization through additional mechanisms, for example, through activation of transducer channels resulting in PDGF-BB–mediated inward current. We performed perforated patch recordings in voltage-clamp mode, holding the cells at −65 mV. Platelet-derived growth factor-BB focally applied onto the cells did not induce a significant inward current in the recorded neurons (n = 9, supplementary Figs. 2A and B, available at http://links.lww.com/PAIN/A758). Subsequent bath perfusion of capsaicin produced a substantial inward current (supplementary Fig. 2A, available at http://links.lww.com/PAIN/A758). These data suggest that PDGF-BB does not mediate depolarization and neuronal firing by direct activation of cation permeable transducer channels. It is noteworthy that although analysis of all recorded neurons (n = 9, supplementary Fig. 2B, available at http://links.lww.com/PAIN/A758) did not show difference in inward current levels before and after application of PDGF, in 4 of 9 cells, PDGF-BB caused a 46 ± 8.5-pA inward current (P = 0.02; one-way analysis of variance on these cells). Such a small PDGF-BB–mediated inward current is consistent with PDGF-BB–induced IM inhibition because blockade of IM, which is outward at resting membrane potential, would likely generate a small inward current.41
Taken together, our results indicate that IM inhibition is a main mechanism underlying PDGF-BB–induced increase in intrinsic neuronal excitability.
3.4. Platelet-derived growth factor-BB–induced inhibition of IM, increased neuronal excitability, and pain hypersensitivity are mediated by the platelet-derived growth factor receptor
It is widely accepted that PDGF promotes cell proliferation, growth, and tissue healing through activation of the PDGFR.50 We therefore examined whether the PDGFR is required for PDGF-BB–mediated neuronal hyperexcitability and pain hypersensitivity. The addition of 10 μM of imatinib, a well-established PDGFR inhibitor,36 to the bath solution prevented the PDGF-BB–mediated blockade of IM in all (n = 7) recorded neurons (Fig. 4A, middle, Figs. 4B and C, see also Fig. 7D). In these neurons, IM was fully inhibited by subsequent application of XE991 (Fig. 4A, right, Fig. 4C, see also Fig. 7D). These data indicate that PDGF-BB inhibits IM in nociceptive DRG neurons through activation of the PDGFR.
Figure 4.
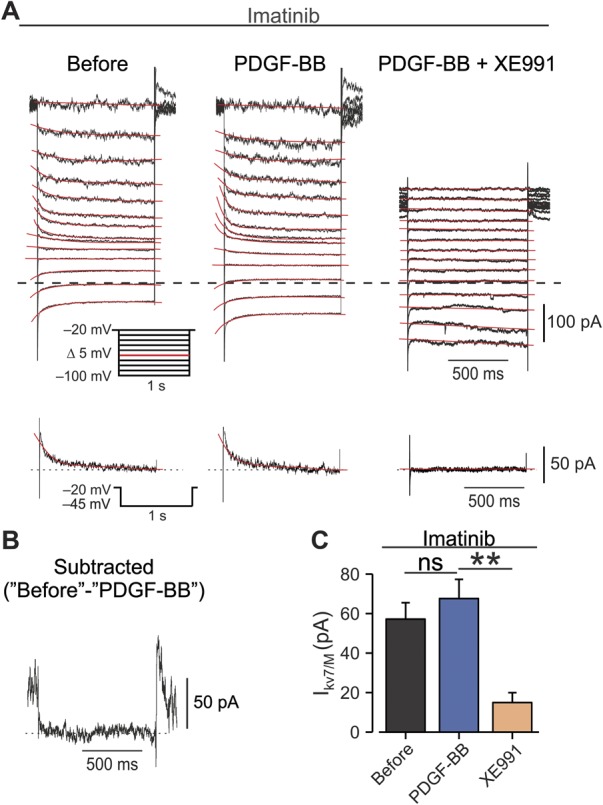
Inhibition of PDGFR prevents PDGF-BB–mediated IM attenuation. (A) Voltage-clamp perforated patch recordings from DRG neurons treated before and during the experiment with extracellular solution containing 10 µM of imatinib. Families of IM evoked by a series of 1-second, 5-mV hyperpolarizing voltage steps from a holding potential of −20 mV (shown in inset). The 3 subpanels show the current responses in cells treated with imatinib, before (left), after focal application of 125 ng/mL of PDGF-BB (middle), and after bath application of 10 μM of XE991 on top of focal application of PDGF-BB (right). The dotted line indicates zero current level. The current response obtained by stepping to −45 mV is shown at the bottom of each subpanel. (B) Subtracted IM trace, evoked by a −45-mV step before application of PDGF-BB from an IM trace evoked by the same step after PDGF-BB. (C) Bar graph depicting mean ± SEM of peak IM amplitude (measured by stepping to −45 mV) in cells treated with 10 µM of imatinib before (gray), 10 minutes after the application of PDGF-BB (blue), and 10 minutes after the bath application of XE991 on top of PDGF-BB (orange). n = 7, ns, not significant, **P < 0.01, RM one-way ANOVA with post hoc Bonferroni test. ANOVA, analysis of variance; DRG, dorsal root ganglion; PDGF, platelet-derived growth factor; RM, repeated-measures.
Furthermore, in cells treated with imatinib, focal continuous application of PDGF-BB affected neither the passive membrane properties of nociceptive neurons nor their excitability (Table 2). In the presence of imatinib, PDGF-BB also failed to induce membrane depolarization (−55.74 ± 1.9 mV vs −55.48 ± 5.69 mV; Figs. 5A and B) or neuronal firing (Fig. 5A). Application of imatinib alone for more than 15 minutes did not alter neuronal excitability in all recorded neurons (Table 2) and did not induce any changes in membrane potential (−55.74 ± 1.9 mV vs −56.21 ± 2.25 mV, Table 2). Collectively, these findings indicate that the PDGFR is necessary to cause the hyperexcitability induced by PDGF-BB application.
Table 2.
Parameters of excitability of imatinib (10 µM)-treated nociceptor-like DRG neurons before and after the application of PDGF (125 ng/mL).
Figure 5.
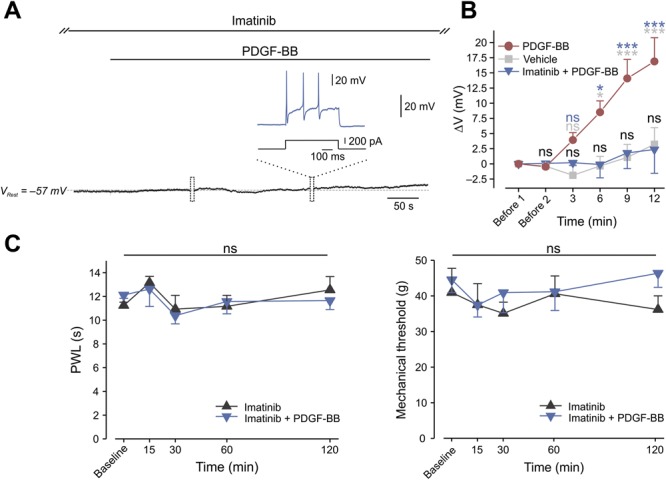
PDGFR is required for PDGF-BB–induced spontaneous firing and hypersensitivity to pain. (A) Representative trace (7 of 7 neurons) of membrane voltage changes in nociceptor-like cultured DRG neurons treated with 10 µM of imatinib. In these conditions, focal application (marked by the horizontal bar) of 125 ng/mL of PDGF-BB did not lead to membrane depolarization. Free-run recording was interrupted (marked by boxes) to examine the excitable properties of the neuron. Dashed lines indicate resting potentials before drug application (−57 mV). All recorded neurons (n = 7) showed no PDGF-BB–induced hyperexcitability, but fired normally in response to a depolarizing current step (inset). (B) Mean ± SEM of changes in resting membrane potential ΔV (Vt − V0) after focal application of PDGF-BB alone (red), vehicle (5 μM of HCl, light gray), or PDGF-BB on cells pretreated with imatinib (blue). Time point “Before 1” indicates the time where V0 values were measured (3 minutes before application of either PDGF-BB or vehicle). Time point “Before 2” indicates the time just before application of either PDGF-BB or vehicle. ns, not significant, *P < 0.05, **P < 0.01; ***P < 0.001; blue asterisks—comparison between “PDGF-BB” and “Vehicle” groups; light gray asterisks—comparison between “PDGF-BB” and “Imatinib + PDGF-BB” groups; black—comparison between “Imatinib + PDGF-BB” and “Vehicle” groups. RM 2-way ANOVA with post hoc Bonferroni, n = 5 cells in each group. The data of “PDGF-BB” and “Vehicle” is presented in Figure 1B and used here for the convenient comparison between these groups and the “Imatinib + PDGF-BB” group. Note that there is no significant difference between the “Imatinib + PDGF-BB” and the vehicle group. (C) Paw withdrawal latency (PWL, radiant heat, left) and mechanical threshold (von Frey, right) after intraplantar injection of 12 µg/mL of PDGF-BB together with 12 ng/g of imatinib as compared to injection of imatinib alone (12 ng/g). n = 6 rats per group, ns, not significant, RM 2-way ANOVA with post hoc Bonferroni. ANOVA, analysis of variance; DRG, dorsal root ganglion; PDGF, platelet-derived growth factor; RM, repeated-measures.
Importantly, we show that subcutaneous injection of imatinib in vivo prevented the PDGF-BB–mediated decreases in both mechanical withdrawal threshold and noxious heat paw withdrawal latencies (Fig. 5C). Injection of imatinib alone did not exert any effects on nocifensive behavior or sensitivity to noxious thermal and mechanical stimuli (Fig. 5C). Taken together, these results suggest that PDGF-BB, by activating the PDGFR, inhibits IM and leads to nociceptive hyperexcitability and pain hypersensitivity.
The abovementioned in vitro experiments on dissociated DRG neurons suggest that PDGF-BB acts on PDGFR expressed by nociceptive neurons. Platelet-derived growth factor-BB, which we used also for in vivo injections (Figs. 1 and 5), primarily activates the PDGF receptor subtype β (PDGFR-β3). Using immunohistochemical staining, we showed that the majority of both nonpeptidergic (isolectin B4 [IB4]-positive) and peptidergic (IB4-negative, calcitonin gene-related peptide [CGRP]-positive) nociceptive neurons coexpress PDGFR-β (Fig. 6A, supplementary Table 2, available at http://links.lww.com/PAIN/A758). Importantly, nociceptive IB4 or CGRP-expressing fibers and bundles in the glabrous skin coexpress PDGFR-β (Fig. 6B). These results, together with the PDGF-BB–mediated effects shown above (Figs. 1–3), imply that PDGF-BB–mediated activation of the PDGFR-β could account for the processes underlying PDGF-BB–induced increase in nociceptive excitability in vitro and pain hypersensitivity in vivo.
Figure 6.
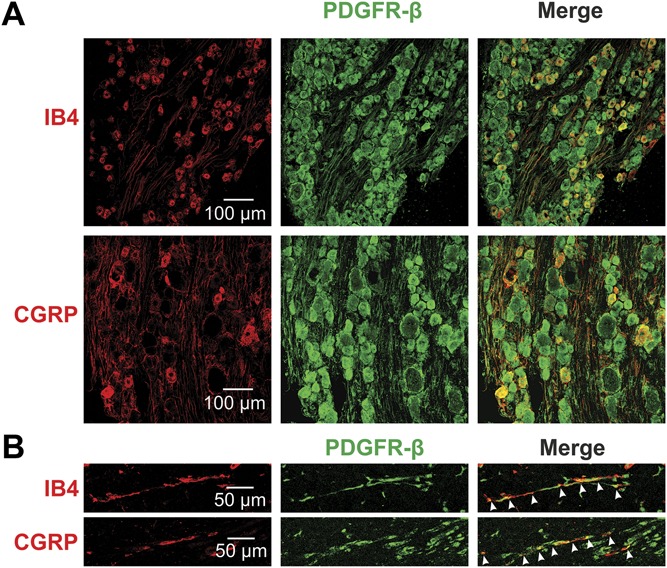
PDGFR-β is expressed in nociceptive DRG neurons and in nociceptive fibers. (A) PDGFR-β is colocalized with markers of nociceptive neurons (IB4, CGRP) shown by immunohistochemistry of rat DRG. (B) Immunohistochemical analysis of rat skin showing the expression of PDGFR-β in fibers also expressing the nociceptive markers IB4 and CGRP. Arrows indicate the double-labeled axons. CGRP, calcitonin gene-related peptide; DRG, dorsal root ganglion.
We then determined which PDGFR-β–mediated signaling cascade was involved in inhibition of Kv7/M channels. It has been demonstrated that in DRG neurons, bradykinin inhibits IM through the activation of phospholipase Cγ (PLCγ).41 In general, activation of PLC facilitates the hydrolysis of PIP2 into DAG and IP3, which in turn promotes the mobilization of Ca2+ from internal stores, resulting in increase in [Ca2+]i. Accordingly, we performed measurements of changes in [Ca2+]i from dissociated DRG neurons to examine whether PDGF-BB could promote PLC activity. To focus on the [Ca2+]i rise from internal stores, we omitted Ca2+ from the extracellular solution. Under these conditions, neither bath application of PDGF-BB (examined in 104 neurons in 3 plates, not shown) nor focal continuous puff application onto individual cells (supplementary Figs. 3A and B, available at http://links.lww.com/PAIN/A758) produced an increase in [Ca2+]i. In the latter conditions, consequent application of thapsigargin, used as a positive control to deplete endoplasmic reticular Ca2+ stores,64 produced a significant increase in [Ca2+]i (supplementary Figs. 3A and B, available at http://links.lww.com/PAIN/A758). Hence, these data show that the application of PDGF-BB does not produce release of Ca2+ from internal stores in DRG neurons and suggest that the PLCγ-IP3 cascade is not likely to mediate PDGF-BB–induced effects. We therefore examined whether another common PDGFR-β–activated signaling cascade, the PI3Kinase (PI3K) pathway,3 was involved in the PDGF-BB–mediated inhibition of Kv7/M channels. We incubated DRG neurons in 20 nM of wortmannin, a specific PI3K inhibitor61 and then examined the effect of application of PDGF-BB on IM. Treatment with wortmannin prevented PDGF-BB–mediated blockade of IM in all (n = 7) recorded neurons (Figs. 7A and B–D middle). In these neurons, IM was inhibited by subsequent application of XE991 (Figs. 7A, C, and D right). These data suggest that the activation of the PDGFR inhibits IM in nociceptive DRG neurons at least in part through activation of the PI3K pathway.
3.5. Platelet-derived growth factor facilitates acute inflammatory pain
Several reports have demonstrated that PDGF is released during tissue inflammation and its receptors are acutely upregulated after inflammation.5,43,68 Therefore, we studied whether PDGF/PDGFR-induced sensitization played a role in the genesis of inflammatory pain. Considering that intraplantar injection of PDGF-BB in vivo produced short-lasting (15 minutes, see Fig. 1C) nocifensive behavior and pain hypersensitivity that lasted about 60 minutes (Fig. 1D), we examined whether PDGF contributed to acute inflammatory pain. We first determined whether PDGF levels were upregulated during the second stage of formalin-induced nocifensive behavior, which reflects formalin-induced acute cutaneous inflammation.1,2,33,63 Intraplantar injection of saline did not induce upregulation of PDGF in the hind paw (supplementary Fig. 4, available at http://links.lww.com/PAIN/A758). Intraplantar injection of formalin (2%, 40 µL) produced the stereotypical 2-phased increase in nocifensive behavior (measured by time spent licking and biting, Figs. 8A and B, gray). Western blot analysis of the injected paw examined 35 minutes after intraplantar injection of 2% formalin, at the time point at which formalin-induced nocifencive behavior reaches its maximal values (Figs. 8A and B, gray), showed high levels of PDGF (supplementary Fig. 4, available at http://links.lww.com/PAIN/A758). These data suggest that PDGF could be released during formalin-induced acute cutaneous inflammation. To strengthen this hypothesis, we examined the effect of PDGF scavenging on the second stage of formalin-induced nocifensive behavior. We hypothesized that if PDGF is released during inflammation, its quenching would prevent activation of PDGFR and therefore may reduce acute inflammatory pain. Considering the observed in vitro and in vivo effects of PDGF-BB, which imply possible involvement of PDGFR-β in the PDGF-mediated effect, we used a protein constructed of the PDGFR-β extracellular domains fused to Fc antibody fragments (PDGFR-β-Fc) to scavenge PDGF. The PDGFR-β-Fc construct has been shown to effectively scavenge released PDGF both in vitro and in vivo.22,67 Indeed, coinjection of PDGFR-β-Fc together with formalin significantly decreased the overall time spent licking and biting in the second phase (Figs. 8A and C), but not in the first phase (Figs. 8A and C). Intraplantar injection of PDGFR-β-Fc (500 ng, 40 µL) alone did not cause nocifensive behavior (Fig. 8A, light green) and did not affect either mechanical paw withdrawal thresholds or paw withdrawal latencies to noxious heat (supplementary Fig. 5, available at http://links.lww.com/PAIN/A758). This decrease in formalin-induced nocifensive behavior due to PDGFR-β-Fc–mediated PDGF scavenging implies that PDGF could be released during acute inflammation and facilitates inflammatory pain.
Figure 8.
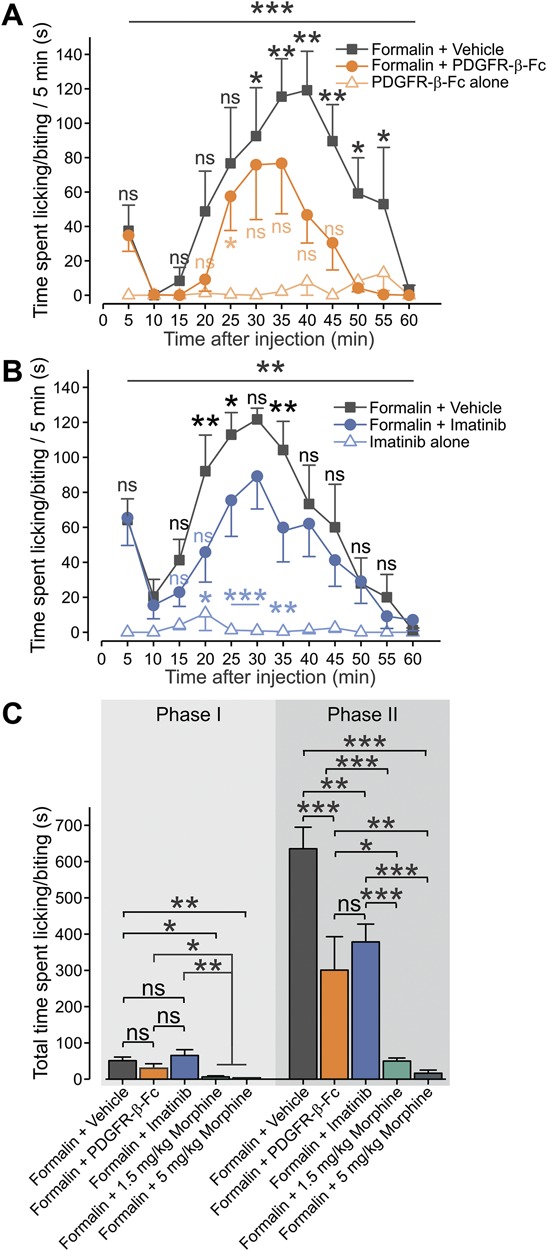
Platelet-derived growth factor scavenging and inhibition of PDGFR reduces formalin-induced inflammatory pain. (A) Mean ± SEM of duration of paw licking and biting per 5 minutes plotted vs time after injection of 2% formalin with vehicle (PBS, gray squares); 2% formalin with 500 ng/40 μL of PDGFR-β-Fc (orange circles) and 500 ng/40 μL of PDGFR-β-Fc alone (light orange triangles). RM two-way ANOVA with post hoc Bonferroni, n = 6 rats per group; black bar and asterisks—comparison between the “Formalin + vehicle” and the “Formalin + PDGFR-β-Fc” groups; light orange bar and asterisks—comparison between the “PDGFR-β-Fc alone” and the “Formalin + PDGFR-β-Fc” groups. (B) Mean ± SEM of duration of paw licking and biting per 5 minutes plotted vs time after injection (at time “0”) of 2% formalin (dark gray); 2% formalin together with 60 ng/g of imatinib (blue) and 60 ng/g of imatinib alone (light blue). RM 2-way ANOVA with post hoc Bonferroni; n = 6 rats per group, ns, not significant; *P < 0.05; **P < 0.01, ***P < 0.001 black bar and asterisks—comparison between the “Formalin + vehicle” and the “Formalin + imatinib” groups; light blue bar and asterisks—comparison between the “Imatinib alone” and the “Formalin + imatinib” groups. (C) Summary of mean ± SEM of total duration of time spent in licking and biting in phase I (0-10 minutes) and phase II (10-60 minutes) after injection of 2% formalin with vehicle (dark gray); 2% formalin together with 500 ng/40 μL of PDGFR-β-Fc (orange); 2% formalin together with 60 ng/g of imatinib (blue); 2% formalin together with 1.5 mg/kg (green) and 5 mg/kg of morphine (dark green) Student t-test; ns, not significant; *P < 0.05, **P < 0.01; ***P < 0.001; n = 6 animals in all groups apart from the “Formalin + vehicle” group containing n = 12 animals. ANOVA, analysis of variance; PDGF, platelet-derived growth factor; RM, repeated-measures.
To further support the idea that PDGF may have a role in inflammatory pain, we examined whether blockade of the PDGFR by imatinib would affect the second phase of the formalin-induced responses (Figs. 8B and C). We found that injection of 60 ng/g of imatinib together with formalin significantly decreased the nocifensive behavior in the second phase of formalin-induced responses (Figs. 8B and C). Considering that imatinib alone does not cause analgesia (Fig. 5C; see also Refs. 22,67), these results suggest that PDGFR activation is an important factor in the genesis of acute inflammatory pain.
It is noteworthy that the effect of sequestration of PDGF or blockade of PDGFR significantly reduces but does not abolish formalin-induced nocifensive behavior (Figs. 8A–C). Inhibition of PDGF signaling caused less analgesia than the systemic administration of either 5 mg/kg or 1.5 mg/kg of morphine (Fig. 8C). Therefore, we suggest that PDGF partially contributes to the formalin-induced inflammatory pain.
We next investigated whether PDGF signaling could also be involved in mediating complete Freund's adjuvant (CFA)-induced subacute inflammatory pain. Injection of imatinib into the CFA-treated paw did not ameliorate thermal or mechanical CFA-induced sensitization measured on days 3 to 5 after CFA (supplementary Fig. 6, available at http://links.lww.com/PAIN/A758), suggesting that PDGF signaling may be important for maintaining acute but not subacute inflammatory pain.
Taken together, our results suggest that during acute inflammation PDGF levels rise and promote sensitization of nociceptors through IM inhibition, thus partially contributing to acute inflammatory pain.
4. Discussion
Here, we show, for the first time, that PDGF facilitates acute inflammatory pain. This facilitation occurs, at least in part, through activation of the PDGFR because its pharmacological inhibition significantly reduces inflammatory pain. Moreover, we demonstrated that PDGF-BB–mediated activation of the PDGFR leads to inhibition of IM through the PI3K signaling cascade, thereby activating nociceptive neurons and enhancing their excitability.
The data we presented from dissociated nociceptor-like neurons show that activation of PDGFR leads to inhibition of IM. This slow, noninactivating K+ current possesses biophysical properties that are tuned to control subthreshold excitability in many types of neurons. In nociceptive neurons, the majority of IM is mediated by activation of KV7.2/3 (KCNQ2/3) channels.23,47,51 These channels are active near resting potential, such that at subthreshold potentials, they produce an outward current, thus clamping the resting potential in a hyperpolarized range.24 Inhibition of IM by either XE991 or proinflammatory mediators such as bradykinin and proteases has been shown to sensitize nociceptive neurons by increasing evoked firing and decreasing the action potential threshold.8,40,41 Moreover, we recently demonstrated that inhibition of IM in nociceptive neurons with XE991 leads to a gradual depolarization followed by a slow onset (4-6 minutes) of ongoing action potential firing.8 These findings suggest that inhibition of IM alone in nociceptive neurons, similar to previous reports in CA1 pyramidal cells58,65 and in spinal motor neurons,42 is sufficient to produce spontaneous firing.8 Importantly, these results imply that PDGF-BB–mediated inhibition of IM could underlie PDGF-BB–induced depolarization and ongoing activity.
There are other potential mechanisms that could explain the PDGF-BB–mediated hyperexcitability that we have observed. We ruled out the possibility that PDGF-BB, like bradykinin or histamine, activates transducer channels7,10,60 by showing that application of PDGF did not produce a substantial inward current. It is also unlikely that PDGF affects voltage-gated sodium channels because in this case, one would expect to see a significant increase in (dV/dt)max,11,28,34 which we did not observe after the application of PDGF-BB (Fig. 2 and Table 1). Moreover, we showed that application of PDGF-BB led to a significant increase in the Rin, suggesting that PDGF-BB blocks ion conductances. It is worth mentioning that the PDGF-BB–mediated increase in Rin was significant even under perforated patch conditions, where the increase in Rin might be masked by the nystatin-mediated decrease due to gradual membrane perforation.55 Platelet-derived growth factor could also increase Rin by blocking Cl– conductance. However, considering the reversal potential of Cl– in nociceptive neurons, inhibition of Cl– conductance would likely lead to hyperpolarization.19 We cannot, however, rule out the possibility that PDGF-BB blocks other potassium or leak conductances or activates sodium TTX-resistant persistent sodium channels (Nav 1.9), or low threshold calcium channels which could contribute to its overall effect on excitability.
The above arguments, taken together with our data showing that inhibition of IM by XE991 prevented the effect of PDGF-BB on nociceptive excitability, highlight IM as the most probable target for PDGF-BB action. Moreover, Linley et al. showed that inhibition of IM in vivo led to thermal and mechanical hyperalgesia and produced nocifensive behavior,40,41 similar to the effect of PDGF-BB in vivo reported here. This suggests that PDGF-BB–mediated inhibition of IM could potentially account for the behavioral effects of PDGF.
The question remains, however, whether PDGF facilitation of inflammatory pain is mediated through PDGF-induced inhibition of IM. Our results showing that (1) imatinib prevented PDGF-BB–mediated inhibition of IM; (2) imatinib prevented the PDGF-BB–mediated increase in nociceptive excitability; (3) imatinib prevented PDGF-BB–mediated pain hypersensitivity; and (4) imatinib reduced formalin-induced inflammatory pain strongly suggest that PDGF-BB facilitates inflammatory pain by IM inhibition.
Our pharmacological results using the PDGFR blocker imatinib indicate that PDGFR is the starting point of the PDGF-mediated effects, which culminate in the blockade of IM. However, imatinib is not completely selective for the PDGFR. It was previously demonstrated that about 0.1 mg/g of imatinib injected intraplantarly in vivo impaired detection of noxious thermal stimuli by nociceptive neurons through inhibiting c-kit receptors.46 Here, we used a 10,000 X lower dose of imatinib and showed that intraplantar injection of 12 to 60 ng/g of imatinib alone did not exert any effects on the nocifensive behavior or sensitivity to noxious thermal and mechanical stimuli (Fig. 5C, see also Fig. 8B).
Recent transcriptome data show that both PDGFR-α and PDGFR-β are expressed in nociceptive neurons.18 Imatinib inhibits both PDGFR-α and PDGFR-β. Our results using PDGF-BB, which primarily activates PDGFR-β, suggest that PDGFR-β is part of the cascade by which PDGF inhibits IM, and leads to nociceptive hyperexcitability and pain hypersensitivity. The fact that PDGF-BB scavenging produces similar effects to imatinib further supports this hypothesis.
We have shown that PDGFR-β–mediated activation of PI3K is required to inhibit IM. PIP2 is required to open Kv7/M channels, whereas activation of GPCRs causes IM inhibition through PLC-induced PIP2 depletion.62,70 Moreover, IP3-induced [Ca2+]i increases35 and PKC activation38,44 have also been implicated in IM inhibition. Our results demonstrating that PDGF-BB did not cause intracellular Ca2+ mobilization suggest that in nociceptors, the PDGFR is not coupled to PLCγ activation. The results showing that wortmannin prevented PDGF-BB–mediated IM inhibition suggest that the PI3K pathway is responsible for the inhibition of IM. However, the exact mechanism by which PI3K signaling regulates Kv7/M channels is unknown and warrants further investigation.
In addition to the prominent role of PDGF and its receptors in development3,50 and cancer,21 several reports previously described that PDGF participates in inflammatory processes.30,49,68 Here, we demonstrated that scavenging of PDGF significantly reduced inflammatory pain after formalin injection. These data indicate that PDGF contributes to acute inflammatory pain. The latter conclusion is further supported by our results showing that the pharmacological inhibition of PDGFR by imatinib also significantly reduces acute formalin-induced nocifensive behavior. The time kinetics of PDGF effects on dissociated neurons in vitro could theoretically parallel time kinetics of the PDGF-mediated increase in pain sensitivity in vivo (Fig. 1). But, could it explain PDGF-mediated facilitation of inflammatory pain? Increases in expression levels of PDGFR, in particular PDGFR-β, starts minutes after injury and persist throughout the inflammatory process.43 Our data showing that PDGF levels are increased 35 minutes after injection of formalin and that scavenging of PDGF reduced pain in the model of formalin-induced inflammation suggest that PDGF is rapidly released during inflammation. This rapid release, followed by activation of PDGFR, which are acutely expressed after inflammation43 could, through inhibition of IM, participate in the generation of formalin-induced inflammatory pain. It is noteworthy that blockade of PDGFR or scavenging of PDGF-BB significantly reduced, but did not abolish, formalin-induced nocifensive behavior, and provided less analgesia than the systemic administration of either 5 mg/kg or 1.5 mg/kg of morphine (Fig. 8C). These data suggest that PDGF-BB partially contributes to formalin-induced inflammatory pain.
Although PDGF was initially discovered in platelets, and platelets have been shown to play a role in nociceptive sensitization,54 there are several other potential sources for release in both neural and nonneural tissues. Platelet-derived growth factor is expressed in DRG neurons26,57 as well as both myelinated and unmyelinated peripheral nerve fibers.26 Platelet-derived growth factor expression has also been demonstrated in keratinocytes.4 Thus, there are a number of potential sources for PDGF release that could mediate the nociceptive sensitization shown here. Additional experiments will be needed to clarify the specific cell types involved.
To the best of our knowledge, this is the first study suggesting that PDGF plays a role in acute inflammatory pain. Previous reports have shown the involvement of PDGFR-α and PDGF in chronic neuropathic or cancer pain.48,69 Platelet-derived growth factor is released from injured spinal nerves and contributes to neuropathic pain.22 However, the biophysical mechanisms underlying the effect of PDGF in neuropathic pain are not known. In a bone cancer pain model, increased PDGF expression in spinal cord neurons and glial cells was observed, and intrathecal injection of siRNA against PDGF reduced thermal and mechanical hyperalgesia.69 Unrelated to PDGF activity, suppression of Kv7/M-current in spinal cord and DRG neurons has been implicated as an important causal factor for bone cancer pain.16,71 Considering these findings, it will be important to determine whether PDGF also modulates cancer pain and neuropathic pain by IM inhibition.
In conclusion, our findings introduce PDGF as a new “spice” in the inflammatory soup. By targeting the nociceptive Kv7/M-channels through the activation of the PI3K cascade, PDGF causes neuronal hyperexcitability and increases inflammatory pain. Moreover, we demonstrated that this effect was reduced by the clinically used anticancer agent imatinib, suggesting the possibility of an additional clinical use of imatinib for the treatment of inflammatory pain. Our results also position nociceptive Kv7/M-channels as a pivotal target for the convergence of inflammatory mediators in the production of pain.
Conflict of interest statement
The authors have no conflict of interest to declare.
Supplementary Material
Acknowledgments
Support is gratefully acknowledged from the Deutsch-Israelische Projectkooperation program of the Deutsche Forschungsgemeinschaft (DIP) grant agreement BI 1665/1-1ZI1172/12 to 1 (O. Barkai, B. Title, B. Katz, S. Lev and A.M. Binshtok); the Israeli Science Foundation—grant agreement 1470/17 (O. Barkai, B. Title, B. Katz, S. Lev and A.M. Binshtok); the Hoffman Leadership program (O. Barkai); and the NIH grant 1R01DA38860 (H.B. Gutstein).
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/A758.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
References
- [1].Abbadie C, Taylor BK, Peterson MA, Basbaum AI. Differential contribution of the two phases of the formalin test to the pattern of c-fos expression in the rat spinal cord: studies with remifentanil and lidocaine. PAIN 1997;69:101–10. [DOI] [PubMed] [Google Scholar]
- [2].Alhadeff AL, Su Z, Hernandez E, Klima ML, Phillips SZ, Holland RA, Guo C, Hantman AW, De Jonghe BC, Betley JN. A neural circuit for the suppression of pain by a competing need state. Cell 2018;173:140–52.e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev 2008;22:1276–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ansel JC, Tiesman JP, Olerud JE, Krueger JG, Krane JF, Tara DC, Shipley GD, Gilbertson D, Usui ML, Hart CE. Human keratinocytes are a major source of cutaneous platelet-derived growth factor. J Clin Invest 1993;92:671–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Antoniades HN, Galanopoulos T, Neville-Golden J, Kiritsy CP, Lynch SE. Injury induces in vivo expression of platelet-derived growth factor (PDGF) and PDGF receptor mRNAs in skin epithelial cells and PDGF mRNA in connective tissue fibroblasts. Proc Natl Acad Sci USA 1991;88:565–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Antoniades HN, Galanopoulos T, Neville-Golden J, Kiritsy CP, Lynch SE. Expression of growth factor and receptor mRNAs in skin epithelial cells following acute cutaneous injury. Am J Pathol 1993;142:1099–110. [PMC free article] [PubMed] [Google Scholar]
- [7].Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, Patapoutian A. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 2004;41:849–57. [DOI] [PubMed] [Google Scholar]
- [8].Barkai O, Goldstein RH, Caspi Y, Katz B, Lev S, Binshtok AM. The role of Kv7/M potassium channels in controlling ectopic firing in nociceptors. Front Mol Neurosci 2017;10:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell 2009;139:267–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 2006;124:1269–82. [DOI] [PubMed] [Google Scholar]
- [11].Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci 2007;8:451–65. [DOI] [PubMed] [Google Scholar]
- [12].Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji RR, Bean BP, Woolf CJ, Samad TA. Nociceptors are interleukin-1beta sensors. J Neurosci 2008;28:14062–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bonnington JK, McNaughton PA. Signalling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. J Physiol 2003;551:433–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 1980;283:673–6. [DOI] [PubMed] [Google Scholar]
- [15].Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol 2009;156:1185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cai J, Fang D, Liu XD, Li S, Ren J, Xing GG. Suppression of KCNQ/M (Kv7) potassium channels in the spinal cord contributes to the sensitization of dorsal horn WDR neurons and pain hypersensitivity in a rat model of bone cancer pain. Oncol Rep 2015;33:1540–50. [DOI] [PubMed] [Google Scholar]
- [17].Cardenas CG, Del Mar LP, Scroggs RS. Variation in serotonergic inhibition of calcium channel currents in four types of rat sensory neurons differentiated by membrane properties. J Neurophysiol 1995;74:1870–9. [DOI] [PubMed] [Google Scholar]
- [18].Chiu IM, Barrett LB, Williams EK, Strochlic DE, Lee S, Weyer AD, Lou S, Bryman GS, Roberson DP, Ghasemlou N, Piccoli C, Ahat E, Wang V, Cobos EJ, Stucky CL, Ma Q, Liberles SD, Woolf CJ. Transcriptional profiling at whole population and single cell levels reveals somatosensory neuron molecular diversity. Elife 2014;3:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cho H, Yang YD, Lee J, Lee B, Kim T, Jang Y, Back SK, Na HS, Harfe BD, Wang F, Raouf R, Wood JN, Oh U. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci 2012;15:1015–21. [DOI] [PubMed] [Google Scholar]
- [20].Cook AD, Christensen AD, Tewari D, McMahon SB, Hamilton JA. Immune cytokines and their receptors in inflammatory pain. Trends Immunol 2018;39:240–55. [DOI] [PubMed] [Google Scholar]
- [21].Demoulin JB, Essaghir A. PDGF receptor signaling networks in normal and cancer cells. Cytokine Growth Factor Rev 2014;25:273–83. [DOI] [PubMed] [Google Scholar]
- [22].Donica CL, Cui Y, Shi S, Gutstein HB. Platelet-derived growth factor receptor-beta antagonism restores morphine analgesic potency against neuropathic pain. PLoS One 2014;9:e97105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Du X, Gamper N. Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Curr Neuropharmacol 2013;11:621–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Du X, Gao H, Jaffe D, Zhang H, Gamper N. M-type K(+) channels in peripheral nociceptive pathways. Br J Pharmacol 2017;175:2158–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Du X, Hao H, Gigout S, Huang D, Yang Y, Li L, Wang C, Sundt D, Jaffe DB, Zhang H, Gamper N. Control of somatic membrane potential in nociceptive neurons and its implications for peripheral nociceptive transmission. PAIN 2014;155:2306–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Eccleston PA, Funa K, Heldin CH. Expression of platelet-derived growth factor (PDGF) and PDGF alpha- and beta-receptors in the peripheral nervous system: an analysis of sciatic nerve and dorsal root ganglia. Dev Biol 1993;155:459–70. [DOI] [PubMed] [Google Scholar]
- [27].Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol 2005;566:689–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gudes S, Barkai O, Caspi Y, Katz B, Lev S, Binshtok AM. The role of slow and persistent TTX-resistant sodium currents in acute tumor necrosis factor-alpha-mediated increase in nociceptors excitability. J Neurophysiol 2015;113:601–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res 1982;250:71–92. [DOI] [PubMed] [Google Scholar]
- [30].He C, Medley SC, Hu T, Hinsdale ME, Lupu F, Virmani R, Olson LE. PDGFRbeta signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat Commun 2015;6:7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol 1988;92:145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron 2007;55:365–76. [DOI] [PubMed] [Google Scholar]
- [33].Hunskaar S, Hole K. The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. PAIN 1987;30:103–14. [DOI] [PubMed] [Google Scholar]
- [34].Jenerick H. Phase plane trajectories of the muscle spike potential. Biophys J 1963;3:363–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jones S, Brown DA, Milligan G, Willer E, Buckley NJ, Caulfield MP. Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and G alpha q/11. Neuron 1995;14:399–405. [DOI] [PubMed] [Google Scholar]
- [36].Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008;26:127–32. [DOI] [PubMed] [Google Scholar]
- [37].Kidd BL, Urban LA. Mechanisms of inflammatory pain. Br J Anaesth 2001;87:3–11. [DOI] [PubMed] [Google Scholar]
- [38].Lee SY, Choi HK, Kim ST, Chung S, Park MK, Cho JH, Ho WK, Cho H. Cholesterol inhibits M-type K+ channels via protein kinase C-dependent phosphorylation in sympathetic neurons. J Biol Chem 2010;285:10939–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Linley JE, Pettinger L, Huang D, Gamper N. M channel enhancers and physiological M channel block. J Physiol 2012;590:793–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Linley JE, Rose K, Patil M, Robertson B, Akopian AN, Gamper N. Inhibition of M current in sensory neurons by exogenous proteases: a signaling pathway mediating inflammatory nociception. J Neurosci 2008;28:11240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu B, Linley JE, Du X, Zhang X, Ooi L, Zhang H, Gamper N. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl- channels. J Clin Invest 2010;120:1240–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lombardo J, Harrington MA. Nonreciprocal mechanisms in up- and downregulation of spinal motoneuron excitability by modulators of KCNQ/Kv7 channels. J Neurophysiol 2016;116:2114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Majesky MW, Reidy MA, Bowen-Pope DF, Hart CE, Wilcox JN, Schwartz SM. PDGF ligand and receptor gene expression during repair of arterial injury. J Cel Biol 1990;111:2149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Marrion NV. M-current suppression by agonist and phorbol ester in bullfrog sympathetic neurons. Pflugers Arch 1994;426:296–303. [DOI] [PubMed] [Google Scholar]
- [45].Masuda J, Tsuda M, Tozaki-Saitoh H, Inoue K. Intrathecal delivery of PDGF produces tactile allodynia through its receptors in spinal microglia. Mol Pain 2009;5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Milenkovic N, Frahm C, Gassmann M, Griffel C, Erdmann B, Birchmeier C, Lewin GR, Garratt AN. Nociceptive tuning by stem cell factor/c-Kit signaling. Neuron 2007;56:893–906. [DOI] [PubMed] [Google Scholar]
- [47].Mucha M, Ooi L, Linley JE, Mordaka P, Dalle C, Robertson B, Gamper N, Wood IC. Transcriptional control of KCNQ channel genes and the regulation of neuronal excitability. J Neurosci 2010;30:13235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Narita M, Usui A, Narita M, Niikura K, Nozaki H, Khotib J, Nagumo Y, Yajima Y, Suzuki T. Protease-activated receptor-1 and platelet-derived growth factor in spinal cord neurons are implicated in neuropathic pain after nerve injury. J Neurosci 2005;25:10000–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Paniagua RT, Sharpe O, Ho PP, Chan SM, Chang A, Higgins JP, Tomooka BH, Thomas FM, Song JJ, Goodman SB, Lee DM, Genovese MC, Utz PJ, Steinman L, Robinson WH. Selective tyrosine kinase inhibition by imatinib mesylate for the treatment of autoimmune arthritis. J Clin Invest 2006;116:2633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Papadopoulos N, Lennartsson J. The PDGF/PDGFR pathway as a drug target. Mol Aspects Med 2018;62:75–88. [DOI] [PubMed] [Google Scholar]
- [51].Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, Dickenson AH, Brown TA, Burbidge SA, Main M, Brown DA. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci 2003;23:7227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Paulsson J, Ehnman M, Östman A. PDGF receptors in tumor biology: prognostic and predictive potential. Future Oncol 2014;10:1695–708. [DOI] [PubMed] [Google Scholar]
- [53].Reuterdahl C, Sundberg C, Rubin K, Funa K, Gerdin B. Tissue localization of beta receptors for platelet-derived growth factor and platelet-derived growth factor B chain during wound repair in humans. J Clin Invest 1993;91:2065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ringkamp M, Schmelz M, Kress M, Allwang M, Ogilvie A, Reeh PW. Activated human platelets in plasma excite nociceptors in rat skin, in vitro. Neurosci Lett 1994;170:103–6. [DOI] [PubMed] [Google Scholar]
- [55].Robinson DW, Cameron WE. Time-dependent changes in input resistance of rat hypoglossal motoneurons associated with whole-cell recording. J Neurophysiol 2000;83:3160–4. [DOI] [PubMed] [Google Scholar]
- [56].Ronchetti S, Migliorati G, Delfino DV. Association of inflammatory mediators with pain perception. Biomed Pharmacother 2017;96:1445–52. [DOI] [PubMed] [Google Scholar]
- [57].Sasahara M, Fries JW, Raines EW, Gown AM, Westrum LE, Frosch MP, Bonthron DT, Ross R, Collins T. PDGF B-chain in neurons of the central nervous system, posterior pituitary, and in a transgenic model. Cell 1991;64:217–27. [DOI] [PubMed] [Google Scholar]
- [58].Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci USA 2008;105:7869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Shi L, Bian X, Qu Z, Ma Z, Zhou Y, Wang K, Jiang H, Xie J. Peptide hormone ghrelin enhances neuronal excitability by inhibition of Kv7/KCNQ channels. Nat Commun 2013;4:1435. [DOI] [PubMed] [Google Scholar]
- [60].Shim WS, Tak MH, Lee MH, Kim M, Kim M, Koo JY, Lee CH, Kim M, Oh U. TRPV1 mediates histamine-induced itching via the activation of phospholipase A2 and 12-lipoxygenase. J Neurosci 2007;27:2331–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Stein AT, Ufret-Vincenty CA, Hua L, Santana LF, Gordon SE. Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J Gen Physiol 2006;128:509–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 2002;35:507–20. [DOI] [PubMed] [Google Scholar]
- [63].Taylor BK, Peterson MA, Basbaum AI. Persistent cardiovascular and behavioral nociceptive responses to subcutaneous formalin require peripheral nerve input. J Neurosci 1995;15:7575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A 1990;87:2466–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tzour A, Leibovich H, Barkai O, Biala Y, Lev S, Yaari Y, Binshtok AM. Kv7/M channels as targets for lipopolysaccharide-induced inflammatory neuronal hyperexcitability. J Physiol 2017;595:713–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 1998;282:1890–3. [DOI] [PubMed] [Google Scholar]
- [67].Wang Y, Barker K, Shi S, Diaz M, Mo B, Gutstein HB. Blockade of PDGFR-β activation eliminates morphine analgesic tolerance. Nat Med 2012;18:385–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev 2003;83:835–70. [DOI] [PubMed] [Google Scholar]
- [69].Xu Y, Liu J, He M, Liu R, Belegu V, Dai P, Liu W, Wang W, Xia QJ, Shang FF, Luo CZ, Zhou X, Liu S, McDonald J, Liu J, Zuo YX, Liu F, Wang TH. Mechanisms of PDGF siRNA-mediated inhibition of bone cancer pain in the spinal cord. Sci Rep 2016;6:27512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhang H, Craciun LC, Mirshahi T, Rohács T, Lopes CM, Jin T, Logothetis DE. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 2003;37:963–75. [DOI] [PubMed] [Google Scholar]
- [71].Zheng Q, Fang D, Liu M, Cai J, Wan Y, Han JS, Xing GG. Suppression of KCNQ/M (Kv7) potassium channels in dorsal root ganglion neurons contributes to the development of bone cancer pain in a rat model. PAIN 2013;154:434–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.