

Supplemental Digital Content is available in the text.
Keywords: angiogenesis, HB-EGF, hypertrophy, VEGF-B, VEGFR-1
Abstract
Background:
Heart failure, which is a major global health problem, is often preceded by pathological cardiac hypertrophy. The expansion of the cardiac vasculature, to maintain adequate supply of oxygen and nutrients, is a key determinant of whether the heart grows in a physiological compensated manner or a pathological decompensated manner. Bidirectional endothelial cell (EC)–cardiomyocyte (CMC) cross talk via cardiokine and angiocrine signaling plays an essential role in the regulation of cardiac growth and homeostasis. Currently, the mechanisms involved in the EC-CMC interaction are not fully understood, and very little is known about the EC-derived signals involved. Understanding how an excess of angiogenesis induces cardiac hypertrophy and how ECs regulate CMC homeostasis could provide novel therapeutic targets for heart failure.
Methods:
Genetic mouse models were used to delete vascular endothelial growth factor (VEGF) receptors, adeno-associated viral vectors to transduce the myocardium, and pharmacological inhibitors to block VEGF and ErbB signaling in vivo. Cell culture experiments were used for mechanistic studies, and quantitative polymerase chain reaction, microarrays, ELISA, and immunohistochemistry were used to analyze the cardiac phenotypes.
Results:
Both EC deletion of VEGF receptor (VEGFR)-1 and adeno-associated viral vector–mediated delivery of the VEGFR1-specific ligands VEGF-B or placental growth factor into the myocardium increased the coronary vasculature and induced CMC hypertrophy in adult mice. The resulting cardiac hypertrophy was physiological, as indicated by preserved cardiac function and exercise capacity and lack of pathological gene activation. These changes were mediated by increased VEGF signaling via endothelial VEGFR2, because the effects of VEGF-B and placental growth factor on both angiogenesis and CMC growth were fully inhibited by treatment with antibodies blocking VEGFR2 or by endothelial deletion of VEGFR2. To identify activated pathways downstream of VEGFR2, whole-genome transcriptomics and secretome analyses were performed, and the Notch and ErbB pathways were shown to be involved in transducing signals for EC-CMC cross talk in response to angiogenesis. Pharmacological or genetic blocking of ErbB signaling also inhibited part of the VEGF-B–induced effects in the heart.
Conclusions:
This study reveals that cross talk between the EC VEGFR2 and CMC ErbB signaling pathways coordinates CMC hypertrophy with angiogenesis, contributing to physiological cardiac growth.
Clinical Perspective.
What Is New?
We show that angiogenesis induces physiological cardiomyocyte hypertrophy via paracrine signaling between endothelial cells and cardiomyocytes.
We demonstrate that the endothelial cell vascular endothelial growth factor receptor 2-Notch and cardiomyocyte ErbB signaling pathways coordinate cardiac hypertrophy and angiogenesis during physiological myocardial growth.
What Are the Clinical Implications?
There are several ongoing studies examining the utility of exogenous ErbB ligands for the treatment of cardiac diseases. Conversely, ErbB inhibitors are widely used in cancer treatment, which has been associated with cardiotoxicity.
Our study highlights the importance of vascular endothelial growth factor receptor and ErbB signaling in determining whether cardiac growth is physiological or pathological.
Editorial, see p 2585
Heart failure is a major worldwide health problem with an increasing socioeconomic burden. Despite advances in heart failure treatment options, there is an urgent need for new therapies that impact not only the symptoms of the disease but also the underlying pathological processes. Cardiac hypertrophy is an adaptive response of cardiomyocytes (CMCs) to physiological stimuli or pathological stress. Pathological hypertrophy occurs in response to sustained cardiac overload (eg, hypertension or aortic stenosis) or after myocardial infarction, and it is often accompanied by increased fibrosis and heart failure.1 Physiological hypertrophy develops in response to regular exercise training or during pregnancy, and it is reversible, unlike pathological cardiac remodeling.2 Thus, a mechanistic understanding of the molecular events that determine whether the heart grows in a physiological or pathological manner could lead to the development of new therapeutic options for the treatment of heart failure.
The physiological growth of the myocardium requires that myocyte growth be matched by a corresponding expansion of the cardiac vasculature to maintain an adequate supply of oxygen and nutrients to the heart.3 In exercise-induced physiological hypertrophy, the heart preserves its oxygen supply by matching the proportional increases in CMC size and the extent of coronary microvasculature.4,5 In heart failure, however, pathological progression is associated with a mismatch between oxygen supply and demand, because the extent of CMC hypertrophy is associated with vascular rarefaction.6 Both heart size and cardiac function are angiogenesis dependent, and disruption of the coordinated tissue growth and angiogenesis in the heart contributes to progression from adaptive cardiac hypertrophy to heart failure.7 Furthermore, the stimulation of vascular growth has been shown to increase cardiac mass in mice and rats,8–10 but the signaling mechanisms that mediate this growth regulation are largely unknown. Endothelial cells (ECs) are the most abundant cell type in the heart in terms of absolute numbers,11 although CMCs are predominant contributors to heart mass. Although ECs are increasingly recognized as regulators of tissue homeostasis and function, there are significant phenotypic differences between ECs in different tissues, and their importance in the heart has been underappreciated.12
Emerging evidence suggests that perturbed cross talk between CMCs and ECs is involved in the pathogenesis of several heart diseases,13 but the mechanisms of how the ECs affect CMC function are not well understood. In other tissues, such as in adult liver and lung, ECs and EC-derived secreted proteins, referred to as angiocrine factors, have been shown to control tissue growth and regeneration.14–16 In the heart, nitric oxide (NO), endothelin-1, neuregulin (Nrg)-1, and apelin, which are produced by ECs, have been shown to regulate the functions of neighboring CMCs.13,17,18 However, the mechanisms that link angiogenesis and myocyte hypertrophy during cardiac growth have not been elucidated. Our previous studies have shown that an excess of vascular endothelial growth factor B (VEGF-B) expression in the heart leads to increased coronary vasculature and mild cardiac hypertrophy.10,19
Members of the vascular endothelial growth factor (VEGF) family, including VEGF, VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF), show distinct patterns of binding to VEGF receptors (VEGFRs) on ECs to differentially regulate blood and lymphatic vessel development and growth.20 VEGF-VEGFR2 signaling is essential for vascular development and maintenance, whereas VEGFR1 acts an antiangiogenic decoy receptor for VEGF and is required for proper vasculature development.21–23 Here we show that indirect activation of VEGFR2 signaling in cardiac ECs induces angiogenesis and angiocrine release of ErbB receptor ligands. In turn, these ligands activate growth signaling in CMCs. Notably, the hypertrophy induced by angiogenic stimuli is reversible and does not progress to heart failure, thus resembling physiological cardiac growth. These findings place VEGFR2-ErbB signaling at the nexus of pathological and physiological cardiac growth.
Methods
Please refer to the online-only Data Supplement for an expanded Methods section. The data, analytical methods, and most of the study materials will be available to other researchers for purposes of reproducing the results or replicating procedures by contacting the corresponding authors. Some of the transgenic mouse lines were produced by other researchers and used under the restrictions of material transfer agreements.
Mouse Models
All animal experiments were approved by the animal care committee appointed by the District of Southern Finland. All mouse lines and their backgrounds are listed in the online-only Data Supplement. The animal numbers for each experiment are provided in the respective figure legends.
Adeno-Associated Viral Vectors
Recombinant adeno-associated viral vectors (AAV; serotype 9) encoding mouse VEGFB186 (mVEGFB186), mouse PlGF (mPlGF), mouse VEGF164 (mVEGF164), or scrambled control sequences (Ctrl) were constructed and amplified as described previously.24
Blocking VEGFR2 and ErbB Signaling
To block VEGF/VEGFR2 signaling, 8-week-old C57BL/6J wild-type (WT) mice were injected with AAV-Ctrl, AAV-mVEGFB186, or AAV-mPlGF and treated with DC101 monoclonal antibody (30 µg/g, Bio X Cell) every 3 to 4 days. To study the effects of ErbB signaling inhibition on VEGF-B–induced cardiac hypertrophy, recombinant AAV9-mErbB4ECD or AAV9-Ctrl was injected together with AAV9-mVEGFB186 into WT C57BL/6J mice. In addition, another group of mice were treated with 25 mg/kg afatinib dimaleate (BIBW2992, catalog number S7810; Selleckchem) by oral gavage for 2 weeks.
Cell Culture
Human cardiac arterial ECs, human cardiac microvascular ECs, human dermal microvascular ECs, and human umbilical vein ECs were treated with recombinant VEGF (100 ng/mL) or PBS for 4 hours, and RNA was collected after stimulation. In another experiment, human cardiac microvascular ECs were cultured with serum-free medium overnight, then treated with VEGF165 (50 ng/mL) for 1, 3, or 6 hours. Culture media were collected and subjected to ELISA analysis of Nrg1-β1 concentration.
Real-Time Quantitative Polymerase Chain Reaction, Immunohistochemical Staining of the Heart, ELISA, and Western Blotting
For the quantitative polymerase chain reaction, complete primer sequences and TaqMan probe set catalog numbers are listed in Tables I and II in the online-only Data Supplement. Antibodies and procedures used in immunohistochemistry, ELISA, and Western blotting are listed in Table III in the online-only Data Supplement.
Microarray Analyses
The quality of RNA was determined with the Bioanalyzer system (Agilent Technologies) and analyzed on genome-wide Illumina Mouse WG-6 v2 Expression BeadChips (Illumina). The microarray data have been submitted to the Gene Expression Omnibus database, under series accession number GSE110532.
Statistical Analysis
The data sets from individual experiments were analyzed by 2-way ANOVA with Holm-Sidak post hoc test or 2-tailed Student t test. P<0.05 was considered statistically significant. The data are presented as mean±SEM. GraphPad Prism 7 software was used for these statistical analyses.
Results
VEGFR1 Deletion From ECs Induces Angiogenesis and CMC Hypertrophy
We and others have shown that overexpression of VEGF-B or PlGF in the heart expands the coronary vasculature and induces CMC hypertrophy.9,10,19,25 To test whether VEGFR1 contributes to this process, we first studied the effects of VEGF-B in VEGFR1-TK−/− mice, which lack the tyrosine kinase (TK) domain of the VEGFR1, thus rendering the receptor unable to signal. Although cardiac vascular density and CMC size were higher in TK−/− knockout mice than in WT control mice, both were significantly increased after AAV-mediated delivery of VEGF-B (Figure IA through ID in the online-only Data Supplement), which indicates that VEGFR1 TK activity is not required for the effects of VEGF-B in the heart.
We then deleted VEGFR1, the receptor for VEGF, VEGF-B, and PlGF, specifically from ECs by administering tamoxifen to 7- to 8-week-old Pdgfb-CreERT2;VEGFR1fl/fl mice (referred to as R1ECΔ/Δ mice). This led to an ≈80% decrease in VEGFR1 mRNA and protein in the heart, as well as reduced levels of soluble extracellular domain of VEGFR1 in serum (Figure IIF through IIH in the online-only Data Supplement). Considering the EC specificity of the VEGFR1 deletion (Figure IIA through IIC in the online-only Data Supplement), much of the remaining VEGFR1 mRNA and protein was attributed to non-EC cell types, such as macrophages.21 We then administered AAV-VEGF-B to both WT and VEGFR1-deleted mice. The WT mice that received AAV-VEGF-B displayed increased coronary vasculature density and cardiac hypertrophy (Figure 1A through 1F), as previously reported.10 EC deletion of VEGFR1 also resulted in vascular and cardiac growth, and the combination of VEGFR1 deletion and AAV-VEGF-B administration further enhanced both (Figure 1A through 1F), without any effect on overall body weight (Figure IID in the online-only Data Supplement). VEGFR2 protein was increased in cardiac tissues of the VEGF-B transduced, VEGFR1 deleted, and VEGFR1 TK−/− mice (Figure 1G; Figure IE through IG in the online-only Data Supplement). VEGF-B overexpressing hearts also showed increased VEGFR2 phosphorylation (Figure 1H and 1I) but no increase in VEGF mRNA (Figure IIE in the online-only Data Supplement). Furthermore, silencing of VEGFR1 in cultured mouse ECs resulted in a significant increase of VEGFR2 mRNA, which suggests an EC-intrinsic response (Figure IH in the online-only Data Supplement).
Figure 1.
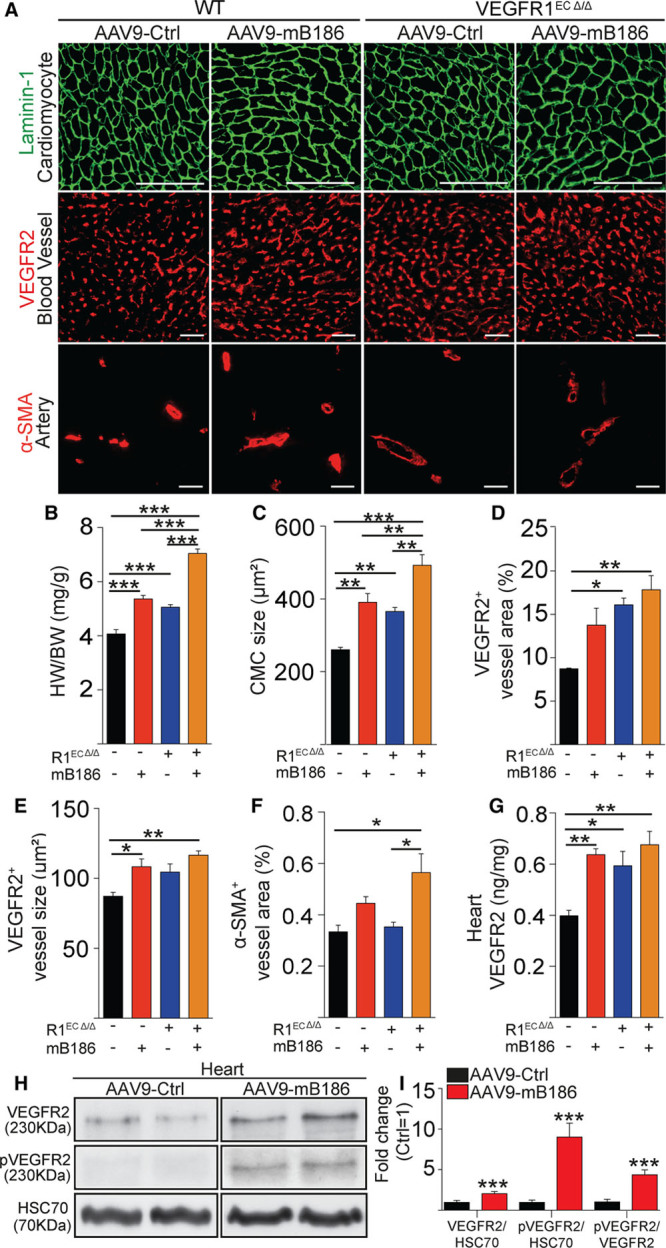
VEGFR1 deletion from endothelial cells induces angiogenesis and cardiomyocyte hypertrophy. A, Representative images of staining for cardiomyocytes (Laminin-1), blood vessels (VEGFR2), and arteries (α-SMA) in AAV-VEGFB186–treated or VEGFR1-deleted hearts (R1ECΔ/Δ). B and C, Quantification of heart weight normalized to body weight and cardiomyocyte size (in µm2). D through F, Quantification of blood vessel area, average capillary size (in µm2), and α-SMA area. G, VEGFR2 protein concentration (in ng/mg) in the heart. H, Western blots of total VEGFR2 and phosphorylated VEGFR2 in VEGF-B–overexpressing and wild-type hearts. Heat shock cognate 70 (HSC70) was used as a loading control. I, Quantification of the Western blot signals is shown as fold change compared with AAV-Ctrl treatment. Data are mean±SEM. AAV9 indicates adeno-associated viral vector serotype 9; CMC, cardiomyocyte; Ctrl, control; HW/BW, heart weight/body weight; mB186, mouse VEGF B-186; pVEGFR2, phosphorylated VEGFR2; α-SMA, α-smooth muscle actin; VEGF-B, vascular endothelial growth factor B; VEGFR2, vascular endothelial growth factor receptor 2; and WT, wild type. Two-way ANOVA (Holm-Sidak test) and Student t test were used, as appropriate; *P<0.05, **P<0.01, ***P<0.001 (N=4 per group). Scale bar, 100 µm.
To compare the effects of VEGF-B overexpression and VEGFR1 deletion in the heart, we performed transcriptomic profiling. AAV-VEGF-B expression, VEGFR1 deletion, or their combination induced significant changes (false discovery rate <0.05) in 497, 212, and 1499 transcripts, respectively. Of the genes upregulated in the VEGFR1-deleted hearts, 73% were also upregulated in the VEGF-B–overexpressing hearts, and 39% of the genes downregulated by VEGFR1 deletion were also found to be decreased by VEGF-B overexpression (Figure 2A). These data suggest that VEGF-B overexpression and endothelial VEGFR1 deletion control similar sets of gene-regulatory pathways to induce vascular growth and cardiac hypertrophy. The transcripts increased in both included Notch ligands (Dll4, Jag1) and receptors (Notch1, Notch4), as well as apelin and APJ, which are recognized to be important for vascular or CMC growth. Stanniocalcin-1 and Esm-1 were the most upregulated genes in all 3 experimental conditions. These results were corroborated by quantitative polymerase chain reaction analysis of samples from an independent experiment (Figure 2B).
Figure 2.
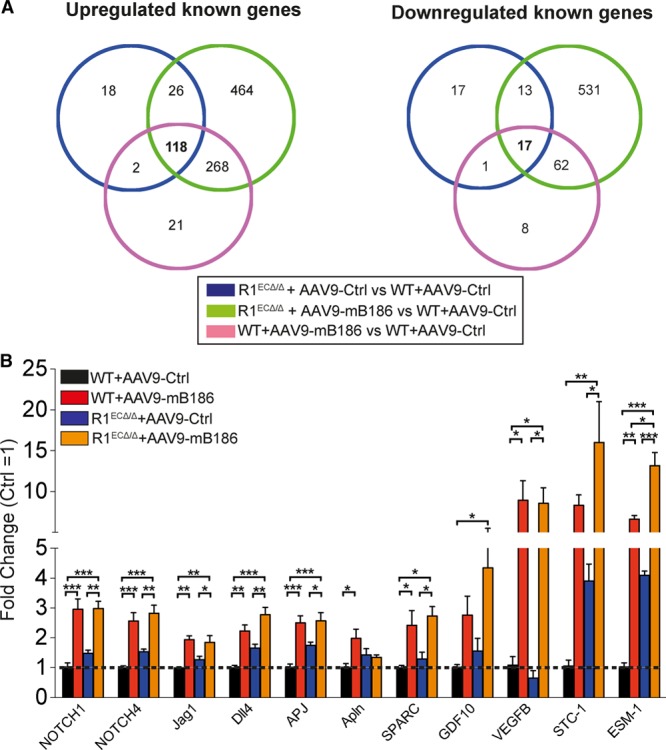
Transcriptomic profiling of VEGFR1-deleted or AAV9-VEGFB186–overexpressing adult mouse heart. A, Venn diagram showing the number of common and unique cardiac gene expression changes in hearts deleted of VEGFR1 (R1ECΔ/Δ) or expressing AAV-VEGFB186 or both, compared with control (WT+AAV9-Ctrl) mice. B, Validation of microarray findings for Notch signaling pathway genes and genes identified by secretome analysis in an independent experiment (normalized to Hprt-1). Data are mean±SEM. AAV9 indicates adeno-associated viral vector serotype 9; Ctrl, control; mB186, mouse VEGF B-186; VEGFR2, vascular endothelial growth factor receptor 2; and WT, wild type. Two-way ANOVA with Holm-Sidak multiple comparison test; *P<0.05, **P<0.01, ***P<0.001 (N=4 per group).
To test whether the effect on cardiac growth was specific for VEGF-B, we administered AAV9-mPlGF2, encoding another VEGFR1 ligand, to WT and R1ECΔ/Δ mice. PlGF induced cardiac hypertrophy in adult mice to a similar extent as VEGF-B (Figure III in the online-only Data Supplement), consistent with previous reports on transgenic mice.9,25 The mice expressing VEGF-B or PlGF displayed no overt health problems, unlike mice treated with a 5-fold lower dose of VEGF.22 We did not observe a significant increase in vascular leakage in various tissues after VEGF-B treatment or VEGFR1 deletion, although their combination resulted in slightly increased leakage which, however, was significantly less than that induced by VEGF (Figure IV in the online-only Data Supplement). Together, these findings indicate that the cardiac effects of VEGFR1 deletion from ECs and the overexpression of a VEGFR1 ligand are similar, in agreement with the concept that VEGFR1 acts as an antiangiogenic decoy receptor.22,23
VEGFR2 Signaling Mediates Angiogenesis-Induced CMC Hypertrophy
Deletion of VEGFR1 or an excess of VEGF-B or PlGF is expected to displace VEGF from VEGFR1 and increase endogenous free VEGF, allowing it to bind to and activate the main angiogenic signaling receptor, VEGFR2.21 To determine whether activation of VEGFR2 was responsible for cardiac hypertrophy in AAV-VEGF-B–treated mice, we first blocked VEGF/VEGFR2 signaling using the anti-VEGFR2 antibody DC101. Strikingly, DC101 treatment, when started concomitantly with the AAV transduction, completely inhibited the VEGF-B–induced cardiac hypertrophy, without affecting VEGF or VEGF-B expression levels (Figure 3A and 3B and Figure VA and VB in the online-only Data Supplement). Next, we assessed whether the VEGF-B–induced cardiac hypertrophy could be reversed by VEGFR2 inhibition. In this experiment, DC101 treatment was started 2 weeks after the AAV injection, when significant hypertrophy was already observed in the VEGF-B–treated mice (Figure 3C). Both the expansion of the coronary vasculature and CMC hypertrophy were completely reversed by DC101, which indicates that angiogenesis-induced cardiac hypertrophy is reversible and mediated by VEGFR2 signaling (Figure 3C through 3F and Figure VC and VD in the online-only Data Supplement). The VEGF-B–induced increase in the mRNAs encoding Pecam1, VEGFRs, APJ, Esm1, and Stc1 (Figure 3G and Figure VE in the online-only Data Supplement) was also reversed by DC101 treatment. Neither VEGF-B or DC101 induced expression of transcripts associated with pathological hypertrophy in the heart (Figure VF in the online-only Data Supplement). AAV-PlGF–induced expansion of the coronary vasculature and CMC hypertrophy were also inhibited by DC101 (Figure VIA through VIE in the online-only Data Supplement). Collectively, the data from VEGFR1 deletion and VEGFR2 signaling inhibition experiments indicate that VEGFR2 activation mediates CMC growth even in the absence of VEGFR1.
Figure 3.
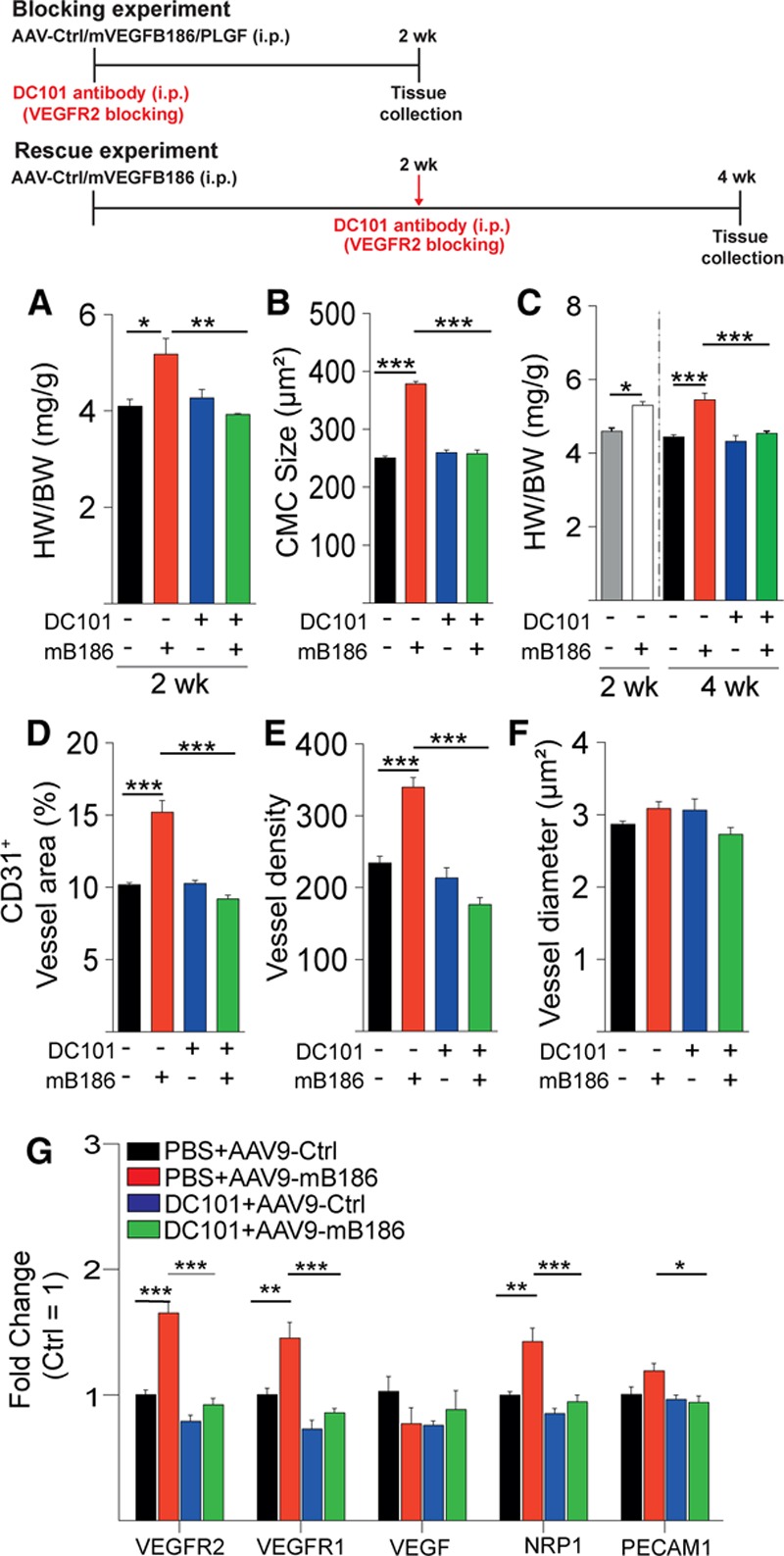
VEGF-B–induced cardiac hypertrophy and vascular growth are inhibited by blocking VEGFR2 signaling. A and B, Heart weight normalized to body weight (HW/BW; in mg/g) and cardiomyocyte (CMC) size (in µm2) in mice injected with AAV-VEGF-B186 or AAV-Ctrl and treated for 2 weeks with VEGFR2-blocking antibody DC101. In A and B, DC101 was started at the same time as the AAV injections (blocking experiment), and in C, 2 weeks after AAV administration, after hypertrophy had developed (rescue experiment; shown as gray and white bars). These 2 groups were analyzed 2 weeks after AAV and before DC101 treatment was started. D through F, Quantification of blood vessel area, density, and average vessel diameter (blocking experiment). G, Quantification of cardiac mRNAs (normalized to Hprt-1). Data are mean±SEM. AAV indicates adeno-associated viral vector; AAV9, AAV serotype 9; Ctrl, control; mB186 and mVEGFB186, mouse VEGF B-186; NRP1, neuropilin 1; PECAM1, platelet and endothelial cell adhesion molecule 1; PLGF, placental growth factor; VEGF-B, vascular endothelial growth factor B; VEGFR2, vascular endothelial growth factor receptor 2; and WT, wild type. Two-way ANOVA (Holm-Sidak test); *P<0.05, **P<0.01, ***P<0.001 (N= 5 per group).
VEGFR2 Regulates EC-CMC Cross Talk
To assess whether the effects of VEGF-B and PlGF on CMCs are mediated by EC-CMC cross talk or by direct activation of VEGFRs in the CMCs, we first analyzed the expression of VEGFRs and their coreceptor, neuropilin-1, in human cardiac cells. VEGFR1 was abundantly expressed in human cardiac microvascular and arterial ECs, and VEGFR2 was mainly expressed in microvascular ECs (Figure 4A through 4C). In contrast, very little if any VEGFR RNA expression was observed in the CMCs or fibroblasts, whereas neuropilin-1 was abundantly expressed in all cell types.
Figure 4.
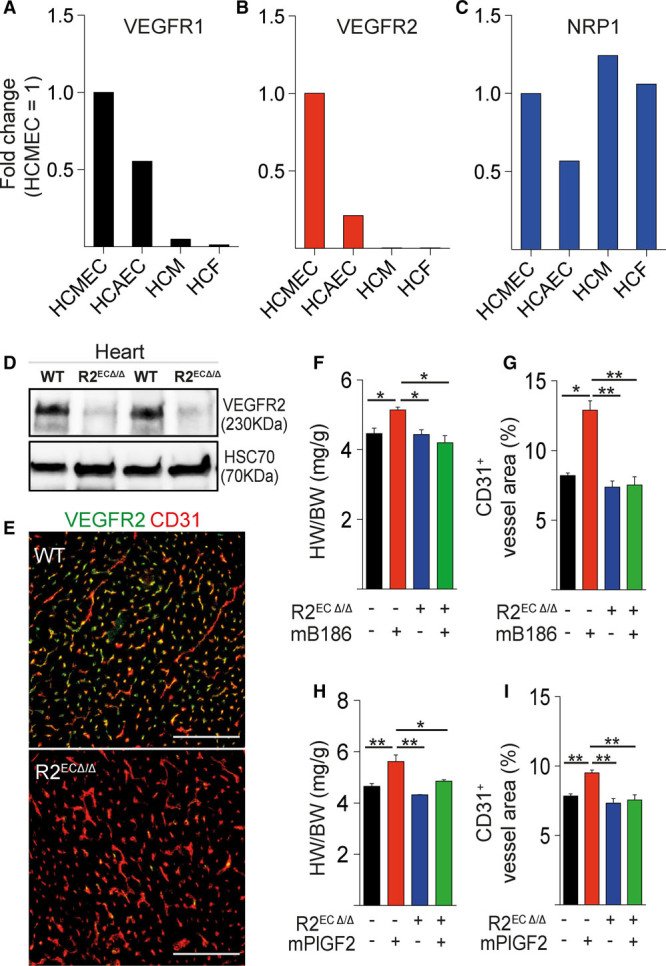
Endothelial VEGFR2 deletion inhibits VEGF-B–induced cardiomyocyte hypertrophy. A through C, Relative VEGFR1, VEGFR2, and NRP1 mRNA expression levels in human coronary microvascular endothelial cells (HCMEC), human coronary arterial endothelial cells (HCAEC), human cardiomyocytes (HCM), and human cardiac fibroblasts (HCF). D and E, VEGFR2 protein levels in R2ECΔ/Δ mouse hearts analyzed by Western blot and immunohistochemistry. Heart to body weight ratio (HW/BW; in mg/g) and blood vessel area (%) are shown in AAV-VEGF-B186 (F and G) and in AAV-PlGF–treated wild-type and R2ECΔ/Δ mice (H and I). Data are mean±SEM. AAV indicates adeno-associated viral vector; HSC70, heat shock cognate 70; mB186, mouse VEGF B-186; mPlGF2, mouse placental growth factor 2; NRP1, neuropilin 1; VEGF-B, vascular endothelial growth factor B; VEGFR1 and VEGFR2, vascular endothelial growth factor receptor 1 and 2; and WT, wild type. Two-way ANOVA (Holm-Sidak test); *P<0.05, **P<0.01, ***P<0.001 (N= 3 per group). Scale bars, 100 µm.
To provide additional evidence that endothelial VEGFR2 signaling mediates CMC growth in a paracrine manner, we treated Cdh5-CreERT2;VEGFR2fl/fl mice with tamoxifen to delete VEGFR2 specifically in the ECs of adult mice (R2ECΔ/Δ mice; Figure 4D and 4E). AAV-VEGF-B and AAV-PlGF were injected into these mice 1 week after VEGFR2 gene deletion. Similar to the DC101 antibody treatment, deletion of VEGFR2 from ECs completely prevented the VEGF-B–induced (Figure 4F and 4G) and PlGF-induced (Figure 4H and 4I) cardiac phenotypes. Deletion of VEGFR1 in CMCs using conditional Myh6-CreERT2 or constitutive αMHC-Cre deleter mice did not alter the cardiac phenotype, in marked contrast to the deletion of VEGFR1 in the ECs (Figure VIIA and VIIB in the online-only Data Supplement). Collectively, these data are consistent with the observation that CMCs express little or no VEGFR1 (Figure 4A and 4B), and they support the hypothesis that VEGFR2 activation in ECs promotes CMC growth. Thus, these data provide multiple lines of evidence in support of the concept that angiogenesis-induced cardiac hypertrophy is mediated by EC-CMC cross talk. Specifically, these data show that VEGFR1 deletion from the ECs, or its occupancy by VEGF-B or PlGF, induces expansion of the coronary vasculature and concomitant CMC growth via increased VEGF-VEGFR2 signaling in ECs.
Paracrine Signaling From ECs to CMCs
VEGFR2 activation has been shown to increase the production of angiocrine factors from ECs to support parenchymal cell regeneration and growth on injury, for example, in the lung or liver.14–16,26 To identify possible angiocrines responsible for CMC growth, we compared the profiles of transcripts encoding secreted proteins in the hearts of VEGFR1-deleted, AAV-VEGF-B–expressing, and control mice. Transcripts encoding a secretion signal peptide that were significantly upregulated by both treatments and their combination were selected for further analysis. Furthermore, we focused only on the transcripts for which increased expression after AAV-VEGF-B injection was inhibited by DC101. Such transcripts included Adam12, Apelin/APJ, Hbegf, Tgfb1, Stc1, Egfl7, Gdf10, Timp3, Angpt2, Esm1, and Klk8 (quantitative polymerase chain reaction data presented in Figure 2B and in Figure VE in the online-only Data Supplement).
Interaction Between VEGFR2 and ErbB Pathways
First, we tested the possible role of apelin and its receptor APJ in VEGF-B–induced hypertrophy by injecting AAV-VEGF-B into mice deficient in the apelin receptor APJ.27 VEGF-B induced similar vascular and cardiac growth in both WT and APJ knockout mice, which suggests that apelin/APJ signaling does not mediate EC-induced CMC growth (Figure VIIIA through VIIID in the online-only Data Supplement).
Next, we focused on Adam12, Hbegf, and Klk8, which all have been linked to ErbB signaling. ErbB receptors and their ligands have been shown to be important for heart development, homeostasis, and regeneration.13,28–30 Adam12 is a protease involved in the shedding of proteins of the epidermal growth factor (EGF) family (eg, HB-EGF [heparin-binding EGF–like growth factor]) from the cell surface,31 which then can bind and activate ErbB receptors on CMCs and induce cardiac growth.30,32 Klk8 (also known as neuropsin), in turn, has been shown to cleave Nrg1 from cell surface, resulting in ErbB4 activation in the brain.33 The upregulation of Adam12, Hbegf, and Klk8 by VEGF-B expression or VEGFR1 deletion suggested that ErbB signaling is responsible for the CMC activation and hypertrophy. Importantly, these changes were fully inhibited by DC101, which indicates that they are downstream of VEGFR2 activation.
To study the interaction between the VEGF and ErbB receptor pathways, human ECs were treated with VEGF. This treatment led to significantly increased Hbegf mRNA levels in human cardiac arterial and microvascular ECs and dermal microvascular ECs (Figure 5A through 5D). In addition, expression of the short form of Adam12 was upregulated after VEGF treatment of human cardiac arterial ECs and human umbilical vein ECs. VEGF treatment of ECs induced the release of Nrg1 into cell culture medium (Figure 5E and Figure IXA and IXB in the online-only Data Supplement), and incubation of CMCs with the conditioned medium from VEGF-treated ECs resulted in increased phosphorylation of Akt, a signaling kinase that promotes myocyte growth,7 at Ser473 (Figure IXC in the online-only Data Supplement). Furthermore, activation of Akt was inhibited by addition of Nrg1 blocking antibody to the culture medium (Figure 5F). These data show that VEGF induces Hbegf and Adam12 production and promotes Nrg1 release from the ECs. Because Nrg1 activates Akt, these data support the hypothesis that ErbB can mediate EC-CMC cross talk (Figure IXD in the online-only Data Supplement).
Figure 5.
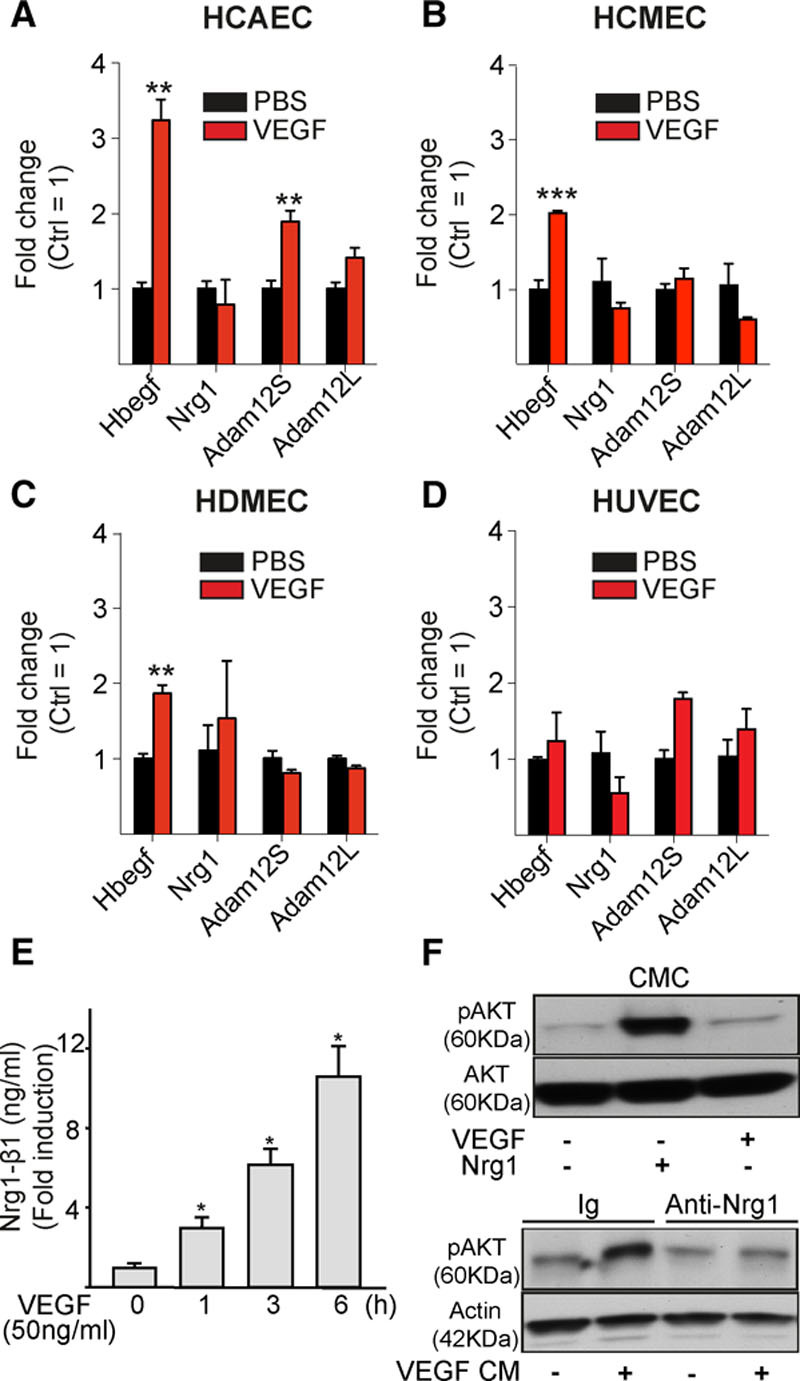
VEGF stimulation induces HB-EGF and ADAM12 mRNA expression and Nrg1 release in endothelial cells. A through D, Human cardiac arterial (HCEAC), cardiac microvascular (HCMEC), dermal microvascular (HDMEC), and umbilical vein endothelial cells (HUVEC) were stimulated with vascular endothelial growth factor (VEGF; 100 ng/mL) for 4 hours, and mRNA expression was analyzed. E, Nrg1 levels in conditioned medium from HCMECs stimulated with VEGF. F, Phosphorylation of Akt in cardiomyocytes (CMC) treated with conditioned medium (CM) from HCMECs stimulated with VEGF. Note that Akt activation is blocked with the anti-Nrg1 antibody. HB-EGF indicates heparin-binding epidermal growth factor–like growth factor; Ig, immunoglobulin; and Nrg1, neuregulin 1. Student t test (A through D) and 1-way ANOVA (E); **P<0.01, ***P<0.001 (N=3 per group).
Next, we designed an AAV vector encoding the extracellular ligand-binding domain of the ErbB4 receptor fused with the immunoglobulin gamma Fc domain (AAV-ErbB4ECD) to trap ErbB ligands in serum and in tissues. WT mice were injected with AAV-VEGF-B together with AAV-ErbB4ECD or a control vector. AAV-VEGF-B treatment induced phosphorylation of EGF receptor and ErbB4 in the heart, and this was attenuated in mice that also received AAV-ErbB4ECD (Figure 6A and 6D through 6F). No effect on the phosphorylation of ErbB3 was observed. HB-EGF protein levels were increased in VEGF-B–expressing hearts, whereas there was no effect on total cardiac Nrg1 or Nrg4 expression (Figure 6B, 6C, 6G, and 6H). However, ErbB4ECD did not alter VEGF-B–induced angiogenesis, which indicates that it only affected signals downstream of VEGFR2 and angiogenesis (Figure XA and XB in the online-only Data Supplement).
Figure 6.
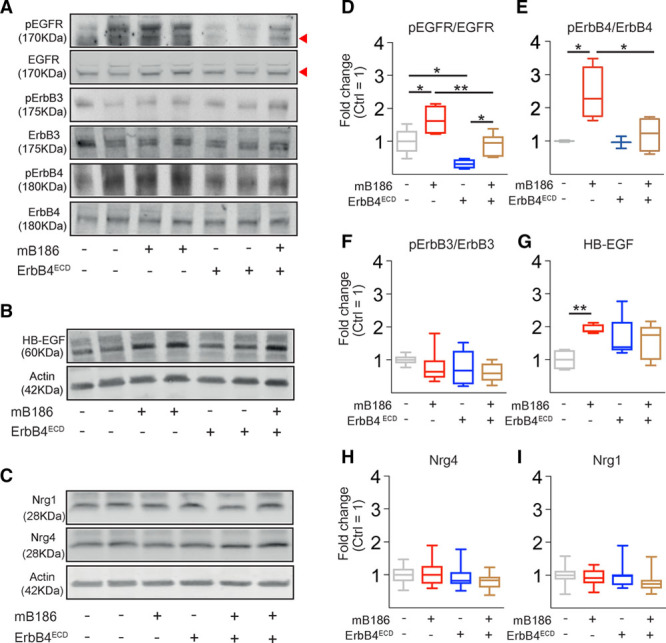
Analysis of ErbB ligands and receptors in the AAV-VEGF-B186 and AAV-ErbB4-ECD–treated mouse heart. A through I, Representative Western blots and quantification of phosphorylation of ErbB receptors normalized to total receptors and expression of HB-EGF, Nrg1, and Nrg4 normalized to β-actin (as fold change, Ctrl=1). For epidermal growth factor receptor (EGFR), the lower 175-kDa band was quantified. Numbers per group: pEGFR, n=4–6; pErbB3, n=7–8; pErbB4, n=3–4; HB-EGF, n=5; and Nrg1 and Nrg 4, n=10–15. AAV indicates adeno-associated viral vector; HB-EGF, heparin-binding epidermal growth factor–like growth factor; mB186, mouse VEGF B-186; Nrg1, neuregulin 1; Nrg4, neuregulin 4; pEGFR, phosphorylated epidermal growth factor receptor; and pErbB3 and pErbB4, phosphorylated ErbB3 and ErbB4. Shapiro-Wilk normality test and Mann–Whitney multiple comparison test; *P<0.05, **P<0.01.
In another experiment, mice were also treated with the EGF receptor/ErbB TK inhibitor afatinib together with AAV-VEGF-B, and the expression of signaling molecules downstream of ErbB was analyzed. The VEGF-B–induced increase in cardiac hypertrophy–associated transcripts, encoding PI3K-p110β, Akt, Carp/Ankrd1, and Tbx3, was blocked, and the VEGF-B–induced decrease of C/EBPβ (CCAAT/enhancer binding protein beta) mRNA was restored by afatinib (Figure 7A). Treatment with DC101 to block VEGFR2 signaling had similar effects (Figure 7B). Importantly, afatinib did not affect the VEGF-B–induced increase of VEGFR2 expression (Figure 7A); thus, its effects were mediated at the level of downstream signaling. Afatinib blocked the VEGF-B–induced phosphorylation of Akt and Erk, and there was a trend toward decreased S6 kinase phosphorylation (Figure 7C and 7D). Notably, downregulation of C/EBPβ is consistent with activation of physiological heart growth.34 Although both AAV-ErbB4ECD and afatinib inhibited the hypertrophy-associated biochemical signals induced by VEGF-B in the heart, their effects on the VEGF-B–induced increase in cardiac mass did not reach statistical significance (Figure XC through XH in the online-only Data Supplement).
Figure 7.

VEGF-B–induced changes in signaling molecules related to cardiac hypertrophy are inhibited by both VEGFR2 and ErbB blocking. A and B, VEGF-B–induced increased transcript expression of PI3K-p110β, Akt, Carp/Ankrd1, and Tbx3 and decreased expression of C/EBPβ in the heart were blocked by both DC101 and afatinib treatment. C through F, Afatinib restored the VEGF-B–induced phosphorylation of Akt and Erk, with a similar trend for S6 kinase (S6K). Data are mean±SEM. AAV9 indicates adeno-associated viral vector serotype 9; Ctrl, control; mB186, mouse VEGF B-186; pErk1/2, phosphorylated Erk1/2; PI3K, phosphoinositide 3-kinase; pS6K, phosphorylated S6 kinase; and VEGF-B, vascular endothelial growth factor B. Two-way ANOVA (Holm-Sidak test); *P<0.05, **P<0.01 (N=4–5 per group).
AAV-VEGF-B Does Not Induce Pathological Cardiac Hypertrophy
To evaluate the long-term effects of hypertrophy in VEGFR1-deleted and VEGF-B–overexpressing mice, we analyzed cardiac function by echocardiography in adult mice 3 weeks, 2.5 months, and 5 months after tamoxifen administration and AAV-VEGF-B treatment. Despite mild cardiac hypertrophy, cardiac function was not significantly altered (Figure XIA and Table IV in the online-only Data Supplement), and there was no change in pathological marker gene expression or in maximal exercise capacity in these mice 5 months after the treatments (Figure XIB and XIC in the online-only Data Supplement). Significantly lower blood pressure was observed in both the VEGFR1-deficient and VEGF-B–overexpressing mice and their combination (Figure XID in the online-only Data Supplement).
Discussion
ECs are emerging as important signaling centers that regulate growth, regeneration, and differentiation of their surrounding cells and tissues. Paracrine (angiocrine) release of molecules from ECs has recently been shown to be fundamentally important for such cross talk in other tissues.35,36 The present findings shed light on the mechanism of how angiogenesis induces physiological CMC growth and highlight the importance of EC-CMC cross talk in cardiac homeostasis. We show that activation of endothelial VEGFR2 signaling in the heart induces vascular growth and contributes to CMC hypertrophy by inducing release of ErbB ligands from ECs, which then act on the CMCs. We propose that angiogenesis-induced hypertrophy exhibits features of physiological hypertrophy, because there was no change in cardiac function, exercise capacity, or the expression of pathological marker transcripts in mice treated with AAV-VEGF-B or in VEGFR1-deleted mice, similar to what we have shown previously in VEGF-B transgenic rats.10 We suspect that this is attributable to the balanced growth of the vasculature and CMCs, whereas in pathological hypertrophy and subsequent heart failure, there is a mismatch that is caused by vascular rarefaction.6 Importantly, both VEGFR1 deletion and AAV-VEGF-B overexpression and their combination decreased blood pressure, which indicates that the effects on cardiac hypertrophy cannot be attributed to increased blood pressure.
VEGF-B and PlGF bind exclusively to VEGFR1, which is considered to act mainly as a decoy receptor that functions to adjust the levels of VEGF, the predominant angiogenic factor, to a physiologically appropriate range.37,38 Recent data also indicate that VEGFR1 is required for the spatial regulation of vascular growth by controlling the formation of anastomoses of vascular sprouts.23 The proposed decoy function of VEGFR1 is supported by our observations that both the deletion of VEGFR1 and the overexpression of its ligands VEGF-B and PlGF produced similar effects on vascular growth and CMC hypertrophy in the heart. Further support for this model comes from the finding that deletion of the TK domain of VEGFR1 was not essential for VEGF-B–induced vascular or cardiac growth. Although our previous studies indicated that constitutive loss of the VEGFR1 TK domain is required for cardiac hypertrophy induced by the VEGF-B167 isoform during mouse development, the present results indicate that TK activity is not essential for VEGF-B–induced cardiac growth in adult mice. Interestingly, we found that both VEGFR1-deficient and TK−/− hearts had increased VEGFR2 levels and that silencing of VEGFR1 in cultured ECs increased VEGFR2 expression. Increased VEGFR2 expression has also been observed recently in ECs of VEGFR3-deleted mice,39 which indicates the EC-intrinsic regulation of VEGFRs. Changes in the global gene expression profiles were highly similar in VEGFR1-deficient and VEGF-B–overexpressing hearts, which demonstrates that both manipulations affected similar angiogenic pathways. Our findings are also consistent with those of Ho et al,40 who showed that global deletion of VEGFR1 using the Rosa26-CreERT2 transgene increases vasculature and VEGFR2 levels in the heart, although possible effects on CMCs or cardiac growth were not reported.
Because we did not find evidence that intracellular VEGFR1 signaling is required for the coronary vascular expansion or cardiac hypertrophy induced by its ligands, we focused on the role of VEGFR2 in EC-CMC cross talk. Our results showed that both VEGFR1 and VEGFR2 were almost exclusively expressed in cardiac ECs, whereas very little, if any, expression was detected in CMCs and cardiac fibroblasts. A recent study using VEGFR1 (Flt1) and VEGFR2 (Flk1) reporter mice demonstrated that VEGFR1 is homogenously expressed in ECs throughout the heart, whereas VEGFR2 showed an epicardial-to-endocardial gradient and was downregulated in quiescent large coronary vessels in adult mice,41 as also indicated by our previous findings.10 Using the VEGFR2-blocking antibody DC101, we showed here that the cardiac hypertrophy induced by VEGF-B or PlGF can be prevented and, importantly, reversed by this inhibition of VEGFR2 signaling. Conditional deletion of VEGFR2 in the vascular endothelium of adult mice provided additional evidence that endothelial VEGFR2 activation is required for vascular expansion and VEGF-B– and PlGF-induced hypertrophy. Although VEGF is known to cause vascular leakage, even when VEGF-B was overexpressed in VEGFR1-deficient mice, we observed only a modest increase in leakage that was several-fold less than what is observed with VEGF treatment. Thus, our combined results suggest that VEGFR1 and its specific ligands fine-tune endogenous VEGF-VEGFR2 signaling in the heart. The resulting physiological vascular growth induces mild CMC hypertrophy (20%–30%) within 1 to 2 weeks, without progression to heart failure.
What are the signals from VEGF-B–stimulated ECs that promote CMC growth? Using whole-genome microarray analyses, we identified upregulated endothelial transcripts that encode a signal peptide for secretion of their protein products. Of the potential candidates, we first tested the role of the apelin/APJ pathway, which has been suggested to regulate pathological heart growth.17 However, we found no difference in AAV-VEGF-B–induced cardiac hypertrophy between WT and APJ-deficient mice. Genetic loss of APJ has been shown to confer resistance to pressure overload–induced myocardial hypertrophy,42 but our results indicated that APJ does not mediate angiogenesis-induced hypertrophy under these experimental conditions.
VEGFR2 activation markedly increased the expression of Adam12 metalloprotease both in vivo and in cultured ECs, and this effect was fully inhibited by the DC101 antibody treatment. Adam12 is known to shed HB-EGF from ECs,31 allowing HB-EGF to bind to and activate ErbB receptors on CMCs to induce myocardial hypertrophy.32 HB-EGF belongs to the EGF family, which also includes neuregulins. Nrg1 and ErbB2 and ErbB4 receptors are essential for cardiac development, and they play a critical role in both healthy and diseased adult heart.28–30 HB-EGF–induced activation of ErbB receptors has been shown to be crucial for normal heart development and function.43,44 AAV-VEGF-B increased the amount of HB-EGF in the heart and induced phosphorylation of EGF and ErbB4 receptors. Furthermore, cell culture studies showed that VEGF stimulation increased the release of Nrg1 from cardiac ECs, and the conditioned medium from ECs induced phosphorylation of Akt in CMCs, which could be blocked by Nrg1 antibodies added to the medium.
A previous report demonstrated that Nrg1 activates mTORC1 (mammalian target of rapamycin complex 1) and Akt in cultured CMCs via ErbB2/ErbB4, which was accompanied by increased glucose uptake and protein synthesis.45 These observations paralleled our previous findings, which demonstrated activation of Akt and mTORC1 and increased glucose uptake in VEGF-B transgenic rat hearts.10 Furthermore, we demonstrated that the AAV-VEGF-B–induced increase in hypertrophy-associated transcripts was restored by blocking the VEGFR2 or ErbB signals. In addition, both DC101 and afatinib inhibited the VEGF-B–induced downregulation of C/EBPβ, which has been shown to be important for the development of physiological hypertrophy.34 Collectively, these findings implicate pathways associated with physiological hypertrophy downstream of angiocrine signaling.
Cardiac overexpression of the extracellular ligand-binding domain of ErbB4, an inhibitory ligand trap, had no effect on VEGF-B–induced angiogenesis but attenuated the phosphorylation of EGF receptor and ErbB4 and had a limited impact on VEGF-B–induced cardiac hypertrophy. Together, the results from cell culture and animal experiments suggest that VEGFR2 activation in ECs leads to ErbB ligand shedding from the ECs and stimulation of their receptor(s) in CMCs. Although this may be one of the links between angiogenesis and CMC activation and growth, it is likely that other signals that are not inhibited by the blocking of ErbB signaling also contribute to the cross talk between ECs and CMCs. For example, NO has been suggested to mediate PlGF-induced hypertrophy, because chemical or genetic inhibition of endothelial NO synthase partially prevented cardiac growth.8,9 Because NO production is increased by VEGFR2 activation, it is possible that increased NO availability and Notch-ErbB pathways downstream of VEGFR2 cooperate to control CMC growth.
The mechanism proposed herein is also supported by studies in other cell types. For example, VEGFR and Notch signaling pathways are well known to interact in both ECs and neural cells.46 In cancer cells and fibroblasts, Notch has been demonstrated to increase the expression of Adam1247 and to induce HB-EGF shedding by Adam12.31 Furthermore, in the developing heart endocardium, Notch increases Hbegf expression, which acts as a paracrine signal for the neighboring myocardium.48 These data collectively support our model, which connects activation of VEGFR2/Notch to increased expression of Adam12 and the release of HB-EGF. One of the genes that was induced by VEGF-B and blocked by DC101 was Klk8 (kallikrein-8/neuropsin). In the hippocampus, Klk8 is required for Nrg1 cleavage and ErbB4 activation,33 and transgenic overexpression of Klk8 has been shown to induce cardiac hypertrophy.49 Thus, the present results suggest that VEGF, VEGF-B, or PlGF secreted by CMCs activate VEGFR2 in cardiac ECs, and this will induce Notch signaling and upregulation of Id1, EphrinB2, Hbegf, Adam12, and Klk8 in ECs. In turn, this leads to the release of ErbB ligands from the ECs, which can then activate ErbB receptors in CMCs. Importantly, inhibition of endothelial VEGFR2, either with antibodies or by genetic ablation, fully blocked these downstream effects. This model is illustrated in Figure 8. Notably, the 2 EGF family ligands, HB-EGF and Nrg1, have distinct receptor binding and expression profiles, and thus, they likely have distinct functions in EC-CMC paracrine cross talk.
Figure 8.
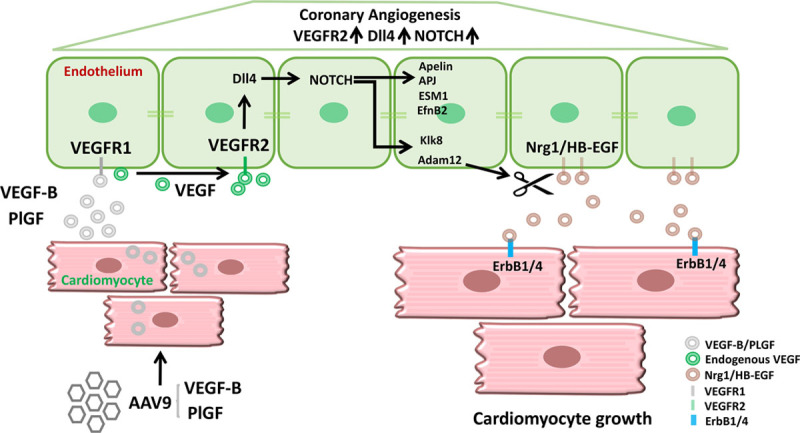
Schematic illustrating endothelial cell to cardiomyocyte cross talk in angiogenesis-induced cardiomyocyte hypertrophy. Adeno-associated viral vector serotype 9 (AAV9) encoding vascular endothelial growth factor B (VEGF-B) or placental growth factor (PlGF) transduces the cardiomyocytes. The secreted VEGF-B and PlGF bind to vascular endothelial growth factor receptor 1 (VEGFR1) in the endothelial cells and increase the bioavailability of endogenous vascular endothelial growth factor (VEGF) to VEGF receptor 2 (VEGFR2). Activation of VEGFR2 in endothelial cells induces the activation of Dll4/NOTCH signaling, which leads to coronary angiogenesis and arteriogenesis. In addition, endothelial cell VEGFR2 activation upregulates the expression of Id1, apelin, APJ, ESM1, EfnB2, Klk8, and Adam12. Adam12 and Klk8–mediated shedding of heparin-binding epidermal growth factor–like growth factor (HB-EGF) and neuregulin 1 (Nrg1) from the endothelial cell surface produces soluble cleaved forms of these proteins. HB-EGF and Nrg1 bind and activate epidermal growth factor receptor 1 (EGFR1 [ErbB1]) and ErbB4 in cardiomyocytes and promote cardiomyocyte growth.
The present results also demonstrate that angiogenesis-induced hypertrophy is physiological, even during prolonged periods of time. AAV-VEGF-B–transduced or VEGFR1-deleted hearts did not show signs of pathological hypertrophy or heart failure, which extends the results we previously obtained in VEGF-B transgenic rats.10 This is likely because of the coordinated growth of both the vasculature and the CMCs by these manipulations. Reversibility is a key hallmark of the physiological hypertrophy induced by exercise or pregnancy. Importantly, by blocking angiogenesis with VEGFR2 antibody, we were able to reverse the AAV-VEGF-B–induced hypertrophy. The present results suggest that vascular growth could be the main trigger of physiological hypertrophy, which makes it fundamentally different from the development of pathological hypertrophy, in which angiogenesis occurs as a compensatory mechanism induced by increased myocardial mass and cardiac hypoxia.
In conclusion, using various genetic and pharmacological animal models and cell culture experiments, we demonstrate that angiogenesis-induced myocardial hypertrophy is mediated via activated VEGFR2 and Notch pathways in ECs, which can induce the release of ligands activating ErbB receptors present in CMCs. The present results further our understanding of the differences between physiological and pathological hypertrophy and highlight the importance of EC-CMC cross talk by identifying a new mechanism for how ECs control the physiological growth of CMCs.
Acknowledgments
We thank Dr Seppo Kaijalainen for cloning the ErbB4ECD vector, Dr Sinem Karaman and Dr Markus Räsänen for discussions and help with the animal experiments, Dr Yoshiaki Kubota for providing the Cdh5-CreERT2 and VEGFR2 floxed mice, Dr Ralf Adams for providing the Pdgfb-CreERT2 mice, and Genentech (Dr Napoleone Ferrara) for providing the VEGFR1 floxed mice. Kirsi Mattinen, Maria Arrano de Kivikko, Tanja Laakkonen, and Tapio Tainola are acknowledged for their excellent technical help. We also thank the Laboratory Animal Center at the University of Helsinki for expert animal care, the Biomedicum Imaging Unit for microscope support, the AAV Gene Transfer and Cell Therapy Core Facility of Biocentrum Finland for the AAV vectors, and Biomedicum Functional Genomics Unit for the microarray experiments. R.K., K.A.H., and M.R. designed and performed the experiments and R.K. and K.A.H. analyzed the data. K.V. and K.E. performed ErbB pathway analyses from in vivo samples and interpreted the data. Y.I. and K.W. designed and performed Nrg1 experiments together with X.P. and D.B.S. in ECs and CMCs. H.K. and N.T. performed the experiments using APJ knockout mice. K.A. designed and supervised the study. R.K., K.A.H., and K.A. wrote the manuscript. All authors have seen, commented on, and accepted the manuscript.
Sources of Funding
Funding for the study was provided by the Jenny and Antti Wihuri Foundation, Novo Nordisk Foundation, Academy of Finland (297245 to Dr Kivelä, 307366 to Dr Alitalo), European Research Council (ERC-2010-AdG-268804 to Dr Alitalo), Marie Curie Actions of European Union Seventh Framework Program (FP7/2007–2013 grant 317250 to Dr Alitalo), the Finnish Foundation for Cardiovascular Research (Dr Kivelä and K.A. Hemanthakumar), the Sigrid Juselius Foundation (Drs Kivelä and Alitalo), the Finnish Cultural Foundation (Dr Kivelä), and the National Institutes of Health (grants HL131006 and HL129120 to Dr Walsh).
Disclosures
None.
Supplementary Material
Footnotes
Dr Kivelä and K.A. Hemanthakumar contributed equally.
Sources of Funding, see page 2583
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/circulationaha.118.036099.
References
- 1.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. doi: 10.1172/JCI62839. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mann N, Rosenzweig A. Can exercise teach us how to treat heart disease? Circulation. 2012;126:2625–2635. doi: 10.1161/CIRCULATIONAHA.111.060376. doi: 10.1161/CIRCULATIONAHA.111.060376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh K, Shiojima I. Cardiac growth and angiogenesis coordinated by intertissue interactions. J Clin Invest. 2007;117:3176–3179. doi: 10.1172/JCI34126. doi: 10.1172/JCI34126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hudlicka O, Brown M, Egginton S. Angiogenesis in skeletal and cardiac muscle. Physiol Rev. 1992;72:369–417. doi: 10.1152/physrev.1992.72.2.369. doi: 10.1152/physrev.1992.72.2.369. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–262. doi: 10.1016/j.yjmcc.2016.06.001. doi: 10.1016/j.yjmcc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–559. doi: 10.1161/CIRCULATIONAHA.114.009625. doi: 10.1161/CIRCULATIONAHA.114.009625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tirziu D, Chorianopoulos E, Moodie KL, Palac RT, Zhuang ZW, Tjwa M, Roncal C, Eriksson U, Fu Q, Elfenbein A, Hall AE, Carmeliet P, Moons L, Simons M. Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J Clin Invest. 2007;117:3188–3197. doi: 10.1172/JCI32024. doi: 10.1172/JCI32024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaba IM, Zhuang ZW, Li N, Jiang Y, Martin KA, Sinusas AJ, Papademetris X, Simons M, Sessa WC, Young LH, Tirziu D. NO triggers RGS4 degradation to coordinate angiogenesis and cardiomyocyte growth. J Clin Invest. 2013;123:1718–1731. doi: 10.1172/JCI65112. doi: 10.1172/JCI65112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kivelä R, Bry M, Robciuc MR, Räsänen M, Taavitsainen M, Silvola JM, Saraste A, Hulmi JJ, Anisimov A, Mäyränpää MI, Lindeman JH, Eklund L, Hellberg S, Hlushchuk R, Zhuang ZW, Simons M, Djonov V, Knuuti J, Mervaala E, Alitalo K. VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol Med. 2014;6:307–321. doi: 10.1002/emmm.201303147. doi: 10.1002/emmm.201303147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD. Revisiting cardiac cellular composition. Circ Res. 2016;118:400–409. doi: 10.1161/CIRCRESAHA.115.307778. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, James D, Ding BS, Schachterle W, Liu Y, Rosenwaks Z, Butler JM, Xiang J, Rafii A, Shido K, Rabbany SY, Elemento O, Rafii S. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell. 2013;26:204–219. doi: 10.1016/j.devcel.2013.06.017. doi: 10.1016/j.devcel.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim SL, Lam CS, Segers VF, Brutsaert DL, De Keulenaer GW. Cardiac endothelium-myocyte interaction: clinical opportunities for new heart failure therapies regardless of ejection fraction. Eur Heart J. 2015;36:2050–2060. doi: 10.1093/eurheartj/ehv132. doi: 10.1093/eurheartj/ehv132. [DOI] [PubMed] [Google Scholar]
- 14.Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, Mittal V, Kobayashi H, Shido K, Lyden D, Sato TN, Rabbany SY, Rafii S. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. 2010;468:310–315. doi: 10.1038/nature09493. doi: 10.1038/nature09493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, Penfold ME, Shido K, Rabbany SY, Rafii S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97–102. doi: 10.1038/nature12681. doi: 10.1038/nature12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, Crystal RG, Simons M, Sato TN, Worgall S, Shido K, Rabbany SY, Rafii S. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell. 2011;147:539–553. doi: 10.1016/j.cell.2011.10.003. doi: 10.1016/j.cell.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamo T, Akazawa H, Komuro I. Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ Res. 2015;117:89–98. doi: 10.1161/CIRCRESAHA.117.305349. doi: 10.1161/CIRCRESAHA.117.305349. [DOI] [PubMed] [Google Scholar]
- 18.Brutsaert DL. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev. 2003;83:59–115. doi: 10.1152/physrev.00017.2002. doi: 10.1152/physrev.00017.2002. [DOI] [PubMed] [Google Scholar]
- 19.Bry M, Kivelä R, Holopainen T, Anisimov A, Tammela T, Soronen J, Silvola J, Saraste A, Jeltsch M, Korpisalo P, Carmeliet P, Lemström KB, Shibuya M, Ylä-Herttuala S, Alhonen L, Mervaala E, Andersson LC, Knuuti J, Alitalo K. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. 2010;122:1725–1733. doi: 10.1161/CIRCULATIONAHA.110.957332. doi: 10.1161/CIRCULATIONAHA.110.957332. [DOI] [PubMed] [Google Scholar]
- 20.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 21.Shibuya M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis. Angiogenesis. 2006;9:225–231. doi: 10.1007/s10456-006-9055-8. doi: 10.1007/s10456-006-9055-8. [DOI] [PubMed] [Google Scholar]
- 22.Robciuc MR, Kivelä R, Williams IM, de Boer JF, van Dijk TH, Elamaa H, Tigistu-Sahle F, Molotkov D, Leppänen VM, Käkelä R, Eklund L, Wasserman DH, Groen AK, Alitalo K. VEGFB/VEGFR1-induced expansion of adipose vasculature counteracts obesity and related metabolic complications. Cell Metab. 2016;23:712–724. doi: 10.1016/j.cmet.2016.03.004. doi: 10.1016/j.cmet.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nesmith JE, Chappell JC, Cluceru JG, Bautch VL. Blood vessel anastomosis is spatially regulated by Flt1 during angiogenesis. Development. 2017;144:889–896. doi: 10.1242/dev.145672. doi: 10.1242/dev.145672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anisimov A, Alitalo A, Korpisalo P, Soronen J, Kaijalainen S, Leppänen VM, Jeltsch M, Ylä-Herttuala S, Alitalo K. Activated forms of VEGF-C and VEGF-D provide improved vascular function in skeletal muscle. Circ Res. 2009;104:1302–1312. doi: 10.1161/CIRCRESAHA.109.197830. doi: 10.1161/CIRCRESAHA.109.197830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Accornero F, van Berlo JH, Benard MJ, Lorenz JN, Carmeliet P, Molkentin JD. Placental growth factor regulates cardiac adaptation and hypertrophy through a paracrine mechanism. Circ Res. 2011;109:272–280. doi: 10.1161/CIRCRESAHA.111.240820. doi: 10.1161/CIRCRESAHA.111.240820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu J, Srivastava K, Wieland M, Runge A, Mogler C, Besemfelder E, Terhardt D, Vogel MJ, Cao L, Korn C, Bartels S, Thomas M, Augustin HG. Endothelial cell-derived angiopoietin-2 controls liver regeneration as a spatiotemporal rheostat. Science. 2014;343:416–419. doi: 10.1126/science.1244880. doi: 10.1126/science.1244880. [DOI] [PubMed] [Google Scholar]
- 27.Ishida J, Hashimoto T, Hashimoto Y, Nishiwaki S, Iguchi T, Harada S, Sugaya T, Matsuzaki H, Yamamoto R, Shiota N, Okunishi H, Kihara M, Umemura S, Sugiyama F, Yagami K, Kasuya Y, Mochizuki N, Fukamizu A. Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J Biol Chem. 2004;279:26274–26279. doi: 10.1074/jbc.M404149200. doi: 10.1074/jbc.M404149200. [DOI] [PubMed] [Google Scholar]
- 28.D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, Lysenko M, Konfino T, Hegesh J, Brenner O, Neeman M, Yarden Y, Leor J, Sarig R, Harvey RP, Tzahor E. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. 2015;17:627–638. doi: 10.1038/ncb3149. doi: 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- 29.Polizzotti BD, Ganapathy B, Walsh S, Choudhury S, Ammanamanchi N, Bennett DG, dos Remedios CG, Haubner BJ, Penninger JM, Kühn B. Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci Transl Med. 2015;7:281ra45. doi: 10.1126/scitranslmed.aaa5171. doi: 10.1126/scitranslmed.aaa5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Odiete O, Hill MF, Sawyer DB. Neuregulin in cardiovascular development and disease. Circ Res. 2012;111:1376–1385. doi: 10.1161/CIRCRESAHA.112.267286. doi: 10.1161/CIRCRESAHA.112.267286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Díaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol. 2013;201:279–292. doi: 10.1083/jcb.201209151. doi: 10.1083/jcb.201209151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, Ohmoto H, Node K, Yoshino K, Ishiguro H, Asanuma H, Sanada S, Matsumura Y, Takeda H, Beppu S, Tada M, Hori M, Higashiyama S. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat Med. 2002;8:35–40. doi: 10.1038/nm0102-35. doi: 10.1038/nm0102-35. [DOI] [PubMed] [Google Scholar]
- 33.Tamura H, Kawata M, Hamaguchi S, Ishikawa Y, Shiosaka S. Processing of neuregulin-1 by neuropsin regulates GABAergic neuron to control neural plasticity of the mouse hippocampus. J Neurosci. 2012;32:12657–12672. doi: 10.1523/JNEUROSCI.2542-12.2012. doi: 10.1523/JNEUROSCI.2542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boström P, Mann N, Wu J, Quintero PA, Plovie ER, Panáková D, Gupta RK, Xiao C, MacRae CA, Rosenzweig A, Spiegelman BM. C/EBPβ controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036. doi: 10.1016/j.cell.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature. 2016;529:316–325. doi: 10.1038/nature17040. doi: 10.1038/nature17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramasamy SK, Kusumbe AP, Adams RH. Regulation of tissue morphogenesis by endothelial cell-derived signals. Trends Cell Biol. 2015;25:148–157. doi: 10.1016/j.tcb.2014.11.007. doi: 10.1016/j.tcb.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 38.Shibuya M. Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem. 2013;153:13–19. doi: 10.1093/jb/mvs136. doi: 10.1093/jb/mvs136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinolainen K, Karaman S, D’Amico G, Tammela T, Sormunen R, Eklund L, Alitalo K, Zarkada G. VEGFR3 modulates vascular permeability by controlling VEGF/VEGFR2 signaling. Circ Res. 2017;120:1414–1425. doi: 10.1161/CIRCRESAHA.116.310477. doi: 10.1161/CIRCRESAHA.116.310477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ho VC, Duan LJ, Cronin C, Liang BT, Fong GH. Elevated vascular endothelial growth factor receptor-2 abundance contributes to increased angiogenesis in vascular endothelial growth factor receptor-1-deficient mice. Circulation. 2012;126:741–752. doi: 10.1161/CIRCULATIONAHA.112.091603. doi: 10.1161/CIRCULATIONAHA.112.091603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurotsu S, Osakabe R, Isomi M, Tamura F, Sadahiro T, Muraoka N, Kojima H, Haginiwa S, Tani H, Nara K, Kubota Y, Ema M, Fukuda K, Suzuki T, Ieda M. Distinct expression patterns of Flk1 and Flt1 in the coronary vascular system during development and after myocardial infarction. Biochem Biophys Res Commun. 2018;495:884–891. doi: 10.1016/j.bbrc.2017.11.094. doi: 10.1016/j.bbrc.2017.11.094. [DOI] [PubMed] [Google Scholar]
- 42.Scimia MC, Hurtado C, Ray S, Metzler S, Wei K, Wang J, Woods CE, Purcell NH, Catalucci D, Akasaka T, Bueno OF, Vlasuk GP, Kaliman P, Bodmer R, Smith LH, Ashley E, Mercola M, Brown JH, Ruiz-Lozano P. APJ acts as a dual receptor in cardiac hypertrophy. Nature. 2012;488:394–398. doi: 10.1038/nature11263. doi: 10.1038/nature11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwamoto R, Yamazaki S, Asakura M, Takashima S, Hasuwa H, Miyado K, Adachi S, Kitakaze M, Hashimoto K, Raab G, Nanba D, Higashiyama S, Hori M, Klagsbrun M, Mekada E. Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc Natl Acad Sci U S A. 2003;100:3221–3226. doi: 10.1073/pnas.0537588100. doi: 10.1073/pnas.0537588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nanba D, Kinugasa Y, Morimoto C, Koizumi M, Yamamura H, Takahashi K, Takakura N, Mekada E, Hashimoto K, Higashiyama S. Loss of HB-EGF in smooth muscle or endothelial cell lineages causes heart malformation. Biochem Biophys Res Commun. 2006;350:315–321. doi: 10.1016/j.bbrc.2006.09.060. doi: 10.1016/j.bbrc.2006.09.060. [DOI] [PubMed] [Google Scholar]
- 45.Pentassuglia L, Heim P, Lebboukh S, Morandi C, Xu L, Brink M. Neuregulin-1β promotes glucose uptake via PI3K/Akt in neonatal rat cardiomyocytes. Am J Physiol Endocrinol Metab. 2016;310:E782–E794. doi: 10.1152/ajpendo.00259.2015. doi: 10.1152/ajpendo.00259.2015. [DOI] [PubMed] [Google Scholar]
- 46.Thomas JL, Baker K, Han J, Calvo C, Nurmi H, Eichmann AC, Alitalo K. Interactions between VEGFR and Notch signaling pathways in endothelial and neural cells. Cell Mol Life Sci. 2013;70:1779–1792. doi: 10.1007/s00018-013-1312-6. doi: 10.1007/s00018-013-1312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Solomon E, Duhachek Muggy S, Sun D, Zolkiewska A. Metalloprotease-disintegrin ADAM12 expression is regulated by Notch signaling via microRNA-29. J Biol Chem. 2011;286:21500–21510. doi: 10.1074/jbc.M110.207951. doi: 10.1074/jbc.M110.207951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacGrogan D, D’Amato G, Travisano S, Martinez-Poveda B, Luxán G, Del Monte-Nieto G, Papoutsi T, Sbroggio M, Bou V, Gomez-Del Arco P, Gómez MJ, Zhou B, Redondo JM, Jiménez-Borreguero LJ, de la Pompa JL. Sequential ligand-dependent notch signaling activation regulates valve primordium formation and morphogenesis. Circ Res. 2016;118:1480–1497. doi: 10.1161/CIRCRESAHA.115.308077. doi: 10.1161/CIRCRESAHA.115.308077. [DOI] [PubMed] [Google Scholar]
- 49.Cao B, Yu Q, Zhao W, Tang Z, Cong B, Du J, Lu J, Zhu X, Ni X. Kallikrein-related peptidase 8 is expressed in myocardium and induces cardiac hypertrophy. Sci Rep. 2016;7:20024. doi: 10.1038/srep20024. doi: 10.1038/srep20024. [DOI] [PMC free article] [PubMed] [Google Scholar]