

Graphical Abstract
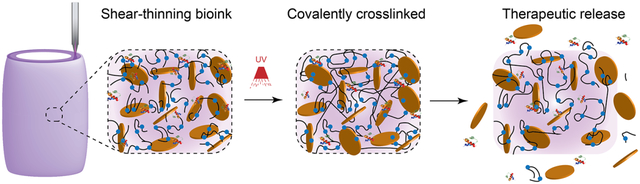
A nanoengineered bioink loaded with therapeutic proteins is designed to direct cell function in a three-dimensional (3D) printed construct. This approach to design biologically active inks to control and direct cell migration can be used to engineer 3D vascularized tissue structure for regenerative medicine.
Keywords: 3D printing, hydrogel bioink, protein therapeutics, nanocomposites, additive manufacturing, two-dimensional (2D) nanomaterials
A nanoengineered bioink loaded with therapeutic proteins is designed to direct cell function in a three-dimensional (3D) printed construct. The bioink is developed from a hydrolytically degradable polymer and two-dimensional (2D) synthetic nanoparticle. The synthesis of poly(ethylene glycol)-dithiothreitol (PEGDTT) via a Michael-like step growth polymerization results in acrylate terminated degradable macromer. The addition of 2D nanosilicates to PEGDTT results in formation of shear-thinning bioinks with high printability and structural fidelity. The mechanical properties, swelling kinetics, and degradation rate of 3D printed constructs can be modulated by changing the ratio of PEG:PEGDTT and nanosilicates concentration. Due to high surface area and charged characteristic of nanosilicates, protein therapeutics can be sequestered in 3D printing structure for prolong duration. Sustained release of pro-angiogenic therapeutics from 3D printed structure, promoted rapid migration of human endothelial umbilical vein cell (HUVEC). This approach to design biologically active inks to control and direct cell behavior can be used to engineer 3D complex tissue structure for regenerative medicine.
1. Introduction
Hydrogels are an integral component for tissue engineering strategies that aim to recapitulate natural tissues and function.[1–4] Polyethylene glycol (PEG) based hydrogels have been evaluated in microfluidics[5], scaffolds for tissue repair[6], and disposable devices as well as cell scaffold matrixes. Use of PEG is advantageous due to low protein adsorption[7], and minimal immunogenicity[8], modifications of physical properties via changes in chain length, end groups, and biofunctionality enhance its usefulness as tissue engineered scaffold. In addition, modification of the degradation profile of PEG enhances the likelihood of adopting PEG as it does not require removal if the matrix is implanted. One such method, has been the introduction of hydrolysable crosslinkers[9] and backbones.[4, 10] To this end, PEG-dithiothreitol (PEGDTT) has previously been developed as a degradable cell scaffold.[11] However, low viscosity of PEGDTT limits the usefulness to pre-cast hydrogels. As tissue engineering progresses from cast hydrogel matrixes to precision deposition via three-dimensional printing, PEG based precursor solutions do not meet the complex mechanical requirements for 3D printing.[12]
Injectable and printable polymer hydrogels with tunable physical properties can be used for a variety of 3D printing and precision deposition processes (i.e. precise delivery of therapeutic protein).[13] Methods to enhance printability in an hydrogel precursor solution include changes in molecular architecture[14], addition of additives such as nanoparticles[13, 15], and/or complete re-design using thermo-responsive hydrogels[16]. Specifically, the combination of synthetic polymers, such as PEG, and nanoparticles give rise to an interesting class of nanocomposite hydrogels that can support cell growth, withstand physiological loadings, and may have enhanced printability compared to polymer hydrogels by themselves.[2, 17, 18]
Two-dimensional (2D) nanosilicates are shown to interact with a range of polymers as well as small molecules and can be used to form a shear-thinning hydrogel for 3D bioprinting.[19] For example, combining nanosilicates with PEG[20, 21], gelatin[22], and kappa-carrageenan[23] result in injectable hydrogels that have been used for cell delivery. Nanosilicates modified the shear-thinning ability and recoverability of polymer solutions permitting use for 3D printing. In addition, these nanosilicate-based hydrogels can be used as vehicles for delivery of protein therapeutics due to charged characteristics. Nanosilicates can act as plug-and-play device to sequester a range of therapeutics for prolong delivery (>3 weeks).[24]
Combination of a degradable polymer with nanoparticle to produce a shear-thinning, printable hydrogel precursor solution with therapeutic holds promise for precision therapeutic delivery.[1] Hydrogel degradation characteristics and subsequent mesh size influence therapeutic diffusion and delivery to surrounding tissues.[3] The fabrication of degradable hydrogels rely on the ester linkage in hydrogel backbone. Previous studies have varied PEG compositions to examine in vivo degradation profiles.[25] For example, PEG has been modified with poly (lactic acid) end groups (PLA) to modulate network degradation[4, 10] and cell adhesion[26] and proliferation.[27] Inclusion of more hydrolytically labile ester groups, as compared to the ether present in PEG backbone, presents an established strategy for degradation.[28] Di-acrylated PEG presents a facile approach for modifying the backbone through which a Michael-like reaction can occur between acrylate and thiol groups present in dithiothreitol (DTT). Through precise control of initial reactants, the acrylate crosslinking functionality can be preserved while creation a hydrolytically labile ester. Controlling polymer chemistry, addition of nanosilicate, and sequestering therapeutics can facilitate fabrication of 3D printable therapeutic bioink.[20, 29]
Herein we describe a simple approach to create a hydrolytically labile, PEG-based bioink that exhibits a rheological profile suitable for precision delivery and 3D printing. Synthesis of hydrolytically labile poly(ethylene glycol)-dithiothreitol (PEGDTT) was achieved through Michael-like addition of dithiothreitol via step growth polymerization. By stoichiometrically imbalancing the reaction towards PEG-diacrylate, the acrylate functionality of the resulting macromer was preserved and the resulting hydrogel solutions were UV curable. The addition of nanosilicates (Laponite XLG) to PEGDTT imparts shear-thinning properties and render it useful for 3D bioprinting applications. The high surface area and charged characteristic of nanosilicates will be used to sequester protein therapeutics for prolong duration within 3D printed structure. Sustained release of pro-angiogenic therapeutics such as vascular endothelial growth factors (VEGF) from 3D printed structure, is expected to promoted rapid migration of human endothelial umbilical vein cell (HUVEC). Through combining PEGDTT with nanosilicates and the inclusion of growth factor, precision deposition of hydrogel solutions was possible, resulting in a facile method for protein therapeutic patterning.
2. Results and Discussion
2.1. Synthesis of shear-thinning bioinks.
Polyethylene glycol diacylate (PEGDA) was reacted overnight with dithiothreitol resulting in a hydrolytically labile macromer (Figure 1a). The reaction of PEGDA with DTT in DCM was efficient as measured by Ellman’s assay, which showed 99.4% of all thiols having been consumed in the reaction. Michael addition reaction between thiol and acrylate groups are widely used as summarized by Nair et al.[30] Specifically, we used a base-catalyzed (triethylamine) Michael addition that facilitates the reaction between the thiol (DTT) and electron-deficient vinyl group (acrylates on PEG). Complete or nearly complete consumption of DTT is highly important within this reaction; DTT has been used to reduce disulfide bonds in proteins and deprotect thiolated DNA.[31] The reaction of PEGDA with DTT forms a stable macromer (Figure 1). Modification of this facile synthesis yields many macromer compositions that dictates degradation rate, mechanical properties (compressive modulus), and swelling properties.[11, 32, 33, 34]
Figure 1.

Synthesis schematic of shear-thinning bioink. (a) A Michael-like addition reaction between poly(ethylene glycol) diacrylate and dithiothreitol to form hydrolytically labile linkages into the polymer chain. Blue dots represent DTT linkages. Confirmation of PEGDTT synthesis via NMR spectroscopy (peaks labeled). Nanosilicates inclusion creating a nanocomposite hydrogel precursor that can be subsequently crosslinked. (b) Shear rate sweep (from 10−3-103 s−1) show the shear thinning characteristics due to addition of nanosilicates to PEGDTT. Power-law parameters determined from shear-rate sweep of bioink. (c) Due to low viscosity of PEGDTT, cannot be used for 3D printing, while addition of nanosilicates render printability of bioink.
We have used a low molecular weight PEGDA (3,350 g/mol) prior to addition of DTT as this molecular weight can be cleared by the body via the kidneys.[35] Hudalla et al. have investigated the PEG to DTT ratio and demonstrated the synthesis process.[11] While Hudulla et al., presents a synthesis in PBS, Cereceres et al. presents a similar synthesis as the one present here.[34] It is suggested by Hudalla et al. that reaction reaches completion within 60 minutes, but due to the difference in solvent (dichloromethane as compared to PBS) it was allowed to complete overnight yielding PEGDTT with 95% acrylation as verified through NMR (Figure 1a, inset). Using NMR and Caruther’s equation, PEGDTT molecular weight is estimated to be between 8,000–12,000 Da. The control for this reaction was achieved by careful dropwise addition of DTT to the PEGDA to allow for complete Michael addition to the PEGDA chains and via a stoichiometric imbalance in favor of PEGDA was to ensure acrylate terminated macromer.
To obtain shear-thinning bioink, we added nanosilicates (4% wt./vol) to PEGDTT. Our earlier studies have shown that 3–4% wt./vol of nanosilicates are required to obtain shear-thinning characteristics.[20, 21] Addition of lower amount (<4% wt/vol) of nanosilicates to PEGDTT will result low viscos sol, while addition of high amount (>4% wt/vol) of nanosilicates will result in stronger gels that required high force for extrusion. Thus, we selected 4% wt/vol of nanosilicates to be added to PEGDTT for bioprinting. Nanoengineered bioinks are created for subsequent testing by exfoliating a double concentration of nanosilicates in photoinitiator solution and then the addition of PEGDTT. To the best of our knowledge, there have been no reported bioink using PEGDTT as the macromer.
Injectability of precursor solutions through a needle is a crucial parameter for 3D printing. High viscosity at low shear rate elucidates if the hydrogel will flow freely when placed on a surface. Ideally, hydrogel precursor solutions should have a decrease in viscosity with an increase in shear rate. PEGDTT is a low viscosity solution across all tested shear rates (Figure 1b). The addition of nanosilicates at 4% wt/vol increases low shear rate viscosity while maintaining low viscosity at higher shear rates.[36] Power-law rheological models were fit to these data and the inclusion of nanosilicates decreases n from ~1 (Newtonian fluid) to ~0.5 (Figure 1b), suggesting that nanosilicates is the cause of shear-thinning behaviors. The ability of nanosilicate addition to PEGDTT on 3D printing is evaluated by extrusion though needle. PEGDTT is not able to retain its shape after printing, while PEGDTT/nSi can be printed in desired shape. In the following section we will evaluate physical and chemical characteristics of these bioink formulations.
2.2. Mechanical stability of bioink and crosslinked 3D structure.
Shear-thinning characteristics of bioink facilitate printing of high-fidelity structure, however covalent crosslinking is need to obtained mechanically robust structure for long-term stability. Bioink formulation (PEGDTT/nSi) demonstrated high printability and the printed structure can be crosslinked after exposure to UV to obtain mechanically stiff network (Figure 2a). To quantitatively determine the printability of bioink formulation, we have designed a peak-hold viscosity tests to mimic the printing process. In peak-hold test, solutions were exposed to sequential shear-rate holds to mimic the rates experienced within the printing apparatus (Figure 2b). The recovery of samples containing nanosilicates was observed while PEGDTT by itself remained fluid. Taken together, these data suggest that PEGDTT/nSi can be used for precision deposition and printing applications.
Figure 2.

3D printing of shear-thinning bioink. (a) Schematic showing interaction of PEGDTT and nanosilicates before and after crosslinking. Inset show printability of PEGDTT/nSi bioink. (b) Schematic of printing process through barrel, needle, and on printing bed. Peak hold experiments to mimic flow during 3D extrusion based printing. Rheological sweeps (frequency and oscillatory stress sweeps) of (c) precursor solutions and (d) crosslinked hydrogels. Note: maroon squares=PEGDTT (10% wt/vol)/nSi (4% wt/vol), red circles=PEG (10% wt/vol)/nSi (4% wt/vol), blue triangles=PEGDTT (10% wt/vol), full colored shapes indicate storage modulus (G’), open shapes indicate loss modulus (G”).
To determine the stability of bioink, storage modulus (G’) and loss modulus (G”) of PEGDTT and PEGDTT/nSi were determined via stress and frequency sweeps (Figure 2c). Storage modulus is a measure of the elastic, or recoverable energy, in the sample while loss modulus is the viscous, or dissipated energy, in the sample. Nanosilicates is gel forming at concentrations above 3% wt./vol when using low ionic solvent.[37] PEGDTT dissolved well in aqueous environments and appears fluidic. With an increase in applied oscillation frequency, PEGDTT storage modulus increased as a result. Addition of nanosilicates results in the solution becoming stiffer and more gel-like as indicated by G’>G”. Both PEG/nSi and PEGDTT/nSi responded similarly to applied oscillation frequency and within a linear viscoelastic region throughout the testing parameters (Figure 2c). Oscillation stress further demonstrated that nanosilicates containing samples are within the linear viscoelastic region. Here, yield stresses were apparent with PEG/nSi having a higher yield stress than PEGDTT/nSi. Yield stresses were 71±11 and 64±15 Pa for PEG/nSi and PEGDTT/nSi, respectfully and are not statistically different.
After 3D printing, hydrogel precursors were exposed to UV light (after 3 min of UV exposure at 365 nm, 7 mW/cm2). Stress and frequency sweeps (Figure 2d) of crosslinked hydrogels increase due to covalent crosslinking that occurred through UV light exposure. Prior to crosslinking, the precursor solution remained stable through nanosilicates-nanosilicates interactions. Terminal acrylate π-bonds are converted to δ-bonds during crosslinking, increasing the overall stiffness of the hydrogel and making it such that they no longer freely flow. PEGDTT/nSi has a lower modulus (G’) than PEGDTT which is contrary to literature. However, inclusion of nanosilicates immediately exposes PEGDTT to a basic environment. While the basic environment is critical for modulation of the degradation, it adversely affects storage modulus. Storage modulus of crosslinked PEGDTT/nSi remained higher than of un-3crosslinked PEGDTT/nSi. Crosslinking is a necessary step for prolonged localization of PEGDTT hydrogels.
2.3. Swelling and degradation of covalently crosslinked hydrogels.
PEG-DTT degrades at the β-thio ester linkage created during the Michael addition and swells considerably due to entropy decrease surrounding free chain movement.[11, 34] Depending on the original molecular weight of PEG used in the reaction and the ratio of PEG:DTT during the reaction, swelling and degradation of hydrogels can be controlled.[11] Here, we focus on one composition of PEG-DTT with a 3:2 (PEG:DTT) ratio during synthesis and the addition of nanosilicates to the matrix. PEG-DA was used as a control hydrogel; furthermore, PEG-DA was incorporated into PEG-DTT hydrogels at 25% weight ratio increments (PEG:PEG-DTT) while maintaining an overall 10% (wt/vol) polymer concentration to demonstrate degradation control after synthesis of macromer (Figure 3a). Samples were 1 mm thick (height) by 5 mm diameter for swelling and degradation studies. Changes in surface to volume ratio of sample, in-fill density, and ratio between sample/solvent can alter the degradation time of 3D printed structure. Nanosilicates (4% wt/vol) were included as experimental treatments. 100% PEG-DTT/nSi samples degraded at an appreciable time scale (7 days). Control hydrogels of PEG-DTT swell and degrade by day 21 where they are completely absent from solution. Inclusion of PEG-DA into the hydrogels resulted in longer lasting hydrogels due to the lack of PEG-DA degradation during the investigated timescale. Therefore, replacing PEG-DTT with PEG-DA can be used to modulate the swelling or degradation kinetics.
Figure 3.

Swelling and degradation of crosslinked nanocomposite hydrogels. (a) Swelling of covalently crosslinked PEGDTT and PEGDTT/nSi hydrogels was monitored over 21 days. The dimensions of the sample used for swelling and degradation studies were 1 mm thick × 5 mm diameter. The ratio of PEG:PEGDTT was modulated to control the physiochemical characteristics of hydrogels (n=5). (b) Effect of PEG:PEGDTT ratio and addition of nanosilicate (4% wt/vol)) on compressive properties of crosslinked hydrogel. PEG:PEGDTT was constant at 10% wt/vol of the solution (n=5). (c) Proposed mechanism of nanoparticle induced degradation of PEGDTT. Adsorbed sodium cation gradually releases from nanosilicates surface resulting in the solution achieving a pH of 9.4 which accelerates PEGDTT degradation. (d) Swelling characteristics of 3D printed structure of PEGDTT (10% wt/vol)/nSi (4% wt/vol) bioink after 1 hr in PBS (n=9, *p<0.05, one-way ANOVA with Tukey post-hoc testing).
During degradation studies, phosphate buffered saline solution (PBS) was replaced every 24 hours. As PBS was replaced, free chains are removed from the samples thereby reducing the swollen weight of samples. Composition of 100% PEG-DTT and 100% PEG-DTT/nSi were ultimately chosen due to the degradation profile. All subsequent experimentation used the 100% PEG-DTT and 100% PEG-DTT/nSi hydrogel compositions due to degradation timescale and potential to release protein growth factors. Compression testing was performed on these formulations to show the ability to change the mechanical properties without changing total polymer concentration (Figure 3b). We have performed these experiments, to demonstrate the modular property of the proposed bioink formulations.
PEGDTT hydrogels containing nanosilicates showed lower degree of swelling compared to PEGDTT hydrogel alone (Figure 3a, b). Previous studies with nanosilicates demonstrate the hydrogels will not swell as much as those not containing nanosilicates.[38] Suggested interactions between the polymer and nanoparticle, hinders swelling of polymer chains; it has been proposed that nanosilicates acts as a physical crosslinker within the hydrogels which generally limits the swelling ability of the material.[39] PEGDTT/nSi hydrogels faster than PEGDTT samples. For example, PEGDTT/nSi was completely degraded within day 7, while PEGDTT was intact till day 7. The addition of PEGDA to PEGDTT/nSi suppresses the degradation rate. In case of PEGDTT:PEGDA(75:25)/nSi, complete degradation was observed by day 21, while PEGDTT:PEGDA(75:25) hydrogels was intact. Accelerated degradation of PEGDTT/nSi is mostly attributed to the chemical composition and cation balance of nanosilicates.
Earlier study have shown that solutions of the nanosilicates will naturally exhibit a pH of 9.4 as described by Thompson and Butterworth.[40] Given that esters are hydrolytically labile, basic solutions exacerbate susceptibility due to large hydroxide concentration. This is a likely explanation for the decreased time interval needed to observe complete degradation of PEGDTT/nSi hydrogels. The ionic structure of nanosilicates evolves with increasing time from having sodium cation adsorbed on the surface of the particle to balance the oxygens that are covalently bound to the silicon to having sodium cation freely in bulk solution (Figure 3c). Sodium cation is in its lowest energy state when surrounded by 5–6 water or hydroxide molecules.[41] Through osmotic pressure, the sodium diffuses away from the surface of nanosilicates, leaving exposed oxygen with two electrons pairs. Stabilization of nanosilicates particles occurs through disassociation of water, forming hydrogen and hydroxide ions. Hydrogen ions will quickly adsorb onto the surface of the particle creating a stable Stern layer.[42] The hydroxide anions will surround the sodium cation present in solution. For every sodium cation released, 4–8 hydrogen ions are needed to stabilize nanosilicates.[43] This imbalance creates a surplus of free hydroxide ions caused pure nanosilicates solutions to evolve in pH and rest at 9.4.[40] Equilibrium pH of nanosilicates (i.e pH 9.4) causes a decrease in time needed for PEGDTT samples to degrade. PBS buffers the solution and prolongs the degradation time compared to pure water. When samples were swollen in water, complete sample degradation occurred in <24 hrs for 1 mm thick, 5 mm diameter samples. Earlier studies have soaked their samples in water overnight to remove salts from hydrogels in order to obtain pure wet and dry weight measurements of samples.[11, 32] Due to the quick erosion time our samples experience in water via the aforementioned pH changes with nanosilicates, we are unable to remove the dissolved salt that are present in our samples. Swelling corresponds with degradation as they are simultaneously occurring.
For 3D printed structures, we also observed similar mechanical, and swelling characteristics (Figure 3d). Several needle gauges were tested and extrudate swell was calculated. 20-gauge needle extrudate swelled 25±9%, while 21-gauge and 23-gauge needle extrudate swelled 47±4% and 148±9%, respectfully (n=9). Volumetric flow rate and printing parameters were kept constant for all experiments. Extrudate swell along with rheological properties such as recovery time of sheared, un-crosslinked precursor solutions and the storage modulus of similar solutions dictate how well a material will print. Here, the print using 20-gauge needle did not collapse upon its own weight nor did it sag.
2.4. Protein Therapeutic loaded 3D printed structure.
Our previous studies have established nanosilicates as a plug-and-play type of therapeutics delivery platform.[44] Nanosilicate have a permanent negative surface charge on each face of the particle and a positive charge along the edges.[2, 18] This facilitate interactions of nanosilicates with a wide range of biomolecules including bone morphogenetic proteins 2 (BMP2), transforming growth factor beta 1 (TGF-β), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF), thus eliminating complex chemical modification.[24, 45] The high surface area and unique surface charge of nanosilicates can results in high-efficiency binding (~100%) and slow release of proteins for prolong duration (>3 weeks).
The ability of nanosilicate to prolong the release of protein therapeutics is leveraged here to load therapeutic protein in 3D printed structure, which is not demonstrated earlier. It is expected that degradation of PEGDTT network will result in release of sequester protein therapeutics (Figure 4a). As a model protein, fluorescein conjugated Bovine serum albumin (FITC-BSA) was used as a large glomerular protein model that can be released from the nanosilicates containing structure. Pre-exfoliated nanosilicates and FTIC-BSA were thoroughly mixed overnight, allowing conjugation to occur. Overtime, the FTIC-BSA is released due to ion exchange and affinity to various ions to adsorb onto nanosilicates surface. As a biological mimic, PBS was used as a sink solution for release of FTIC-BSA; the release was monitored for 28 days. Both images and quantification, showed that 3D printed structure is able to release the sequestered protein over the 7 days and entire protein within 28 days (Figure 4b, c). (Note: sample fabrication size was changed compared to swelling studies in order for samples to maintain structure for 28 days).
Figure 4.

Printing therapeutics in 3D. (a) The high surface area and charged characteristics of nanosilicates are able to sequester protein therapeutics within 3D printing structure. The degradation of printed network result in release of therapeutics. (b) The release of fluorescently labeled protein from 3D printed structure was monitored over 28 days in PEGDTT (10% wt/vol)/nSi (4% wt/vol) hydrogels (no PEG-DA in formulation). Printed structure dimensions ~ 10 mm diameter by 15 mm height (significantly larger mass than used in degradation studies) (c) Sustained release of sequester therapeutics was observed during initial time period, while complete protein was released within 28 days in PEGDTT (10% wt/vol)/nSi (4% wt/vol) hydrogels (n=3, *p<0.05, one-way ANOVA with Tukey post-hoc testing).
The physiological stability of hydrogel network directly influences the release of entrapped protein. By controlling degradation characteristic (via PEG:PEGDTT ratio), release of entrapped protein can be controlled. Alternatively, decreasing the macromer molecular weight could increase the overall time for releasing protein since more covalent crosslinks would be present, resulting in a decrease in mesh size. Fabrication size changes at a constant PEG-DTT/nSi concentrations will not alter crosslink density however is will influence degradation time due to an increase or decrease in overall number of bonds for degradation. An increase in nanosilicates concentration should delay release of protein or small molecule therapeutic at the increase of overall faster degradation of PEGDTT hydrogel. These approaches can be used to modulate the release kinetics of entrapped protein depending on targeted biomedical applications.
2.5. Activity of released protein therapeutic from 3D printing structures
Therapeutic efficacy of VEGF and FGF released from 3D printed structure was measured by the migration of human umbilical vein endothethial cells (HUVECs) across a transwell membrane. Nanosilicates was loaded with VEGF and FGF in similar fashion as albumin. 3D printed structure were placed on the bottom of a well and HUVECs were seeded on the top of transwell inserts (Figure 5a). Cells were stained for nuclei and actin via DAPI and Phalloidin respectively. Cell staining will determine the activity of growth factors, as HUVECs will only migrate through the transwell if the released growth factors are biological active. Samples that did not contain growth factor (negative control) showed little to no cell migration. PEGDTT/nSi control was significantly different from no administered growth factor negative control (p=0.0010). When the samples containing growth factor and not containing growth factor are examined together, it is possible the nanosilicates potentially acts as an angiogenic agent. The PEGDTT/nSi containing growth factors induced similar response to the positive control of exogenously delivered growth factor (Figure 5b, c). PEGDTT/nSi and exogenously delivery growth factor had no difference in total number of migrating cells. PEGDTT was significantly different from exogenous control (n=5, p=0.0044) and PEGDTT/nSi (n=5, p=0.0248). As such, delivery of angiogenic growth factors could be used within PEGDTT scaffolds to promote migration of endothelial cells in wounds.
Figure 5.

Bioactivity of released protein therapeutics from 3D printed structure. (a) Schematic of invasion assay (b)Actin and nuclei staining of migrate HUVECs across transwell. Addition of growth factor to PEGDTT/nSi influences cell migration. SEM images of deposited extracellular matrix (outlined in white) due to cell migration. (c) Quantification of invading cells (n=5, *p<0.05, one-way ANOVA with Tukey post-hoc testing). (d) Live/dead image showing high viability (~85%) of encapsulated cells within 3D printed structure. (e) 3D bioprinted scaffolds loaded with HUVECs stained via cell tracker. (f) VEGF loaded PEGDTT/nSi bioink direct migration of HUVECs encapsulated in surrounding GelMA hydrogels (day 7). Cells migrated towards the PEGDTT/nSi due to the bioavailabilty of VEGF.
Further scanning electron microscopy (SEM) images of the transverse side (non-cell seeded side) of the transwell insert suggest extracellular matrix deposition caused by migratory HUVECs on the growth factor containing scaffolds. Sample without growth factor contained no amounts of deposited extracellular matrix. SEM images coupled with actin/nuclei staining suggested that conjugated VEGF and FGF to nanosilicates retained bioactivity once released.
To demonstrate the potential of bioink for cell studies, we encapsulated HUVECs within the bioink formulation (PEGDTT/nSi). After the 3D printing process, the encapsulated cells showed high cell viability (~85%) (Figure 5d). We also stained these cell via cell tracker and printed cell-laden bioink (Figure 5e). To show the ability of growth factors loaded bioink to direct cell migration, we first printed PEGDTT/nSi loaded with VEGF and then encapsulated the printed structure in gelatin methacrylate (GelMA) loaded with HUVECs (Figure 5f). The migration of cells was monitor using florescence microscopes on day 1 and 7. A uniform dispersion of cells was observed on day 1 in GelMA region. After 7 days, HUVECs migrated towards PEGDTT/nSi structure loaded with VEGF. These results suggest release of bioactive growth factor and migration of HUVECs along a chemotaxis. Promoting retained bioactivity of growth factors could reduce the amount of growth factors to be administered and reduce potentially negative side-effects of supra-physiological growth factor doses. Overall, this work demonstrated that we can control and direct cell migration and potentially obtain vascularized structure by printing therapeutic protein in 3D.
3. Conclusion
Shear-thinning bioink with the ability to sequester and release therapeutic proteins from 3D printed structure have been introduced. The bioink consist of degradable PEGDTT and 2D nanosilicates. The mechanical properties, swelling kinetics, and degradation rate can be modulated via the amount of PEGDA and nanosilicates. Using the cation exchange capacity of nanosilicates, growth factors were sequestered and released from the 3D printed structure. The activity of released protein therapeutics from 3D structure were verified via migration of cells. Overall, this study provides proof of principle to print protein therapeutics in 3D that can be used to control and direct cell functions.
4. Experimental Section
Materials.
Nanosilicates (Laponite XLG), procured from BYK Additives and Instruments, was dried in the oven at 100°C for 4h to ensure limited environmental water swelling of particles. Poly (ethylene glycol) was dried before acrylate modification using procedures previously reported.[46] In short, 20 g PEG (3.4kDa) was dissolved in dichloromethane along with triethylamine (Sigma). Acryloyl chloride (Alfa Aesar) was added dropwise to the solution on ice and stirred for 24 hours. After washing, the solution was precipitated into diethyl ether and dried over vacuum. 1H NMR *300 MHz, CDCl3, δ): 3.62 (s, 297H; -OCH2CH2), 5.81 (dd, 2H, J= 10.5 and 1.2 Hz; -CH=CH2), 6.40 (dd, 2H, J = 17.3 and 1.5 Hz; -CH=CH2) confirmed diacrylation of PEG3.4kDa. PEG-Dithiothreitol (PEGDTT) was synthesized as reported by Cereceres et al.[34] PEG3.4kDa was dissolved in dichloromethane with triethylamine. DTT was dissolved in dichloromethane and added dropwise to the solution. Molar ratio of reactants were as follows PEG:DTT:TEA::3:2:0.9 to ensure a acrylate terminated macromer. Elman’s assay was used to measure free thiol groups after reaction completion. 1H NMR *300 MHz was used to approximate macromer molecule weight, using DTT backbone as the standard.
Bioink formulation.
The desired amount nanosilicates (4% wt/vol) and polymer was dispersed into 18 MΩ water (pH=7.4) and vortexed vigorously for at least 2 minutes. Due to previously reported degradation profile for PEGDTT, PEG3.4kDa was added into the polymer mix at 25% increments though total polymer concentration was kept constant at 10% wt/vol. Samples were identified as percentage (%) of PEGDTT and with or without nanosilicates (PEGDTT/nSi). As such, 10 experimental samples were used for evaluation of: compressive modulus and swelling. At the conclusion of swelling experiments evaluation was conducted to reduce the number of samples for rheology, therapeutic incorporation, and cell studies. Samples were photocrosslinked via the inclusion of 0.3% wt/vol IGRAcure2959 and exposure to 365 nm UV light for 90 seconds at an intensity of 7.0 mW/cm2.
3D Printing.
PEGDTT ± nanosilicates constructs were fabricated utilizing a HYREL System 30M 3D printer. Solution was loaded into a HYREL VOL-25 extruder (HYREL L.L.C., Norcross, GA) equipped with a luer lock adapter and 23 gauge blunted stainless steel needle (Jensen Global Inc, Santa Barbara, CA). Once connected to the printer, constructs were modeled in Solidworks 3D CAD Design, exported as an STL file, and imported into Slic3r version 1.2.9. Overall, this process converts the Solidworks design into layer-by-layer instructions for the printer, or G-code. The G-code files are then imported into HYREL’s proprietary software (Repetrel Rev2.828) and printed at room temperature onto glass slides. Upon completion, glass slides were placed under a UV lamp and photo-crosslinked for 150 seconds at an intensity of 25 mw/cm2.
Hydrogel Swelling.
Samples were cut into 5 mm circles (1 mm thick) and allowed to swelling in 1mL PBS at 37°C for 1, 3, 5, 7, 14, or 21 days with PBS replaced daily for the first 7 days and every-other day thereafter. Weight measurements were taken before swelling (W0) and after swelling (Ws). Comparing the wet weight to dry weight, swelling (%) was calculated according as (Ws - W0)/W0
Mechanical Analysis.
An ADMET eXpert 7600 Single Column Testing System equipped with 25 lb load cell was used for compression testing. Strain rate of 1 mm/min was used to compress the samples 50% of original height. The compressive modulus was calculated and plotted versus the sample composition. All samples were used for mechanical testing to ensure proper synthesis and trends with inclusion of PEG were being observed.
Rheological Analysis.
A Discovery Hybrid Rheometer 2 (DHR-2) (TA Instruments) with attached 40 mm parallel plate at gap height of 0.25 mm and 25°C was used for all experiments. Rotational shear rate sweeps were executed between 10−3-103 s−1 to determine the power law region. Power-law parameter n (flow behavior index) and K (flow consistency index) were calculated using TRIOS software (TA Instruments). Rotational time sweeps were executed at three different shear rates (s−1) in sequential order: 10−2 (60 s), 3000 (5s), 10−2 (120s) to determine shear recovery of solutions.[20] Time to 80% recovery was manually observed/calculated.[47] Oscillatory shear stress sweeps between 10−1-103 performed at 1 Hz and frequency sweeps between 100-102 performed at 10 Pa to further validate yield points and investigate dependence on frequency.
Therapeutic incorporation.
Fluorescein conjugated Bovine serum albumin (FITC-BSA) was used as a model protein. Nanosilicates were allowed to exfoliate for 24 hours. FITC-BSA was added to the solution such that final concentration was 140ng/1 mL. After overnight incorporation, polymer was added to the solutions and 100 μL gels were formed. Samples were placed in 1 mL of PBS at 37 °C and supernatant removed at Day 1, 3, 7 and 21. FTIC-BSA release was measured using fluorescence well plate reader (TECAN Infinite® 200 PRO, Ex/Em: 494 nm/520 nm). Similarly, vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) were incorporated into hydrogel samples for in vitro testing.
In vitro Studies.
To test therapeutic efficacy, migration of HUVECs through 8 um transwell inserts was monitored. Samples containing 40 ng/mL VEGF and 400 ng/mL FGF were UV crosslinked and cut out to obtain 8 mm circles, 1 mm thick. Positive control consisted of exogenously delivery VEGF (0.5 uL/mL) and FGF (2 uL/mL). Experimental negative controls were composed of PEGDTT and PEGDTT/nSi. Human umbilical vein endothethial cells (HUVECs) were seeded on top of transwell at a density of 10,000 cells/mL and allowed to adhere. Wells were then inserted into appropriate treatments, allowed to proceed for 24 hours, then fixed with 10% neutral buffered formalin. Subsequently cells were treated with 1X Triton X for 30 min, washed with PBS and then DAPI was added for 5 min. Cells were washed and Phalloidin was added for 20 min. All well were them imaged under a Nikon Eclipse TE2000-S Microscope at 20X. Afterwards, gold plating of wells was completed and a JEOL (JCM-5000: Benchtop SEM) scanning electron microscope (SEM) was used to visual deposited extra cellular matrix.
Statistical Analysis.
For rheology data sets, the data was not pre-processed and a representative curve are shown for each prescribed composition. Sample sizes for other data sets are as follows: swelling %: n =3, Compressive modulus: n=3, Extrudate swell: n = 9, cumulative release: n=5. Determination of statistical significance between multiple groups was completed via analysis of variance (ANOVA) with Tukey post-hoc testing method. Significant p-values were considered <0.05 unless otherwise noted. All analysis was completed in GraphPad Prism (San Diego, CA).
Acknowledgements
AKG would like to acknowledge financial support from the National Science Foundation (CBET 1705852), and the National Institute of Health (DP2 EB026265). The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Contributor Information
Charles W. Peak, Biomedical Engineering, Dwight Look College of Engineering, Texas A&M University, College Station, TX 77843
Kanwar Abhay Singh, Biomedical Engineering, Dwight Look College of Engineering, Texas A&M University, College Station, TX 77843.
Mu’ath Adlouni, Biomedical Engineering, Dwight Look College of Engineering, Texas A&M University, College Station, TX 77843.
Jeffrey Chen, Biomedical Engineering, Dwight Look College of Engineering, Texas A&M University, College Station, TX 77843.
Akhilesh K. Gaharwar, Biomedical Engineering, Dwight Look College of Engineering, Texas A&M University, College Station, TX 77843 Material Science and Engineering, Dwight Look College of Engineering, Texas A&M University, College Station, TX 77843; Center for Remote Health Technologies and Systems, Texas A&M University, College Station, TX 77843.
References
- [1].Drury JL, Mooney DJ, Biomaterials 2003, 24, 4337. [DOI] [PubMed] [Google Scholar]
- [2].Gaharwar AK, Peppas NA, Khademhosseini A, Biotechnology and Bioengineering 2014, 111, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hoare TR, Kohane DS, Polymer 2008, 49, 1993. [Google Scholar]
- [4].Peak CW, Nagar S, Watts RD, Schmidt G, Macromolecules 2014, 47, 6408. [Google Scholar]
- [5].Zhan W, Seong GH, Crooks RM, Anal. Chem 2002, 74, 4647; [DOI] [PubMed] [Google Scholar]; Cuchiara MP, Allen ACB, Chen TM, Miller JS, West JL, Biomaterials 2010, 31, 5491. [DOI] [PubMed] [Google Scholar]
- [6].Zhu JM, Biomaterials 2010, 31, 4639; [DOI] [PMC free article] [PubMed] [Google Scholar]; Zustiak SP, Leach JB, Biomacromolecules 2010, 11, 1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Michel R, Pasche S, Textor M, Castner DG, Langmuir : the ACS journal of surfaces and colloids 2005, 21, 12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schellekens H, Hennink WE, Brinks V, Pharm Res-Dord 2013, 30, 1729. [DOI] [PubMed] [Google Scholar]
- [9].Lee WK, Ichi T, Ooya T, Yamamoto T, Katoh M, Yui N, J. Biomed. Mater. Res. Part A 2003, 67A, 1087. [DOI] [PubMed] [Google Scholar]
- [10].Dai XS, Chen X, Yang L, Foster S, Coury AJ, Jozefiak TH, Acta Biomater. 2011, 7, 1965. [DOI] [PubMed] [Google Scholar]
- [11].Hudalla GA, Eng TS, Murphy WL, Biomacromolecules 2008, 9, 842. [DOI] [PubMed] [Google Scholar]
- [12].Li HJ, Liu SJ, Li L, International Journal of Bioprinting 2016, 2, 54. [Google Scholar]
- [13].Chimene D, Lennox KK, Kaunas RR, Gaharwar AK, Ann Biomed Eng 2016, 44, 2090. [DOI] [PubMed] [Google Scholar]
- [14].Pekkanen AM, Mondschein RJ, Williams CB, Long TE, Biomacromolecules 2017, 18, 2669. [DOI] [PubMed] [Google Scholar]
- [15].Murphy SV, Atala A, Nature Biotechnology 2014, 32, 773; [DOI] [PubMed] [Google Scholar]; Jaiswal MK, Carrow JK, Gentry JL, Gupta J, Altangerel N, Scully M, Gaharwar AK, Advanced Materials 2017, 29, 1702037. [DOI] [PubMed] [Google Scholar]
- [16].Suntornnond R, An J, Chua CK, Macromolecular Materials and Engineering 2017, 302, n/a; [Google Scholar]; Chimene D, Peak C, Gentry J, Carrow J, Cross L, Mondragon E, Cardoso G, Kaunas R, Gaharwar A, ACS Applied Materials & Interfaces 2018, 10, 9957. [DOI] [PubMed] [Google Scholar]
- [17].Guarino V, Gloria A, De Santis R, Ambrosio L, Biomedical Applications of Hydrogels Handbook 2010, 227; [Google Scholar]; Jin R, Dijkstra PJ, Biomedical Applications of Hydrogels Handbook 2010, 203; [Google Scholar]; Murosaki T, Gong JP, Biomedical Applications of Hydrogels Handbook 2010, 285. [Google Scholar]
- [18].Carrow JK, Gaharwar AK, Macromolecular Chemistry and Physics 2015, 216, 248. [Google Scholar]
- [19].Gaharwar AK, Cross LM, Peak CW, Gold KA, Carrow JK, Brokesh A, Singh KA, Advanced Materials 2019, DOI 10.1002/adma.201900332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Peak CW, Stein J, Gold KA, Gaharwar AK, Langmuir 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Peak CW, Carrow JK, Thakur A, Singh A, Gaharwar AK, Cell Mol Bioeng 2015, 8, 404. [Google Scholar]
- [22].Xavier JR, Thakur T, Desai P, Jaiswal MK, Sears N, Cosgriff-Hernandez E, Kaunas R, Gaharwar AK, Acs Nano 2015, 9, 3109; [DOI] [PubMed] [Google Scholar]; Sheikhi A, Afewerki S, Oklu R, Gaharwar AK, Khademhosseini A, Biomaterials Science 2018, 6, 2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Thakur A, Jaiswal MK, Peak CW, Carrow JK, Gentry J, Dolatshahi-Pirouz A, Gaharwar AK, Nanoscale 2016, 8, 12362; [DOI] [PubMed] [Google Scholar]; Wilson SA, Cross LM, Peak CW, Gaharwar AK, ACS Applied Materials & Interfaces 2017, 9, 43449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lokhande G, Carrow JK, Thakur T, Xavier JR, Parani M, Bayless KJ, Gaharwar AK, Acta Biomater. 2018, 70, 35; [DOI] [PMC free article] [PubMed] [Google Scholar]; Howell DW, Peak CW, Bayless KJ, Gaharwar AK, Advanced Biosystems 2018, 1800092. [Google Scholar]
- [25].Browning MB, Cosgriff-Hernandez E, Biomacromolecules 2012, 13, 779; [DOI] [PubMed] [Google Scholar]; Browning MB, Cereceres SN, Luong PT, Cosgriff-Hernandez EM, Journal of biomedical materials research. Part A 2014, 102, 4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Guarino V, Gloria A, De Santis R, Ambrosio L, Composite Hydrogels for Scaffold Design, Tissue Engineering, and Prostheses, 2010. [Google Scholar]
- [27].Cushing MC, Anseth KS, Science 2007, 316, 1133. [DOI] [PubMed] [Google Scholar]
- [28].Rashkov I, Manolova N, Li SM, Espartero JL, Vert M, Macromolecules 1996, 29, 50; [Google Scholar]; Bryant SJ, Anseth KS, Journal of Biomedical Materials Research Part A 2003, 64A, 70. [DOI] [PubMed] [Google Scholar]
- [29].Jalili NA, Jaiswal MK, Peak CW, Cross LM, Gaharwar AK, Nanoscale 2017, 9, 15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nair DP, Podgórski M, Chatani S, Gong T, Xi W, Fenoli CR, Bowman CN, Chemistry of Materials 2014, 26, 724. [Google Scholar]
- [31].Konigsberg W, in Methods in Enzymology, Vol. 25, Academic Press, 1972, 185. [DOI] [PubMed] [Google Scholar]
- [32].Parlato M, Johnson A, Hudalla GA, Murphy WL, Acta Biomater. 2013, 9, 9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Parlato M, Reichert S, Barney N, Murphy WL, Macromolecular Bioscience 2014, 14, 687; [DOI] [PMC free article] [PubMed] [Google Scholar]; Whitely M, Cereceres S, Dhavalikar P, Salhadar K, Wilems T, Smith B, Mikos A, Cosgriff-Hernandez E, Biomaterials 2018, 185, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cereceres S, Touchet T, Browning MB, Smith C, Rivera J, Hook M, Whitfield-Cargile C, Russell B, Cosgriff-Hernandez E, Advances in Wound Care 2015, 4, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yamaoka T, Tabata Y, Ikada Y, Journal of Pharmaceutical Sciences 1994, 83, 601. [DOI] [PubMed] [Google Scholar]
- [36].Peak CW, Stein J, Gold KA, Gaharwar AK, Langmuir 2018, 34, 917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ruzicka B, Zaccarelli E, Soft Matter 2011, 7, 1268. [Google Scholar]
- [38].Takeno H, Kimura Y, Nakamura W, Gels 2017, 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wu CJ, Gaharwar AK, Chan BK, Schmidt G, Macromolecules 2011, 44, 8215. [Google Scholar]
- [40].Thompson DW, Butterworth JT, Colloid Interface Sci. J 1992, 151, 236. [Google Scholar]
- [41].Mähler J, Persson I, Inorganic Chemistry 2012, 51, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].de Carvalho R, Skipper NT, Journal of Chemical Physics 2001, 114, 3727. [Google Scholar]
- [43].Lockhart NC, Colloid Interface Sci. J 1980, 74, 509. [Google Scholar]
- [44].Lokhande G, Carrow JK, Thakur T, Xavier JR, Parani M, Bayless KJ, Gaharwar AK, Acta Biomaterialia 2018, 10.1016/j.actbio.2017.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cross LM, Carrow JK, Ding X, Singh K-A, Gaharwar AK, ACS Applied Materials & Interfaces 2019, 10.1021/acsami.8b17733n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gaharwar AK, Dammu SA, Canter JM, Wu CJ, Schmidt G, Biomacromolecules 2011, 12, 1641. [DOI] [PubMed] [Google Scholar]
- [47].Sommer MR, Alison L, Minas C, Tervoort E, Ruhs PA, Studart AR, Soft Matter 2017, 13, 1794. [DOI] [PubMed] [Google Scholar]