

Abstract
STUDY QUESTION
Is there an increased prevalence of male microchimerism in women with Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome, as evidence of fetal exposure to blood and anti-Müllerian hormone (AMH) from a (vanished) male co-twin resulting in regression of the Müllerian duct derivatives?
SUMMARY ANSWER
Predominant absence of male microchimerism in adult women with MRKH syndrome does not support our hypothesis that intrauterine blood exchange with a (vanished) male co-twin is the pathophysiological mechanism.
WHAT IS KNOWN ALREADY
The etiology of MRKH is unclear. Research on the phenotype analogous condition in cattle (freemartinism) has yielded the hypothesis that Müllerian duct development is inhibited by exposure to AMH in utero. In cattle, the male co-twin has been identified as the source for AMH, which is transferred via placental blood exchange. In human twins, a similar exchange of cellular material has been documented by detection of chimerism, but it is unknown whether this has clinical consequences.
STUDY DESIGN, SIZE, DURATION
An observational case–control study was performed to compare the presence of male microchimerism in women with MRKH syndrome and control women. Through recruitment via the Dutch patients’ association of women with MRKH (comprising 300 members who were informed by email or regular mail), we enrolled 96 patients between January 2017 and July 2017. The control group consisted of 100 women who reported never having been pregnant.
PARTICIPANTS/MATERIALS, SETTING, METHODS
After written informed consent, peripheral blood samples were obtained by venipuncture, and genomic DNA was extracted. Male microchimerism was detected by Y-chromosome–specific real-time quantitative PCR, with use of DYS14 marker. Possible other sources for microchimerism, for example older brothers, were evaluated using questionnaire data.
MAIN RESULTS AND THE ROLE OF CHANCE
The final analysis included 194 women: 95 women with MRKH syndrome with a mean age of 40.9 years and 99 control women with a mean age of 30.2 years. In total, 54 women (56.8%) were identified as having typical MRKH syndrome, and 41 women (43.2%) were identified as having atypical MRKH syndrome (when extra-genital malformations were present). The prevalence of male microchimerism was significantly higher in the control group than in the MRKH group (17.2% versus 5.3%, P = 0.009). After correcting for age, women in the control group were 5.8 times more likely to have male microchimerism (odds ratio 5.84 (CI 1.59–21.47), P = 0.008). The mean concentration of male microchimerism in the positive samples was 56.0 male genome equivalent per 1 000 000 cells. The prevalence of male microchimerism was similar in women with typical MRKH syndrome and atypical MRKH syndrome (5.6% versus 4.9%, P = 0.884). There were no differences between women with or without microchimerism in occurrence of alternative sources of XY cells, such as older brothers, previous blood transfusion, or history of sexual intercourse.
LIMITATIONS, REASON FOR CAUTION
We are not able to draw definitive conclusions regarding the occurrence of AMH exchange during embryologic development in women with MRKH syndrome. Our subject population includes all adult women and therefore is reliant on long-term prevalence of microchimerism. Moreover, we have only tested blood, and, theoretically, the cells may have grafted anywhere in the body during development. It must also be considered that the exchange of AMH may occur without the transfusion of XY cells and therefore cannot be discovered by chimerism detection.
WIDER IMPLICATIONS OF THE FINDINGS
This is the first study to test the theory that freemartinism causes the MRKH syndrome in humans. The study aimed to test the presence of male microchimerism in women with MRKH syndrome as a reflection of early fetal exposure to blood and AMH from a male (vanished) co-twin. We found that male microchimerism was only present in 5.3% of the women with MRKH syndrome, a significantly lower percentage than in the control group (17.2%). Our results do not provide evidence for an increased male microchimerism in adult women with MRKH as a product of intrauterine blood exchange. However, the significant difference in favor of the control group is of interest to the ongoing discussion on microchimeric cell transfer and the possible sources of XY cells.
STUDY FUNDING/COMPETING INTEREST(S)
None.
TRIAL REGISTRATION NUMBER
Dutch trial register, NTR5961.
Keywords: Mayer–Rokitansky–Küster–Hauser syndrome, Müllerian aplasia, microchimerism, etiology, freemartinism
Introduction
Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome is characterized by congenital aplasia of the uterus and the upper part of the vagina. It affects around 1 in 5000 females (Herlin et al., 2016). The diagnosis is usually made in adolescence after the presentation of primary amenorrhea. Further examination reveals vaginal agenesis with absence of the uterus, normal secondary sex characteristics, and normal female 46,XX karyotype (Choussein et al., 2017). It may also be associated with renal and/or skeletal malformations, classified as atypical MRKH syndrome (Oppelt et al., 2006; Rall et al., 2015).
The etiology of the MRKH syndrome is unclear (Patnaik et al., 2015). Embryological evidence supports the hypothesis that this syndrome occurs as a result of failure of Müllerian duct development. In the normal male embryo, the Sertoli cells of the testes produce anti-Müllerian hormone (AMH), resulting in regression of the Müllerian duct. This inhibitory action of AMH on the Müllerian duct starts during the fifth week of pregnancy and is progressive during the critical time window of uterine development (Taguchi et al., 1984). Genetic activation of AMH or its receptor has been implicated as a cause of MRKH syndrome but without any supporting results (Oppelt et al., 2005; Resendes et al., 2001).
In cattle, a similar phenotypical syndrome to MRKH exists: a so-called freemartin. In this infertile female calf, the absence of Müllerian structures occurs due to intrauterine AMH exposure, originating from a male co-twin (Vigier et al., 1984). Vascular connections in the placenta transport AMH from the male to the female calf (Lillie, 1916). As a result of this intrauterine blood exchange via the placenta, the freemartins are ‘chimeras’; in addition to the normal XX cells, they have an extra XY cell line, originating from their co-twin. The term chimerism means that two genetically different cell lines are present in one individual, originating from more than one zygote.
In humans, intrauterine cell trafficking between twins can also result in chimerism (Peters et al., 2017; van Dijk et al., 1996). It is unknown whether this has clinical consequences. Bogdanova et al. (2010) reported a possible case of ‘human freemartinism’ in a female twin with aplasia of the uterus. In this female, blood exchange via the placenta with her brother had resulted in male chimerism (the presence of XY cells) (Bogdanova et al., 2010). In addition, a vanishing twin can leave its traces in the form of microchimerism, in which a second cell population is present at variable concentrations in the surviving fetus (de Bellefon et al., 2010).
It should also be mentioned that fetal–maternal exchange in a normal pregnancy is a well-known source for chimeric cells (Lo et al., 1996). Fetal microchimerism refers to the phenomenon of fetal cells entering the maternal circulation during pregnancy. As the exchange between fetus and mother is bidirectional, the presence of maternal cells in the circulation of their children is called maternal chimerism. Chimerism can also occur following transplantation or blood transfusion (Lee et al., 1999).
There have been no studies to date to examine the presence of male chimerism in women with the MRKH syndrome. The main purpose of the research reported here was to study whether the etiology associated with freemartinism could be the cause of the MRKH syndrome in humans. We hypothesized that a male—possibly vanished—co-twin was present during the early embryological development of women with MRKH syndrome, resulting in exchange of AMH via placental vascular connections. Therefore, we investigated the presence of male microchimerism (as a result of cell trafficking from male twin to female twin) in MRKH patients and compared these results with control women.
Materials and Methods
Study subjects
This observational case–control study compared the presence of male microchimerism in women with the MRKH syndrome and control women. Through recruitment via the Dutch patients’ association of women with MRKH (‘Stichting MRK-vrouwen’), we enrolled 96 women diagnosed with the MRKH syndrome (in total, the 300 members were informed by email or regular mail and the participation rate was 32%). All participants were 18 years or older and provided written informed consent prior to enrollment. Between January 2017 and July 2017, blood samples were obtained by venipuncture, by one researcher (H.E.P.). The blood samples were collected in EDTA vacutainer® tubes. All participants completed a questionnaire, comprising questions about demographic information, medical history, MRKH diagnosis, and family history.
The control group comprised women who volunteered to participate in an earlier study at our hospital. This study evaluated reproductive functioning in female childhood cancer survivors (DCOG LATER-VEVO study, NL15106.029.06) (Overbeek et al., 2012; van den Berg et al., 2018). For this study, women who did not have a history of cancer in the past were included as control subjects. Phenotypic data were collected between 2008 and 2014 by questionnaires, and biological material was collected via blood sampling. Out of the 390 women who provided biological material for the study, we selected 100 women who reported to have never been pregnant for the control group.
MRKH diagnosis
The MRKH diagnosis of the participants was confirmed by contacting the general practitioner or gynecologist, to retrieve the detailed information about the diagnosis based on physical examination, imaging or laparoscopy results. The patients were identified as having typical MRKH syndrome (also referred to as type 1) in case of no known other malformations and as atypical (type 2) MRKH syndrome when renal and/or skeletal malformations were present (Oppelt et al., 2006).
Preparation of samples
Genomic DNA was extracted from peripheral venous blood according to standard procedures. The extracted DNA was stored at −80°C until further processing. The DNA concentration ranged from 2.48 to 261 ng/μL (mean 77.47 ng/μL). The A260/A280 ratio ranged from 1.33 to 2.46 (mean 1.85).
Analysis of male microchimerism
Real-time quantitative PCR (qPCR) was performed using a QuantStudio™ 7 Flex system (Applied Biosystems, Waltham, MA, USA). The DNA sequence utilized for detection of male genome targets a Y-chromosome specific region, DYS14. Amplification primers and probe sequences for DYS14 have been described previously (Lambert et al., 2002). Also, the rationale for using this region as a Y-chromosome target is detailed (Lambert et al., 2002). As a measure of the total input quantity of genomic mass per reaction, qPCR of the β-globin gene was measured in parallel. Primers and probes for β-globin are also previously described (Lo et al., 1998). All genome equivalent (GEq) conversions utilized a conversion factor of 6.6 pg DNA per cell (Saiki et al., 1988). Male GEq was reported per 1 million cells and calculated using the following equation:
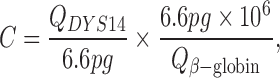 |
where C = concentration of male GEq per 1 million cells, QDYS14 = mean mass of DYS14 (ng) determined by qPCR, and Qβ-Globin = mean mass of β-globin (ng) determined by qPCR.
Amplification measurement was accomplished using a dual-labeled minor groove binder probe with a 5′-bound fluorescent dye reporter [6-carboxyfluorescein (FAM)] and a 3′-bound non-fluorescent quencher. While both are bound to the probe, the fluorescent signature of the reporter is quenched. Taq DNA polymerase 5′-3′ exonuclease activity during the extension phase of PCR causes cleavage of the probe, releasing the FAM reporter (Heid et al., 1996; Holland et al., 1991). Once separated from the quencher, the reporter will emit its fluorescence (Heid et al., 1996; Lambert et al., 2002). This fluorescent emission is detected by the Quantstudio™ 7 Flex system and analyzed by Quantstudio™ Real-Time PCR software (v 1.1) (Applied Biosystems, Waltham, MA, USA).
Calibration standards for the DYS14 assay were produced by diluting known male DNA in female DNA to a total concentration of 66 ng/μL, with subsequent 10-fold dilutions ranging from 66 ng to 0.0066 ng male genome (mean r2 = 0.994, n = 11). Calibration standards for the β-globin assay were produced by 10-fold serial dilution of DNA, ranging from 500 ng—0.5 ng (mean r2 = 0.997, n = 11). All standards were measured in triplicate on each reaction plate simultaneously with subject reactions. Also included on each reaction plate were positive and negative control subjects for male genome diluted in female DNA and a no template control (NTC) for each assay. The negative controls and NTC tested consistently negative during all experiments.
Performance of the assay was measured for reproducibility and linearity by using female samples spiked with male DNA. A series of 10-fold dilutions of male DNA in female DNA demonstrated target specificity of the DYS14 assay. The measured male GEq of our validation controls demonstrated excellent linear regression with a correlation coefficient of 0.998. Reproducibility was measured using three samples with mean male GEqs per million of 1594.64 ± 137.41 [coefficient of variation (CV) = 8.62], 10.50 ± 2.31 (CV = 22.02), and 6.72 ± 1.82 (CV = 27.14).
The qPCR reactions were set up in a 10 uL reaction on 384-well reaction plates. The target genome input of blood-extracted DNA was 66 ng per reaction, thus achieving 10 000 genomes per reaction. Each reaction also included 5.0 μL TaqMan® Fast Advanced Master Mix (Applied Biosystems, Waltham, MA, USA), 0.5 μL 20× custom assay (Applied Biosystems, Waltham, MA, USA) including a dual-labeled probe as well as forward and reverse primers. Each sample simultaneously tested 12 aliquots for DYS14 and in duplicate for β-globin on the same reaction plate. The amplification thermal cycling initiated with 50°C for 2 min then denaturation at 95°C for 2 min then 46 cycles of 95°C for 15 s and 56°C for 1 min.
All qPCR reactions and analyses were performed by a scientist who was blinded to MRKH status. Results were reported as male GEq per 1 million cells, calculated by using the mean measure of DYS14 and β-globin for each subject. Male microchimerism was defined as a quantifiable measure of DYS14 in any of 12 aliquots tested per sample.
Precautions against contamination
We utilized strict precautions while handling these samples to prevent contamination. Standard precautions for a PCR reaction set up were followed (Kwok and Higuchi, 1989). Separate pre-amplification and post-amplification areas were strictly adhered to. All handling of samples was done inside a class II biosafety cabinet, which pulls contaminated air from the work surface and exterior environment through a high efficiency particulate air (HEPA) filter to sterilize before returning to the work surface. All pipetting was carried out using aerosol-resistant filtered pipette tips. Each reaction plate included multiple NTC wells for each assay. After each use and preparation of each plate, the biosafety cabinet was thoroughly cleaned using a system of 10% bleach, cleaned with deionized water, followed by DNAZap™ (Invitrogen, Waltham, MA, USA) as per manufacturer’s directions, two cycles of deionized water cleaning and 1 h of an ultraviolet lamp.
Statistical analysis
For sample size calculation, we assumed 25% microchimerism in the control group [according to results in previous studies (Yan et al., 2005)], and we assumed that in at least 50% of the adult women with MRKH syndrome, male microchimerism would still be detectable in the blood. With a significance level (alpha) of 5% and a power of 95%, we calculated a sample size of 91 women in both groups. Anticipating a 5% margin of error, in total, 192 women needed to be included.
Not normally distributed variables were compared using the Mann–Whitney U test. For categorical variables, the chi-squared (χ2) or Fisher’s exact test was used as appropriate. We used logistic regression to examine the association between microchimerism and MRKH syndrome by calculating the odds ratio (OR). Age (years) was included as a covariate as it may affect the presence of microchimerism. P < 0.05 was considered statistically significant. The statistical analysis was performed using SPSS 22.0 (SPSS, Chicago, IL, USA).
Ethics
The study protocol has been approved by the Institutional Review Board of the VU Medical Center, Amsterdam, the Netherlands, on 5 January 2017 (METC VUmc 2016.374). The trial was registered in the Dutch National Trial Registry (trial registration number NTR5961).
Results
For this study, 96 women were included in the MRKH group, and 100 control women were selected who reported to have never been pregnant. After collection of all available information, we excluded one woman in the patient group because, in retrospect, she did not have the MRKH syndrome. In the control group, one subject was excluded due to the limited amount of extracted DNA available for the microchimerism analysis. The final analysis included 194 women.
Patient characteristics
All women in the patient group reported having been diagnosed with the MRKH syndrome, with a mean time since diagnosis of 25.5 years (range 2–64 years). The mean age at the time of diagnosis was 16.5 years (range 6–26 years). In five women (5.3%), confirmation of the MRKH diagnosis by medical records could not be achieved. In total, 54 women (56.8%) were identified as having typical MRKH syndrome, and 41 women (43.2%) were identified as having atypical MRKH syndrome. For the women with atypical MRKH syndrome, 28 out of 41 (53.7%) had a renal malformation, 18 (24.4%) had a skeletal malformation, and 9 (22.0%) had combined malformations (Table I). Of the participants in the patient group, three (3.2%) reported a positive family history for MRKH syndrome: two sisters were both participants in this study, and one woman reported an affected cousin (daughter of sister of father of participant). Two women with MRKH syndrome were part of dizygotic twin pairs, one with a twin brother and one with a twin sister, the latter twin sister being unaffected for the MRKH syndrome. All pregnancies resulting in the birth of a woman with MRKH syndrome were conceived naturally. The mean age of the mother at the time of birth of the women with MRKH syndrome was 28.7 years (range 18–42 years). All women in the control group reported having reached menarche and had never been pregnant. No women in the control group were part of a twin pair.
Table I.
Reported malformations in 41 women with atypical Mayer–Rokitansky–Küster–Hauser syndrome.
| Malformations | |
|---|---|
| Renal (28/41)* | (n) |
| Unilateral renal agenesis | 15 |
| Pelvic kidney | 3 |
| Hypoplastic kidney | 3 |
| Horse shoe kidney | 2 |
| Abnormal position | 2 |
| Malrotation | 1 |
| Cirrhotic kidney | 1 |
| Duplex kidney | 1 |
| Skeletal (18/41)* | |
| Scoliosis | 13 |
| Cervical ribs | 1 |
| Arm agenesis | 1 |
| Scoliosis and fusion of vertebrae | 1 |
| KFS, SD, and scoliosis | 1 |
| Spina bifida, KFS, SD, and scoliosis | 1 |
| Other (4/41)* | |
| Hearing loss | 2 |
| Scaphoid hypoplasia and hearing loss | 1 |
| Clubfoot | 1 |
*Combined malformations in nine women. KFS, Klippel–Feil syndrome (cervical vertebral fusion); SD, Sprengel’s deformity (high scapula).
Male microchimerism
In total, 194 women were included in the analyses concerning male microchimerism (Table II). In the total group, 22 out of 194 (11.3%) women tested positive for male microchimerism. The prevalence of male microchimerism was significantly higher in the control group than in the MRKH group (17.2% versus 5.3%, P = 0.009) (Fig. 1). After correcting for age, women in the control group were 5.8 times more likely to have male microchimerism (OR 5.84 (95% CI 1.59–21.47), P = 0.008). The mean concentration of male microchimerism in the positive samples was 56.0 male GEq per 1 000 000 cells (Fig. 2). The mean concentration of male microchimerism was significantly higher in the control group than in the MRKH group (P = 0.007). The prevalence of male microchimerism was similar in women with typical MRKH syndrome and atypical MRKH syndrome (5.6% versus 4.9%, P > 0.999).
Table II.
Male microchimerism in study subjects.
| Total group (n = 194) | MRKH (n = 95) | Control (n = 99) | P-value |
|---|---|---|---|
| Age (years) | 40.9 (15.5) | 30.2 (6.6) | — |
| BMI (kg/m2) | 24.3 (5.3) | 22.8 (3.5) | — |
| Presence of male microchimerism | 5 (5.3%) | 17 (17.2%) | 0.009a |
| ORc: 0.27 (CI 0.10–0.76) ORd: 0.17 (CI 0.05–0.63) | |||
| Concentration of male microchimerism (GEq*) | |||
| Positive samples (n = 22) | 2.3 (1.5) | 71.8 (127.0) | 0.055b |
| Total group | 0.1 (0.6) | 12.3 (58.1) | 0.007b |
| Selection of study subjects with no older brother (n = 134) | MRKH (n = 63) | Control (n = 71) | P-value |
| Age (years) | 38.1 (12.9) | 30.5 (6.5) | — |
| Presence of male microchimerism | 3 (4.8%) | 10 (14.1%) | 0.069a |
| ORc: 0.31 (CI 0.08–1.16) ORd:0.18 (CI 0.03–0.90) | |||
| Concentration of male microchimerism (GEq*) | |||
| Positive samples (n = 13) | 1.3 (0.3) | 27.3 (67.1) | 0.018b |
| Total group | 0.1 (0.3) | 3.9 (25.9) | 0.053b |
Data are mean (SD) or n (%). MRKH, Mayer–Rokitansky–Küster–Hauser syndrome.
*Male genome equivalents (GEq) per 1 000 000 female cells
aChi-squared test
bMann–Whitney U test
cUnadjusted odds ratio (95% confidence interval), logistic regression
dOdds ratio (95% confidence interval), adjusted for age
Figure 1.
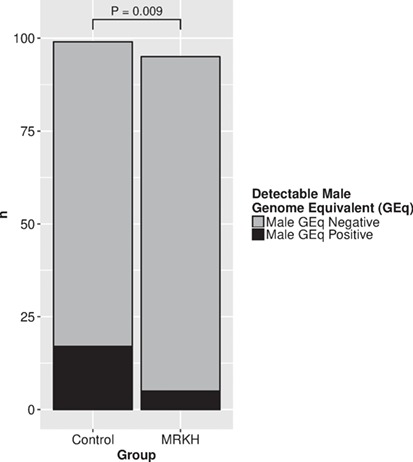
Number of subjects with male microchimerism in blood in control women and women with Mayer–Rokitansky–Küster–Hauser syndrome.
MRKH, Mayer-Rokitansky-Küster-Hauser; statistical test used: chi-squared test.
Figure 2.
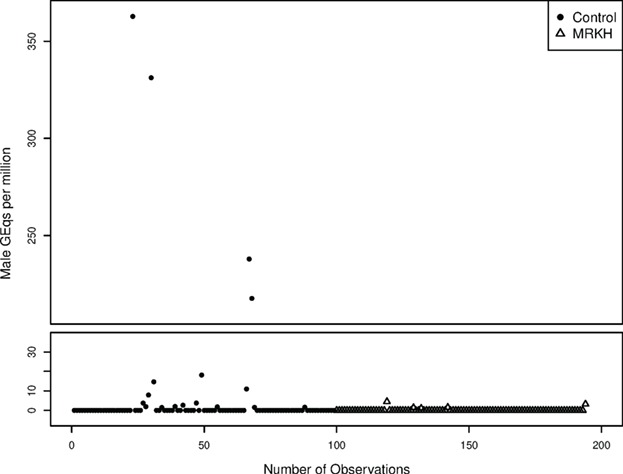
Calculated male GEq per 1 million cells in blood, for control women and women with MRKH syndrome.
To account for a possible older brother as a source for chimerism (Dierselhuis et al., 2012), we made a subgroup analysis. From the total group, 134 women (69.1%) had no older brother (Table II). Also in this subgroup, the prevalence and concentration of male microchimerism were higher in the controls than in the MRKH group.
Table III presents possible sources for male microchimerism. No significant difference was found for the prevalence of male microchimerism in the group with an older brother as opposed to the group without (P = 0.263). In the MRKH group, five women reported having received a blood transfusion; these women all tested negative for male microchimerism. The prevalence of male microchimerism wassimilar when comparing the group with and without blood transfusion (P > 0.999). In the control group, eight women reported never having had sexual intercourse; of these women, one tested positive for male microchimerism. The prevalence of male microchimerism was similar when comparing the group with and without sexual intercourse (P > 0.999).
Table III.
Identifying additional sources for male microchimerism.
| Male microchimerism positive (n = 22) | Male microchimerism negative (n = 172) | P-value | |
|---|---|---|---|
| Age (years) | 35.3 (11.6)+ | 35.4 (13.1) | — |
| Study group | |||
| MRKH | 5 (22.7%) | 90 (52.3%) | 0.009a |
| Control | 17 (77.3%) | 82 (47.7%) | |
| Older brother | |||
| yes | 9 (40.9%) | 50 (29.1%) | 0.263a |
| no | 13 (59.1%) | 121 (70.3%)# | |
| Blood transfusion* | |||
| yes | 0 | 5 (5.6%) | >0.999b |
| no | 5 (100%) | 85 (94.4%) | |
| Sexual intercourse** | |||
| yes | 16 (94.2%) | 75 (91.5%) | >0.999b |
| no | 1 (5.9%) | 7 (8.5%) |
Data are mean (SD) or n (%).
*Data missing for control women; this question was only asked in questionnaire for MRKH women.
**Data missing for MRKH women; this question was only asked in questionnaire for control women.
+Mean (SD) age in women with MRKH syndrome who tested positive for microchimerism = 49.9 (10.9) years; age in control women = 31.0 (7.9) years.
#One MRKH woman with a twin brother tested negative for male microchimerism.
aChi-squared test
bFisher’s exact test
Discussion
MRKH is a condition that has been well characterized yet the etiology has remained elusive (Fontana et al., 2017). Research of the phenotype analogous condition in cattle, freemartinism, has yielded the hypothesis that uterine development is inhibited by exposure to AMH in utero (Vigier et al., 1984). In cattle, the male co-twin has been identified as the source of AMH, which is transferred via placental blood exchange. In humans, a similar exchange of cellular material has been documented between twins by detection of blood chimerism (Peters et al., 2017; van Dijk et al., 1996). We sought to explain fetal exposure to blood and AMH from a male (vanished) co-twin in women with MRKH by the measure of male blood microchimerism.
The use of a male genome target has been previously implemented in other studies of chimerism including fetal microchimerism (Lambert et al., 2002). However, to our knowledge, there are no studies to date that have explored the presence of male microchimerism in women with MRKH. Our approach utilized a qPCR technique that was able to demonstrate exceptional target specificity and sensitivity for male genome. In this study of 194 women, 5 of 95 women (5.3%) with MRKH demonstrated male microchimerism, while the control group demonstrated male microchimerism in 17 of 99 (17.2%). This difference shows a significantly lower frequency in detection of male microchimerism for women with MRKH (P = 0.009). Similarly, when subjects with older brothers are removed, the percentage of male microchimerism in controls and cases are 14.1% and 4.8%, respectively. Our results do not support increased male microchimerism in adult women with MRKH as a consequence of intrauterine blood exchange.
Although these results contradict our underlying hypothesis of inadvertent AMH exposure in early pregnancy as the pathophysiological mechanism, it is too early to definitively conclude that occurrence of AMH exchange during early embryologic development in MRKH was not involved. There are several limitations to our approach that must be considered. First, this method relies on the detection of male genome in these subjects and is therefore contingent on the occurrence of cell grafting as a consequence of this exchange. There are several factors that are essential to effective grafting of cellular material in a host in order to evade immune targeting and thrive (Ichinohe, 2010). It is possible for intrauterine cellular exchange to occur passively between twins and not result in a persistent chimerism. Similarly, our subject population includes all adult women and therefore is reliant on long-term prevalence of the male (XY) cell population to be detected at this stage of life. These XY cells may be eliminated or never graft in the host body, thereby shortening the timespan where detection is achievable. This technique exclusively examines the blood-derived genome, resulting in a limited scope of microchimerism detection and is not a global representation of chimerism throughout the individual. Owing to the proposed transfusion mechanism, it is most plausible that a male cell population may be localized in the blood; however, the cells may have grafted anywhere in the body during development (Korbling et al., 2002). It must also be considered that the exchange of AMH may occur without the transfusion of XY cells and therefore cannot be discovered by chimerism detection.
Our finding of male microchimerism in a larger percentage of women (17.2%) in the control group without a history of pregnancy is in line with findings from the literature. Prior studies demonstrated male microchimerism being present in 13.6% of adolescent girls (Muller et al., 2015) and 13.3% of healthy null gravid women (Yan et al., 2005). Potential sources for the male cells are considered to be transfusion (Utter et al., 2007), older brothers (or discontinued male pregnancies from their mother) (Dierselhuis et al., 2012), unrecognized male miscarriages (Yan et al., 2005), vanishing twins (de Bellefon et al., 2010), or possibly sexual intercourse without pregnancy (Muller et al., 2015). The significant difference in our study, in favor of the control group, suggests that a substantial proportion of the microchimerism could be explained by unrecognized pregnancies or the harboring of microchimeric cells after sexual intercourse. Moreover, the higher concentration in the control group could reflect a larger transfusion of microchimeric cells by these sources. In our study, there were no differences in occurrence of older brothers, previous blood transfusion, or history of sexual intercourse between women with or without microchimerism, both in participants and controls. We are unable to rule out the potential for microchimerism resulting from an unrecognized pregnancy or a vanishing twin.
The findings in women with MRKH illustrate that this population has a decreased prevalence of male microchimerism relative to others. This significant difference demonstrates that women with MRKH may serve as a suitable control group in chimerism research, in part due to the certainty that they have no history of pregnancy. Still, three women with MRKH and no older brother had detectable microchimerism, which may have been obtained through several mechanisms including a vanishing twin or miscarriage in their mother.
Considering the several potential sources of XY cells, our results from the control population demonstrate that there may be a larger prevalence of microchimerism in the general population, which requires further investigation. In general, there is increasing interest in the clinical consequences of microchimerism (Gammill and Harrington, 2017; Kinder et al., 2017). It has been associated with various autoimmune disorders (Nelson, 2012), but possible beneficial consequences have also been described. Microchimeric cells could replace injured cells in diseased tissues (Kinder et al., 2017). Recently, it has been suggested that naturally acquired microchimerism has ‘protective effects in promoting success of future pregnancies’ (Kinder et al., 2015).
In summary, we hypothesized that women with MRKH syndrome had an unrecognized male co-twin that was lost early in gestation (a vanishing twin) and have investigated possible fetal exposure to male blood by measurement of male microchimerism. We are led to the conclusion that women with MRKH syndrome are not evident (micro)chimeras. However, we are not able to draw definitive conclusions regarding the occurrence of AMH exchange. Further research should involve the presence of MRKH syndrome in a large cohort of girls with a twin brother and in girls born after a pregnancy complicated by vanishing twin syndrome. Additional research on twin chimerism is needed to study the true prevalence of intrauterine blood exchange between dizygotic twins.
Acknowledgements
We thank all participating women and the patients’ association of women with MRKH (‘Stichting MRK-vrouwen’). We thank T.J. Korsen (research nurse) and R.J. van Schooten (Genome Diagnostics VUmc) for their contributions in study organization.
Authors’ roles
Study design, study organization, data collection, data analysis, and manuscript draft were performed by H.E.P., supervised by C.B.L.; analysis of male microchimerism and manuscript draft were performed by B.N.J., supervised by E.A.E and G.E.D.; D.M. organized DNA isolation and data analysis; A.O. M.H.B., E.D.B, and F.E.L. collected data of control group; supervision of study design and manuscript draft were performed by J.J.M.L.D., V.M., M.O.V., D.I.B.; all authors had read the manuscript before submission.
Funding
None.
Conflict of interest
None.
References
- de, Bellefon LM, Heiman P, Kanaan SB, Azzouz DF, Rak JM, Martin M, Roudier J, Roufosse F, Lambert NC. Cells from a vanished twin as a source of microchimerism 40 years later. Chimerism 2010;1:56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanova N, Siebers U, Kelsch R, Markoff A, Ropke A, Exeler R, Tsokas J, Wieacker P. Blood chimerism in a girl with Down syndrome and possible freemartin effect leading to aplasia of the Mullerian derivatives. Hum Reprod 2010;25:1339–1343. [DOI] [PubMed] [Google Scholar]
- Choussein S, Nasioudis D, Schizas D, Economopoulos KP. Mullerian dysgenesis: a critical review of the literature. Arch Gynecol Obstet 2017;295:1369–1381. [DOI] [PubMed] [Google Scholar]
- Dierselhuis MP, Blokland EC, Pool J, Schrama E, Scherjon SA, Goulmy E. Transmaternal cell flow leads to antigen-experienced cord blood. Blood 2012;120:505–510. [DOI] [PubMed] [Google Scholar]
- Fontana L, Gentilin B, Fedele L, Gervasini C, Miozzo M. Genetics of Mayer–Rokitansky–Kuster–Hauser (MRKH) syndrome. Clin Genet 2017;91:233–246. [DOI] [PubMed] [Google Scholar]
- Gammill HS, Harrington WE. Microchimerism: defining and redefining the prepregnancy context—a review. Placenta 2017;60:130–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res 1996;6:986–994. [DOI] [PubMed] [Google Scholar]
- Herlin M, Bjorn AM, Rasmussen M, Trolle B, Petersen MB. Prevalence and patient characteristics of Mayer–Rokitansky–Kuster–Hauser syndrome: a nationwide registry-based study. Hum Reprod 2016;31:2384–2390. [DOI] [PubMed] [Google Scholar]
- Holland PM, Abramson RD, Watson R, Gelfand DH. Detection of specific polymerase chain reaction product by utilizing the 5′---3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc Natl Acad Sci U S A 1991;88:7276–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T. Long-term feto-maternal microchimerism revisited: microchimerism and tolerance in hematopoietic stem cell transplantation. Chimerism 2010;1:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinder JM, Jiang TT, Ertelt JM, Xin L, Strong BS, Shaaban AF, Way SS. Cross-generational reproductive fitness enforced by microchimeric maternal cells. Cell 2015;162:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinder JM, Stelzer IA, Arck PC, Way SS. Immunological implications of pregnancy-induced microchimerism. Nat Rev Immunol 2017;17:483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbling M, Katz RL, Khanna A, Ruifrok AC, Rondon G, Albitar M, Champlin RE, Estrov Z. Hepatocytes and epithelial cells of donor origin in recipients of peripheral-blood stem cells. N Engl J Med 2002;346:738–746. [DOI] [PubMed] [Google Scholar]
- Kwok S, Higuchi R. Avoiding false positives with PCR. Nature 1989;339:237–238. [DOI] [PubMed] [Google Scholar]
- Lambert NC, Lo YM, Erickson TD, Tylee TS, Guthrie KA, Furst DE, Nelson JL. Male microchimerism in healthy women and women with scleroderma: cells or circulating DNA? A quantitative answer. Blood 2002;100:2845–2851. [DOI] [PubMed] [Google Scholar]
- Lee TH, Paglieroni T, Ohto H, Holland PV, Busch MP. Survival of donor leukocyte subpopulations in immunocompetent transfusion recipients: frequent long-term microchimerism in severe trauma patients. Blood 1999;93:3127–3139. [PubMed] [Google Scholar]
- Lillie FR. The theory of the free-martin. Science 1916;43:611–613. [DOI] [PubMed] [Google Scholar]
- Lo YM, Lo ES, Watson N, Noakes L, Sargent IL, Thilaganathan B, Wainscoat JS. Two-way cell traffic between mother and fetus: biologic and clinical implications. Blood 1996;88:4390–4395. [PubMed] [Google Scholar]
- Lo YM, Tein MS, Lau TK, Haines CJ, Leung TN, Poon PM, Wainscoat JS, Johnson PJ, Chang AM, Hjelm NM. Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet 1998;62:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller AC, Jakobsen MA, Barington T, Vaag AA, Grunnet LG, Olsen SF, Kamper-Jorgensen M. Microchimerism of male origin in a cohort of Danish girls. Chimerism 2015;6:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JL. The otherness of self: microchimerism in health and disease. Trends Immunol 2012;33:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppelt P, Renner SP, Kellermann A, Brucker S, Hauser GA, Ludwig KS, Strissel PL, Strick R, Wallwiener D, Beckmann MW. Clinical aspects of Mayer–Rokitansky–Kuester–Hauser syndrome: recommendations for clinical diagnosis and staging. Hum Reprod 2006;21:792–797. [DOI] [PubMed] [Google Scholar]
- Oppelt P, Strissel PL, Kellermann A, Seeber S, Humeny A, Beckmann MW, Strick R. DNA sequence variations of the entire anti-Mullerian hormone (AMH) gene promoter and AMH protein expression in patients with the Mayer–Rokitanski–Kuster–Hauser syndrome. Hum Reprod 2005;20:149–157. [DOI] [PubMed] [Google Scholar]
- Overbeek A, van den Berg MH, Kremer LC, van den Heuvel-Eibrink MM, Tissing WJ, Loonen JJ, Versluys B, Bresters D, Kaspers GJ, Lambalk CB et al. A nationwide study on reproductive function, ovarian reserve, and risk of premature menopause in female survivors of childhood cancer: design and methodological challenges. BMC Cancer 2012;12:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik SS, Brazile B, Dandolu V, Ryan PL, Liao J. Mayer–Rokitansky–Kuster–Hauser (MRKH) syndrome: a historical perspective. Gene 2015;555:33–40. [DOI] [PubMed] [Google Scholar]
- Peters HE, Konig TE, Verhoeven MO, Schats R, Mijatovic V, Ket JC, Lambalk CB. Unusual twinning resulting in chimerism: a systematic review on monochorionic dizygotic twins. Twin Res Hum Genet 2017;20:161–168. [DOI] [PubMed] [Google Scholar]
- Rall K, Eisenbeis S, Henninger V, Henes M, Wallwiener D, Bonin M, Brucker S. Typical and atypical associated findings in a group of 346 patients with Mayer–Rokitansky–Kuester–Hauser syndrome. J Pediatr Adolesc Gynecol 2015;28:362–368. [DOI] [PubMed] [Google Scholar]
- Resendes BL, Sohn SH, Stelling JR, Tineo R, Davis AJ, Gray MR, Reindollar RH. Role for anti-Mullerian hormone in congenital absence of the uterus and vagina. Am J Med Genet 2001;98:129–136. [DOI] [PubMed] [Google Scholar]
- Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988;239: 487–491. [DOI] [PubMed] [Google Scholar]
- Taguchi O, Cunha GR, Lawrence WD, Robboy SJ. Timing and irreversibility of Mullerian duct inhibition in the embryonic reproductive-tract of the human male. Dev Biol 1984;106:394–398. [DOI] [PubMed] [Google Scholar]
- Utter GH, Reed WF, Lee TH, Busch MP. Transfusion-associated microchimerism. Vox Sang 2007;93:188–195. [DOI] [PubMed] [Google Scholar]
- van den Berg MH, Overbeek A, Lambalk CB, GJL K, Bresters D, van den Heuvel-Eibrink MM, Kremer LC, Loonen JJ, van der Pal HJ, Ronckers CM et al. Long-term effects of childhood cancer treatment on hormonal and ultrasound markers of ovarian reserve. Hum Reprod 2018;33:1474–1488. [DOI] [PubMed] [Google Scholar]
- van Dijk BA, Boomsma DI, de Man AJ. Blood group chimerism in human multiple births is not rare. Am J Med Genet 1996;61:264–268. [DOI] [PubMed] [Google Scholar]
- Vigier B, Tran D, Legeai L, Bezard J, Josso N. Origin of anti-Mullerian hormone in bovine freemartin fetuses. J Reprod Fertil 1984;70:473–479. [DOI] [PubMed] [Google Scholar]
- Yan Z, Lambert NC, Guthrie KA, Porter AJ, Loubiere LS, Madeleine MM, Stevens AM, Hermes HM, Nelson JL. Male microchimerism in women without sons: quantitative assessment and correlation with pregnancy history. Am J Med 2005;118:899–906. [DOI] [PubMed] [Google Scholar]