

Abstract
Protein phosphatase PstP is conserved throughout the Actinobacteria in a genetic locus related to cell wall synthesis and cell division. In many Actinobacteria it is the sole annotated serine threonine protein phosphatase to counter the activity of multiple serine threonine protein kinases. We used transcriptional knockdown, electron microscopy and comparative phosphoproteomics to investigate the putative dual functions of PstP as a specific regulator of cell division and as a global regulator of protein phosphorylation. Comparative phosphoproteomics in the early stages of PstP depletion showed hyperphosphorylation of protein kinases and their substrates, confirming PstP as a negative regulator of kinase activity and global serine and threonine phosphorylation. Analysis of the 838 phosphorylation sites that changed significantly, suggested that PstP may regulate diverse phosphoproteins, preferentially at phosphothreonine near acidic residues, near the protein termini, and within membrane associated proteins. Increased phosphorylation of the activation loop of protein kinase B (PknB) and of the essential PknB substrate CwlM offer possible explanations for the requirement for pstP for growth and for cell wall defects when PstP was depleted.
Subject terms: Kinases, Bacterial physiology, Proteomics
Introduction
Serine/threonine phosphorylation in bacteria is a ubiquitous reversible signal that controls cell cycle events, virulence, and responses to environmental cues1. Phosphoserine and phosphothreonine are chemically stable, and the transient, reversible nature of the signal relies on the activity of protein phosphatases as well as protein turnover. The conserved Actinobacterial regulator GarA is an example of a stable phosphoprotein that is replaced by protein turnover since its buried phosphothreonine resists phosphatase activity2, while the autophosphorylation sites in the activation loops of protein kinases are examples of short-lived phosphorylation signals, since these exposed sites are dephosphorylated rapidly by protein phosphatase3,4.
Serine threonine (S/T) phosphatases are greatly outnumbered by S/T kinases in bacterial and eukaryotic genomes. Human protein phosphatases tend to have lower substrate specificity than protein kinases, and more substrates. Substrate selection is driven by subcellular localisation and interaction with regulatory partners5,6. Studies of purified bacterial S/T phosphatases suggest that these are also multi-specific1,7, for example recombinant Mycobacterium tuberculosis PstP dephosphorylates itself, protein kinases PknB and PknA and phosphoproteins PbpA and GarA3,4,8. These in vitro studies necessarily lack any regulatory proteins and use non-physiological protein concentrations. Phosphatase knockout in Enterococcus faecium has provided evidence of substrate selectivity in cells9.
The ratio of S/T kinase to S/T phosphatase genes is particularly high in Actinobacterial genomes. Non-pathogenic M. smegmatis has 18 S/T kinases, one transmembrane and one soluble S/T phosphatase. The function of the soluble S/T phosphatase MspP (MSMEG_1928) is unknown, although its activity has been demonstrated in vitro10. Only the transmembrane S/T phosphatase PstP is conserved between M. smegmatis and M. tuberculosis (91% identity in the phosphatase domain), and this genetic locus is conserved throughout the Actinobacteria (Fig. 1) and is associated with cell division11. pstP is cotranscribed with genes involved in peptidoglycan synthesis rodA and pbpA12,13 and there is weak positive correlation of transcription with protein kinases pknA and pknB14,15. The kinase genes have separate transcriptional start sites16 and regulate cell growth and cell division17. fhaA and fhaB, also in this locus, encode phosphothreonine recognition proteins that regulate cell growth and cell division18,19.
Figure 1.
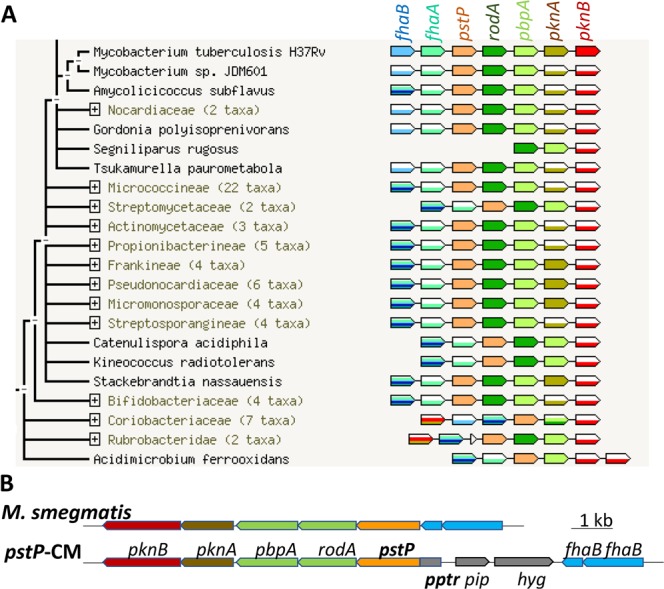
The pstP operon is conserved in the Actinobacteria, and was disrupted in this study to make a conditional mutant strain of M. smegmatis, pstP-CM. (A) PstP is encoded in a cluster of seven genes that are related to cell wall synthesis and protein phosphorylation. The genetic linkage of pstP, rodA, penicillin binding protein A (PbpA), and PASTA-kinase is conserved throughout the Actinobacteria. (B) To generate pstP-CM, homologous recombination was used to replace the endogenous pstP promoter with the pristinamycin-inducible promoter pptr and add the transcriptional repressor pip. In the absence of the inducer pristinamycin, Pip binds to pptr to repress transcription.
The gene cluster pknB-fhaA is conserved in a subset of the Firmicutes as well as the Actinobacteria, while the link between pstP and pknB is conserved in all Firmicutes and Actinobacteria (Fig. S1). PknB homologues, characterised by periplasmic PASTA domains (Penicillin-binding protein And Serine Threonine kinase Associated20), regulate many events in the cell cycle, as demonstrated by diverse phenotypes caused by gene disruption in Bacillus subtilis, Staphylococcus aureus, Corynebacterium glutamicum, Streptomyces coelicolor, M. tuberculosis and Streptococcus pneumoniae21. The mechanisms by which PknB regulates cell division are being elucidated and some may be organism-specific, for example PknB regulates peptidoglycan biosynthesis in M. tuberculosis by phosphorylation of several key enzymes and regulators including the regulatory protein CwlM22,23 and proposed lipid II flippase18.
PstP phosphatase function is also implicated in regulation of cell wall biosynthesis or cell division: a group B Streptococcus knockout had increased phosphorylation of cell division proteins24, S. pneumoniae knockout had defects in cell size and location of peptidoglycan synthesis25, S. aureus knockout had thickened cell wall26, and C. glutamicum knockout was slow-growing with pleiomorphic bulging cells. While this study was in progress, PstP was shown to be essential in M. tuberculosis and Mycobacterium smemgatis because depletion of PstP led to cell wall defects27. However, the cognate substrates of PstP and the mechanism by which it regulates cell cycle processes were unknown.
We hypothesised that PstP might play two roles: as a global negative regulator of protein S/T phosphorylation, and as a specific regulator of PknB and PknA by dephosphorylating enzymes and regulators of cell wall biosynthesis and cell division. We characterised M. smegmatis during PstP depletion, measuring changes in growth and viability, cell morphology and the phosphoproteome. These changes clarified the phosphoproteins regulated by PstP and support both roles.
Results
Knockdown of pstP-operon prevented growth of M.smegmatis
To gain insight into the cellular processes regulated by PstP, a conditional mutant was constructed. The pstP promoter was replaced with the pristinamycin inducible promoter (Fig. 1B) using the method of Forti et al.28. The resulting conditional mutant strain, pstP-CM, was expected to express pstP only in the presence of inducer. Based on the demonstration of pstP essentiality using the Tet-OFF system, pstP-CM was predicted to depend on inducer for growth on agar and liquid medium, as seen in Fig. 2A. Integration of a second copy of pstP at the attP site restored the ability to grow without inducer (Fig. 2A).
Figure 2.
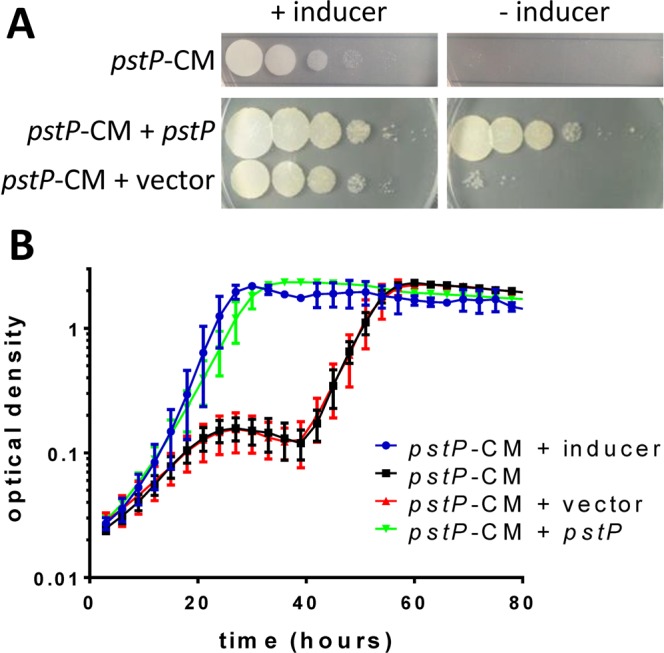
Knockdown of pstP prevented growth of M. smegmatis. Conditional mutant strain (pstP-CM), in which the pstP operon was under the control of the pristinamycin promoter, was only able to grow when the inducer pristinamycin was included in the medium. (A) Knockdown of the pstP operon by omission of inducer led to a 104-fold drop in the number of colonies on 7H10 agar. Vector-borne pstP, but not control vector, restored the ability of the conditional mutant to form colonies in the absence of inducer. The three strains were cultured in liquid medium in the presence of inducer, washed, and serial 10-fold dilutions were spotted on plates with the appropriate antibiotics. (B) Knockdown of the pstP operon by removal of inducer led to cessation of growth in 7H9 broth, then a decline in optical density, followed by emergence of suppressor mutants. Vector-borne pstP, but not control vector, restored the ability of the conditional mutant to grow in the absence of inducer. Error bars show the standard deviation of four cultures. Graphs are representative of three independent experiments.
Knockdown of the pstP operon by removal of inducer led to cessation of growth in liquid medium, followed by a decline in optical density, suggestive of lysis (Fig. 2B). The low number of colony forming units after 22–32 hours of knockdown (Fig. S2) suggests loss of viability. After prolonged knockdown (40 hours, Fig. 2B) growth resumed, presumably due to the selection of second site mutations (suppressor mutations) since subsequent cultures without inducer lacked the prolonged lag phase.
Growth curves and light microscopy were used to identify the earliest time at which knockdown of the pstP operon led to changes in growth and morphology without loss of viability. During the first six hours, representing 2–3 generation times, growth appeared similar between induced and knockdown conditions (Fig. S3), but bulging cells began to appear (Fig. S4), which became more common after 24–30 hours of knockdown. These results led to the choice of six hours knockdown to characterise the proteomic responses to PstP depletion and 30 hours knockdown to characterise the changes in morphology.
Withdrawal of inducer led to knockdown of pstP and pbpA expression
To characterise protein expression changes resulting from knockdown, the total proteome was compared after 6 hours culture with/without inducer (Fig. S5). 2794 proteins were detected, representing 42% of the proteome, which is similar coverage to previous proteomic characterisation of M. smegmatis (2550 proteins29). Only five proteins showed significant changes in abundance (Table 1). PstP and PbpA both decreased 5.7-fold (Table 1, Fig. S5), confirming successful knockdown and confirming co-transcription of these genes (and therefore presumably rodA, although RodA was not detected in the proteome in either condition, likely due to its multiple transmembrane helices). Protein kinases PknA and PknB decreased to a lesser extent, confirming the presence of additional promoter(s) downstream of pstP that contribute to their expression. The conditional mutant strain pstP-CM is therefore considered a triple knockdown of pstP, rodA and pbpA. Reintroduction of pstP restored growth (Fig. 2) despite knockdown of rodA and pbpA. This suggests that the essential kinases pknA and pknB were expressed independently from pstP. The soluble phosphatase MspP was detected in the proteome, though less abundant than PstP, and its abundance did not change during knockdown conditions.
Table 1.
Comparative proteomics identified five proteins that were significantly less abundant (>2 fold change, p < 0.05) after conditional knockdown of the pstP operon.
| Protein name/function | Gene number MSMEG_ | Decrease in abundance (fold change) |
|---|---|---|
| Ribosomal protein L35 | 3792 | 6.55 |
| PstP protein phosphatase | 0033 | 5.76 |
| PbpA penicillin-binding protein A | 0031 | 5.72 |
| Conserved transmembrane protein | 6099 | 3.87 |
| Short-chain dehydrogenase/reductase | 3302 | 2.49 |
The lack of change in global protein levels after 6-hours knockdown (Fig. S5) suggests that this time point is appropriate to investigate changes in protein phosphorylation in response to PstP depletion before widespread changes in gene expression.
pstP-operon knockdown led to changes in the cell wall appearance
Transmission electron microscopy was used to characterise the effects of knockdown of the pstP operon on cell morphology, revealing a thickening of the cell wall, septal defects, rougher cell surface and accumulation of intracellular lipid bodies (Figs 3A,B, S6 and S7). Scanning electron microscopy revealed a reduction in cell length as well as bulging cells and evidence of lysis (Fig. 3C). Reintroduction of pstP restored the cell wall thickness and morphology of the septa (Figs 3A,B, S6 and S7), suggesting that these defects were due to loss of pstP, and that the regulatory role(s) of PstP might encompass cell wall synthesis or cell division. Other aspects of the phenotype were partially complemented (surface roughness and lipid bodies, Figs 3A,B, S6 and S7) or not complemented (cell length, Figs 3C and S8), indicating the influence of knockdown of other genes in the pstP gene cluster in the control of these features. Indeed, knockdown of rodA or pbpA have each been found to perturb cell length and cell wall thickness in M. tuberculosis12, while Tet-OFF knockdown of pstP resulted in elongation of M. tuberculosis and a multiseptum phenotype in M. smegmatis27. Multiseptate cells were not abundant after pstP-operon knockdown, highlighting the interdependence of gene function within this cluster, which means that conditional mutants constructed by different methods were not equivalent.
Figure 3.

Knockdown of the pstP operon led to changes in the cell wall and cell shape. Knockdown for 30 hours led to changes in pstP-CM compared to wild type M. smegmatis, and some of these changes were reversed in the complemented strain pstP-CM + pstP. Cells were imaged by transmission electron microscopy (A,B) and scanning electron microscopy (C). PstP knockdown led to thicker cell wall (black arrow), thicker septum (white arrow), and bulging cells with lysis (white arrowhead). These defects were complemented by reintroduction of pstP. Knockdown of the pstP operon also led to rough cell surface (black arrowhead) and lipid bodies (L) that were partially complemented by reintroduction of pstP, and reduced cell length (quantified in Fig. S8) that was not complemented by reintroduction of pstP. Images within a single row are shown at the same scale, which is indicated by a bar in the right hand image: 200 nm for TEM (A,B) and 2.5 μm for SEM (C). Images are representative of at least twelve fields of view: additional fields for transmission electron microscopy are presented in Figs S6 and S7, and cell lengths for 100 cells are presented in Fig. S8.
pstP-operon knockdown led to global increases in protein phosphorylation
Knockdown of PstP was expected to increase the level of phosphorylation of PstP substrates. Increased protein threonine phosphorylation during knockdown was detected using a phosphothreonine specific antibody (Fig. 4A), confirming that PstP negatively regulates protein phosphorylation on threonine in M. smegmatis. A phosphoprotein of approximately 40 kDa was notable for its abundance and striking increase in phosphorylation during PstP depletion (Fig. 4A). Based on the abundance, molecular weight and fold-change in the phosphoproteome, this protein is tentatively identified as the menaquinone biosynthetic enzyme MenB (Supplementary Table S1).
Figure 4.

PstP knockdown led to increased protein phosphorylation. (A) Removal of inducer led to a global increase in the abundance of protein threonine phosphorylation. Lysates from pstP-CM grown for 6 hours in the presence (+) or absence (−) of inducer were normalized for protein concentration by Coomassie staining (right), blotted and probed with anti-phosphothreonine antibody (left). The increase in protein threonine phosphorylation was non-uniform: a differentially enriched phosphoprotein is marked with a red arrow. (B) The increase in signal upon anti-pThr probing was significant (densitometry and students t test, p < 0.05; data are the mean and standard deviation of 3 independent replicates). (C) Comparative phosphoproteomics identified phosphosites that changed significantly upon knockdown. Dashed lines indicate thresholds for significance: >2 fold-change and p < 0.05.
Comparative phosphoproteomics allowed identification of differentially phosphorylated proteins during knockdown of the pstP operon. We identified 1693 phosphosites on 894 proteins in total. Almost half the phosphosites (838) were significantly more abundant in the knockdown condition (Fig. 4B and supplementary files). The wide-scale increase in phosphopeptide abundance (Fig. 4) without large changes in protein expression (Fig. S5) confirm the role of PstP as a global negative regulator of S/T phosphorylation in M. smegmatis.
Profiling the phosphopeptides that changed upon PstP knockdown could clarify the functional classes of proteins that are regulated by PstP and give an indication of the primary sequence motifs of phosphosites that are regulated by PstP. Phosphothreonine (pT) was the most abundant phosphosite (Fig. 5A), as seen in Mycobacteria and other bacteria30. Furthermore, pT was significantly enriched in the phosphosites that changed in response to knockdown (Fig. 5A), fitting with the observed in vitro activity of M. tuberculosis PstP towards pT and pS3.
Figure 5.

Sequence and functional analysis of phosphosites. (A) Threonine was the most frequent site of phosphorylation. (B) Knockdown of the pstP operon led to a differential increase in phosphorylation at pThr. (C,D) Sequence motifs surrounding pThr and pSer sites with highest increase during knockdown (>8-fold change). (E) pstP knockdown led to a differential increase in phosphorylation on proteins annotated as transmembrane or membrane associated.
Motif analysis of the global phosphoproteome revealed a preference for acidic residues on either side of the phosphorylation site (Fig. 5B), which is similar to the in vitro specificity of M. tuberculosis protein kinases31. Furthermore, amongst the phosphosites changed during knockdown there was a bias towards phosphorylation sites near the N-terminus (10% of the changed phosphopeptides were phosphorylated at position two, and 20% within the first 12 residues, a significant difference compared to the positions of unchanged phosphosites). The greater frequency and more labile nature of phosphorylation near the protein termini is expected from the more exposed nature of the termini compared to residues in defined protein folds12, making them more accessible to kinases and phosphatases.
Protein localisation is also likely to influence substrate access to PstP. Membrane proteins and membrane associated proteins were significantly enriched amongst the differentially phosphorylated proteins, fitting with the membrane localisation of PstP (Fig. 5C).
The functional classes of proteins that are differentially phosphorylated could reveal specific cellular processes that are regulated by PstP. However, half of all phosphopeptides were increased upon knockdown, representing all functional classes (supporting information). The ten phosphoproteins with the greatest increase in abundance (Table 2) include enzymes involved in vitamin biosynthesis, central carbon metabolism and transcription.
Table 2.
Comparative phosphoproteomics identified phosphosites that were more abundant upon pstP knockdown.
| Protein name/function | Gene name MSMEG | Peptide (phosphosite in bold) | p-site | Fold change |
|---|---|---|---|---|
| Cytochrome c oxidase subunit 3 CtaE | 4260 | TSAVGTSGTAITISR | S3 | 750 |
| Menaquinone biosynthesis MenB | 1075 | TGYQYASGETAETVDPAR | T117 | 720 |
| Transaminase (biotin metabolism) | 2450 | TAIQESTLLPNGLTVDTAK | T2 | 131 |
| PEP carboxylase Ppc | 3097 | ADSNDTALEPFGSVQR | T7 | 126 |
| isopentenyl diphosphate synthesis IspG | 2580 | TSIGLGMPAPPAPTLAPR | S3 | 114 |
| Heparin binding heamagglutinin-like | 0919 | VETYTDQAVELTQEALGTVASQTR | T144 | 111 |
| RNA polymerase RpoC | 1368 | NIQVQPTEEAR | T1276 | 104 |
| Conserved hypothetical | 3032 | HGLAADDDRADTDVFAAVSANGAQEHTDTTQTTPLENPDQPR | T159 | 92 |
| ATP synthase epsilon chain AtpC | 4935 | IAVDGGFLSVTEETVR | T73 | 91 |
| Uncharacterised | 6336 | TSDLPASPDQPEPTHGMPPPPPAPGKPK | T2 | 90 |
| methyltransferase | 1049 | TEIRDAADPAPNPHATAEEVEAAMHDSK | T2 | 87 |
| transketolase | 3103 | LVGDTGEIVSIEHYGESADDKTLFR | T674 | 81 |
The phosphosites with greatest fold-increase (p < 0.05) are listed and the full dataset is available as supporting information.
PstP knockdown has direct and indirect routes to increase protein phosphorylation (Fig. S9). Notably, PstP curtails or reverses PknB activation by dephosphorylating the activation loop3,32, and this is likely the case for other kinases33. Conversely, PknB and PknA phosphorylate PstP to increase its activity13, suggesting a possible homeostatic mechanism (Fig. 6). It was therefore important to determine which of the 18 M. smegmatis S/T protein kinases became activated during PstP knockdown, since kinase activation would influence the phosphoproteome. Phosphorylation sites were detected in seven of the kinases (Table 3). Fourteen phosphosites were significantly more abundant in the knockdown condition, while only one site on PknG was significantly less abundant. Phosphopeptides from PknB and PknD had the greatest fold change, notably including the active site loop of PknB (Table 3), thus the changes in the phosphoproteome reflect both decreased PstP activity and the ensuing increase in activity of PknB and other kinases.
Figure 6.

Negative feedback between PknB and PstP allow a transient response to stimulation. 1 Transient stimulation of PknB. 2 PknB autophosphorylates becoming activated. 3 Activated PknB phosphorylates its substrates. 4 Activated PknB phosphorylates PstP, activating PstP. 5 Activated PstP dephosphorylates PknB, returning it to the unactivated form. 6 Activated PstP dephosphorylates PknB substrates (including itself). PknB and PstP oppose each other, so that the response to PknB stimulation is transient. When PstP is knocked down there is no negative regulator to counter the positive feedback of PknB autoactivation, so transient stimuli lead to lasting PknB activation and widespread hyperphosphorylation of PknB substrates. Structures deposited in the PDB are used in this scheme (1O6Y, 2KUI, 2CM1), although the structural changes that occur during signalling are unknown. Similar feedback loops may exist for other Mycobacterial protein kinases.
Table 3.
Phosphorylation sites on Ser/Thr protein kinases that were significantly changed during pstP knockdown.
| Kinase | Peptide (phosphosite in bold) | p-site | Fold change |
|---|---|---|---|
|
PknB MSMEG_0028 |
ALADTGNSVTQTAAVIGTAQYLSPEQAR | T166 | 49.5 |
| ALADTGNSVTQTAAVIGTAQYLSPEQAR | T179* | 36.1 | |
| AGAATQDMPVPRPAGYSK | T309 | 5.8 | |
| VLTDAER | T289 | 5.5 | |
|
PknD MSMEG_4366 A0R0F3 |
RTPTPPAPAAVAAPAVR | T333 | 32.0 |
| TPTPPAPAAVAAPAVR | T335 | 6.9 | |
| QFSELYPNPEHTGFTPYPPAYPSVPAPPR | Y319 | −2.1 | |
|
PknG MSMEG_0786 |
GTVVTEAYDQVTMATR | T75 | 18.5 |
| PMATQAVYRPEFDDTDGTSR | T55 | −3.1 | |
|
PknA MSMEG_0027 A0QNG2 |
RPPRPNQAPTLGR | T288 | 4.2 |
| RPFTGDGALTVAMK | T224 | 2.5 | |
| DVKPGNILITPTGQVK | T152 | 2.4 | |
| RPPRPNQAPTLGR | T288 | 4.2 | |
|
PknF MSMEG_3677 A0QYJ2 |
LGTDADVADPDATR | T290 | 3.2 |
| SPFHHSNAAVVISK | S221 | 3.0 | |
| PknL MSMEG_4243 A0R032 | FHNRPQPDLETTAEVAPPPPAPATVPR | T316 | 2.3 |
| FHNRPQPDLETTAEVAPPPPAPATVPR | T317 | 2.3 |
* indicates phosphosites in the kinase activation loop.
To test whether higher PknB activity could account for some of the increased phosphorylation seen during knockdown of the pstP operon, we searched our phosphoproteome dataset for substrates of PknB identified in previous proteome-wide studies22,23,34. Phosphosites in CwlM, MurJ (MviN), the transporter MSMEG_1642 (Rv1747-homologue), FabG, SigH and EspI were increased >16-fold during knockdown. CwlM, MviN and Rv1747 are involved in synthesis, regulation and export of cell wall components. Phosphorylation-dependent changes in their activity could contribute to the cell wall thickening seen by electron microscopy (Fig. 3A). Thus prolonged activation of PknB is a plausible contributor to the increased phosphorylation during knockdown of the pstP operon. However, the changes in phosphorylation are likely to result from a combination of factors, especially taking into consideration that additional proteins that are not known substrates of PknB were also hyperphosphorylated in the knockdown condition.
Only seven phosphopeptides were significantly decreased in the knockdown condition (Table 4), and these derived from proteins involved in cell wall synthesis and transmembrane transport. The greatest decrease in phosphorylation was at T369 of MSMEG_0317, which is a transmembrane protein involved in lipomannan maturation and lipoarabinomannan synthesis in C. glutamicum (NCgl2760) and which is essential in both M. tuberculosis (Rv0227) and M. smegmatis35. It is tempting to speculate that this reflects a role for PstP in control of cell wall processes, fitting with the changes in morphology observed during prolonged PstP knockdown (Fig. 3). PstP itself was one of the proteins with decreased phosphorylation in the knockdown sample. The change in phosphopeptide abundance (19.3- and 8.0- fold) exceeded the change in protein abundance (5.7-fold), providing support for previous findings that PstP may itself be regulated by phosphorylation13.
Table 4.
Phosphosites that decreased in abundance upon pstP knockdown (threshold >2-fold change; p < 0.05).
| Protein name/function | Gene name MSMEG_ | Peptide (phosphosite in bold) | p-site | Fold change |
|---|---|---|---|---|
| Lipomannan elongation | 0317 | TESTLIDPGLDTADHGFFDTQGIQVPGAEAK | T369 | 1534 |
| EspI regulator of ESX secretion | 0067 | LFHSSEPGKPVDDATITVDR | S11 | 74.9 |
| PstP | 0033 | ITAEEAHSHPQR | T146 | 19.3 |
| PstP | 0033 | ITAEEAHSHPQR | S152 | 8.0 |
| Peptidoglycan binding LysM | 5526 | VIEAPELDDLNDTDDLEPVRLDIPSEFLADAR | T52 | 7.5 |
| PEP protein phosphotransferase PtsI | 0088 | TLDAGSDKPLK | S322 | 4.1 |
| PknG | 0786 | PMATQAVYRPEFDDTDGTSR | T55 | 3.1 |
Discussion
PstP is required for normal cell wall synthesis and cell division, as demonstrated by the cell wall and morphological changes with loss of viability upon knockdown of the pstP operon. Differential phosphorylation of cell division-related proteins during PstP knockdown revealed possible mechanisms by which PstP might regulate peptidoglycan synthesis by dephoshorylating PknB and CwlM, as well as other cell cycle related proteins including Rv0227, MurJ (MviN) and FhaA. Further work will be required to characterise the specific effects of PstP dephosphorylation on the activity of each. The soluble S/T phosphatase MspP, which shares 38% amino acid identity with the catalytic domain of PstP, remained undisrupted in the knockdown (mspP is in a distant genetic locus and there was no significant change in protein abundance) thus MspP cannot substitute for the function of PstP. No MspP phosphosites were identified during phosphoproteome analysis.
Western blot and phosphoproteomics suggest additional, broader roles for PstP. The changes in phosphorylation upon knockdown of the pstP operon surpassed those observed in previous phosphoproteomic comparisons of gene disrupted bacteria in the number and proportion of proteins that were differentially phosphorylated and in the magnitude of the fold-change in the phosphopeptides. The outnumbering of PstP by 18 S/T kinases implies that PstP depletion could affect the concentration of many phosphoproteins but the high fold-change values was unexpected given the mild and brief period of PstP depletion: by 6 hours PstP concentration fell 5.7-fold while 52 phosphopeptides increased >32-fold and globally 50% of phosphosites were significantly changed with 22% increased by over 8-fold. By comparison, knockout of S/T kinases or phosphatase in other bacteria led to fewer significant changes and lower fold-change: 8 out of 64 sites in B. subtilis S/T phosphatase knockout, 55/603 in Mycobacterium bovis pknG knockout36 and 19/456 in M. tuberculosis pknB knockdown23. A possible explanation for the large, extensive and rapid changes in phosphorylation emerged from examining differential phosphorylation of protein kinases. PknB had the greatest increase in phosphorylation, including at threonines in and near the activation loop (49-fold increase in pT166 and 36-fold increase in pT179). In vitro evidence suggests that PknB and PstP regulate each other3,8. Loss of PstP would lead to prolonged PknB activation32, and potentially to positive feedback from uncontrolled autoactivation of PknB (Fig. 6). The dramatic and extensive changes in the phosphoproteome that arise from PstP disruption would fit with this model. Thus, differential phosphorylation in the proteome during PstP knockdown could result from a combination of prolonged PknB activation causing increased phosphorylation of PknB substrates, as well as slower dephosphorylation of PstP substrates. The observed substrate preferences (Fig. 5) may reflect a combination of the preferences of PknB and PstP. PknB was the only kinase whose activation loop was detected in the phosphoproteome, but we cannot exclude that PstP normally curtails activation of other kinases. Remembering that knockdown of the pstP operon is eventually lethal, it is likely that the massive up-regulation of phosphoproteins is due to non-physiological conditions.
The phosphoproteomic and genetic evidence linking the function of PstP and PknB provides a mechanism by which PstP can regulate cell wall synthesis by dephosphorylation of the substrates of PknB (and PknA) and by dephosphorylation of PknB itself to reduce activation. Supporting this explanation are similarities between the morphological defects that result from PstP-depletion and PknB overexpression: both result in widened, bulging, pleiomorphic cells (Fig. 3 and17,27) albeit with possible confounding effects from the growth medium, growth phase and polar effects12. Similarly PstP overexpression and PknB depletion both result in longer cells23,27. Overall, the non-uniform increase in protein phosphorylation upon knockdown of the pstP operon demonstrates that the role of PstP is more complex than general dampening or reversal of the effects of protein kinases. Phosphorylation changes upon knockdown were enhanced at certain motifs and on certain substrates. A study of the PstP homologue in Enterococcus faecium also found evidence of substrate selectivity when recombinant phosphatase was used to dephosphorylate whole cell extracts9.
In summary, PstP is a regulator of cell wall synthesis and cell division and is also a global regulator of protein S/T phosphorylation thus influencing all cellular processes. The first role in cell division may occur directly by the action of PstP of the enzymes and regulators of cell wall synthesis or indirectly by specifically controlling the activity of PknB, or by both mechanisms. The general role in global regulation may also occur directly through substrate dephosphorylation, or indirectly by specific dephosphorylation of the activation loops of kinases, or by both routes.
Methods
Bacterial strains and culture conditions
M. smegmatis mc2155 was routinely cultured at 37 °C on Middlebrook 7H9 (Becton, Dickinson and Company) liquid media supplemented with 10% (v/v) Albumin-Dextrose Complex (0.5% bovine serum albumin, 0.2% dextrose, 0.085% NaCl), 0.2% (v/v) glycerol and Tween 80, 0.05% (w/v). Antibiotics were used as follows: the conditional mutant pstP-CM was routinely cultured in the presence of hygromycin (50 μg/ml) and pristinamycin (2 μg/ml), and pstP-CM transformed with pMV306 or derivatives was routinely cultured in the presence of hygromycin, pristinamycin and kanamycin (30 μg/ml).
Gene cloning
pstP with the cognate promoter was PCR amplified from M. smegmatis genomic DNA and cloned in the KpnI and HindIII sites of pMV30637. [aaaaGGTACCggtgacaagcagcccgcttcac, aaaaAAGCTtcatgacaccgcccggcag]
Generation of conditional knockout and complemented strains
The 5′ end of pstP was PCR including the ribosome-binding site (402 bp) was PCR amplified from M. smegmatis genomic DNA [actCCATGGaggagcagaatgaccctcgttct, AcaGCATGCttggtgatctgtgtcagctcacc] and cloned under the control of the pristinamycin inducible promoter in the NcoI and SpeI sites of pAZI947928 in order to disrupt the genomic pstP locus by homologous recombination. The sequenced plasmid was transformed into M. smegmatis by electroporation and selection with hygromycin. The site of recombination was confirmed by PCR [aggagcagaatgaccctcgttct, acacgttcgtgcagaccctcgt].
Determining the effects of knockdown on growth on solid and liquid medium
M. smegmatis pstP-CM was cultured in Middlebrook 7H9 broth with appropriate antibiotics (hygromycin for pstP-CM; hygromycin and kanamycin for pstP-CM transformed with pMV306 or pMV306-pstP). Cells were washed in 7H9 without inducer, serially diluted, and spotted in parallel onto 7H10 plates with/without inducer. Plates were incubated at 37 °C for three days and used to take photographic images. Images are representative of 3 independent experiments.
To determine the effects of knockdown of the pstP operon on growth in liquid medium, cultures were prepared and washed as above, and used to inoculate parallel cultures with/without inducer. The initial optical density was 0.05. Cultures were grown at 37 °C with shaking in a BioScreen (Fig. 2) or conical flasks (supplementary figures), and optical density was monitored.
Transmission Electron Microscopy (TEM)
M. smegmatis strains were seeded at OD 0.001 in 7H9 with/without inducer and incubated at 37 °C with shaking for 30 hours. Bacterial cells were harvested and washed in PBS then fixed in 2.5% glutaraldehyde in PBS. An aliquot was saved for scanning electron microscopy (see below) and the remainder were pelleted in 3% agar, cooled, and cut into 1 mm3 cubes. Agar cubes were treated with 1% w/v aqueous osmium tetroxide, dehydrated through an ethanol series, and embedded in Spurr’s Resin. Samples were sectioned to a thickness of approximately 70 nm using a Reichert Ultracut E ultramicrotome and collected onto copper mesh grids. Dry grids were submerged briefly in methanol before staining in 2% (w/v) uranyl acetate for 30 minutes followed by Reynolds lead citrate for 5 minutes. Samples were viewed on a JEOL JEM-1400 TEM with an accelerating voltage of 100 kV. Images were collected using a Megaview III digital camera with iTEM software.
Scanning Electron Microscopy (SEM)
An aliquot of glutaraldehyde-fixed cells (10 µl of each strain) was applied to a Poly-L-lysine treated glass slide, then further treated with 0.5% (w/v) aqueous osmium tetroxide and dehydrated through ethanol and anhydrous hexamethyldisilazane. Dried slides were mounted onto aluminium stubs and sputtercoated with gold/palladium in a Quorum Q150TES coating unit. Bacteria were viewed on a Hitachi S3000H SEM with an accelerating voltage of 10 kV. The length of 100 cells of each strain was measured using ImageJ and differences between the strains were analysed by Mann Whitney test.
Preparation of cell extract for proteomic and Western blot analysis
Exponential phase pstP-CM was washed and used to seed parallel 200 ml cultures with and without inducer at initial optical density of 0.1. Cultures were incubated at 37 °C with shaking and harvested after 6 hours. Replicate cultures were used for Western blotting and for proteomic analysis. Cells were resuspended in extraction buffer (ammonium bicarbonate 100 mM, NaCl 1 M, Urea 8 M, DTT 20 mM, PhosSTOP phosphatase inhibitors and cOmplete protease inhibitors (Roche)). Cells were disrupted using a Minilys personal homogenizer (Bertin instruments) using glass beads (Sigma) and cell debris was removed by centrifugation at 20,000 g for 30 minutes. The supernatant was concentrated using 1000 kDa molecular weight cut-off filters and protein concentration estimated using Qubit assay (Thermofisher). Protein concentration was normalized before Western blot analysis, and normalization was verified by parallel SDS PAGE with Coomassie blue staining. Polyclonal phosphothreonine antibody was purchased from Cell Signalling and used according to the manufacturer’s instructions.
Comparative proteomics and phosphoproteomics
M. smegmatis pstP-CM cell extracts were reduced and alkylated in extraction buffer containing 5 mM Tris(2-carboxyethyl) phosphine hydrochloride (TCEP) and 10 mM chloroacetamide (CAA), for 30 min at room temperature. Proteins were precipitated using the methanol-chloroform method38 then dissolved in 20 mM HEPES-Trypsin-LysC (1:100) and digested for 16–17hrs.
The tryptic peptides concentration was determined using Qubit. Part of the peptide sample (95%) was used for phosphoproteomics analysis and the remainder (5%) for proteome analysis. The peptides were dried using a SpeedVac concentrator and then resuspended in 80% acetonitrile, 5% trifluoroacetic acid (TFA). For the phosphoproteomics analysis, phosphopeptides were enriched using Titansphere (10 µm or 5 µm, 500 mg; 5020-75010 GL Sciences Inc. Japan) according to the manufacturer’s instructions. Dried peptides were resuspended in a buffer containing 2% acetonitrile and 0.1% TFA prior to analysis by mass spectrometry.
Reversed phase chromatography used an Acclaim PepMap µ-precolumn cartridge 300 µm i.d. × 5 mm, 5 μm, 100 Å and an Acclaim PepMap RSLC 75 µm i.d. × 50 pstP-CM, 2 µm, 100 Å (Thermo Scientific) using 0.1% formic acid and acetonitrile containing 0.1% formic acid. Eluted peptides were converted to gas-phase ions by means of electrospray ionization and analysed on a Thermo Orbitrap Fusion (Thermo Scientific).
MS raw files were analyzed by MaxQuant software version 1.5.5.339 and peptide lists were searched by the Andromeda Search Engine40 against the M. smegmatis Uniprot database (Proteome IF UP000000757, 6601 proteins). Bioinformatics and statistical analyses were performed in the Perseus software (version 1.5.5.3). Confidently assigned phosphorylation sites with localization probabilities greater than 75% and Score difference greater than 5 were defined as Class I sites and were used as site localization cutoff from the Phospho (STY)Sites table generated by MaxQuant analysis. Missing data points were imputed by creating a normal distribution with a width of 0.3 and a downshift of 1.8. Student’s T-tests for statistical significance were performed with permutation-based FDR correction threshold of 0.05. PRIDE accession number: PXD011805.
Supplementary information
Acknowledgements
Funding was provided by the BBSRC (BB/P001513/1) and the Indonesian Ministry of Education and Culture Direktorat Jenderal Pendidikan Tinggi (DIKTI). We would like to thank the University of Leicester Core Biotechnology Services Electron Microscopy Facility and Dr Maria Viskaduraki, Bioinformatics and Biostatistics Hub, for advice and suggestions on this work.
Author Contributions
H.M.O. wrote the manuscript text, all authors prepared figures and reviewed the manuscript.
Data Availability
The datasets generated during the current study are available in PRIDE, accession number: PXD011805. https://www.ebi.ac.uk/pride/archive/login.
Competing Interests
Paul Ajuh is a director and shareholder in Gemini Biosciences Limited, Liverpool, UK. Other authors declare no competing interests
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-44841-9.
References
- 1.Garcia-Garcia T, et al. Role of Protein Phosphorylation in the Regulation of Cell Cycle and DNA-Related Processes in Bacteria. Front Microbiol. 2016;7:184. doi: 10.3389/fmicb.2016.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rieck B, et al. PknG senses amino acid availability to control metabolism and virulence of Mycobacterium tuberculosis. PLoS pathogens. 2017;13:e1006399. doi: 10.1371/journal.ppat.1006399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boitel B, et al. PknB kinase activity is regulated by phosphorylation in two Thr residues and dephosphorylation by PstP, the cognate phospho-Ser/Thr phosphatase, in Mycobacterium tuberculosis. Mol Microbiol. 2003;49:1493–1508. doi: 10.1046/j.1365-2958.2003.03657.x. [DOI] [PubMed] [Google Scholar]
- 4.England P, et al. The FHA-containing protein GarA acts as a phosphorylation-dependent molecular switch in mycobacterial signaling. FEBS Lett. 2009;583:301–307. doi: 10.1016/j.febslet.2008.12.036. [DOI] [PubMed] [Google Scholar]
- 5.Chiang DY, Heck AJ, Dobrev D, Wehrens XH. Regulating the regulator: Insights into the cardiac protein phosphatase 1 interactome. J Mol Cell Cardiol. 2016;101:165–172. doi: 10.1016/j.yjmcc.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oberoi J, et al. Structural and functional basis of protein phosphatase 5 substrate specificity. Proc Natl Acad Sci USA. 2016;113:9009–9014. doi: 10.1073/pnas.1603059113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pereira SF, Goss L, Dworkin J. Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol Mol Biol Rev. 2011;75:192–212. doi: 10.1128/MMBR.00042-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chopra P, et al. Phosphoprotein phosphatase of Mycobacterium tuberculosis dephosphorylates serine-threonine kinases PknA and PknB. Biochem Biophys Res Commun. 2003;311:112–120. doi: 10.1016/j.bbrc.2003.09.173. [DOI] [PubMed] [Google Scholar]
- 9.Sacco E, et al. Serine/threonine protein phosphatase-mediated control of the peptidoglycan cross-linking L,D-transpeptidase pathway in Enterococcus faecium. MBio. 2014;5:e01446–01414. doi: 10.1128/mBio.01446-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellinzoni M, Wehenkel A, Shepard W, Alzari PM. Insights into the catalytic mechanism of PPM Ser/Thr phosphatases from the atomic resolution structures of a mycobacterial enzyme. Structure. 2007;15:863–872. doi: 10.1016/j.str.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez P, et al. The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J Bacteriol. 2006;188:7778–7784. doi: 10.1128/JB.00963-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arora D, Chawla Y, Malakar B, Singh A, Nandicoori VK. The transpeptidase PbpA and noncanonical transglycosylase RodA of Mycobacterium tuberculosis play important roles in regulating bacterial cell lengths. J Biol Chem. 2018;293:6497–6516. doi: 10.1074/jbc.M117.811190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sajid A, et al. Phosphorylation of Mycobacterium tuberculosis Ser/Thr phosphatase by PknA and PknB. PLoS One. 2011;6:e17871. doi: 10.1371/journal.pone.0017871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galagan JE, et al. TB database 2010: overview and update. Tuberculosis (Edinb) 2010;90:225–235. doi: 10.1016/j.tube.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Roback P, et al. A predicted operon map for Mycobacterium tuberculosis. Nucleic Acids Res. 2007;35:5085–5095. doi: 10.1093/nar/gkm518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cortes T, et al. Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep. 2013;5:1121–1131. doi: 10.1016/j.celrep.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang CM, et al. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev. 2005;19:1692–1704. doi: 10.1101/gad.1311105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gee CL, et al. A phosphorylated pseudokinase complex controls cell wall synthesis in mycobacteria. Sci Signal. 2012;5:ra7. doi: 10.1126/scisignal.2002525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Viswanathan, G., Yadav, S., Joshi, S. V., Raghunand, T. R. & Jass, J. Insights into the function of FhaA, a cell division-associated protein in mycobacteria. FEMS Microbiol Lett364, 10.1093/femsle/fnw294 (2017). [DOI] [PubMed]
- 20.Yeats C, Finn RD, Bateman A. The PASTA domain: a beta-lactam-binding domain. Trends Biochem Sci. 2002;27:438. doi: 10.1016/S0968-0004(02)02164-3. [DOI] [PubMed] [Google Scholar]
- 21.Manuse S, Fleurie A, Zucchini L, Lesterlin C, Grangeasse C. Role of eukaryotic-like serine/threonine kinases in bacterial cell division and morphogenesis. FEMS Microbiol Rev. 2016;40:41–56. doi: 10.1093/femsre/fuv041. [DOI] [PubMed] [Google Scholar]
- 22.Boutte, C. C. et al. A cytoplasmic peptidoglycan amidase homologue controls mycobacterial cell wall synthesis. Elife5, 10.7554/eLife.14590 (2016). [DOI] [PMC free article] [PubMed]
- 23.Turapov O, et al. Two Faces of CwlM, an Essential PknB Substrate, in Mycobacterium tuberculosis. Cell Rep. 2018;25:57–67 e55. doi: 10.1016/j.celrep.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burnside K, et al. Serine/threonine phosphatase Stp1 mediates post-transcriptional regulation of hemolysin, autolysis, and virulence of group B Streptococcus. J Biol Chem. 2011;286:44197–44210. doi: 10.1074/jbc.M111.313486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ulrych A, et al. Characterization of pneumococcal Ser/Thr protein phosphatase phpP mutant and identification of a novel PhpP substrate, putative RNA binding protein Jag. BMC Microbiol. 2016;16:247. doi: 10.1186/s12866-016-0865-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cameron DR, et al. Serine/threonine phosphatase Stp1 contributes to reduced susceptibility to vancomycin and virulence in Staphylococcus aureus. J Infect Dis. 2012;205:1677–1687. doi: 10.1093/infdis/jis252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma AK, et al. Serine/Threonine Protein Phosphatase PstP of Mycobacterium tuberculosis Is Necessary for Accurate Cell Division and Survival of Pathogen. J Biol Chem. 2016;291:24215–24230. doi: 10.1074/jbc.M116.754531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forti F, Crosta A, Ghisotti D. Pristinamycin-inducible gene regulation in mycobacteria. J Biotechnol. 2009;140:270–277. doi: 10.1016/j.jbiotec.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Wang R, Marcotte EM. The proteomic response of Mycobacterium smegmatis to anti-tuberculosis drugs suggests targeted pathways. J Proteome Res. 2008;7:855–865. doi: 10.1021/pr0703066. [DOI] [PubMed] [Google Scholar]
- 30.Macek B, et al. The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol Cell Proteomics. 2007;6:697–707. doi: 10.1074/mcp.M600464-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Prisic S, et al. Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc Natl Acad Sci USA. 2010;107:7521–7526. doi: 10.1073/pnas.0913482107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lombana TN, et al. Allosteric activation mechanism of the Mycobacterium tuberculosis receptor Ser/Thr protein kinase, PknB. Structure. 2010;18:1667–1677. doi: 10.1016/j.str.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenstein AE, Echols N, Lombana TN, King DS, Alber T. Allosteric activation by dimerization of the PknD receptor Ser/Thr protein kinase from Mycobacterium tuberculosis. J Biol Chem. 2007;282:11427–11435. doi: 10.1074/jbc.M610193200. [DOI] [PubMed] [Google Scholar]
- 34.Carette, X. et al. Multisystem Analysis of Mycobacterium tuberculosis Reveals Kinase-Dependent Remodeling of the Pathogen-Environment Interface. MBio9, 10.1128/mBio.02333-17 (2018). [DOI] [PMC free article] [PubMed]
- 35.Cashmore TJ, et al. Identification of a Membrane Protein Required for Lipomannan Maturation and Lipoarabinomannan Synthesis in Corynebacterineae. J Biol Chem. 2017;292:4976–4986. doi: 10.1074/jbc.M116.772202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakedi KC, et al. Identification of Novel Physiological Substrates of Mycobacterium bovis BCG Protein Kinase G (PknG) by Label-free Quantitative Phosphoproteomics. Mol Cell Proteomics. 2018;17:1365–1377. doi: 10.1074/mcp.RA118.000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stover CK, et al. New use of BCG for recombinant vaccines. Nature. 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- 38.Mirza SP, Halligan BD, Greene AS, Olivier M. Improved method for the analysis of membrane proteins by mass spectrometry. Physiol Genomics. 2007;30:89–94. doi: 10.1152/physiolgenomics.00279.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 40.Cox J, et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available in PRIDE, accession number: PXD011805. https://www.ebi.ac.uk/pride/archive/login.