

Abstract
Background
Molecular imaging such as positron emission tomography (PET) and single-photon emission computed tomography (SPECT) can provide the crucial pharmacokinetic-pharmacodynamic information of a drug non-invasively at an early stage of clinical drug development. Nevertheless, not much has been known how molecular imaging has been actually used in drug development studies.
Methods
We searched PubMed using such keywords as molecular imaging, PET, SPECT, drug development, and new drug, or any combination of those to select papers in English, published from January 1, 1990, to December 31, 2015. The information about the publication year, therapeutic area of a drug candidate, drug development phase, and imaging modality and utility of imaging were extracted.
Results
Of 10,264 papers initially screened, 208 papers met the eligibility criteria. The more recent the publication year, the bigger the number of papers, particularly since 2010. The two major therapeutic areas using molecular imaging to develop drugs were oncology (47.6%) and the central nervous system (CNS, 36.5%), in which efficacy (63.5%) and proof-of-concept through either receptor occupancy (RO) or other than RO (29.7%), respectively, were the primary utility of molecular imaging. PET was used 4.7 times more frequently than SPECT. Molecular imaging was most frequently used in phase I clinical trials (40.8%), whereas it was employed rarely in phase 0 or exploratory IND studies (1.4%).
Conclusions
The present study confirmed the trend that molecular imaging has been more actively employed in recent clinical drug development studies although its adoption was rather slow and rare in phase 0 studies.
Electronic supplementary material
The online version of this article (10.1007/s13139-019-00593-y) contains supplementary material, which is available to authorized users.
Keywords: Molecular imaging, Drug development, PET, SPECT
Introduction
Molecular imaging is “the visualization, characterization, and measurement of biological processes at the molecular and cellular levels in humans and other living systems” [1], whereas anatomical imaging visualizes morphologically noticeable changes by disease typically using magnetic resonance imaging (MRI) and computed tomography (CT) [2]. Molecular imaging can provide functional information even before the pathological process disrupts anatomical integrity, which is not easily obtained by anatomical imaging techniques. This unique feature of molecular imaging allows for early diagnosis of a disease, which also helps monitor a patient’s outcome to treatment. For example, positron emission tomography (PET) with [18F] fluorodeoxyglucose (FDG) has been widely used to diagnose a variety of cancers, while single-photon emission computed tomography (SPECT) with [99mTc] methoxyisobutylisonitrile can evaluate myocardial perfusion in patients with coronary artery disease [3]. In addition, [18F] FDG and [18F] fluorothymidine have been used to monitor response to tyrosine kinase inhibitors in lung cancer patients [4]. Furthermore, the mechanism of unknown pathophysiology can be studied using molecular imaging, which helps eventually identify new targets [5], enabling the development of novel drugs [6], diagnostics [7], and gene therapies [8]. PET and SPECT are the two most frequently used imaging modalities in molecular imaging. PET uses positron-emitting radioisotopes, whereas SPECT reconstructs images by electron capture and/or gamma emission of radioisotopes [9]. Both PET and SPECT are highly sensitive, and they have been widely used in clinical settings thanks to the availability of various tracers and radioligands with a good depth of penetration [10].
Drug development is a lengthy, risky, and costly task. The average cost to develop and obtain marketing approval for a new drug is currently estimated to be $2.558 billion (average out-of-pocket costs of $1.395 billion and time costs of $1.163 billion) [11]. Therefore, early termination of a drug candidate that is not likely to succeed at later stages makes the overall drug development program more efficient and economical [12]. To this end, molecular imaging can be used to visualize the biological activity of a test article or lack thereof even in an initial stage of drug development [13]. Furthermore, molecular imaging can non-invasively evaluate if a new drug candidate is distributed to its site(s) of action in humans and how tightly and extensively the candidate is bound to its receptor [14].
However, to the best of our knowledge, no investigation systematically evaluated how molecular imaging has been utilized in clinical drug development studies. Based on this understanding, the objectives of the present study were (1) to evaluate the employment of molecular imaging in clinical drug development, and (2) to identify factors associated with the choice of molecular imaging modality in clinical drug development studies. To achieve these objectives, we systematically reviewed papers that have reported the results of clinical drug development studies with molecular imaging over the last 25 years.
Materials and Methods
Literature Search
To identify eligible papers, PubMed was searched using the following keywords: molecular imaging, PET (or positron emission tomography), SPECT (or single-photon emission computed tomography), drug development, new drug, pharmacokinetics, pharmacodynamics, receptor occupancy, microdosing, or any combination of the keywords. Additional papers were also identified from the references listed in the review papers of molecular imaging in drug development. Only original papers published in English from January 1, 1990, to December 31, 2015, were eligible, and papers reporting the results of a new tracer development were excluded. Studies that were conducted after the study drug had been approved by the regulatory agency were also removed from the final study database because these studies were unlikely to have contributed to the regulatory approval of the study drug.
Data Extraction
Using the final study database, the following information were extracted: publication year, therapeutic area of the drug candidate (oncology, the central nervous system or CNS, cardiovascular, gastrointestinal, antibiotics, and others), clinical drug development phase (phases 0 through III, others, and unknown), imaging modality (PET, PET/CT, PET/MRI, SPECT, SPECT/CT, SEPCT/MRI, PET/SPECT combined, and PET/SPECT/CT combined), and utility of imaging (efficacy, proof-of-concept through receptor occupancy, proof-of-concept other than through receptor occupancy, and pharmacokinetics; multiple choice was allowed). If a paper presented a ClinicalTrials.gov identifier number, the extracted data were double-checked with the information obtained at www.clinicaltrials.gov. Three authors (HS, KJ, and HL) independently cross-checked for concurrence, and any differences were discussed until an agreement was reached.
Statistical Analysis
Data were summarized using descriptive statistics. Chi-square or Fisher’s exact test (in case of any cell with an expected number < 5) was performed to analyze whether the distribution of a categorical covariate was significantly different from that of another. The SAS statistical software (version 9.4, SAS Institute Inc., Cary, NC, USA) was used for the statistical analysis, and a two-tailed p value < 0.05 was considered significant.
Results
Literature Search and Selection of Papers
A total of 10,264 potentially relevant papers were screened, 208 of which were found eligible to be included in the final study database. Because three papers reported more than one imaging modality, the total number of imaging modalities was 211.
Publication Year and Therapeutic Area (Table 1)
Table 1.
Number of papers reporting the results of a drug development study with molecular imaging by therapeutic area and publication year
| Therapeutic area | Publication year* | |||||||
|---|---|---|---|---|---|---|---|---|
| 1990–1994 | 1995–1999 | 2000–2004 | 2005–2009 | 2010–2014 | 2015 | Total | p value | |
| Oncology | 0 (0.0) | 0 (0.0) | 16 (59.3) | 14 (32.6) | 60 (68.2) | 9 (52.9) | 99 (47.6) | |
| CNS | 9 (75.0) | 18 (85.7) | 5 (18.5) | 19 (44.2) | 21 (23.9) | 4 (23.4) | 76 (36.5) | |
| Cardiovascular | 1 (8.3) | 1 (4.8) | 5 (18.5) | 7 (16.3) | 5 (5.7) | 3 (17.6) | 22 (10.6) | |
| Gastrointestinal | 0 (0.0) | 0 (0.0) | 1 (3.7) | 1 (2.3) | 1 (1.1) | 0 (0.0) | 3 (1.4) | |
| Antibiotics | 2 (16.7) | 1(4.8) | 0 (0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 3 (1.4) | |
| Others | 0 (0) | 1 (4.8) | 0 (0) | 2 (4.7) | 1 (1.1) | 1(5.9) | 5 (2.4) | |
| Total | 12 (100) | 21 (100) | 27 (100) | 43 (100) | 88 (100) | 17 (100) | 208 (100) | <0.0001 |
Frequency (column percentage) is shown
CNS, central nervous system
*5-year epoch except for 2015, for which 1-year data were presented
Overall, the more recent the publication year, the greater the number of papers reporting the results of a clinical drug development study with molecular imaging. This trend was apparent particularly after 2010, i.e., the number of papers published in 2010–2014 has more than doubled than that in 2005–2009 (n = 88 vs. 43).
Molecular imaging was most commonly used in the development of anticancer drugs (n = 99, 47.6%), followed by drugs targeting the CNS (n = 76, 36.5%), whereas the development of antibiotics (n = 3, 1.4%) and drugs to treat gastrointestinal diseases (n = 3, 1.4%) barely used molecular imaging. The percentage that oncology occupied in each 5-year epoch has drastically increased since the early 2000s, whereas the proportion represented by the CNS in the same period has appeared to decrease. Collectively, the proportion taken by each therapeutic area was statistically significantly different by 5-year epoch of publication (p < 0.0001, Table 1).
Imaging Modality
PET (n = 174, 82.5%) was used 4.7 times more frequently than SPECT (n = 37, 17.5%), and both PET and SPECT were used alone more commonly than with CT or MRI (Fig. 1). When PET and SPECT were used in combination with CT or MRI, CT was the preferred imaging modality (22.3% vs. 15.2% for CT and MRI, respectively, in combination with PET; 1.9% vs. 0.9% for CT and MRI in combination with SPECT, Fig. 1).
Fig. 1.
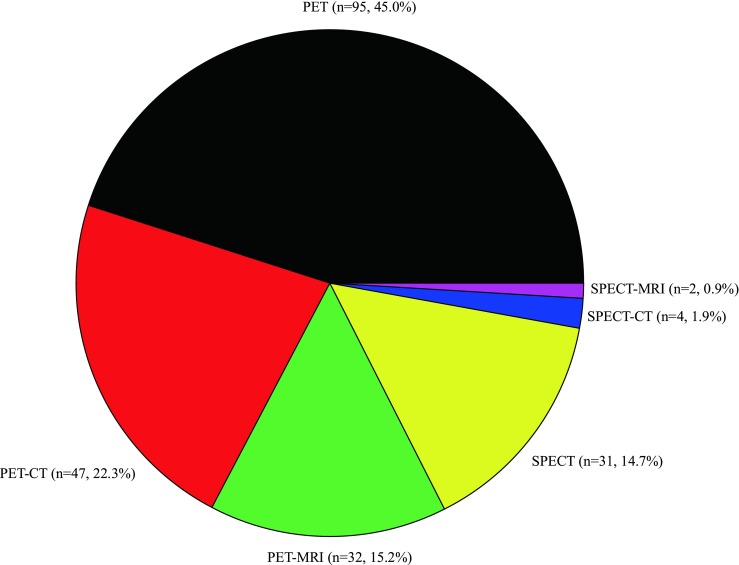
Imaging modality used in clinical drug development studies
The use of imaging modality was significantly different by therapeutic area (p < 0.0001, Table 2). Namely, PET was used more frequently than SPECT in the development of anticancer agents and CNS-targeting drugs, while cardiovascular (CV) and gastrointestinal drug development studies used SPECT more frequently than PET.
Table 2.
Imaging modality by therapeutic area
| Imaging modality | Therapeutic area | |||||||
|---|---|---|---|---|---|---|---|---|
| Oncology | CNS | Cardiovascular | Gastrointestinal | Antibiotics | Others | Total | p value | |
| PET | 96 (94.1) | 62 (81.6) | 10 (45.5) | 0 (0.0) | 3 (100.0) | 3 (60.0) | 174 (82.5) | |
| SPECT | 6 (5.9) | 14 (18.4) | 12 (54.5) | 3 (100.0) | 0 (0.0) | 2 (40.0) | 37 (17.5) | |
| Total | 102 (100.0) | 76 (100.0) | 22 (100.0) | 3 (100.0) | 3 (100.0) | 5 (100.0) | 211 (100.0) | < 0.0001 |
Frequency (column percentage) is shown. The total number of imaging modalities was 211 because three papers reported more than one imaging modality
PET, positron emission tomography; SPECT, single-photon emission computed tomography; CNS, central nervous system
Utility of Molecular Imaging
In general, the majority (63.5%) of molecular imaging studies has been conducted to evaluate efficacy, and assessment of proof-of-concept (POC) either through receptor occupancy (RO) or other than by RO was the second common utility of molecular imaging (29.7%, Table 3). However, this trend was reversed for CNS-targeting drug development, where molecular imaging was used most commonly to evaluate POC (69.1%, Table 3). Molecular imaging was infrequently used to obtain pharmacokinetic information (6.8%, Table 3).
Table 3.
Therapeutic area by utility of molecular imaging
| Therapeutic area | Utility of molecular imaging | |||||
|---|---|---|---|---|---|---|
| Efficacy | POC/RO | POC/non-RO | PK | Total | p value | |
| Oncology | 90 (84.9) | 1 (0.9) | 4 (3.8) | 11 (10.4) | 106 (100.0) | |
| CNS | 22 (27.2) | 49 (60.5) | 7 (8.6) | 3 (3.7) | 81 (100.0) | |
| Cardiovascular | 21 (91.3) | 0 (0.0) | 2 (8.7) | 0 (0.0) | 23 (100.0) | |
| Gastrointestinal | 3 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 3 (100.0) | |
| Antibiotics | 2 (66.7) | 0 (0.0) | 0 (0.0) | 1 (33.3) | 3 (100.0) | |
| Others | 3 (50.0) | 0 (0.0) | 3 (50.0) | 0 (0.0) | 6 (100.0) | |
| Total | 141 (63.5) | 50 (22.5) | 16 (7.2) | 15 (6.8) | 222 (100.0) | < 0.0001 |
Frequency (row percentage) is shown. The total number was 222 because multiple choice was allowed in a single paper
POC/RO, proof-of-concept based on receptor occupancy; POC/non-RO, proof-of-concept other than through receptor occupancy; PK, pharmacokinetics; CNS, central nervous system
Clinical Drug Development Phase
After excluding the unknown category, molecular imaging studies were most frequently conducted in phase I (40.8%), followed by phase II (20.9%), whereas phase 0 and III clinical trials adopting molecular imaging were rare (1.4% and 1.9%, respectively, Table 4). This trend has been observed in all therapeutic areas except for cardiovascular drugs, where phase II clinical trials employed molecular imaging most frequently (50.0%, Table 5). It was also interesting to note that SPECT was used most frequently in phase II (24.3%), although clinical drug development phase was unknown in almost half of the studies with SPECT (48.6%, Table 4).
Table 4.
Imaging modality by clinical drug development phase
| Imaging modality | Clinical drug development phase | |||||||
|---|---|---|---|---|---|---|---|---|
| Phase 0 | Phase I | Phase II | Phase III | Others | Unknown | Total | p value | |
| PET | 3 (1.7) | 80 (46.0) | 35 (20.1) | 2 (1.1) | 10 (5.7) | 44 (25.3) | 174 (100.0) | |
| SPECT | 0 (0.0) | 6 (16.2) | 9 (24.3) | 2 (5.4) | 2 (5.4) | 18 (48.6) | 37 (100.0) | |
| Total | 3 (1.4) | 86 (40.8) | 44 (20.9) | 4 (1.9) | 12 (5.7) | 62 (29.4) | 211 (100.0) | 0.0184 |
Frequency (row percentage) is shown. The total number of imaging modalities was 211 because three papers reported more than one imaging modality
PET, positron emission tomography; SPECT, single-photon emission computed tomography
Table 5.
Clinical drug development phase by therapeutic area that used molecular imaging
| Clinical drug development phase | Therapeutic area | |||||||
|---|---|---|---|---|---|---|---|---|
| Oncology | CNS | Cardiovascular | Gastrointestinal | Antibiotics | Others | Total | p value | |
| Phase 0 | 2 (2.0) | 1 (1.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 3 (1.4) | |
| Phase I | 66 (66.7) | 14 (18.4) | 3 (13.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 83 (39.9) | |
| Phase II | 27 (27.3) | 4 (5.3) | 11 (50.0) | 1 (33.3) | 0 (0.0) | 1 (20.0) | 44 (21.2) | |
| Phase III | 0 (0.0) | 4 (5.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 4 (1.9) | |
| Others | 1 (1.0) | 9 (11.8) | 1 (4.5) | 0 (0.0) | 0 (0.0) | 1 (20.0) | 12 (5.8) | |
| Unknown | 3 (3.0) | 44 (57.9) | 7 (31.8) | 2 (66.7) | 3 (100.0) | 3 (60.0) | 62 (29.8) | |
| Total | 99 (100.0) | 76 (100.0) | 22 (100.0) | 3 (100.0) | 3 (100.0) | 5 (100.0) | 208 (100.0) | < 0.0001 |
Frequency (column percentage) is shown
CNS, central nervous system
Imaging Tracers
A total of 61 tracers were used, i.e., 43 for PET and 18 for SPECT, of which [18F] FDG (n = 90, 50.8%) and [99mTc] sestamibi (n = 8, 22.2%) were the most frequently used ones for PET and SPECT, respectively (Supplementary Tables 1 and 2).
Discussion
Our results demonstrated that molecular imaging has been employed in clinical drug development studies more actively in recent years than in the past. For example, the number of papers reporting the results of a clinical drug development study with molecular imaging has doubled in the most recent 5-year epoch (i.e., 2010–2014) than in the previous years of 2005–2009 (Table 1). This is certainly a welcome finding, given that the US Food and Drug Administration stressed advancing the use of new imaging techniques in drug development, particularly as a product development tool [15]. However, it is uncertain if this trend will continue in the coming years because the number of papers with molecular imaging in clinical drug development has been slightly reduced in two recent years in a row (n = 20 and 17, respectively, in 2014 and 2015) after reaching the peak in 2013 (n = 24, data not shown). Although no clear explanation is available or it could be just a chance finding due to year-to-year variation, the reduction in the number of papers reporting the results of a molecular imaging study in recent years may have reflected the latest decrease in spending in research and development by pharmaceutical companies [16].
The largest number of studies with molecular imaging came from anticancer drug development, which has become even more apparent in the most recent 5-year epoch (Table 1). For example, RAD001 was tested in patients with non-small cell lung cancer, where [18F]FDG-PET was used to assess the efficacy (i.e., metabolic activity or tumor response) by quantifying the reduction in maximum standardized uptake value (SUVmax) [17]. RAD001 or everolimus was first approved by the US FDA in March 2009 in patients with advanced kidney cancer. The utility of [18F]FDG-PET to assess the efficacy of an anticancer agent has been well established. PET is known to be more sensitive in evaluating early response to treatment than anatomical imaging modalities such as CT, which has been traditionally used to assess the response to anticancer treatment [18]. For example, PET was consistently more accurate than CT for detecting or excluding nodal disease in patients with non-small-cell lung cancer, resulting in a much higher sensitivity (84% vs. 57% for PET vs. CT, respectively) and specificity (89% vs. 82% for PET vs. CT, respectively) for staging the mediastinum [19].
Drugs targeting the CNS comprised the second largest therapeutic area in clinical drug development studies with molecular imaging (Tables 2 and 3). It is also noteworthy that most drug development studies with molecular imaging prior to the 2000s were conducted in the CNS area (81.8%). This is probably because more CNS-targeting drugs were approved than anticancer agents in those early years; for example, the relative proportion of CNS drugs approved in the 1980s was almost twice as big as that of anticancer drugs (~ 10% vs. ~ 5%) [20]. In any case, PET and SPECT are useful tools to determine the pharmacokinetic and pharmacodynamic properties of CNS-targeting drugs as exemplified in the study of YKP1358, a novel antipsychotic agent [21]. In that study, Lim et al. successfully developed a pharmacokinetic-pharmacodynamic model in healthy volunteers based on the plasma drug concentration and receptor occupancy data of YKP1358 in the brain using PET, which guided the authors in determining a range of effective doses for further clinical trials in patients. This approach was practical and useful because doses effective for patients with psychotic diseases are generally too toxic to be tested in healthy volunteers. Therefore, a well-understood pharmacokinetic-pharmacodynamic relationship in healthy volunteers obtained from molecular imaging along with advanced modeling analysis can be extrapolated to patients to predict the efficacy directly related to receptor occupancy, even with doses not tested in healthy volunteers. Furthermore, molecular imaging can increase the understanding of CNS-targeting drug action, not necessarily by RO mechanism. For example, Wagner et al. used PET/MRI to evaluate the function of P-glycoprotein in the blood–brain barrier non-competitively inhibited by tariquidar [22]. Additionally, Shah et al. used PET/CT to determine whether MSDC-0160, an mTOT-modulating insulin sensitizer, would affect glucose metabolism in patients with mild Alzheimer’s disease [23].
PET has been used much more frequently than SPECT in clinical drug development studies with molecular imaging (Fig. 1, Tables 2 and 4). SPECT has been widely adopted in clinical practice because the half-life of the SPECT isotopes is considerably longer, and the cost to obtain images is lower than PET [24]. However, the use of SPECT in clinical drug development has fallen behind that of PET probably because of lower image resolution. Furthermore, PET is at least tenfold more sensitive than SPECT, and positron-emitting isotopes such as 11C, 13N, 15O, and 18F which replace a hydrogen atom in a molecule are the elements found in nearly every biomolecule so it can directly label molecules without interfering with their biological activity [25, 26]. These advantages of PET, further strengthened by its lower variability [27], have made PET a more preferred imaging modality than SPECT, particularly in the development of anticancer drugs and drugs targeting the CNS (Table 2). On the other hand, SPECT has been more frequently used than PET in the development of cardiovascular and gastrointestinal drugs (Table 2). The adequacy of such tracers as 201TI, [99mTc] sestamibi, and [99mTC] pertechnetate to image myocardial perfusion is well documented [28]. Additionally, gastric volume was successfully determined using SPECT with [99mTc] sestamibi and [99mTC] pertechnetate instead of invasive gastric barostat [29–31].
Both PET and SPECT were used alone more commonly than with other imaging modalities such as CT and MRI (Fig. 1). Although additional anatomical information can be obtained by co-registering PET or SPECT images with CT or MRI images using software approaches [32], co-registered images may not be good enough for internal abdominal organs that can move independently between scans even with the same stance [33]. To overcome this shortfall in image co-registration, multi-modality, which can acquire both functional (PET or SPECT) and anatomical (CT or MRI) images sequentially with a single scanner, has been developed [32].
CT was preferred to MRI as a co-registration modality for PET and SPECT (Fig. 1). The combination of PET and CT has already shown great value in clinical applications [34]. When compared with only anatomical imaging obtained by stand-alone CT, additional quantitative functional information gained by PET-CT not only allows for a more accurate diagnosis of a disease and response evaluation to treatment, but also enables optimizing treatment protocols. However, increased radiation to achieve good quality of the image in hybrid PET-CT is not negligible (~ 25 mSv) [35], and the quality of the image can be degraded by a subject’s voluntary or involuntary movement because PET and CT scans cannot be obtained simultaneously. On the other hand, MRI combined with PET does not engender an additional radiation dose because MRI does not emit ionizing radiation [36]. Furthermore, PET-MRI is capable of motion correction due to synchronous data collection from the two modalities, not to mention its excellent spatial resolution. Despite these advantages, however, the use of PET-MRI occupied only 15.2% of clinical drug development studies with molecular imaging in our results (Fig. 1) partly because of higher cost. The strong magnetic field and the radiofrequency from MRI are also likely to interfere with PET detectors, which can increase the noise level of MRI [37]. Therefore, these technical problems of PET-MRI should be overcome before we see its more frequent use in clinical drug development. The additional information that can be obtained by hybrid SPECT-CT or SPECT-MRI is not as useful as PET-CT or PET-MRI despite their increased costs, which may explain why those hybrid modalities were rarely used (Fig. 1). Of course, this trend might be reverted if improved attenuation correction and added value by hybrid imaging can be materialized [38].
It was rather surprising to find that only a minor fraction (6.8%, Table 3) of molecular imaging studies was performed to evaluate the pharmacokinetics (PK) of a drug candidate although drug concentrations in the target tissue can be continuously and non-invasively determined using real-time distribution imaging data [39]. The paucity of PK studies with molecular imaging may be attributed to the difficulty in labeling the drug candidate sufficient for imaging and additional regulatory burden to show the labeled drug is safe enough to be administered in humans.
Molecular imaging has been used most frequently in phase I clinical trials (Tables 4 and 5). Molecular imaging adopted in early stages of the clinical drug development program can reduce the time and costs for drug development [12, 40–43]. However, our results also showed that phase 0 or commonly known as exploratory investigational new drug (eIND) studies, in which a non-pharmacologic minute or micro dose is administered [44], comprised only 1.4% of the molecular imaging drug development studies we found (Tables 4 and 5). The phase 0 study using molecular imaging can rather easily assess the pharmacokinetic and pharmacodynamic properties of a new drug candidate without much animal toxicology data, greatly contributing to early, therefore better, attrition of compounds that are unlikely to be developed as a drug. For example, diazepam, midazolam, and ZK253 showed consistent and comparable pharmacokinetic profiles and oral bioavailability between the microdose and therapeutic dose studies [45]. Furthermore, in a microdose study with N-[2-(dimethylamino)ethyl]acridine-4-carboxamide (DACA) used as a cytotoxic agent, the investigators successfully observed not only the pharmacokinetic profile, but also the concentrations in tumor and tissue, which have implications for predicting activity and toxicity of DACA [46]. Difficulty in synthesizing a radiolabeled drug candidate at a study site, concern about non-linear pharmacokinetics at the higher doses, and the lack of regulatory guidance are major obstacles to overcome before more phase 0 molecular imaging studies are to be conducted. Furthermore, cultural and psychological resistance among drug development scientists to innovative technologies, partly based on a failure to recognize the potential benefits of microdose phase 0 studies, needs to be adequately addressed [47].
The present study had several limitations. Our study database might not be complete although we searched relevant papers in a systematic and comprehensive manner, complemented by additional search of individual papers listed in the review papers. Misclassification is another limitation, particularly with regard to clinical drug development phase because ~ 30% of the papers were classified as “unknown.” However, this would not affect the study conclusion much because it is hard to believe that the information on development phase was differentially missing by imaging modality (Table 4) or therapeutic area (Table 5). Despite these potential limitations, the present study successfully documented the active adoption of molecular imaging in clinical drug development studies, particularly since the 2000s, and associated factors for the first time to the best of our knowledge.
Conclusions
Molecular imaging has been rapidly employed in the early stage of clinical drug development, particularly in oncology and CNS-targeting drugs, although its adoption was rather slow in phase 0 studies. Molecular imaging can play an important role in optimizing drug development by studying the efficacy, POC, and pharmacokinetics of a drug candidate in a non-invasive manner. To facilitate the adoption of more molecular imaging in clinical drug development studies, technical issues such as co-registration of images for better resolution and efficient and practical radiolabeling should be adequately addressed. Furthermore, regulatory guidelines need to be provided, particularly to standardize imaging data such that the whole steps of image acquisition, processing, transfer, and archival are clearly defined.
Electronic Supplementary Material
(DOCX 27 kb)
Funding
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI14C1072).
Data Availability
The database that contains the design specifics of the molecular imaging drug development studies we reviewed in the present study can be accessed and downloaded at http://www.bioimaging.or.kr.
Compliance with Ethical Standards
Conflicts of Interest
Hyeomin Son, Kyungho Jang, Heechan Lee, Sang Eun Kim, Keon Wook Kang, and Howard Lee certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hyeomin Son and Kyungho Jang contributed equally to this work.
References
- 1.Mankoff DA. A definition of molecular imaging. J Nucl Med 2007;48:18N, 21N. [PubMed]
- 2.McDermott S, Kilcoyne A. Molecular imaging-its current role in cancer. QJM. 2016;109:295–299. doi: 10.1093/qjmed/hcv141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filipiak-Strzecka D, Kowalczyk E, Hamala P, Kot N, Kasprzak JD, Kusmierek J, et al. Long-term prognostic value of inducible and resting perfusion defects detected by single-photon emission computed tomography in the era of wide availability of coronary revascularization. Clin Physiol Funct Imaging. 2013;33:218–223. doi: 10.1111/cpf.12016. [DOI] [PubMed] [Google Scholar]
- 4.Teng FF, Meng X, Sun XD, Yu JM. New strategy for monitoring targeted therapy: molecular imaging. Int J Nanomedicine. 2013;8:3703–3713. doi: 10.2147/IJN.S51264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oh P, Li Y, Yu J, Durr E, Krasinska KM, Carver LA, et al. Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature. 2004;429:629–635. doi: 10.1038/nature02580. [DOI] [PubMed] [Google Scholar]
- 6.Gross S, Piwnica-Worms D. Molecular imaging strategies for drug discovery and development. Curr Opin Chem Biol. 2006;10:334–342. doi: 10.1016/j.cbpa.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 7.Rollo FD. Molecular imaging: an overview and clinical applications. Radiol Manage. 2003;25:28–32. [PubMed] [Google Scholar]
- 8.Shah K, Jacobs A, Breakefield XO, Weissleder R. Molecular imaging of gene therapy for cancer. Gene Ther. 2004;11:1175–1187. doi: 10.1038/sj.gt.3302278. [DOI] [PubMed] [Google Scholar]
- 9.Galban CJ, Galban S, Van Dort ME, Luker GD, Bhojani MS, Rehemtulla A, et al. Applications of molecular imaging. Prog Mol Biol Transl Sci. 2010;95:237–298. doi: 10.1016/B978-0-12-385071-3.00009-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaffer FA, Weissleder R. Molecular imaging in the clinical arena. JAMA. 2005;293:855–862. doi: 10.1001/jama.293.7.855. [DOI] [PubMed] [Google Scholar]
- 11.DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ. 2016;47:20–33. doi: 10.1016/j.jhealeco.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Molecular imaging in drug development. Nat Rev Drug Discov. 2008;7:591–607. doi: 10.1038/nrd2290. [DOI] [PubMed] [Google Scholar]
- 13.Josephs D, Spicer J, O'Doherty M. Molecular imaging in clinical trials. Target Oncol. 2009;4:151–168. doi: 10.1007/s11523-009-0117-x. [DOI] [PubMed] [Google Scholar]
- 14.Uppoor RS, Mummaneni P, Cooper E, Pien HH, Sorensen AG, Collins J, et al. The use of imaging in the early development of neuropharmacological drugs: a survey of approved NDAs. Clin Pharmacol Ther. 2008;84:69–74. doi: 10.1038/sj.clpt.6100422. [DOI] [PubMed] [Google Scholar]
- 15.Food and Drug Administration of USA. Critical path opportunities initiated during 2006. 2006. http://wayback.archive-it.org/7993/20180125075636/https://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/default.htm. Accessed 06 Jan 2018.
- 16.Pharmaceutical Research and Manufacturers of America (2015) Biopharmaceutical research industry profile. 2015. http://phrma-docs.phrma.org/sites/default/files/pdf/2015_phrma_profile.pdf. Accessed 06 Dec 2017.
- 17.Owonikoko TK, Ramalingam SS, Miller DL, Force SD, Sica GL, Mendel J, et al. A translational, Pharmacodynamic, and pharmacokinetic phase IB clinical study of everolimus in resectable non-small cell lung cancer. Clin Cancer Res. 2015;21:1859–1868. doi: 10.1158/1078-0432.CCR-14-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dimitrakopoulou-Strauss A. PET-based molecular imaging in personalized oncology: potential of the assessment of therapeutic outcome. Future Oncol. 2015;11:1083–1091. doi: 10.2217/fon.15.28. [DOI] [PubMed] [Google Scholar]
- 19.Toloza EM, Harpole L, McCrory DC. Noninvasive staging of non-small cell lung cancer: a review of the current evidence. Chest. 2003;123:137S–146S. doi: 10.1378/chest.123.1_suppl.137S. [DOI] [PubMed] [Google Scholar]
- 20.Tufts Center for the Study of Drug Development. Outlook 2015. 2015. https://static1.squarespace.com/static/5a9eb0c8e2ccd1158288d8dc/t/5aa2fd18f9619a2463540b42/1520631066097/Outlook-2015.pdf. Accessed 08 Jan 2018.
- 21.Lim KS, Kwon JS, Jang IJ, Jeong JM, Lee JS, Kim HW, et al. Modeling of brain D2 receptor occupancy-plasma concentration relationships with a novel antipsychotic, YKP1358, using serial PET scans in healthy volunteers. Clin Pharmacol Ther. 2007;81:252–258. doi: 10.1038/sj.clpt.6100049. [DOI] [PubMed] [Google Scholar]
- 22.Wagner CC, Bauer M, Karch R, Feurstein T, Kopp S, Chiba P, et al. A pilot study to assess the efficacy of tariquidar to inhibit P-glycoprotein at the human blood-brain barrier with (R)-11C-verapamil and PET. J Nucl Med. 2009;50:1954–1961. doi: 10.2967/jnumed.109.063289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah RC, Matthews DC, Andrews RD, Capuano AW, Fleischman DA, VanderLugt JT, et al. An evaluation of MSDC-0160, a prototype mTOT modulating insulin sensitizer, in patients with mild Alzheimer’s disease. Curr Alzheimer Res. 2014;11:564–573. doi: 10.2174/1567205011666140616113406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pimlott SL, Sutherland A. Molecular tracers for the PET and SPECT imaging of disease. Chem Soc Rev. 2011;40:149–162. doi: 10.1039/B922628C. [DOI] [PubMed] [Google Scholar]
- 25.Paans AM, van Waarde A, Elsinga PH, Willemsen AT, Vaalburg W. Positron emission tomography: the conceptual idea using a multidisciplinary approach. Methods. 2002;27:195–207. doi: 10.1016/S1046-2023(02)00075-0. [DOI] [PubMed] [Google Scholar]
- 26.Miller PW, Long NJ, Vilar R, Gee AD. Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew Chem Int Ed Eng. 2008;47:8998–9033. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 27.Catafau AM, Bullich S, Nucci G, Burgess C, Gray F, Merlo-Pich E, et al. Contribution of SPECT measurements of D2 and 5-HT2A occupancy to the clinical development of the antipsychotic SB-773812. J Nucl Med. 2011;52:526–534. doi: 10.2967/jnumed.110.081885. [DOI] [PubMed] [Google Scholar]
- 28.Min JJ, Gambhir SS. Gene therapy progress and prospects: noninvasive imaging of gene therapy in living subjects. Gene Ther. 2004;11:115–125. doi: 10.1038/sj.gt.3302191. [DOI] [PubMed] [Google Scholar]
- 29.Camilleri M, Vazquez-Roque M, Iturrino J, Boldingh A, Burton D, McKinzie S, et al. Effect of a glucagon-like peptide 1 analog, ROSE-010, on GI motor functions in female patients with constipation-predominant irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol. 2012;303:G120–G128. doi: 10.1152/ajpgi.00076.2012. [DOI] [PubMed] [Google Scholar]
- 30.Camilleri M, Bharucha AE, Ueno R, Burton D, Thomforde GM, Baxter K, et al. Effect of a selective chloride channel activator, lubiprostone, on gastrointestinal transit, gastric sensory, and motor functions in healthy volunteers. Am J Physiol Gastrointest Liver Physiol. 2006;290:G942–G947. doi: 10.1152/ajpgi.00264.2005. [DOI] [PubMed] [Google Scholar]
- 31.Delgado-Aros S, Chial HJ, Cremonini F, Ferber I, McKinzie S, Burton DD, et al. Effects of asimadoline, a kappa-opioid agonist, on satiation and postprandial symptoms in health. Aliment Pharmacol Ther. 2003;18:507–514. doi: 10.1046/j.1365-2036.2003.01670.x. [DOI] [PubMed] [Google Scholar]
- 32.Townsend DW, Cherry SR. Combining anatomy and function: the path to true image fusion. Eur Radiol. 2001;11:1968–1974. doi: 10.1007/s003300101007. [DOI] [PubMed] [Google Scholar]
- 33.Beyer T, Townsend DW, Brun T, Kinahan PE, Charron M, Roddy R, et al. A combined PET/CT scanner for clinical oncology. J Nucl Med. 2000;41:1369–1379. [PubMed] [Google Scholar]
- 34.Pichler BJ, Judenhofer MS, Pfannenberg C. Multimodal imaging approaches: PET/CT and PET/MRI. Handb Exp Pharmacol. 2008:109–32. [DOI] [PubMed]
- 35.Brix G, Lechel U, Glatting G, Ziegler SI, Munzing W, Muller SP, et al. Radiation exposure of patients undergoing whole-body dual-modality 18F-FDG PET/CT examinations. J Nucl Med. 2005;46:608–613. [PubMed] [Google Scholar]
- 36.de Rosales RT. Potential clinical applications of bimodal PET-MRI or SPECT-MRI agents. J Label Compd Radiopharm. 2014;57:298–303. doi: 10.1002/jlcr.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sauter AW, Wehrl HF, Kolb A, Judenhofer MS, Pichler BJ. Combined PET/MRI: one step further in multimodality imaging. Trends Mol Med. 2010;16:508–515. doi: 10.1016/j.molmed.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 38.Madsen MT. Recent advances in SPECT imaging. J Nucl Med. 2007;48:661–673. doi: 10.2967/jnumed.106.032680. [DOI] [PubMed] [Google Scholar]
- 39.Fischman AJ, Alpert NM, Rubin RH. Pharmacokinetic imaging: a noninvasive method for determining drug distribution and action. Clin Pharmacokinet. 2002;41:581–602. doi: 10.2165/00003088-200241080-00003. [DOI] [PubMed] [Google Scholar]
- 40.Wagner CC, Langer O. Approaches using molecular imaging technology -- use of PET in clinical microdose studies. Adv Drug Deliv Rev. 2011;63:539–546. doi: 10.1016/j.addr.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aboagye EO, Price PM, Jones T. In vivo pharmacokinetics and pharmacodynamics in drug development using positron-emission tomography. Drug Discov Today. 2001;6:293–302. doi: 10.1016/S1359-6446(01)01684-1. [DOI] [PubMed] [Google Scholar]
- 42.Rudin M, Weissleder R. Molecular imaging in drug discovery and development. Nat Rev Drug Discov. 2003;2:123–131. doi: 10.1038/nrd1007. [DOI] [PubMed] [Google Scholar]
- 43.Gomes CM, Abrunhosa AJ, Ramos P, Pauwels EK. Molecular imaging with SPECT as a tool for drug development. Adv Drug Deliv Rev. 2011;63:547–554. doi: 10.1016/j.addr.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 44.Kummar S, Rubinstein L, Kinders R, Parchment RE, Gutierrez ME, Murgo AJ, et al. Phase 0 clinical trials: conceptions and misconceptions. Cancer J. 2008;14:133–137. doi: 10.1097/PPO.0b013e318172d6f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lappin G, Kuhnz W, Jochemsen R, Kneer J, Chaudhary A, Oosterhuis B, et al. Use of microdosing to predict pharmacokinetics at the therapeutic dose: experience with 5 drugs. Clin Pharmacol Ther. 2006;80:203–215. doi: 10.1016/j.clpt.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 46.Saleem A, Harte RJ, Matthews JC, Osman S, Brady F, Luthra SK, et al. Pharmacokinetic evaluation of N-[2-(dimethylamino)ethyl]acridine-4-carboxamide in patients by positron emission tomography. J Clin Oncol. 2001;19:1421–1429. doi: 10.1200/JCO.2001.19.5.1421. [DOI] [PubMed] [Google Scholar]
- 47.Garner RC, Lappin G. The phase 0 microdosing concept. Br J Clin Pharmacol. 2006;61:367–370. doi: 10.1111/j.1365-2125.2006.02575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 27 kb)
Data Availability Statement
The database that contains the design specifics of the molecular imaging drug development studies we reviewed in the present study can be accessed and downloaded at http://www.bioimaging.or.kr.