

Abstract
Background
Previous work, by us and others, has shown that mammalian galectins-1 have a growth-inhibitory activity for mammalian cells which is apparently independent of their β-galactoside binding site.
Results
We have made recombinant human galectin-1 as a bacterial fusion protein with an N-terminal hexahistidine tag. This protein displays both haemagglutination and growth-inhibitory activities, even in the presence of the hexahistidine tag. Site-directed mutagenesis of this protein has confirmed the independent nature of the protein sites responsible for the two biological activities. Mutant proteins were created, which displayed each activity in the absence of the other.
Conclusions
Human galectin-1 possesses a growth-inhibitory site, which is not part of the β-galactoside binding site. A surface loop, comprising amino acid residues 25–30, and joining two internal β-strands, forms part of the growth-inhibitory site. This region is relatively close to the N-terminus of the protein, and N-terminal substitutions or extensions also affect growth-inhibitory activity. Further experiments will be necessary to fully define this site.
Background
It is now widely accepted that several galectins, and galectin-1 in particular, have biological functions over and above their obvious role in the binding or cross-linking of β-galactoside-containing glycans [1-3]. The concept of a galactose-binding protein as a negative growth regulator was first seriously proposed when a protein from mouse fibroblast cultures, the murine homologue of galectin-1, was shown to have both properties [4]. These workers went on to demonstrate that growth-inhibitory activity of this mGBP was present even when it bound a glycan at its galactose-binding site [5], suggesting that the two properties were independent functions of galectin-1. In fact, evidence for a growth-inhibitory phase could be seen in several earlier reports of the mitogenic activity of galectins from other species [6-8]. There was some evidence for a similar activity associated with human galectin-1, and it was suggested that proteolysis of secreted galectin-1 could account in part for the action of a growth-related cell-surface proteinase [9].
In an attempt to test this hypothesis, recombinant human galectin-1 was prepared as a bacterial fusion protein with glutathione-S-transferase [10]. Both the fusion protein, and the galectin-1 derived from it by proteolysis and re-purification, functioned as multivalent lectins, and displayed mitogenic activity, which was inhibited by lactose. Growth-inhibitory activity was not a property of the intact GST-galectin fusion protein, but only of the cleaved and purified galectin-1, and lactose did not inhibit this activity. It seemed likely that the GST domain of the fusion protein may sterically restrict access to a growth-inhibitory site on the galectin domain, but it clearly did not have a similar steric effect upon the galactose-binding site. These observations strengthened the case for independent galactose-binding and growth-inhibitory sites on the galectin-1 molecule.
Specific mutagenesis of the recombinant galectin molecule is an obvious way of confirming this conclusion. Consideration of both tertiary structure and mutagenesis studies suggests that histidine-45, asparagine-47, arginine-49, tryptophan-69, glutamate-72 and arginine-74 are all involved in, or influence, sugar binding [11-14]. (In numbering galectin-1 amino acid residues, we have found it more convenient to number from the N-terminal methionine, rather than to use the convention of Abbott and Feizi [15], who numbered from alanine-2, which is the N-terminus of the mature, natural protein). It should thus be straightforward to make a substitution mutant of galectin-1, in which galactose-binding activity is greatly reduced, or totally ablated. The assay of growth-inhibitory activity in such a mutant should indicate the existence, or otherwise, of a separate growth-inhibitory site. The earlier mutagenesis studies also suggest, however, that the yield and/or solubility of bacterial mutant galectins may often be low [11,14]. Our own experience was of poor solubility and yield for mutated or truncated GST-galectin fusion proteins (A. Cameron, K.Y. Chung and KS, unpublished observations). In attempting this study, we therefore thought it worthwhile to adopt another expression vector. The ProEX system produces fusion proteins with a short N-terminal extension, containing a hexahistidine sequence for nickel ion chelation chromatography [16]. This was chosen to produce recombinant proteins that were not dependent on β-galactoside binding for their purification, and were at the same time smaller, to minimise problems due to poor solubility and to steric interference between domains.
It may also be possible to identify a growth-inhibitory site directly by site-specific mutagenesis. The steric interference with growth-inhibitory activity, exerted by the GST domain attached to the N-terminus of galectin-1 in the fusion protein, does offer some insight into the possible location of such a site. Consideration of the three-dimensional structure of galectin-1 [12,13], in conjunction with that observation, suggested additional sites for mutagenesis. The structure bears a strong resemblance to that of galectin-2 [17]. Each monomer has a β-sandwich structure, comprising two antiparallel β-sheets of six (S1–S6) and five (F1–F5) strands, respectively. The β-strands are joined by turns or relatively short loops; almost 70% of the amino acids are in β-strands. The galactose-binding site is comprised of residues in the S4, S5, S6 and F3 strands. The N- and C-termini of the monomer are located close together, on the opposite side of the structure from the galactose-binding site. The surface that incorporates both termini is the contact region in the self-association of two monomers. Whilst it seems unlikely that the growth-inhibitory site could be directly on this "dimerisation interface", it may be close to it, as deduced from the steric effects of N-terminal extension. We identified the surface loops of the structure, located close to this interface, and distant from the galactose-binding site, and chose convenient amino acids within them for mutagenesis.
Using this rationale, we have created mutant proteins, which displayed either galactoside-binding or growth-inhibitory activites in the virtual absence of the other. A surface loop, comprising amino acid residues 25–30, and joining two internal β-strands, forms part of the growth-inhibitory site. This region is relatively close to the N-terminus of the protein, and N-terminal substitutions or extensions also affect growth-inhibitory activity.
Results and discussion
Figure 1 summarises SDS-PAGE analysis of several of the galectin-1 preparations. The hexahis-galectin-1 fusion protein corresponds to a protein band at about 16 kDa, and most preparations contain traces of a dimer at about 32 kDa, as was previously observed with GST-galectin-1 [10]. Despite the altered expression system, many mutant fusion proteins were still produced in low yields. In most cases where the yield of a mutant was low, there was a strong protein band at 16 kDa in the bacterial pellet, probably indicating protein precipitation following expression (not shown). As noted below, several of the mutants tended to precipitate slowly at the higher concentrations used for testing cell growth inhibition, and we also noted that some of these proteins precipitated even from much more dilute solutions after prolonged storage at -20°C. Table 1 summarises our findings with each of the mutants of galectin-1. Two of the mutant proteins (R49G and D47N) were produced in such low yields that complete experimental investigation was not feasible, but the partial findings were of some interest. In addition to these positive cases, we also created cDNAs corresponding to the mutants V6D and A28D, but in neither case could we detect expressed protein in E. coli DH5α.
Figure 1.

Electrophoresis of Galectin-1 Preparations SDS-PAGE analysis of 15% gels of recombinant human galectin-1 preparations A a) marker proteins b) bacterial lysate containing M1R mutant galectin-1 c) M1R galectin-1, purified by affinity chromatography d) normal galectin-1, purified by affinity chromatography B e) marker proteins f) purified D27N galectin-1 g) purified K29M galectin-1 h) purified K29T galectin-1
Table 1.
Summary of Galectin-1 Mutations
| Mutation | Yield (mg/l culture) | Haemagglutination. Endpoint (μg/ml) | Growth Inhibition I50 (μg/ml) |
| None | 8.0 | 1 | 5 |
| R49G* | 0.1 | 1 | nd |
| N47D* | 0.1 | 100 | nd |
| A28R* | 0.3 | - | >200 |
| D103A | 9.2 | 1 | 20 |
| P79R | 0.6 | 500 | 4 |
| C131S | 0.5 | - | 13 |
| K29M | 8.0 | 1 | - |
| K29T | 8.2 | 1 | - |
| D27N | 0.8 | 1 | 170 |
| M1R | 10.4 | 1 | - |
| M1R (thrombin) | n/a | 1 | 24 |
Yields and Biological Activities of Recombinant Galectin-1 Derivatives For each mutation in galectin-1, the yield of protein per litre of bacterial culture, the endpoint of the haemagglutination titration (in μg/ml) and the concentration giving 50% inhibition of fibroblast growth (I50, in μg/ml) are listed. The three entries marked * are for low-yield mutations, for which full analysis was not feasible. The final row contains data for the M1R mutant following thrombin cleavage and repurification.
One hope was that, with a relatively small fusion peptide tag, the recombinant hexahis-galectin-1 (wild-type) would display full biological activities without the need for proteolytic cleavage and protein re-purification. This was indeed the case. The endpoint of the haemagglutination titration was 1 μg/ml for the hexahis-galectin-1, compared to 1.3 μg/ml for the GST-galectin 1 fusion protein. It should also be noted that the growth-inhibitory activity seen here (I50 = 5 μg/ml) was about 12–15 times greater than with the recombinant galectin-1 cleaved from a GST-galectin 1 fusion protein by thrombin [10], and roughly comparable with that of natural human galectin-1 [9]. It is still, however, about tenfold less active than recombinant mouse or human galectin expressed in COS-1 cells [4,22]. The different growth-inhibitory activities of our two recombinant galectins-1 may indicate that the chemical nature of the N-terminal extensions may have a greater effect on this activity than their size, though a very large extension, such as the entire GST molecule, was clearly inhibitory. Although the linker peptide of hexahis-galectin-1 contains a renin-sensitive site for proteolysis, renin action was not tested experimentally, as it would still leave an N-terminal tripeptide extension.
Arginine-49 is located in the S4 β-strand, in the scheme of Lobsanov et al.[18]. To our knowledge, it has not been modified in galectin-1 before now, but it has been shown to be located at the galactose-binding site, interacting with the 4-hydroxyl of the galactose residue, in the three-dimensional structures proposed by Bourne et al.[12] and Liao et al.[13]. We thus expected that the R49G mutant galectin-1 would show reduced, or at least altered lectin activity, but in fact it was not significantly different from that of the control recombinant galectin-1. Because the yield of soluble protein was low, no further work was done with this mutant. A D47N mutation, also in the S4 β-strand, has been previously reported as lectin-negative [14]. We were able to confirm this finding, but, again, the yield of soluble protein was too low to carry out further experiments.
In the cases of two mutant proteins with reduced haemagglutination activity, additional experiments were done, to distinguish between a failure to bind to erythrocyte surface β-galactoside binding sites, and a reduction in erythrocyte cross-linking, either of which would result in reduced haemagglutination. Pretreatment with saturating concentrations of the mutant galectin-1 was followed by a haemagglutination titration with wild-type galectin-1. For both the P79R and the C131S mutants, this procedure resulted in agglutination at approximately the same endpoint (1 μg/ml) as with wild-type galectin-1 in a control experiment. If the mutant galectin-1 was binding to the erythrocyte surface, but failing to cross-link erythrocytes, we would have expected to see reduced haemagglutination with the wild-type galectin, as the mutant would block binding sites. Thus, in each case, the mutations disrupt the β-galactoside binding site, rather than the dimerisation of the galectin-1. This finding was confirmed by failure of the mutant protein in lectin-affinity chromatography (not shown). The dimeric nature of several of the mutant galectins-1, including P79R, is also shown by gel filtration (Figure 2). Lysozyme (14.5kDa) was used as a standard, which would co-elute with the galectin-1 monomer, but it is clear that all of the galectin preparations are substantially bigger under non-denaturing conditions.
Figure 2.

Gel Filtration of Recombinant Galectin-1 and Variants. Galectin-1 preparations purified by metal chelation chromatography were subjected to gel filtration on a Sephadex G-50 gel filtration column (60 × 1 cm; equilibrated in 30 mM Tris-HCl buffer pH 7.5). Fractions of 2 ml were collected, and absorbances at 280 nm measured. ▴ Normal galectin-1 ▪ N46 D mutant 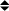 P79R mutant
P79R mutant  Bacterial lysozyme.
Bacterial lysozyme.
The P79R mutation occurs in a conserved, surface loop, between the S6 and F3 β-strands. It is relatively distant from the galactose-binding site, but is closer to the dimerisation interface of galectin-1 [12,13]. It was thus somewhat surprising that this mutant protein had a greatly reduced haemagglutinin activity, over two orders of magnitude below the control. This reduction was due to a loss of affinity for glycans, rather than to loss of cross-linking ability. Equally surprising was the observation that P79R retained full antiproliferative activity. Even if the substitution affects glycan binding as a consequence of a generally disruptive effect on tertiary structure, the retention of growth-inhibitory activity is significant. This is a clear confirmation that the galactose-binding and growth-inhibitory sites of galectin-1 are largely or wholly independent. It may be noteworthy that the elimination of the galactose-binding activity does not seem to result in more productive binding at the growth-inhibitory site.
The C131S mutation is also located close to the dimerisation interface, but in a surface β-strand (F1), rather than a loop. This galectin-1 variant was produced in low yield, and was totally devoid of haemagglutinin activity, but retained most of the growth-inhibitory potential of the wild-type. The loss of haemagglutinin activity is apparently caused by disruption of the β-galactoside-binding site. An earlier analysis of a C131S substitution confirmed the low yield of protein, but reported a relatively normal lactose binding value [14]. Substitutions in the extreme N-terminal region have been reported to disrupt hamster galectin-1 dimerisation, but concomitant substitutions in the C-terminus (F127, I129 and V132) led to a loss of lactose-binding affinity [23].
Cysteine-131 can potentially form a disulphide bond with C2, but failure to make this, and other disulphide bonds (C17 – C89 and C43 – C61), favours lectin activity [11]. A human galectin-1 variant with all six cysteines replaced by serines retains haemagglutinating activity [24]. It has been shown that the oxidative loss of lectin activity correlates with the formation of disulphide bonds [25], and two more recent reports suggest that some biological activities are associated only with this structural form [24,26]. In the present case, the ability to demonstrate lectin and growth-inhibitory activity with identical galectin-1 samples argues against the proposal that the growth-inhibitory activity is a property only of the oxidized, disulphide-bonded form.
The D103A mutation is located in a surface loop, connecting adjacent antiparallel β-strands (F4–F5), but is distant from both the dimerisation interface and the galactose-binding site. This substitution did not appear to affect the lectin function of galectin-1 (Table 1), though growth-inhibitory activity was somewhat reduced, and the titration of this activity was obscured by precipitation at higher concentrations. This latter finding was somewhat surprising, in view of the very high yield of this protein (8 mg/l) in the bacterial expression system.
Following our failure to detect expression of an A28D mutant (in another surface loop, F2–S3), we were able to express an A28R mutant, though the A28R protein was in low yield, and impure following Ni-NDA affinity chromatography, and we were unable to further purify it by Sephadex gel filtration or galactosyl-agarose affinity chromatography (not shown). An unknown protein of about 25 kDa was a persistent contaminant. The data reported in Table 1 was obtained with a protein sample containing 50–60% of the galectin-1 component, as estimated from the SDS-PAGE result. Again, the mutation affected galactose binding, but, in this case, growth-inhibitory activity was also greatly reduced. Despite the reservations imposed by the low yield and impurity of the sample, this observation offered a clue to the possible localization of the growth-inhibitory site, and we decided to make further mutational substitutions in this region of galectin-1.
The K29M mutation, also in the F2–S3 loop, resulted in a galectin that retained its galactose-binding activity. Testing of growth-inhibitory activity was initially obscured by the precipitation of this protein at high concentrations (above 100 μg/ml), but it became clear that it had totally lost its ability to inhibit fibroblast growth. Whilst this radical substitution, of a moderately hydrophobic amino acid for a charged residue, clearly had an effect upon the solubility of the protein, (though, again, not on protein expression) the complete retention of lectin activity argued for the maintenance of overall three-dimensional structure. The K29T mutant, with a less radical substitution, was also a normal lectin, but equally devoid of growth-inhibitory activity (see Table 1). As a further check upon the importance of this part of the galectin-1 molecule to its growth-inhibitory activity, a D27N mutant was made and tested, and demonstrated full lectin activity, but significantly reduced growth-inhibitory activity (Table 1).
If the D27 – K29 region of galectin-1 is part of a growth-inhibitory site, then the effect of N-terminal modifications on growth-inhibitory activity may be explicable, as the "natural" amino terminus is within 8–10 Angstroms of these amino acids [12]. It seems possible that the reduction of this activity, seen in both of our recombinant galectin types, hexahis- and GST-derived, may be due to modification of the N-terminus. As already discussed, it cannot simply be a steric restriction by the extended N-termini, as this would have a more marked effect on hexahis-galectin-1, whereas the reverse is true. To further investigate this region, we designed a forward PCR primer intended to substitute methionine-1 with an arginine (M1R). The M1R fusion protein was expressed in high yield, and was fully functional as a haemagglutinin, but was devoid of growth-inhibitory activity (Table 1). Rather than allowing it to substitute for the terminal amino group, the positioning of the positively-charged arginyl side-chain acts to inhibit the action of the growth-inhibitory site, which helps confirm that the N-terminal region of the normal protein is close to, or part of this site.
It also seemed possible that thrombin might remove the N-terminal 14-residue peptide (hexahis-linker-arginine) from the M1R recombinant protein, and thus re-create the natural, methionine N-terminus. Thrombin treatment and repurification of the protein resulted in a fraction that did not bind to the Ni-NDA-agarose affinity column, so had lost the N-terminal hexahis region. It retained haemagglutinin activity, and had also recovered growth-inhibitory activity, though was not as effective as the wild-type galectin-1. It is possible that this reduction in growth-inhibitory activity (I50 = 24 cf 5 for wild-type galectin-1) reflects the effect of other thrombin cleavages within the molecule, as was seen previously with the GST-derived galectin-1 [10]. In this case, N-terminal amino acid sequencing, following removal of the GST with thrombin revealed a minor component corresponding to the sequence beginning at F50. We deduced that this could be due to a thrombin cleavage at R49-F50. This protein was indistinguishable from intact recombinant galectin-1 in SDS-PAGE. In the present case, we have not examined this experimentally.
Figure 3, derived from a structural model [12], shows the part of the protein (shaded region c) that has been demonstrated to have a role in the growth-inhibitory activity. The β-galactoside-binding site (region a), and the dimerisation interface (region b) are also indicated. The elements of the growth-inhibitory site that have so far been identified would all be contained within a truncated galectin-1, containing the first 35 residues (the S1, F2 and S3 β-strands, and connecting loops), in addition to the fusion tag. Although we were able to amplify this part of the galectin-1 cDNA, and clone it into the ProEx vector, we were unable to detect any expression in E. coli (not shown).
Figure 3.

Location of Active Sites within the Galectin-1 Structure This figure is adapted from one designed to show the three-dimensional structure of galectin-1 (Bourne et al., 1994). The three shaded boxes indicate regions involved in glycan binding (a), dimerisation (b), and cellular growth inhibition (c).
Fusion proteins with an N-terminal tag have given valuable clues to the nature of the growth-inhibitory site, but, for further elucidation of the involvement of the N-terminus, it would be better in the future to use C-terminally-tagged proteins, or to express galectin-1 without any tag, and to use glycan-agarose affinity chromatography for purification of the recombinant proteins. The extreme C-terminus is also part of the dimerisation interface, but points in the opposite direction to the N-terminus (see Figure 3), so C-terminal extensions may not affect the growth-inhibitory site, though, as already noted, C-terminal substitutions may inhibit β-galactoside binding [22].
The D27 – K29 region forms part of a DAKSF sequence (residues 27–31) that is totally conserved in the mammalian galectins-1 [27], but is totally different from the corresponding residues in other galectins [28], for which growth-inhibitory properties have not been reported. However, there is no strong argument here for the evolutionary conservation of a functional part of the galectin-1 structure. Most of the sequences of the mammalian galectins-1 are strongly conserved throughout, whereas, when we consider the whole galectin family, the interstrand loops, and even the β-strands not involved in galactose binding, are not strongly conserved.
Conclusions
In summary, we have shown that we can ablate the β-galactoside binding activity of human galectin-1 by mutation, with no effect on the cellular growth-inhibitory activity, confirming that these are two independent functions of the protein. By further mutational studies, we have at least partially defined the amino acids that make up the growth-inhibitory site in galectin-1. A surface loop, comprising amino acid residues 25–30, and joining two internal β-strands, contains amino acids which contribute to the growth-inhibitory site. This region is relatively close to the N-terminus of the protein, and N-terminal substitutions or extensions also affect growth-inhibitory activity. Further mutational studies may be necessary to fully define the growth-inhibitory site.
The ProEX expression system offers some advantages in the production of bacterial recombinant galectin-1. Our original interest in galectin-1 was as a putative substrate for a growth-regulatory, cell-surface proteinase [9]. Inhibition of the proteinase led to an accumulation of endogenous galectin-1 in the culture medium. The inhibitory action of this galectin-1 could, at least in part, account for the growth-inhibitory effect observed when the proteinase was inhibited. The fact that the recombinant hexahis-galectin-1 has a similar growth-inhibitory activity to the natural protein means that it can be used more realistically in future experiments in this area.
Some of these mutants may enable us to assess the significance of each galectin-1 function to tumour growth and metastasis. We have shown that HEp-2 tumour cells do not express galectin-1, but respond to exogenous galectin-1 as a growth inhibitor [10]. Transfection of HEp-2 cells with each galectin-1 gene variant will permit the study of effects on growth and metastasis, both in vitro, and then in vivo. The galectin-1 variant, P79R, which retains growth-inhibitory activity and reasonable solubility, but has little lectin activity, may be the starting-point for the creation of an effective antitumour agent. It would not have the mitogenic activity seen with native galectin-1 [10], nor would it be involved in non-productive binding to galactosides in the extracellular matrix. This galectin-1 variant could also be used to identify the cellular receptor responsible for the growth-inhibitory effect, since binding to cellular glycans will be minimised.
Materials and methods
Cloning and mutation of the galectin-1 gene
Unless otherwise noted, molecular biology reagents were purchased from Gibco-BRL and all other reagents were from Sigma. The plasmid expression vector ProEX was purchased as part of a kit, which also included the corresponding nickel-containing affinity matrix. We used standard protocols for techniques such as DNA restriction and ligation, bacterial transformation, and agarose gel electrophoresis [18].
Galectin-1 cDNA was re-amplified from human fibroblast cDNA, using the GRP3 and GRP4 primers, as previously described [10]. Specific mutations were introduced using the Overlap Extension PCR technique [19], which uses complementary forward and reverse oligonucleotide primers incorporating the intended substitution. Primers were used as specified in Table 1, in conjunction with the GRP3 and 4 primers. For mutations close to either end of the galectin-1 gene, extended and modified versions of GRP 3 or 4 were used. MasterMix (Qiagen; 25 μl) was used as a source of buffer, nucleotides and polymerase for the PCR reaction, together with 20 pmol of each primer and 2 μg template DNA in a final volume of 50 μl, containing 1 mM Mg Cl2. Thirty cycles of PCR were performed, with 30 s at 95°C for denaturation, 90 s at 55°C for annealing, and 60 s at 72°C for polymerase extension. In some cases, the 55°C annealing temperature was inappropriate for particular overlapping primers, and was modified to suit the primer composition, usually to 45–47°C. The amplified product was run on a 1.0% agarose horizontal gel in 40 mM Tris-acetate, pH 8.0, containing 1 mM EDTA buffer, cut out of the gel and purified using the Rapid Extraction protocol (Gibco-BRL). The product was restricted with BamH I and SalI restriction enzymes and ligated using T4 DNA ligase into the ProEX HTb vector restricted with the same enzymes, then transformed into competent E. coli DH 5α cells, as previously described [10]. After plating on ampicillin-L-broth agar culture plates, to select plasmid-bearing clones, PCR was used to screen cloned colonies for transformants, using part of each colony as a source of the template, with the GRP 3 and 4 primers, and using agarose electrophoresis to check for the presence of a 450-bp galectin-1 band [10]. Following PCR amplification and cloning of normal and of putative mutated galectin-1 cDNAs, each was sequenced bidirectionally by a modified Sanger technique using a 377 Sequencer (ABI) in the Sequencing Unit of the School of Biological Sciences, to confirm the expected mutated sequence, and to check the integrity of the rest of the cDNA.
Table 2.
Oligonucleotide Primers used for Overlap Extension
| Mutants | Primers |
| M1R |
M1R: CTG GGA TCC AGG GCT TGT GGT CTG GTC GRP4: ACGCGTC GAC TCA GTC AAA GGC CAC ACA |
| V6D |
V6D: CTG GGA TCC ATG GCT TGT GGT CTG GAC GCC AGC AAC GRP4: ACGCGTC GAC TCA GTC AAA GGC CAC ACA |
| D27N |
D26N-F: GTG GCT CCT AAC GCT AAG AGC D26N-R: GCT CTT AGC GTT AGG AGC CAC |
| A28D |
A28D-F: GCT CCT GAC GAT AAG AGC TTC A28D-R: GAA GCT CTT ATC GTC AGG AGC |
| A28R |
A28R-F: GCT CCT GAC CGT AAG AGC TTC A28R-R: GAA GCT CTT ACG GTC AGG TTC |
| K29M |
K29M-F: CCT GAC GCT ATG AGC TTC GTG K29M-R: CAC GAA GCT CAT AGC GTC AGG |
| K29T |
K29T-F: CCT GAC GCT ACG AGC TTC GTG K29T-R: CAC GAA GCT CGT AGC GTC AGG |
| N47D |
N47D-F: CTG CAC TTC GAC CCT CGC TTC N47D-R: GAA GCG AGG GTC GAA GTG CAG |
| P79R |
P79R-F: GCT GTC TTT CGC TTC CAG CCT P79R-R: AGC CTG GAA GCG AAA GAC AGC |
| D103A |
D103A-F: AAG CTG CCA GCT GGA TAC GAA D103-R: TTC GTA TCC AGC TGG CAG CTT |
| C131S |
GRP3: CTG GGA TCC ATG GCT TGT GGT CTG GTC C131S-R: ACGCGTCGACTCA GTC AAA GGC CAC AGA TTT GAT CTT |
| R49G |
R49G-F: TTC AAC CCT GGC TTC AAC GCC. R49G-R: GGC GTT GAA GCC AGG GTT GAA. |
Forward primer for galectin-1 geneGRP3: CTG GGATCC ATG GCT TGT GGT CTG GTC (BamH I site is bold and underlined) Reverse primer for galectin-1 geneGRP4: ACGC CTCGAC TCA GTC AAA GGC CAC ACA (SaI I site is bold and underlined). Mutagenesis in Galectin-1 In the case of internal mutations, the primers listed were used in conjunction with the 5' and 3' primers (GRP3 and GRP4), to make two, overlapping galectin-1 cDNA fragments, which then were annealed and extended to create galectin-1 cDNA with the intended substitution. In the case of substitutions near the ends of the molecule, a modified version of GRP 3 or 4 was used, in conjunction with the other, normal primer, for conventional PCR amplification of the cDNA.
Protein expression and characterisation
Transformant colonies having the insert were innoculated in 3 ml L-broth/ampicillin medium and grown at 37°C in a shaking incubator until the absorbance at 600 nm reached 0.8. At this stage the cultures were induced for 3 h with 0.6 mM IPTG, and then microfuged for 10 min at 5000 rpm. The cell pellets were boiled in gel loading buffer, and applied to a 15% SDS polyacrylamide gel (Biorad), to test for expression of the fusion protein. For large-scale expression, 2 litre cultures were grown and induced under the same conditions. Where protein yields were low, cultures of up to 8 litres were used. The cultures were centrifuged in a Sorvall GSA rotor at 5,500 rpm for 10 min to collect cells and the pellet was resuspended in 10 ml of ice-cold sodium phosphate-buffered isotonic saline (PBS) and extensively sonicated at 4°C to lyse the cells. The lysate was centrifuged (15,000 rpm in Sorvall SS34 rotor for 20 min) and the supernatant was used for affinity chromatography.
For affinity purification of the recombinant fusion proteins, a Ni-NDA-agarose column (4 ml) was equilibrated in 20 mM Tris-HCl buffer, pH 8.5, containing 100 mM KCl, 5 mM 2-mercaptoethanol, 10% (v/v) glycerol and 20 mM imidazole at room temperature. The culture supernatant was applied at a flow rate of 0.5 ml/min. The column was then washed with the equilibration buffer until the A280 of the eluant was stable, and then with 10 ml of 20 mM Tris-HCl buffer, pH 8.5, containing 100 mM KCl, 5 mM 2-mercaptoethanol and 10% (v/v) glycerol, and then with a further 10 ml of equilibration buffer. The recombinant protein was eluted with 20 ml of 20 mM Tris-HCl buffer, pH 8.5, containing 100 mM KCl, 5 mM 2-mercaptoethanol, 10% (v/v) glycerol and 100 mM imidazole [16]. Protein-containing fractions were pooled, desalted by overnight dialysis against distilled water at 4°C, and freeze-dried. Protein purity was checked by electrophoresis in SDS-PAGE, as already described.
To a recombinant protein with an introduced arginine residue at the N-terminus (mutation M1R), thrombin (1%, w/w) was added, under the same conditions as were previously used to cleave GST-galectin-1 fusion proteins [10]. The resulting protein was purified by gel filtration on a Sephadex G-50 gel filtration column (60 × 1 cm; equilibrated in 30 mM Tris-HCl buffer pH 7.5), to remove small peptides, and was then re-chromatographed on the Ni-NDA-agarose column, to test for the presence of the N-terminal hexahistidine tag.
Erythrocyte agglutination by recombinant lectins was tested using freshly-trypsinised rabbit erythrocytes [20]. Agglutination experiments were carried out with serial threefold dilutions of the purified fusion protein in ceramic tiles. Each well contained 100 μl of 1% bovine serum albumin in 0.15 M NaCl, 100 μl of 0.15 M NaCl and 100 μl of erythrocyte suspension, and 100 μl protein solution (1 mg.ml-1 in phosphate-buffered saline) was used to start the serial dilution. Visual identification of the endpoint of the titration of agglutination was confirmed by microscopic detection of agglutinated erythrocytes.
Erythrocyte agglutination by wild-type hexahis-galectin-1 (unmodified, apart from the fusion tag) was maximal at concentrations of 100–300 μg.ml-1, and declined at higher concentrations. Where agglutination was not observed with a mutant galectin-1, trypsinised rabbit erythrocytes were incubated with the protein at 250 μg.ml-1 for 30 min, washed several times with saline, and then used for agglutination testing with wild-type galectin-1, as described above, so as to distinguish between a failure of glycan binding, or of cross-linking by the mutant protein.
Tissue culture
WI 3 8 or GM 1604A are normal human diploid fibroblast strains previously used by us in many experiments on growth-related proteolysis. The conditions for maintenance and culture of cells have been previously described, as have assays for inhibition of cell proliferation in microwell culture plates, using an ELISA spectrophotometer to measure fixed and stained cells [21]. In cell proliferation assays, the hexahis-galectin fusion proteins were added to normal culture media at known concentrations. For each assay series, the galectin-1 concentration was plotted semi-logarithmically against percentage of cell growth inhibition [10], to determine the concentration causing 50% growth inhibition (I50).
Acknowledgments
Acknowledgements
This work was supported by the University of Auckland Research Committee, through the Staff Research Fund. The Animal Resources Unit supplied rabbit erythrocytes. We wish to thank Linda Adams and Julia Mount, for technical assistance in the early part of this work, and Andrew Cameron and Kong Wai Chung, for preliminary attempts to make galectin-1 mutants. Libby Hitchings and, particularly, Dr Clyde Smith, helped with molecular graphics, and Dr Cristina Weinberg continued to take an interest.
Contributor Information
Ken Scott, Email: k.scott@auckland.ac.nz.
Jialiang Zhang, Email: Herbert.Zhang@neuronz.com.
References
- Perillo NL, Marcus ME, Baum LG. Galectins: versatile modulators of cell adhesion, cell proliferation and cell death. J Mol Med. 1998;76:402–412. doi: 10.1007/s001090050232. [DOI] [PubMed] [Google Scholar]
- Cooper DNW, Barondes SH. God must love galectins: He made so many of them. Glycobiology. 1999;9:979–984. doi: 10.1093/glycob/9.10.979. [DOI] [PubMed] [Google Scholar]
- Liu FT. Galectins: a new family of regulators of inflammation. Clin Immunol. 2000;97:79–88. doi: 10.1006/clim.2000.4912. [DOI] [PubMed] [Google Scholar]
- Wells V, Mallucci L. Identification of an autocrine negative growth factor: mouse β-galactoside-binding protein is a cytostatic factor and cell growth regulator. Cell. 1991;64:91–97. doi: 10.1016/0092-8674(91)90211-g. [DOI] [PubMed] [Google Scholar]
- Wells V, Mallucci L. Molecular expression of the negative growth factor murine β-galactoside binding protein (mGBP) Biochim Biophys Acta. 1992;1121:239–244. doi: 10.1016/0167-4838(92)90152-4. [DOI] [PubMed] [Google Scholar]
- Lipsick JS, Beyer EC, Barondes SH, Kaplan NO. Lectins from chicken tissues are mitogenic for thy-1 negative murine spleen cells. Biochem Biophys Res Comm. 1980;97:56–61. doi: 10.1016/s0006-291x(80)80133-1. [DOI] [PubMed] [Google Scholar]
- Pitts MJ, Yang DCH. Mitogenicity and binding properties of β-galactoside-binding lectin from chickembryo kidney. Biochem J. 1981;195:435–439. doi: 10.1042/bj1950435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford GL, Harris-Hooker S. Stimulation of vascular cell proliferation by β-galactoside specific lectins. FASEB J. 1990;4:2912–2918. doi: 10.1096/fasebj.4.11.2379767. [DOI] [PubMed] [Google Scholar]
- Manilal S, Scott GK, Tse CA. Inhibition of an endogenous growth-related proteinase enhances the recovery of a negative growth regulator from cultured human cells. Cell Biol Intl. 1993;17:317–323. doi: 10.1006/cbir.1993.1067. [DOI] [PubMed] [Google Scholar]
- Adams L, Scott GK, Weinberg CS. Biphasic modulation of cell growth by recombinant human galectin-1. Biochim Biophys Acta. 1996;1312:137–144. doi: 10.1016/0167-4889(96)00031-6. [DOI] [PubMed] [Google Scholar]
- Abbott WM, Feizi T. Soluble 14 kDa β-galactoside-specific bovine lectin. J Biol Chem. 1991;266:5552–5557. [PubMed] [Google Scholar]
- Bourne Y, Bolgiano B, Liao DI, Strecker G, Cantau P, Herzberg O, Feizi T, Cambillau C. Crosslinking of mammalian lectin (galectin-1) by complex biantennary saccharides. Nature Structural Biology. 1994;1:863–870. doi: 10.1038/nsb1294-863. [DOI] [PubMed] [Google Scholar]
- Liao DI, Kapadia G, Ahmed H, Vasta GR, Herzberg O. Structure of S-lectin, a developmentally regulated vertebrate β-galactoside-binding protein. Proc Natl Acad Sci. 1994;91:1428–1432. doi: 10.1073/pnas.91.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi J, Kasai. KI. Effect of amino acid substitution by site-directed mutagenesis on the carbohydrate recognition and stability of human 14 kDa β-galactoside-binding lectin. J Biol Chem. 1991;266:648–23. [PubMed] [Google Scholar]
- Abbott WM, Feizi T. Evidence that the 14 kDa soluble β-galactoside-binding lectin in man is encoded by a single gene. Biochem J. 1989;259:291–294. doi: 10.1042/bj2590291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SK, Evans DB, Vosters AF, Mcquade TJ, Tarpley WG. Metal affinity chromatography of recombinant HIV-1 reverse transcriptase containing a human renin cleavable metal binding domain. Biotech Appl Biochem. 1991;14:69–81. [PubMed] [Google Scholar]
- Lobsanov YD, Gitt MA, Leffler H, Barondes SH, Rini JM. X-ray crystal structure of the human dimeric S-lac lectin, L-14-II, in complex with lactose at 2.9 A resolution. J Biol Chem. 1993;268:27034–27038. doi: 10.2210/pdb1hlc/pdb. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch F, Maniatis T, eds . Cold Spring Harbor Laboratory, 2. Cold Spring Harbor Press; 1989. Molecular cloning; a laboratory manual. [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Lis H, Sharon N. Soy bean (Glycine max) agglutinin. Methods Enzymol. 1972;28:360–368. [Google Scholar]
- Scott GK, Tse CA. Modulation of cell proliferation by protein proteinase inhibitors; a new analytical approach. Biol Chem Hoppe-Seyler. 1988;369 Suppl 5:131–135. [PubMed] [Google Scholar]
- Wells V, Davies D, Mallucci L. Cell cycle arrest and induction of apoptosis by β galactoside binding protein (βGBP) in human mammary cancer cells. A potential new approach to cancer control. Eur J Cancer. 1999;35:978–983. doi: 10.1016/S0959-8049(99)00020-9. [DOI] [PubMed] [Google Scholar]
- Cho M, Cummings RC. Characterisation of monomeric forms of galectin-1 generated by site-directed mutagenesis. Biochemistry. 1996;35:13081–13088. doi: 10.1021/bi961181d. [DOI] [PubMed] [Google Scholar]
- Inagaki Y, Sohma Y, Horie H, Nozawa R, Kadoya T. Oxidised galectin-1 promotes axonal regeneration in peripheral nerves but does not possess lectin properties. Eur J Biochem. 2000;267:2955–2964. doi: 10.1046/j.1432-1033.2000.01311.x. [DOI] [PubMed] [Google Scholar]
- Tracey BM, Feizi T, Abbott WM, Carruthers RA, Green BN, Lawson AM. Subunit molecular mass assignment of 14, 654 Da to the soluble β-galactoside-binding lectin from bovine heart muscle and demonstration of intramolecular disulfide bonding associated with oxidative inactivation. J Biol Chem. 1992;267:10342–10347. [PubMed] [Google Scholar]
- Yamaoka K, Ingendoh A, Tsubuki S, Nagai Y, Sanai Y. Structural and functional characterisation of a novel tumour-derived rat galectin-1 having transforming growth factor (TGF) activity: the relationship between intramolecular disufide bridges and TGF activity. J Biochem (Tokyo) 1996;119:878–886. doi: 10.1093/oxfordjournals.jbchem.a021325. [DOI] [PubMed] [Google Scholar]
- Iglesias MM, Rabinovich GA, Ivanovic V, Sotomayor C, Wolfenstein-Todel C. Galectin-1 from ovine placenta. Amino-acid sequence, physicochemical properties and implications in T-cell death. Eur J Biochem. 1998;252:400–407. doi: 10.1046/j.1432-1327.1998.2520400.x. [DOI] [PubMed] [Google Scholar]
- Barondes SH, Cooper DN, Gitt MA, Leffler H. Galectins: structure and function of a large family of animal lectins. J Biol Chem. 1994;269:20807–20810. [PubMed] [Google Scholar]