

Abstract
Today, –omics analyses, including the systematic cataloging of messenger RNA and microRNA sequences or DNA methylation patterns in a cell population, organ, or tissue sample, allow for an unbiased, comprehensive genome-level analysis of complex diseases, offering a large advantage over earlier “candidate” gene or pathway analyses. A primary goal in the analysis of these high-throughput assays is the detection of those features among several thousand that differ between different groups of samples. In the context of oral biology, our group has successfully utilized –omics technology to identify key molecules and pathways in different diagnostic entities of periodontal disease.
A major issue when inferring biological information from high-throughput –omics studies is the fact that the sheer volume of high-dimensional data generated by contemporary technology is not appropriately analyzed using common statistical methods employed in the biomedical sciences.
In this chapter, we outline a robust and well-accepted bioinformatics workflow for the initial analysis of –omics data generated using microarrays or next-generation sequencing technology using open-source tools. Starting with quality control measures and necessary preprocessing steps for data originating from different –omics technologies, we next outline a differential expression analysis pipeline that can be used for data from both microarray and sequencing experiments, and offers the possibility to account for random or fixed effects. Finally, we present an overview of the possibilities for a functional analysis of the obtained data.
Keywords: Periodontal disease, Gene expression, Transcriptome, microRNA, DNA methylation, Microarray, Next-generation sequencing, Gingiva, Differential expression analysis, Functional groups
1. Introduction
–omics analyses such as the whole-genome assessments using microarrays or next-generation sequencing outlined in Chapter 18 generate a large number of observations in relatively few samples. It is generally of major interest to assess which of these features differ between subgroups of samples defined a priori on the basis of relevant characteristics, e.g., clinical diagnosis, experimental treatment, etc. When performing these differential expression analyses of –omics data, the researcher is inevitably confronted with the fact that “high-dimensional” data sets are difficult to analyze using traditional statistical approaches. Specifically, the analysis needs to account for thousands of statistical tests performed simultaneously. Additional corrections may be necessary for specific features of clinical samples. The amount of resulting data generated requires unprecedented computational resources in terms of processing power, memory, and disk space.
Our group has considerable experience in the analysis of high-throughput datasets in the context of periodontal infections, e.g., the expression profiles or periodontal health and disease [1–8] or experimental gingivitis [9].
This chapter describes how to process the raw data provided by a core facility after hybridization with microarrays or massively parallel sequencing. We elaborate on typical quality assessments and preprocessing steps, and then proceed to a common differential expression analysis workflow using the R/Bioconductor framework [10] and the limma library [11, 12]. Importantly, both microarray-based expression values and gene counts created based on sequencing results—after transformation into continuous values—can be used as source data for this workflow. Using limma, it is possible to perform a differential expression analysis correcting for both random effects—such as the individual subject in cases where several biologically and statistically dependent samples originate from the same individual—and fixed effects, e.g., the study center, the surgeon harvesting a biopsy, race and ethnicity of the subject, or the level of disease severity of the particular tissue sample as a continuous variable. In a similar fashion, the library allows not only the assessment of differential expression in two or more defined groups, but also the identification of genes that differ significantly in relation to continuous variables, such as periodontal probing depth, or levels of subgingival periodontal bacteria.
It is important to realize that there is a wealth of software packages available for the analysis of both array and sequencing datasets, both from commercial providers and open source software. In this chapter, we have opted to use open source software that is both universally accessible and well established in the field, which we have experience using in studies of periodontal cells and tissues. However, given the rapid evolvement in this field, future modifications of the workflow are likely.
In the next chapter of this series (see Chapter 20 by Kebschull et al. of this volume), we expand the basic analyses described here by using machine learning algorithms on high-throughput data, both for purposes of supervised classification of a priori labeled samples, and for unsupervised discovery of new classes.
2. Materials
2.1. Hardware
For microarray analysis: A computer with x86–64 compatible processor(s) running either Linux or Windows or Mac OS X. RAM >4 GB, about 1 TB free hard drive space.
For next-generation sequencing data analysis: A computer with x86–64 compatible processor(s) running Linux with as many processor cores as possible (See Notes 1, 13), RAM >32GB, and several TB of free hard drive space.
2.2. Software
- The R statistical environment, including the Bioconductor framework, and the following libraries.
-
(a)minfi
-
(b)illuminaio
-
(c)IlluminaHumanMethylation450kanno.ilmn12.hg19
-
(d)affy
-
(e)Rsubreads
-
(f)edgeR
-
(g)limma
-
(h)sva
-
(i)statmod
-
(a)
(Optional, but highly recommended) An integrated programming environment (IDE) for R, e.g., RStudio, or a programming editor, e.g., GNU Emacs/ESS.
(Optional, but highly recommended) A version control system, e.g., git
FastQC software http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
STAR aligner software [13, 14] https://github.com/alexdo-bin/STAR.
Trimmomatic software [15] http://www.usadellab.org/cms/?page=trimmomatic.
GSEA software [16] http://software.broadinstitute.org/gsea/index.jsp.
Cytoscape [17] http://www.cytoscape.org/.
Enrichment Map (Cytoscape plugin) [18] http://www.bader-lab.org/Software/EnrichmentMap.
ErmineJ software [19] http://erminej.chibi.ubc.ca/.
2.3. Manifests, Annotations, Genome Files
Manifest file for the HT-12 bead arrays from Illumina’s website (http://support.illumina.com/array/array_kits/humanht-12_ v4_expression_beadchip_kit/downloads.html).
The manifest for the methylation arrays is part of the IlluminaHumanMethylation450kanno.ilmn12.hg19 R package.
Genome files, e.g., from Ensembl http://ftp.ensembl.org/pub/release84/fasta/homo_sapiens/dna/Homo_sapiens. GRCh38.dna.primary_assembly.fa.gz.
Matching annotation files, e.g., from Ensembl ftp://ftp.ensembl.org/pub/release84/gtf/homo_sapiens/Homo_sapiens.GRCh38.84.gtf.gz.
2.4. Targets File
Tab-delimited text (*.txt) or comma-separated text (*.csv) file.
One row per sample.
- Has all technical information.
-
(a)Lab identifier.
-
(b)Array number (for microarray data).
-
(c)Position on bead array (for microarray data).
-
(d)Batch information.
-
(e)Possibly also quality information (yield, RIN, etc.).
-
(a)
- And all phenotypic information of possible value, e.g. (in case of gingival tissue biopsies).
-
(a)Demographics (age, gender, race, and ethnicity of study subject).
-
(b)Diagnosis.
-
(c)Systemic conditions.
-
(d)Local measures of disease at the biopsy site (periodontal probing depth, clinical attachment level, subgingival levels of periodontal bacteria associated with the tissue biopsy).
-
(a)
2.5. Raw Data
- From microarray experiment.
-
(a)*.idat files for all arrays run.
-
(a)
- From Next-Generation Sequencing experiment.
-
(a)*.fastq files for all sequenced samples, de-multiplexed and adaptor-trimmed by core facility.
-
(a)
3. Methods
3.1. Preprocessing of Array Data
3.1.1. HT-12 Expression Arrays
-
(a)
In R, set working directory, and load the limma and the illuminaio libraries.
>setwd(“~/projects/ht12”) >library(limma) >library(illuminaio)
-
(b)
Place *.bgx manifest file for the HT-12 bead arrays from Illumina’s website (http://support.illumina.com/array/array_kits/humanht-12_v4_expression_beadchip_kit/down-loads.html) and all *.idat source files in a directory, and read them into R using limma’s read.idat function.
> idatfiles = dir(pattern=“idat”) > bgxfile = dir(pattern=“bgx”) > raw <- read.idat(idatfiles, bgxfile)
-
(c)
For quality control, plot the average signal intensities for regular and control probes on the arrays. Very dim arrays are indicative of suboptimal hybridization and should be removed (Fig. 1).
Fig. 1.
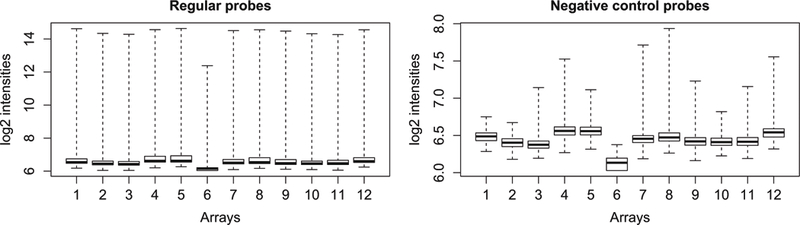
Boxplots of HT-12 expression array signal intensity before normalization. Note that the very dim array #6 should be removed
> pdf(“boxplots_preNorm.pdf”) > par(mfrow=c(1,2)) > boxplot(log2(raw$E[raw$genes$Status==“reg ular”,]),range=0, xlab=“Arrays”,ylab=“log2 intensities”, main=\ “Regular probes”) > boxplot(log2(raw$E[raw$genes$Status==“neg ative”,]),range=0, xlab=“Arrays”,ylab=“log2 intensities”, main\ =“Negative control probes”) > dev.off()
-
(d)
To support these observations, check for the proportion of probes with an “expressed” call—they should be fairly similar.
>propexp <- propexpr(raw)
-
(e)
Read the targets file.
# required for the read.AnnotatedDataFrame # function > library(affy) > targets <- read.AnnotatedDataFrame(file=“t argets.csv”, header=TRUE) > targets <- pData(targets)
-
(f)
Normalize data using quantile normalization (see Note 2).
> y <- neqc(raw)
-
(g)
Explore similarities and dissimilarities of the samples using multidimensional scaling (MDS) plots or hierarchical clustering (and different labels, using variables from the targets file). These plots allow (1) to explore the data for broad differences in expression related to the different variables, (2) to identify possible batch effects, and (3) to detect, in combination with the measures introduced above, arrays that did not hybridize as planned.
> library(limma) > pdf(“MDSplots.pdf”) > par(mfrow=c(1,2)) > plotMDS(y,gene.selection=“common”, labels=variable1) > plotMDS(y,gene.selection=“common”, labels=variable2) > dev.off() > pdf(“hierClust.pdf”) > par(mfrow=c(1,2)) > d = dist(t(y$E)) > plot(hclust(d), labels = variable1) > plot(hclust(d), labels = variable2) > dev.off()
3.1.2. 450k Methylation Arrays
Gather the *.idat source files for all samples, and place a targets file (as *.csv) in the source directory.
- To load the data into R using the minfi library [20],
# load libraries > library(minfi) > library(IlluminaHumanMethylation450kanno. ilmn12.hg19) > library(IlluminaHumanMethylation450kmanif est) # set working directory and load targets file > setwd(“~/projects/methylation”) > workDir <- “~/projects/methylation” > targets <- read.450k.sheet(workDir) # read raw data > RGset <- read.450k.exp(targets = targets)
- Check for bad arrays using the detection p-value (see Note 3):
# get detection p vals > detP <- detectionP(RGset) > failed <- detP > 0.01 > failed <- colMeans(failed) > pData(RGset) -> pDataRGSet > names(failed) <- pDataRGSet$Sample_Name > write.table(failed, file=“failed.txt”, sep=“\t”)
- Generate an extensive quality control report for all samples:
> qcReport(RGset, pdf = “qcReport.pdf”)
- Generate beanplots for all samples (see Note 4):
> pdf(file=“Beanplot.pdf”, 5, 20) > densityBeanPlot(RGset, sampNames = pDataRGSet$Sample_Name) > dev.off()
- Preprocess and additional quality control using the minfiQC function (see Note 5):
# normalize raw data > MSet.Ill <- preprocessIllumina(RGset, bg.correct=TRUE) # get information on arrays minfi thinks that are good or bad > QCout <- minfiQC(MSet.Ill) > pdf(“QC.pdf”) > plotQC(QCout$qc) > dev.off()
Convert to beta:
> ratioSet <- ratioConvert(MSet.Ill, what = “both”, keepCN = TRUE) > MSet.Ill.genome <- mapToGenome(ratioSet) #get beta values for each CpG island (rows) and label the columns with the sample names from the targets file > beta.Ill <- getBeta(MSet.Ill.genome) > colnames(beta.Ill) <- targets$Sample_Name
3.2. Preprocessing of Sequencing Data
3.2.1. RNA Seq Data
Quality Control of the Reads
-
(a)
The raw reads are usually provided as FASTQ files.
-
(b)
First, a general quality assessment of the millions of raw reads per sample should be performed. We recommend the FastQC software, a standalone Java program available at http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. FastQC lists the number and length of reads and their quality encoding, and visualizes and judges several quality parameters (Fig. 2a, b). The results can be viewed using a standard web browser. Please note that not all failures and warnings displayed by FastQC are actually problematic for RNA Seq data (see Note 6–9).
Fig. 2.
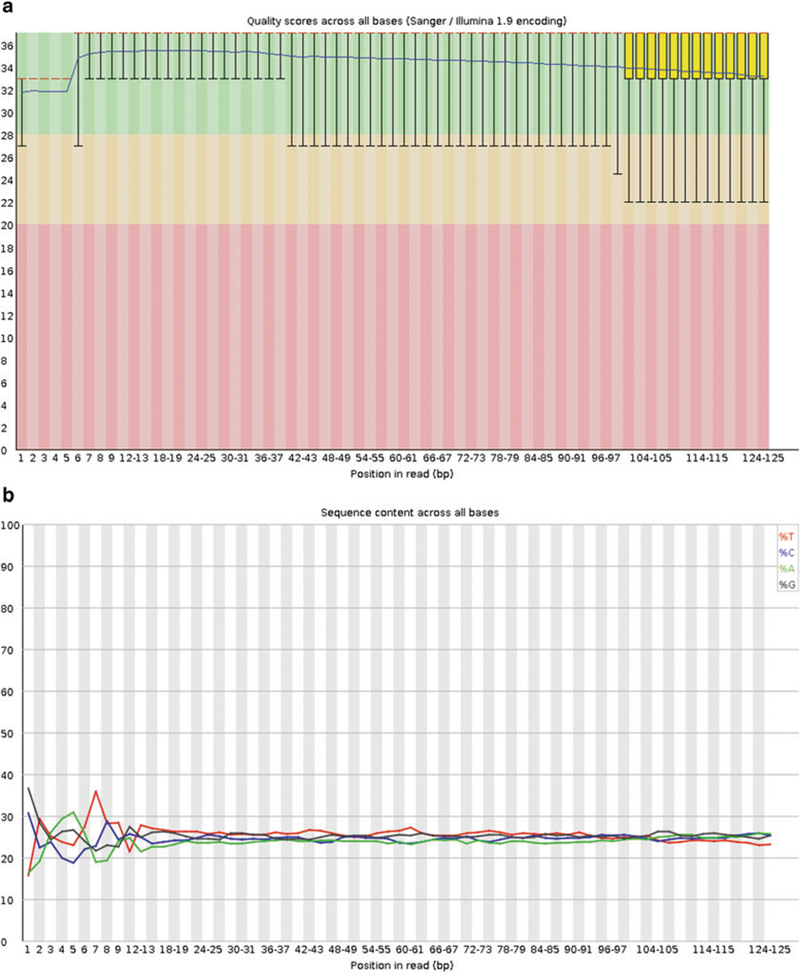
FastQC examples: (a) Per base sequence quality. Note how the quality of the base calls decreases toward the end of the reads. (b) Per base sequence content. For each position in the read, the percentage of the four bases is plotted. Note the bias in the beginning of the read, a typical phenomenon for Illumina RNA Seq data caused by random hexamer priming
> fastqc sample1_read1.fq.gz
Preprocessing
-
(a)
Filtering removes entire reads below a certain quality threshold (see Note 10). We recommend the trimmomatic program http://www.usadellab.org/cms/?page=trimmomatic, a standalone Java application, because it can filter paired end reads and is multithreaded, i.e., fast [15]. The following command runs trimmomatic on a paired end sample, and produces four output files, two paired ones where the initial pairs are still intact after filtering, and two unpaired files containing the data from broken pairs.
> java –jar trimmomatic-0.36.jar PE – threads 24 –phred33 sample1_read1.fq.gz sample1_read2.fq.gz output_paired_read1. fq.gz output_unpaired_read1.fq.gz output_ paired_read2.fq.gz output_unpaired_read2. fq.gz AVGQUAL:25
-
(b)
Trimming removes bases from the end of the reads, based on a given length and/or based on a quality threshold. The following command trims bases from the 3’-end of the reads that are below 25, and eventually filters the whole read when it gets too short by the trimming.
> java –jar trimmomatic-0.3 6.jar PE –threads 24 -phred33 sample1_read1.fq.gz sample1_ read2.fq.gz paired1.fq.gz unpaired1.fq.gz paired2.fq.gz unpaired2.fq.gz TRAILING:25 MINLEN:7 5
In addition, in case FastQC reports adapter contaminations, trimmomatic can remove those using the following option (see Note 11).
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10
-
(c)
Repeat FastQC evaluation to assess whether the preprocessing steps were successful.
Alignment to Reference Genome
-
(a)
The reads are aligned to the genome using the splice-aware and very fast STAR aligner [13].
-
(b)
STAR needs at least 32GB of memory for human genome alignments (see Notes 1, 12, and 15).
-
(c)
STAR is able to take advantage of multiple processing cores of the computer’s processor(s). The number of cores to use is up to 100 % of all present physical cores, or—on more recent machines that allow hyperthreading—up to 200 %. Select the number of parallel processes using the --runThreadN <NThreads > option (see Note 13).
-
(d)
The alignment workflow consists of two steps, (1) the generation of genome index files, and (2) the mapping of the user’s reads to the genome.
-
(e)
Generation of index files
Create directory ./genome in STAR directory, and place the latest ENSEMBL genome sequence in this directory.
> mkdir genome > cd genome > wget http://ftp.ensembl.org/pub/release-84/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz > gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
Create index using STAR (space requirements ~30 GB).
> STAR --runThreadN 24 --runMode genomeGenerate --genomeDir ./ --genomeFastaFiles ./Homo_sapiens. GRCh38.dna.primary_assembly.fa
-
(f)
Mapping of reads.
Download annotation GTF file from the Ensembl ftp server and place it in the ./genome folder.
> wget ftp://ftp.ensembl.org/pub/release-84/gtf/homo_sapiens/Homo_sapiens.GRCh38.84.gtf.gz
Change to the source data directory containing the FASTQ files and map using STAR and the previously generated index. Specify where your genome index is located, how many cores to use, and (for compressed source files) to use zcat instead of cat to decompress on the fly (see Notes 14–18).
> STAR --runThreadN 24 --genomeDir ~/bin/STAR/genome --sjdbGTFfile ~/bin/STAR/genome/Homo_sapiens.GRCh38.84.gtf.gz --readFilesIn ./samplel-readl.fq.gz ./sample1-read2.fq.gz--readFilesCommand zcat --outFilterType BySJout --outFilterMultimapNmax 20 --align-SJoverhangMin 8 -- alignSJDBoverhangMin 1 --outFilterMismatchNmax 999 --alignlntronMin 20 --alignlntronMax 10000 --alignMatesGap-Max 1000000 --genomeLoad LoadAndKeep --out- ReadsUnmapped Fastx
The alignment produces the following files.
Log.final.out—summary mapping statistics.
Log.out—detailed log of the run, can be used for troubleshooting.
Aligned.out.sam—main results file, all aligned reads in SAM format.
SJ.out.tab—splice junctions.
Small RNA Seq Data
-
(a)
The SAM file that was produced by STAR is analyzed using the featureCounts function (included in the Rsubreads R library) [21] to assign reads to genes.
> library(Rsubreads) > counts <- featureCounts(files=“sample1.sam”, annot.ext=“ ~/bin/STAR/genome/ Homo_ sapiens.GRCh3 8.8 4.gtf”, isPairedEnd=TRUE, isGTFAnnotationFile=TRUE, strandSpecific=2, nthreads=2 4, useMetaFeatures=TRUE)
-
(b)
The output of the featureCounts function, the object counts, is a list containing a data frame with annotation information for all genes and a matrix with raw gene counts for each input library.
-
(c)
For a normalization of gene counts using a scale normalization approach, we use the TMM normalization method [22] implemented in the DGEList function of the edgeR package in R.
> library(edgeR) > dge <- DGEList(counts=counts) > dge <- calcNormFactors(dge)
-
(d)
Subsequently, the read counts should be transformed using the voom function from the limma library in R/Bioconductor [23] (see Notes 19 and 20).
> norm <- voom(dge, design, plot=F)
-
(e)
The normalized data can be explored using clustering or MDS plots as described above for array datasets (Subheading 3.1, step 7) to assess whether replicates cluster together, and whether there are obvious batch effects that need to be corrected (see below).
- Alignment.
- (a)
- Quantification.
-
(a)Normalized counts can be acquired using featureCounts, normalized and transformed to continuous data using voom, as detailed above.
3.3. Differential Expression Analysis
-
(a)
Limma. This protocol uses limma, a well-established and powerful R package originally developed for the analysis of microarrays for data from all of the aforementioned techniques (see Note 25–28).
-
(b)
Batch effect correction(optional). In case the preliminary analysis using unsupervised clustering or MDS plots performed during the preprocessing steps provides evidence for a batch effect, this effect can be corrected before the actual differential expression analysis. A typical batch effect would cause sequential samples processed on a single day/array/flow cell, etc., to cluster together, rather than replicates.
To perform a batch correction, we recommend the ComBat procedure implemented in the R package sva.
> library(sva) # read information about batches and the class difference of interest from the targets file > batch <- targets$batch > target <- targets$target # build model matrix > mod <- model.matrix(~as.factor(target), data=data) # correct data for batches using ComBat procedure > data_combat <- ComBat(dat=data, batch=batch, mod=mod, numCoves=NULL, par. prior=TRUE)
-
(c)
Generate a table for the experimental design, using data from the targets file.
# load the limma library > library(limma) > condition <- factor(targets$condition) > design <- model.matrix(~0 + condition) > colnames(design) <- levels(condition) # if the need arises to correct this comparison of ‘condition’ for a covariate, it can easily be added to the design (fixed factor) # design <- model.matrix(~0 + condition + covariatel) # colnames(design) <- c(levels(condition), “covariatel”)
-
(d)
Introduction of a blocking factor to correct for multiple samples from the same individual (random factor).
> library(statmod) > corfit <- duplicateCorrelation(data_combat, design, block=targets$patient)
-
(e)
Fit a model to the data.
> fit=lmFit(data_combat, design, block= targets$patient, correlation=corfit$consensus) # make contrasts, e.g. to compare healthy and diseased samples (assuming that the variable condition has the levels ‘diseased’ and ‘healthy’) > contr <- makeContrasts(healthDisease = diseased - control, levels=design) #fit to model fit2=contrasts.fit(fit, contrasts=contr) fit2=eBayes(fit2) # if needed, add annotation step here # make lists of differentially expressed genes between entities, with correction for multiple testing using the Benjamini Hochberg False Discovery Rate [24] > healthDisease <- topTable(fit2, coef=1, number=Inf, p.value=0.05, sort.by=“logFC”, adjust.method=“BH”, lfc=0.25) # write list to file > write.table(healthDisease, file=“results_ disease_health.txt”, sep=“\t”)
3.4. Functional Analysis
There are several open source software packages that, based on a differential expression analysis as described above, generate lists and/or networks of functional groups enriched in the experimental conditions. Here, we outline how to format the results from the differential expression analysis to run basic functional analyses in ermine [19] or GSEA [16] coupled to visualization using the EnrichmentMap plugin [18] in Cytoscape [17]. An example of the comparison of enriched functional groups in different clinical conditions is in Fig. 3.
Fig. 3.
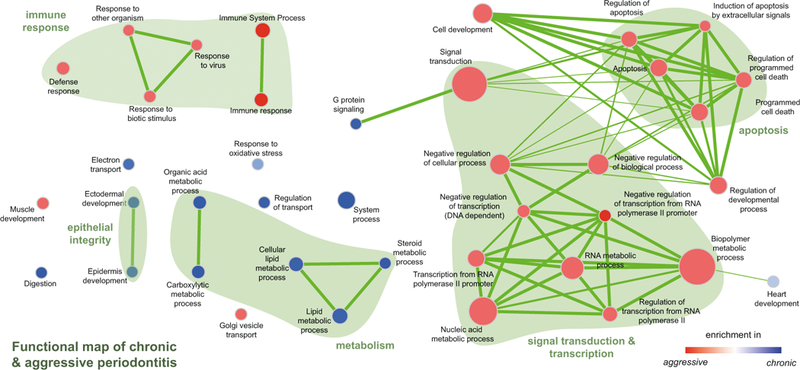
Visualization of GSEA results using the Enrichment Map plugin in Cytoscape. Reprinted from [4] with permission from Sage. Visualization of gene sets significantly enriched in diseased gingival tissues from patients with chronic or aggressive periodontitis. Gene sets are depicted as nodes in a network. Color describes the disease entity (red for AP and blue for CP), and the color intensity represents the degree of enrichment. The size of the node represents the size of the enriched gene set, and the thickness of the connectors stands for the degree of overlap between the nodes [18]
-
(a)
Prepare a ranked list of features. Rank all genes by t-value.
-
(b)
Use these data for Gene Set Enrichment Analysis (GSEA) using the GSEAPreRanked function (when using the GSEA graphical user interface, this function can be found in the “tools” pull-down menu).
-
(c)
Import the results into Cytoscape following this tutorial http://www.baderlab.org/Software/EnrichmentMap/Tutorial.
-
(d)
(Alternatively) use ranked list in ErmineJ.
3.5. Upload to Repositories
Most journals require the submission of the raw and/or processed data from high-throughput experiments to online repositories.
Repositories exist in the US as well as in Europe, with differences in the accepted data formats.
- Array repositories.
-
(a)The Gene Expression Omnibus (GEO, http://ncbi.nlm.nih.gov/geo) at NIH.
-
(b)ArrayExpress (http://www.ebi.ac.uk/arrayexpress) at the European Bioinformatics Institute.
-
(a)
- Sequencing data repositories.
-
(a)The Sequence-Read-Archive (SRA, http://ncbi.nlm.nig.gov/sra) stores raw data and alignment information from Illumina sequencers and other machines.
-
(b)In contrast, the Gene Expression Omnibus (GEO, http://ncbi.nlm.nih.gov/geo) holds processed sequence data files.
-
(c)The European repository ArrayExpress only accepts submissions that include the raw data plus meta data. Only the meta data will be stored at ArrayExpress, the raw data will be deposited at the SRA of the European Nucleotide Archive (http://ebi.ac.uk/ena).
-
(a)
4. Notes
As an alternative to the use of local hardware, cloud computing providers such as Amazon Web Services, Microsoft Azure, or Google offer platforms that allow for a very flexible utilization for bioinformatics workflows.
HT-12 arrays feature 12 samples per slide, thereby reducing batch effects in comparison to array systems that only load a single sample per chip, e.g., from Affymetrix. On the other hand, the design with several arrays per slide may well be reason for additional statistical concern. For example, for the related Sentrix-6 Expression BeadChips (for murine samples), a separate normalization routine was proposed to address the specifics of the design [25].
The percentage of failed probes on bead arrays with good quality is by far less than 1 %. Arrays with higher failure rate should possibly be excluded.
The beanplots should show a pronounced U-shape, with a lot of signal at 0 and 1, indicating unmethylated and hypermethylated regions. Plots with an inverse behavior should be excluded from further analysis.
The minfiQC function provides a very quick overview of what sample could be “bad” and should be scrutinized.
FastQC typically flags the Per base sequence content assessment as a failure with Illumina RNA Seq data. The considerable bias seen in the first bases (Fig. 2b) is caused by random hexamer priming [26].
In an ideal human RNA Seq experiment, the GC content should follow a normal distribution with a single peak at the mean GC content of the human organism. Deviations from this shape (Fig. 2c) are indicative of a contamination, possibly by rRNA or other contaminants—leading to peaks on the right-hand side. Additional peaks on the very left are often caused by sequencing of poly-A tails. In the case of clinical samples from human gingiva, a very considerable source of contamination of the library is oral microorganisms. In this case, sequences from microbes are often found among the over-represented sequences, and can be tested by BLASTing at http://blast.ncbi.nlm.nih.gov/Blast.cgi
If contamination with rRNA is suspected, tools such as SortMeRna can be used to remove it [27]. Some groups generally recommend filtering of rRNA reads, because varying proportions of them in the libraries will not be detectable by measures as the percentage of aligned reads, and thereby introduce a bias.
In contrast to the situation with DNA sequencing, sequence duplicates will inherently be found in RNA Seq studies, because of obvious different expression levels for different genes. It is therefore not recommended to remove duplicates.
Base quality in FASTQ files is expressed in the Phred scale that is the log10 of the probability that a base call was wrong multiplied by −10, e.g., for a one in twenty chance (5 % = 0.05), the score would be 13, for one in one hundred, 20, for one in one thousand 30. Phred scores usually range from 0 to 40 and are encoded, to save disk space, by a single ASCII character. In recent FASTQ files, the Sanger encoding is used, with the 33rd ASCII character representing a score of zero, while FASTQ files containing older Illumina data may well be encoded differently. FastQC can detect the encoding used. Generally, the quality of base calls decreases toward the end of the read (see FastQC’s Per base sequence quality graph, Fig. 2a). It is recommended to aim for the majority of reads to have a mean phred score of 25 or higher (better). Reads with bad quality base calls can be addressed either by filtering—removing the entire read—or trimming, where the lower quality ends of the reads can be removed. The latter procedure preserves a read that can later be aligned.
Note that the order of the options given to trimmomatic in the command line matters—adapter clipping should be done before all other steps to avoid disguising adapters by trimming.
Alignment of NGS data is computationally intensive, the STAR aligner uses a lot of RAM to provide considerable speedups in comparison to older software like Tophat.
Consider to limit the number of threads to ~80 % of those available in the system to allow for other processes to be able to run efficiently.
One big advantage of the STAR aligner is the so called soft clipping functionality. In contrast to other aligners like bowtie that try to align a read end-to-end, STAR performs a local alignment base-by-base, until a threshold of mismatches is reached. Thereby, adapters, poor quality sequencing tails at the end of reads can be removed.
Using the --genomeLoad option, STAR can share the genome index data stored in the main memory between several annotation processes (shared memory concept), reducing the footprint of the aligner when used for several samples in parallel.
If you suspect microbial contamination in your samples, the nonaligned reads can be preserved using the –outReadsUn-mapped Fastx option and tested for alignment with nontarget species.
As an alternative to the alignment to the genome, the reads could also theoretically be aligned to a transcriptome, e.g., after an assembly with Trinity [28] following this protocol [29].
A new trend in RNA-Seq analysis is the use of pseudoalignment engines, such as Kallisto, that can quantify abundances of transcripts without the need for alignment [30], thereby massively reducing the computational demands of the analysis workflow.
The voom transformation routine assumes that genes with zero or very low counts were removed after featureCount by filtering.
The voom function also allows performing microarray-style normalization functions, such as quantile normalization. This is recommended only for very noisy samples.
The main fraction of reads in small RNA sequencing should be around 20–24 nucleotides in length (corresponding to the miRNA fraction). The raw reads, however, include the adapter sequences that need to be removed before alignment—due to the short target sequence, an alignment would not be possible in most cases. After clipping of the adapters, the length distribution should show a peak at the about 20 nt.
There exists a plethora of other aligners for small RNA sequencing workflows, the most common alternative to STAR is bow-tie. However, bowtie seems to be very susceptible to not or not completely trimmed adapters.
Aligner will miss the target regions, seed region is only 6–8 nt.
As an alternative to aligning the short RNA reads to the genome, an alignment to the miRBase database of known miRNAs is possible [31].
It is beyond the possibilities of this chapter to address all features and possibilities of the limma package. However, the reader is encouraged to download the latest version of the very through limma manual from https://www.bioconductor.org/pack-ages/3.3/bioc/vignettes/limma/inst/doc/usersguide.pdf.
Array weights in limma allow to account for microarrays with varying quality, e.g., from human samples, by assigning different weights. It is generally recommended to utilize weights in situations with difficult samples, sparse source materials, and the observation of varying quality. Conversely, when using material from very well controlled cell culture systems, weights need not be used.
In RNA Seq experiments, a combination of the array weights strategy for individual samples and the weighting method used by voom is possible to correct “outlier” samples. This method is implemented by the voomWithQualityWeights function in limma.
This workflow describes how to perform a differential expression analysis of RNA Seq data based on gene counts. Still, the sequencing data also allow for more detailed analyses, e.g., a differential splicing analysis. This analysis can be performed with minor changes to the workflow described herein by simply changing the focus of the featureCounts function from “gene” to “exon” by setting useMetaFeatures=FALSE.
Acknowledgments
This work was supported by grants from the German Society for Periodontology (DG PARO) and the German Society for Oral and Maxillo-Facial Sciences (DGZMK) to M.K. and by grants from NIH/NIDCR (DE015649, DE021820 and DE024735) and by an unrestricted gift from Colgate-Palmolive Inc. to author P.N.P.
References
- 1.Kebschull M, Demmer RT, Grun B, Guarnieri P, Pavlidis P, Papapanou PN (2014) Gingival tissue transcriptomes identify distinct periodontitis phenotypes. J Dent Res 93:459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nowak M, Kramer B, Haupt M, Papapanou PN, Kebschull J, Hoffmann P, Schmidt-Wolf IG, Jepsen S, Brossart P, Perner S, Kebschull M (2013) Activation of invariant NK T cells in periodontitis lesions. J Immunol 190:2282–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kramer B, Kebschull M, Nowak M, Demmer RT, Haupt M, Korner C, Perner S, Jepsen S, Nattermann J, Papapanou PN (2013) Role of the NK cell-activating receptor CRACC in periodontitis. Infect Immun 81:690–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kebschull M, Guarnieri P, Demmer RT, Boulesteix AL, Pavlidis P, Papapanou PN (2013) Molecular differences between chronic and aggressive periodontitis. J Dent Res 92:1081–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stoecklin-Wasmer C, Guarnieri P, Celenti R, Demmer RT, Kebschull M, Papapanou PN (2012) MicroRNAs and their target genes in gingival tissues. J Dent Res 91:934–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kebschull M, Papapanou PN (2010) The use of gene arrays in deciphering the pathobiology of periodontal diseases. Methods Mol Biol 666:385–393 [DOI] [PubMed] [Google Scholar]
- 7.Papapanou PN, Behle JH, Kebschull M, Celenti R, Wolf DL, Handfield M, Pavlidis P, Demmer RT (2009) Subgingival bacterial colonization profiles correlate with gingival tissue gene expression. BMC Microbiol 9:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demmer RT, Behle JH, Wolf DL, Handfield M, Kebschull M, Celenti R, Pavlidis P, Papapanou PN (2008) Transcriptomes in healthy and diseased gingival tissues. J Periodontol 79:2112–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joensson D, Ramberg P, Demmer RT, Kebschull M, Dahlen G, Papapanou PN (2011) Gingival tissue transcriptomes in experimental gingivitis. J Clin Periodontol 38:599–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ritchie ME, Diyagama D, Neilson J, van Laar R, Dobrovic A, Holloway A, Smyth GK (2006) Empirical array quality weights in the analysis of microarray data. BMC Bioinformatics 7:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (20l3) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobin A, Gingeras TR (2015) Mapping RNA-seq reads with STAR. Curr Protoc Bioinformatics 51:11.14.11–19. doi: 10.1002/0471250953.bi1114s51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30:2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merico D, Isserlin R, Bader GD (2011) Visualizing gene-set enrichment results using the cytoscape plug-in enrichment map. Methods Mol Biol 781:257–277 [DOI] [PubMed] [Google Scholar]
- 19.Gillis J, Mistry M, Pavlidis P (2010) Gene function analysis in complex data sets using ErmineJ. Nat Protoc 5:1148–1159 [DOI] [PubMed] [Google Scholar]
- 20.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA (2014) Minfi: a flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics 30:1363–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao Y, Smyth GK, Shi W (2014) feature-Counts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930 [DOI] [PubMed] [Google Scholar]
- 22.Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Law CW, Chen Y, Shi W, Smyth GK (2014) Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc 57:289–300 [Google Scholar]
- 25.Shi W, Banerjee A, Ritchie ME, Gerondakis S, Smyth GK (2009) Illumina WG-6 Bead Chip strips should be normalized separately. BMC Bioinformatics 10:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hansen KD, Brenner SE, Dudoit S (2010) Biases in illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res 38, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kopylova E, Noe L, Touzet H (2012) SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28:3211–3217 [DOI] [PubMed] [Google Scholar]
- 28.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, Macmanes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, Leduc RD, Friedman N, Regev A (2013) De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bray NL, Pimentel H, Melsted P, Pachter L (2016) Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34:525–527 [DOI] [PubMed] [Google Scholar]
- 31.Kozomara A, Griffiths-Jones S (2014) miR- Base: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42(Database issue):D68–D73 [DOI] [PMC free article] [PubMed] [Google Scholar]