

Abstract
Omics analyses, including the systematic cataloging of messenger RNA and microRNA sequences or DNA methylation patterns in a cell population, organ or tissue sample, are powerful means of generating comprehensive genome-level data sets on complex diseases. We have systematically assessed the transcriptome, miRNome and methylome of gingival tissues from subjects with different diagnostic entities of periodontal disease, and studied the transcriptome of primary cells ex vivo, or in vitro after infection with periodontal pathogens. Our data further our understanding of the pathobiology of periodontal diseases and indicate that the gingival -omes translate into discernible phenotypic characteristics and possibly support an alternative, “molecular” classification of periodontitis.
Here, we outline the laboratory steps required for the processing of periodontal cells and tissues for -omics analyses using current microarrays or next-generation sequencing technology.
Keywords: Periodontal disease, Gene expression, Transcriptome, MicroRNA, DNA methylation, Microarray, Next-generation sequencing, Gingiva
1. Introduction
After decades of research confined to the study of “candidate” single molecules or pathways, technologies available today allow for an unbiased, systematic evaluation of biological information in cells or tissues of interest on a large or genome-wide scale, and the associated underlying biology. These approaches are collectively referenced using the suffix “-omics,” e.g., “gen-omics” for the analysis of the Genome, “transcript-omics” for the analysis of (transcribed) messenger RNA, “miRN-omics” for the study of micro RNAs, and “methyl-omics” for the genome-wide assessment of DNA methylation.
-omics studies are a powerful means of generating comprehensive genome-level data sets on complex diseases and have provided enormous insights mostly in cancer research [1–3], but also in other conditions such as muscular dystrophy [4], Alzheimer’s disease and dementia [5, 6], rheumatologic disorders [7, 8], and asthma [9, 10].
We have adopted an omics-based approach in the study of the pathobiology of periodontal and peri-implant diseases, starting with the transcriptome and expanding by integrating other -omes (Fig. 1). Specifically, we examined gingival tissue transcriptomes in clinically healthy and periodontitis-affected gingival tissues [11], in experimental gingivitis [12], in chronic and aggressive periodontitis [13], and in peri-implantitis [unpublished data]. The data were used to assess the relationship of gene expression with the levels of subgingival periodontal bacteria [14, 15], and to test whether the clinical entities of chronic and aggressive periodontitis were also reflected by characteristic differences on the transcriptome level [13]. These studies identified molecules or pathways with a possible role in aggressive periodontitis that were then subject to a more focused characterization, i.e., the activation of natural killer cells by CRACC [16], and the differential activation of invariant natural killer T-cells in chronic and aggressive periodontitis [17]. Subsequently, utilizing unsupervised clustering of the transcriptomic datasets, we uncovered novel, “molecular” classes of periodontitis that also differed in their clinical and microbiological phenotype [18]. The gingival transcriptomes were supplemented by the miRNome [19] and methylome (unpublished data).
Fig. 1.
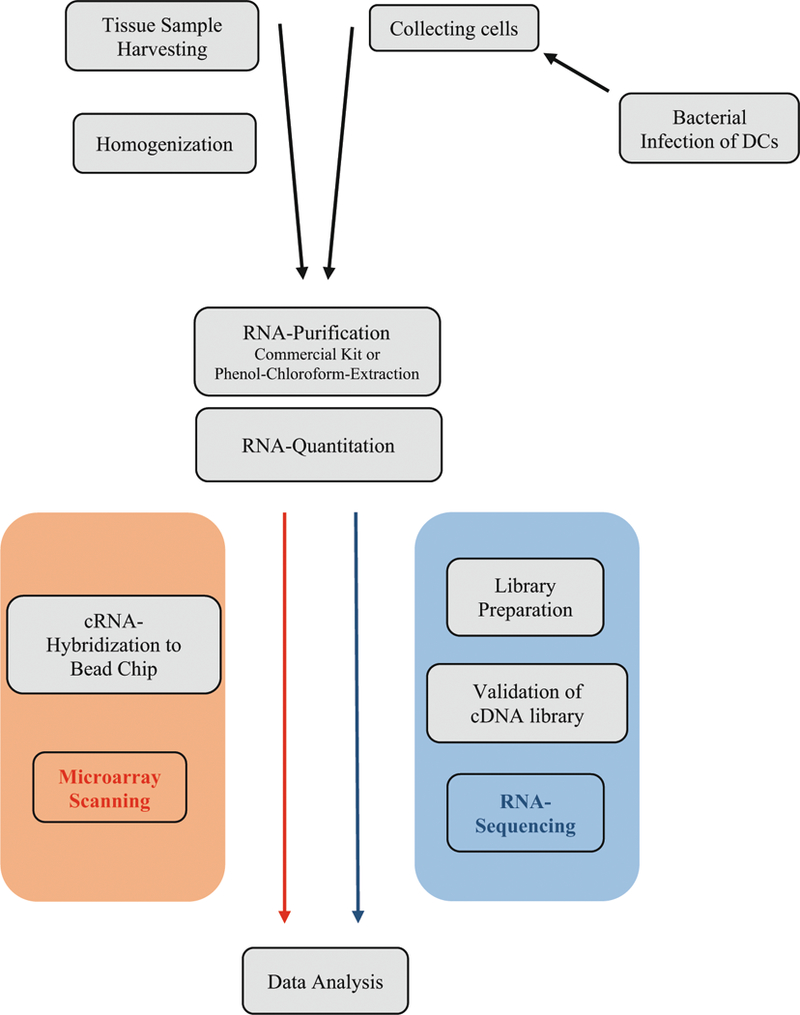
Workflow in a typical gene expression experiment of gingival tissue or infected dendritic cells
In another set of studies, we examined whether comprehensive periodontal therapy may induce changes in gene expression of peripheral blood mononuclear cells, focusing on the potential of therapy to promote an antiatherogenic phenotype [20].
We propose that the -omics-based study of gingival or mucosal tissues, primary cells isolated from subjects with periodontal disease, or cell culture material from model systems designed to mimic periodontitis will allow an enhanced understanding of the pathobiology of the periodontal diseases, inform the design of subsequent studies, and eventually lead to an improved diagnosis and therapy.
Herein, we provide a detailed description of the necessary laboratory steps in order to process gingival tissue samples, peripheral blood samples and material from a cell culture model or periodontal infections in view of hybridization with full-genome microarrays or analysis by next-generation sequencing. We have focused on the description of the procedures that will likely be performed by the oral biology researchers themselves—note that RNA labeling and hybridization to microarrays or the construction of sequencing libraries are routinely performed in core facilities or by solution providers.
In the following chapters, we provide information on the basic analysis steps for data from microarray and sequencing experiments (see Chapter 19 by Kebschull et al., this volume), and on the use of machine learning tools for the supervised and unsupervised analysis of -omics data (see Chapter 20 by Kebschull et al., this volume).
2. Materials
2.1. Source Materials
2.1.1. Tissue Samples
Gingival/Mucosal Tissue Harvesting and Processing
-
(a)
RNAlater (Ambion, Houston, TX, USA, #AM7021).
-
(b)
Eppendorf Biopure Safe-lock 1.5 mL tubes (Eppendorf, Germany, #22600028) (see Note 1).
Tissue Disruption and Homogenization
-
(a)
AllPrep DNA/RNA/Protein Kit, Qiagen (Germany).
-
(b)
Eppendorf Tubes® 5.0 mL, PCR clean (Eppendorf, Germany; #0030119460).
-
(c)
2-Mercaptoethanol ≥99.0%RLT-Lysis-Buffer, AllPrep DNA/RNA/Protein Kit, Qiagen (Germany).
-
(d)
Homogenizer, e.g., Miccra D-1 (ART Prozess- & Labortechnik GmbH & Co. KG, Germany).
-
(e)
DNase/RNase-free distilled water.
-
(f)
Eppendorf Safe-Lock Tubes, 2.0 mL (Eppendorf, Germany, #0030120094).
-
(g)
For alternative protocol (see Subheading 2.2, item b): TRIzol Reagent (Invitrogen, Carlsbad, CA, USA; #15596–018).
2.1.2. Primary Mononuclear Cells Isolated from Patient Blood
Blood Collection
-
(a)
Standard phlebotomy set, e.g., Vacutainer Safety-Lok Blood Collection Set (Becton Dickinson, #367283).
-
(b)
Vacutainer CPT Cell Preparation Tubes 8 mL (BD, #362753).
Blood Cell Separation
-
(a)
Cooled centrifuge with releasable brake, e.g., Centrifuge 5702R (Eppendorf).
-
(b)
50 mL Falcon tubes.
-
(c)
Phosphate-buffered saline without Ca2+/Mg2+ (Mediatech, Manassas, VA, USA; #21–031-CV).
-
(d)
Hemocytometer, e.g., improved Neubauer bright-line (Hausser Scientific, Hersham, PA, USA; #1492).
-
(e)
MACS separation columns (Miltenyi Biotech, Auburn, CA, USA; #130–042-401).
-
(f)
MACS multistand (Miltenyi; #007331).
-
(g)
AutoMACS rinsing solution, pH 7.2 (Miltenyi, #130–091-222).
-
(h)MACS microbeads (Miltenyi).
-
•CD4 (#120–000-440).
-
•CD14 (#120–000-305).
-
•
2.1.3. Cultured Cells
Primary Culture
-
(a)
Dulbecco’s Modified Eagle Medium, DMEM, (Invitrogen, Germany, #41965–039).
-
(b)
Fetal Bovine Serum (Invitrogen, Germany, #26140–079).
-
(c)
Penicillin-streptomycin (10,000 U/mL) (Invitrogen, Germany, #15140–122).
-
(d)
rGM-CSF, 50 μg, (ImmunoTools, Friesoythe, Germany # 12343125).
-
(e)
Corning® CellBIND® 24-Well Plates, (Corning, NY, USA, #3337).
Cell Harvesting and Processing
-
(a)
Trypsin-EDTA (0.05%), phenol red, (Invitrogen, Germany #25300–054).
-
(b)
Eppendorf Biopure Safe-lock 1.5 mL tubes (Eppendorf, Germany, #22600028).
-
(c)
Corning® Small Cell Scraper (Corning, NY, USA, #3010).
-
(d)
Cooled microcentrifUge, e.g., Centrifuge 5415R (Eppendorf, Hamburg, Germany).
2.2. Extraction and Purification of Nucleic Acids (and Protein)
2.2.1. Purification by a Commercial Kit
-
(a)
Cooled microcentrifuge, e.g., Centrifuge 5415R (Eppendorf, Hamburg, Germany).
-
(b)
Absolute ethanol.
-
(c)
2-Mercaptoethanol ≥99.0 %.
-
(d)
AllPrep DNA/RNA/Protein Mini Kit (Qiagen, Germany, #80004).
-
(e)
RNeasy MinElute Cleanup Kit (Qiagen, Germany, #74204).
-
(f)
Eppendorf Tubes® 5.0 mL, PCR clean.
2.2.2. Alternative Protocol
-
(a)
Trizol Reagent (Invitrogen, Carlsbad, CA, USA; #15596–018).
-
(b)
Chloroform.
-
(c)
Cooled microcentrifuge, e.g., Centrifuge 5415R (Eppendorf, Hamburg, Germany).
-
(d)
Ethanol 99.5 % mol. biol. grade.
-
(e)
Ethanol 75 %.
-
(f)
Glycogen (Invitrogen; #10814–010) adjusted to 5 μg/mL with nuclease-free water (Invitrogen, #10977–015).
-
(g)
0.1 M sodium citrate in 10 % ethanol.
-
(h)
8 mM NaOH.
-
(i)
Isopropyl alcohol.
-
(j)
0.3 M Guanidine hydrochloride in 95 % ethanol.
-
(k)
1 % SDS.
-
(l)
RNeasy Mini kit (Qiagen, Valencia, CA, USA; #74104).
2.3. Quantitation and Purity Assessment
Spectrophotometer, e.g., NanoDrop ND1000 (Thermo Scientific, Wilmington, DE, USA).
Agilent 2100 Bioanalyzer (Agilent Technologies Inc., CA, USA).
Agilent RNA 6000 Nano Kit (Agilent Technologies Inc., CA, USA, #5067–1511).
2.4. High-Throughput Analysis
- Microarray platforms.
-
(a)Access to a microarray core facility for hybridization of the samples to Illumina microarrays (ask for site-specific instructions for sample preparation, etc.).
-
(a)
- RNA expression profiling using Illumina HumanHT-12 v4 Expression BeadChips.
-
(a)TargetAmp-Nano Labeling Kit for Illumina Expression BeadChip (TAN07924, Epicentre).
-
(b)Illumina HumanHT-12 v4 BeadChips (BD-103–0204).
-
(a)
- DNA Methylation profiling using Illumina Infinium HumanMethylationEPIC BeadChips.
-
(a)Zymo EZ DNA Methylation Kit (D5001, Zymo Research).
-
(b)Illumina Infinium HumanMethylationEPIC BeadChip (WG-317–1002).
-
(a)
- Next-generation sequencing.
-
(a)Access to a NGS core facility for the analysis of the samples on Illumina sequencing machines (ask for site-specific instructions for sample preparation, etc.).
-
•RNA Sequencing (Illumina platform).
-
•
-
(b)Illumina TruSeq RNA Sample Prep Kit v2 (Illumina, CA, USA, #RS-122–2001, RS-122–2002).
-
(c)Illumina NextSeq500 or HiSeq2500/3000/4000 System (Illumina, CA, USA).
-
•Small RNA Sequencing (Illumina platform).
-
•
-
(d)Illumina TruSeq Small RNA Library Prep Kit (#RS-200–0012).
-
(e)Illumina NextSeq500 or HiSeq2500/3000/4000 System (Illumina, CA, USA).
-
(a)
3. Methods
3.1. Source Materials
3.1.1. Tissue sampies
Gingival/Mucosal Tissue Harvesting and Processing
Tissue Disruption and Homogenization
For purification of total RNA of a high number of samples in parallel we prefer using a commercial kit, e.g., AllPrep DNA/RNA/Protein Kit, Qiagen (see Note 3). The following steps refer to the manufacturer’s instructions. If you prefer the alternative method by phenol–chloroform extraction (see Note 4), add 1 mL of TRIzol reagent per 100 mg of sample to your sample and proceed with step c.
Prepare lysis buffer before starting the homogenization: add 10 μL of 2-mercaptoethanol in 1 mL of RLT buffer.
Weigh the frozen tissue sample with a precision balance and transfer to a 5 mL, precooled polypropylene tube. Work quickly to avoid thawing during weighing. Add the manufacturer’s recommended amount of lysis buffer to each sample and store on ice.
Afterwards, thoroughly homogenize the tissue (three episodes of 20 s each at full speed see Notes 5–7). Clean tip with RNase-free distilled water after each sample to avoid cross-contamination.
Aliquot the homogenized sample in 2 mL nuclease-free tubes (for phenol–chloroform extraction: divide into two tubes), take one aliquot for further processing and freeze the others at −80 °C to keep them as “backup.” For Column-Extraction: take care to not overload the binding capacity of the columns by using more than the recommended amount of starting material/lysate for further purification.
3.1.2. Primary Mononuclear Cells Isolated from Patient Blood
Blood Collection
Phlebotomize according to standard protocols and sample approximately 8 mL of blood each into each of four Vacutainer CPT tubes (see Note 8).
Blood Cell Separation
Centrifuge tubes for 15 min at 1000 × g with centrifuge brake turned off (see Note 9).
Carefully collect the white layer of peripheral blood monocytic cells using a 5 mL pipette and place in 50 mL Falcon tube.
Wash with 50 mL of ice-cold PBS (10 min, 300 × g, 4 °C), remove supernatant by aspiration.
Wash with 15 mL of ice-cold PBS (10 min, 300 × g, 4 °C).
Resuspend pellet in 10 mL of ice-cold PBS, count the cells with the hemocytometer.
Centrifuge (5 min, 300 × g, 4 °C), resuspend in PBS to a density of 107 cells/80 μL, keep on ice.
Add 20 μL of CD4 (or CD14 beads, respectively) per 80 μL of cell suspension, incubate on ice for 15 min.
Wash with 10 mL of ice-cold PBS, centrifuge (5 min, 300 × g, 4 °C), resuspend in 500 μL of ice-cold PBS.
Place MACS column in multi-stand, wash column twice with 5 mL of autoMACS solution.
Apply cell suspension to MACS column, wash twice with 5 mL of autoMACS, collect flow-through and label tube as CD14−.
Remove MACS column from separator stand, place in 15 mL Falcon tube and elute twice with 5 mL of ice-cold autoMACS using the plunger provided and label tube as CD14+.
Using the CD14− cell suspension, proceed accordingly for CD4 (beginning from step 7).
Pellet each cell population by centrifugation (10 min, 300 × g, 4 °C).
Add 0.5 mL of lysis buffer (Qiagen) or (for alternative protocol) TRIzol reagent to pellet (approximately 3 volumes of pellet), mix well by pipetting. Samples can be stored at −80 °C, if needed (see Note 10).
3.1.3. Cultured Cells
Primary Culture
-
1.
Generate dendritic cells from mouse bone marrow progenitor cells as described elsewhere [17].
-
2.
Resuspend prepared and washed cells in DMEM (+10 % FCS, + 1 % P/S) supplemented with 20 ng/mL rGM-CSF and seed 1 × 106 cells/mL per well into a 24-well plate. Incubate for 6 days (37 °C, 5 % CO2). Wash with 1 mL DMEM (+5 % FCS, without antibiotics) and replace culture medium every 2 days.
-
3.
For further processing, cells should be 70–80 % confluent, otherwise repeat washing step and incubate for 2 more days.
-
4.For challenge with a periodontal pathogen.
-
–Remove supernatant and wash cells with 1 mL of DMEM (+5 % FCS, without antibiotics). Add 200 μL trypsin–EDTA and incubate plate at 37 °C for approximately 5 min. Stop trypsinization with 600 μL of DMEM (+5 % FCS, without antibiotics) and transfer cell suspension in a 1.5 mL poly-propylene tube. Centrifuge for 10 min at 280 × g
-
–For infection, resuspend pelleted cells in DMEM (+5 % FCS, without antibiotics). Adjust cell concentration adapted to your MOI (multiplicity of infection) and seed 500 μL cell suspension in a 24-well plate.
-
–
-
–
Scrape 2–3 bacterial colonies (see Note 11) off the agar plate and resupend in DMEM (+5 % FCS, without antibiotics). Adjust cell concentration with a spectrophotometer (OD 0.7 ≙ 109 bacteria cells) according to your MOI. Add to the 24 well plate and incubate for 24 h at 37 °C, 5 % CO2.
Cell Harvesting and Processing
Subsequently, remove supernatant and wash cells three times with DMEM (+10 % FCS, +1 % P/S). Collect cells with a cell scraper and transfer into a 1.5 mL polypropylene tube. Centrifuge for 10 min at 280 × g.
Add 500 μL of Qiagen lysis buffer or TRIzol reagent and carry on with Subheading 3.4.2. Please note that you use half the declared amount of substances for RNA extraction (see Note 12).
3.2. Extraction and Purification of Nucleic Acids (and Protein)
3.2.1. Purification by a Commercial Kit
-
(a)
Centrifuge the lysate at 13,000 × g for 5 min, at 2–8 °C to remove insoluble material. For extraction of different fractions strictly follow the instructions of the manual. Work as quickly as possible in a clean workspace (see Notes 13 and 14).
-
(b)
Prepare all buffers before starting purification: 8 mg DTT per 1 mL of ALO, the recommended amount of ethanol (96–100 %) in Buffer RPE, AW1, and AW2.
-
(c)
Place columns in a new 2 mL collection tube.
-
(d)
Transfer supernatant to an AllPrep DNA spin column and spin for 30 s at ≥10,000 × g. Store columns placed in a new 2 mL collection tube at 4 °C for later DNA purification.
-
(e)
Start with RNA-purification: Add the recommended amount of ethanol (96–100 %) to the flow-through and mix gently by pipetting up and down. Transfer 700 μL to a RNeasy spin column, centrifuge for 15 s (≥10,000 × g) and repeat this step until the entire sample has passed through the membrane. Collect the flow-through for later protein/miRNA purification.
-
(f)
Wash column first with 700 μL of Buffer RW1 and afterwards with 500 μL of RPE (spin 15 s at ≥10,000 × g) . Discard the flow-through after each centrifugation step. Lastly, add 500 of RPE and centrifuge for 2 min to dry the membrane. For eliminating all disturbing remains of buffer, spin additionally in a new collection tube for 1 min at ≥10,000 × g.
-
(g)
Place column in a clean 1.5 mL collection tube and elute RNA by adding 30–50 μL RNase-free water, spin for 1 min at ≥10,000 × g.
-
(h)
miRNA-Purification: add 1 volume of Buffer APP to the flow-through from step e. Mix well and incubate at room temperature. Stop here and follow the instructions of the supplementary protocol for the miRNA purification by using the RNeasy MinElute Cleanup Kit, Qiagen (see Note 15).
-
(i)
Centrifuge for 10 min at full speed. Store the pellet for later protein precipitation. Transfer the supernatant in a 5 mL tube and add 1 volume of ethanol (100 %). Mix well by pipetting. Transfer sample step by step to the RNeasy MinElute spin column, centrifuge for 15 s at ≥10,000 × g and discard the flow-through after each step.
-
(j)
Place column in a new 2 mL collection (ube and wash the membrane with 500 μL of Buffer RPE and subsequently with 500 μL of 80 % ethanol, discard the flow-through.
-
(k)
Place again in a new 2 mL collection tube, open the lid and centrifuge at full speed for 5 min. Discard the flow-through.
-
(l)
Place the column in a new 1.5 mL tube and elute miRNA with 14 μL RNase-free water and spin 1 min. at full speed. Pay attention that you add the water directly to the center of the membrane.
-
(m)
For protein precipitation: add 500 μL of 70 % ethanol to the pellet of step i, centrifuge for 1 min at full speed and decant supernatant. Dry the pellet for 5–10 min at room temperature.
-
(n)
Dissolve pellet in 100 μL ALO (see Note 16).
3.2.2. Alternative Protocol (Phenol–Chloroform Extraction)
-
(a)
Add 100 μL of chloroform to the sample (volume 500 μL in a fume hood (see Note 17), shake vigorously for approximately 15 s, vortex for 1 min and incubate at room temperature for 2 min. Spin (15 min, 12,000 × g, 4 °C) to separate the aqueous and organic layers.
-
(b)
Carefully transfer the upper, colorless aqueous phase containing the RNA into a new 1.5-mL tube using a pipette with a 1 mL tip, add 4 μL of glycogen (5 μg/mL) and 250 μL of ethanol (see Note 18). Mix by shaking for 15 s, incubate for 10 min on ice and spin (10 min, 12,000 × g, 4 °C). From this point on, all work can be carried out on a laboratory bench, ideally devoted to RNA work only. Use of a hood further reduces the risk of contamination with nucleases. Clean work-space and instruments with RNase Zap according to the manufacturer’s instructions.
-
(c)
Keep the whitish interphase and the red phenol–chloroform phase for the isolation of DNA and protein (overnight storage at 4 °C possible).
-
(d)
Remove supernatant by pouring, wash the white RNA pellet (should be clearly visible) with 500 μL of 80 % ethanol (freshly prepared from 100 % ethanol and RNase free water), spin (10 min, 7500 × g, 4 °C).
-
(e)
Remove supernatant by pipetting, invert the tube to allow the pellet to dry (for approximately 10 min) (see Notes 18–20).
-
(f)
Resuspend the pellet carefully in 100 μL of RNase free water (see Note 21). The extracted total RNA can be stored at −80 °C at this point, if needed.
-
(g)
To ensure high quality of the obtained RNA, further purify using the Qiagen RNeasy Mini Kit. To ensure sufficient RNA concentration for subsequent reactions, the volume of RNase free water used to elute the RNA after the column purification should be based on the type of tissue sampled and the pellet size after the precipitation, (e.g., 20 μL for smaller tissue samples and monocytes/lymphocytes, 40 μL for larger tissue samples).
-
(h)
Prior to the column cleanup, the sample contains total RNA including microRNAs.
-
(i)
After the extraction of RNA, the lower phase containing DNA and proteins is processed. First, remove all remaining aequeous phase.
-
(j)
Add 150μL of 100 % ethanol, invert to mix, incubate for 2 min at RT, spin to pellet the precipitated DNA (2000 × g, 5 min, 4 °C).
-
(k)
Remove supernatant for protein isolation, if desired.
-
(l)
Wash DNA pellet with 500 μL of 0.1 M sodium citrate/ethanol solution. Incubate for at least 30 min at RT, invert from time to time to mix. Spin (2000 × g, 5 min, 4 °C), remove supernatant, repeat wash with sodium citrate.
-
(m)
After removing the sodium citrate supernatant, add 1 mL of 75 % ethanol, incubate for 20 min at RT with occasional mixing by inversion. Spin (2000 × g, 5 min, 4 °C), remove supernatant.
-
(n)
Dry pellet (RT, 5–10 min see Notes 22 and 23). Resuspend in 8 mM NaOH (about 500 μL for 50 mg of tissue or per 1 × 107 cells). Spin down insoluble material (12,000 × g, 10 min, 4 °C), transfer DNA to new tube. For long-term storage, adjust pH to 7–8 with HEPES.
-
(o)
For subsequent protein isolation, add 750 μL of isopropanol to the supernatant collected after DNA precipitation (step 3.2.2 (k)), incubate (10 min, RT), spin (12,000 × g, 10 min, 4 °C) to pellet the protein.
-
(p)
Remove supernatant, wash pellet with 0.3 M guanidine hydrochloride in 95 % ethanol for 20 min at RT. Spin (7500 × g, 5 min, 4 °C), remove wash solution, repeat wash twice.
-
(q)
Add 2 mL of 100 % ethanol to pellet, incubate for 20 min at RT, spin (7500 × g, 5 min, 4 °C), remove supernatant, air-dry protein pellet.
-
(r)
Resuspend protein pellet in 1 % SDS. Sample can be warmed up to 50 °C to support dissolving of the pellet (see Notes 24 and 25). Spin down insoluble material (10,000 × g, 10 min, 4 °C), transfer supernatant to new tube.
3.3. Quantitation and Purity Assessment
After purification measure the quality and quantity of the obtained total RNA and DNA by spectrophotometric analysis. Typical yields of a 30 mg gingival tissue sample (input for column extraction) are approximately 40–50 μg of total RNA, of smaller tissue samples 20–40 μg. Yields of phenol–chloroform extraction are expected between 80 and 160 μg from larger tissue samples and 20–60 μg from smaller tissue samples. The 260–280 ratio is typically between 1.9 and 2.1 (see Notes 26 and 27). The samples can be stored at −80 °C.
Additionally, a further quality check based on a chip-electrophoresis, e.g., with the Agilent 2100 Bioanalyzer, is useful to exclude degraded RNA samples, phenol, or salt traces, to assure the comparison of the samples as well as the reproducibility of the following experiments, such as next-generation sequencing. A successful library preparation and the following sequencing strongly depend on the purity of the RNA. Furthermore, unnecessary expenses and effort can be reduced that way. Corresponding to the 18S and 28S ribosomal subunits, clean RNA samples should have two well-defined, sharp peaks and a 28S–18S rRNA ratio of 2:1. The RNA integrity number (RIN) of 8–10 defines respectable quality for further processing (cf. Fig. 2). (see Note 28).
Protein quantity is assessed by Bradford assay (see Note 29).
Fig. 2.
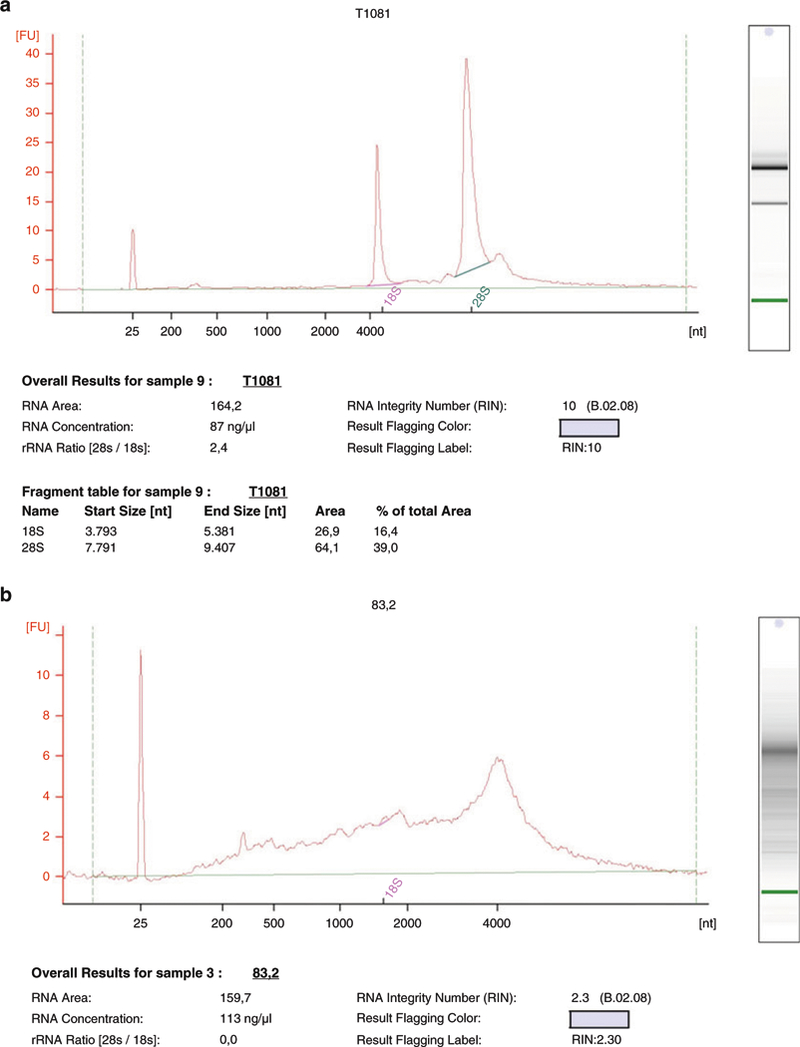
Example for RNA-quantitation by agilent 2100 bioanalyzer (a) Two sharp peaks for the 18S and 28S subunit are visible. The RNA integrity number (RIN) of 10 indicates a clean and undegraded sample of high quality, which is useful for further processing. (b) Degraded RNA-sample. High background and absent peaks represent an inadequate quality which is reflected in a low RIN, here being 2.3
3.4. High-Throughput Analysis
3.4.1. Microarray Platforms
RNA Expression Profiling Using Illumina Human-HT12 v4 Expression BeadChips
Use a minimum of 50 ng (or up to 500 ng) of good quality (RIN > 7) total RNA as input for the TargetAmp Nano labeling kit to produce biotinylated antisense RNA that can be hybridized to the BeadChips. This step is usually automated in the core facility (seeNotes 30 and 31).
Hybridize aRNA with the bead arrays in the core facility.
DNA Methylation Profiling Using Illumina Infinium HumanMethylationEPIC BeadChips
Perform bisulfite conversion of unmethylated cytosines into uracil, whilst methylated cytosines remain unchanged, using 500 ng of genomic DNA and the EZ kit (see Note 32). This step is usually automated in the core facility.
Denature DNA and perform isothermal amplification and subsequent fragmentation. This step is usually automated in the core facility.
Hybridize the fragmented, bisulfite-converted DNA to bead chips using an automated slide processor in the core facility.
3.4.2. Next-Generation Sequencing
RNA Sequencing (Illumina Platform)
Concerning your experimental application, the decision for one of the available sequencing technologies should be well-considered, e.g., the Illumina TruSeq RNA Sample Preparation Kit v2 for preparing the templates sequenced by the Illumina NExtSeq500 orHiSeq2500/3000/4000 (see Note 33).
Dilute purified, high-quality total RNA in distilled nuclease-free water to obtain an amount of 200 ng to a final volume of 50 μL and subsequently follow the sample preparation protocol very closely (see Notes 34–36).
Before pooling the libraries, a supplementary quality check is highly advisable, e.g., with the Agilent 2200 TapeStation System (see Note 37). A sole band of approximately 260 bp is expected in a pure, prepared sample.
Perform paired end RNA sequencing reaction in Illumina HiSeq machine in the core facility.
The recommended number of paired-end reads for gene expression profiling is at least 30 million.
Small RNA Sequencing (Illumina Platform) (See Notes 38 and 39)
When using total RNA including the small RNA fractions, upto 1 μg (in 5 of nuclease-free water) of material can be used as input for the TruSeq library prep kit. If using already purified small RNAs, 10–50 ng are recommended.
Perform library preparation following the manufacturer’s recommendations. This step is usually automated in the core facility.
Perform single-read RNA sequencing reaction in Illumina HiSeq machine in the core facility.
The recommended number of reads depends on the application. For miRNA expression profiling, 1–2 million single-end reads are considered sufficient. For the discovery of novel small RNAs, significantly higher read numbers (10–20 million) are required.
4. Notes
Barcoded tubes can simplify sample identification and storage of a high number of samples, e.g., Cryo.s™ Greiner Bio-One, Germany, #F071080.
Alternatively, the samples can be snap-frozen in liquid nitrogen chair-side and subsequently directly transferred to −80 °C. Keep in mind that handling liquid nitrogen imposes a safety hazard in a clinical setting. Thus, as RNAlater treatment has proven to reliably preserve sample RNA, we prefer to use RNAlater over liquid nitrogen. In addition, keeping RNA stable for up to several days at ambient temperature, it also allows shipment of tissue samples from different study centers to a processing center.
We prefer the simultaneous extraction of total RNA, genomic DNA and protein from the same piece of tissue to allow for the analysis of different -omes in the same samples. Of course, there are kits for solely extracting total RNA fractions available as well.
The organic extraction by phenol-chloroform is a commonly used method to isolate total RNA from tissues and is very cost-efficient. However, it is more time-consuming and needs more practical knowledge to obtain a high amount of intact and purified RNA of comparable quality. As phenol has nearly the same absorbance spectrum of RNA contaminations are difficult to detect and require additional quality checks. Phenol (included in TRIzol reagent) is toxic by inhalation, and chloroform is considered a potential carcinogen. Thus, a fume hood (e.g., Safeaire (Fisher Hamilton, Two Rivers, WI, USA) or good ventilation and appropriate personal safety measures (gloves, safety glasses, protective clothing) are imperative when handling these components. Furthermore, phenol-containing waste must be collected and disposed of separately in many countries.
The complete disintegration of the tissue samples by homogenization is crucial to obtain optimal RNA yields. Residual pieces lead to a clogging of the column pores. Check for remaining intact tissue particles approximately 2 min after homogenization. If needed, continue to process the sample until completely homogenized.
We advise against the utilization of a sonicator for the lysis of tissue samples since the considerable heat generated by this device can result in enhanced RNA degradation.
This protocol is optimized for rather large and fibrous tissue samples (i.e., interdental gingival papillae). To reliably process considerably smaller samples, we recommend the use of a mortar and pestle to finely pulverize the sample after shock-freezing in liquid nitrogen. The pulverized sample can be resuspended in lysis buffer and further processed according to the manufacturer’s instructions.
Instead of using BD Vacutainer CPT tubes already containing Ficoll for cell separation, heparin whole blood can also be diluted 1:1 with PBS and combined with 3 mL of Ficoll in a 15-mL Falcon tube.
Centrifugation without brake is critical for the preparation of peripheral blood monocytic cells (PBMC). Check in advance, as not all standard centrifuges bear this feature.
The isolation of monocytes and lymphocytes should be performed from freshly collected blood. If needed, the blood sample can be stored at 4 °C for several hours before processing, but the sample should not be frozen. In case of emergency, the cells in the white layer isolated by gradient centrifugation (peripheral blood monocytic cells, PBMC) can be frozen in standard cell culture freezing medium [e.g., RPMI (Gibco/Invitrogen) + 10 % fetal bovine serum (Gibco) + 10 % DMSO (Sigma-Aldrich, St. Louis, MO, USA)], but a significant loss in RNA yield must be expected.
The infection of murine DCs by a periodontal pathogen is a typical cell culture model for periodontal infections that allows to test the influence of specific genes and pathways using cells isolated from knockout or transgenic animals. Alternatively, periodontal cells of human origin could be used, possibly after modification using transfection or viral transduction with shRNA.
Cells can also by lysed directly on the plate using 1 mL TRIzol or lysis buffer for a 100 mm plate.
When processing the samples, it is highly advisable to wear gloves (and change them frequently) to avoid contaminations with exogenous nucleases. Further, we recommend the use of certified nuclease-free plasticware and filtered tips. Surfaces and instruments should be treated with an RNase removal fluid, e.g., RNase Away (Sigma Aldrich, #83931). If possible, all RNA-related work should be carried out in dedicated work-space, preferably a hood or a PCR enclosure (e.g., Labconco PCR enclosure, Labconco, Kansas City, MI, USA). The samples should be placed on ice at all times, except when specifically instructed otherwise. Instead of using ice, we found the use of laptop coolers (e.g., Nalgene, Fisher, Rochester, NY, USA) more convenient.
Pay attention that you strictly use the final concentrations of buffers and alcohol as described in the protocol of the kit. Alterations could disturb a successful binding of the nucleic acids to the column.
If you are interested in simultaneously purifying a fraction of miRNA, you could combine the AllPrep DNA/RNA/Protein Mini Kit with RNeasy MinElute Cleanup Kit (Qiagen, Germany) with a supplementary protocol described by the manufacturer. The purified fraction contains miRNA as well as other small RNAs. Of course you can continue with step 3.2.1 (m) if you do not need a fraction of small RNA.
If protein is still insoluble, increase amount of Buffer ALO. Alternatively solve the pellet in 5 % (w/v) SDS or in 8 M urea. Sonication of the sample (5–10 s, 60 %, cooling between each cycle) may be helpful.
Phenol (included in TRIzol reagent) is toxic by inhalation, and chloroform is considered a potential carcinogen. Thus, a fume hood (e.g., Safeaire (Fisher Hamilton, Two Rivers, WI, USA) or good ventilation and appropriate personal safety measures (gloves, safety glasses, protective clothing) are imperative when handling these components.
The precipitation can also be carried out with isopropanol, resulting in a lower salt content of the pellet. However, we recommend the use of ethanol, since isopropanol pellets are more difficult to see and handle. The salts are subsequently removed by the column-based purification step. Furthermore, the use of round-bottomed 2-mL tubes (instead of 1.5-mL) improves the visibility and handling of the obtained pellet.
Pay attention not to lose the pellet when inverting the tube.
Do not overdry the RNA pellet, as a completely dried out pellet is transparent and far more difficult to see. It may also fail to dissolve thoroughly in subsequent steps.
Caution should be taken to completely dissolve the nucleic acid pellet after the drying step by vigorous pipetting for approximately 1 min/sample.
DNA stored in sodium citrate/ethanol solution can be stored at RT for up to two hours, DNA in 75 % ethanol up to several months at 4 °C.
Do not overdry DNA pellet, e.g., by SpeedVac as it becomes very difficult to get the DNA in solution again which normally goes along with DNA degradation.
Sometimes, the protein pellet is difficult to dissolve in 1 % SDS. Alternatively, Hummon and coworkers proposed several alternative solvents, e.g.,10 M urea, 2 % diethylamine, or 1 % SDS and 62.5 mM sarkosyl at pH 8.0–8.8 [21].
- Samples can/should be stored at −80 °C.
-
(a)After harvesting (drained tissue sample or homogenized sample in TRIzol reagent).
-
(b)After extraction of total RNA, miRNA, DNA (DNA samples can be stored at 4 and −20 °C) and protein (protein samples in SDS can be stored at −20 °C).
-
(c)To collect a number of samples over time. Simultaneous processing of 6–8 samples has proven to be safe and efficient.
-
(a)
If the A260–A280 ratio is not in the range of 1.9–2.1 after total RNA isolation, consider re-cleaning the sample with the Qiagen kit. A ratio lower than 2.0 indicates a contamination of protein or phenol. The A260–A230 ratio should also be calculated to check for a contamination by chaotropic salts and organic compounds.
To increase the nucleic acid concentration in the sample, we recommend to use a vacuum centrifuge (e.g., Vacufuge, Eppendorf, Hamburg, Germany) at 4 °C to pellet the nucleic acid and resuspend it in an appropriate volume of RNase-free water.
An alternative measure to judge the quality of the total RNA preparations is to run a formaldehyde 1 % agarose gel and check the 28S rRNA band (~4.5 kB) and 18S rRNA band (~1.9 kB). The 28S band should be twice the intensity of the 18S band.
For Bradford assays for protein quantification, the total SDS concentration must be <0.1 %.
If RNA yields are significantly lower than 50 ng, the TargetAmp Pico or another kit performing a double Eberwine reaction during the labeling process can be utilized. Whilst the Eberwine reaction is a robust amplification, it still generates a 3’-bias with a significantly smaller size distribution, especially when performed twice [22].
The size distribution of the generated biotinylated aRNA can be checked using a Bioanalyzer.
The bisulfite conversion step is very sensitive to DNA quality and phenol contamination of the input DNA. Consider a column cleanup step.
Several library kits and platforms for RNA sequencing are available on the currently growing market; further technologies are e.g.,SOLiD/Applied Biosystems, Ion Torrent NGS/Thermo Fisher Scientific or Roche 454 Life Science. Focused on a specific question, a sequencing experiment should be thoroughly designed. For example, the choice of sequencing depth should comply with the experimental aim; for experiments with gingival tissue, we recommend a sequencing depth of >50 million paired-end reads to ensure an adequate sensitivity for lower gene expression levels.
Work with a multichannel pipette and high precision to avoid unnecessary manipulations which would add up from step to step. If you use Illumina preparation kits for the first time we suggest to start with an input of 12 samples.
For handling beads, there are some aspects to consider in order to purify samples well from rRNA contained therein and in order to avoid an additional loss of nucleic acid: The magnetic beads should be warmed up to room temperature and vortexed thoroughly immediately before each handling. Use a suitable magnetic stand for your 96-well PCR plate. After incubation on the magnetic stand, make sure that the fluid is completely clear and that a compact pellet has formed before you continue. Be careful not to disturb the magnetic beads with tips while pipetting.
Stops are possible after second strand synthesize, end repair, adapter ligation, and the enrichment of DNA fragments. Covered samples can be stored at −20 °C for up to 7 days.
-
We do not recommend a routine quantitation with qPCR as suggested in the manufacturer’s instructions. Though representing a reliable help for the implementation of sequencing methods in a laboratory, processing many samples simultaneously is rather inconvenient and costly.
Alternatively, another reliable and cheap method for quantitation is a measurement by fluorescence, e.g., with the Qubit dsDNA Broad-Range (BR) Assay Kit (Thermo Fisher Scientific, USA).
Most comments for RNA sequencing (see above) are also valid for small RNA sequencing applications.
Safe stopping points are after the reverse transcription and amplification step, and after the normalization of the libraries to 2 nM. Samples at these points can be stored for up to 7 days at −20 °C.
Acknowledgments
This work was supported by grants from the German Society for Periodontology (DG PARO) and the German Society for Oral and Maxillofacial Sciences (DGZMK) to M.K. and by grants from NIH/NIDCR (DE015649, DE021820, and DE024735) and by an unrestricted gift from Colgate-Palmolive Inc. to P.N.P.
References
- 1.Chung CH, Bernard PS, Perou CM (2002) Molecular portraits and the family tree of cancer. Nat Genet 32(Suppl):533–540 [DOI] [PubMed] [Google Scholar]
- 2.Quackenbush J (2006) Microarray analysis and tumor classification. N Engl J Med 354:2463–2472 [DOI] [PubMed] [Google Scholar]
- 3.Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, Gupta S, Moore J, Wrobel MJ, Lerner J, Reich M, Chan JA, Glickman JN, Ikeda K, Hashimoto M, Watanabe G, Daidone MG, Roayaie S, Schwartz M, Thung S, Salvesen HB, Gabriel S, Mazzaferro V, Bruix J, Friedman SL, Kumada H, Llovet JM, Golub TR (2008) Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med 359:1995–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haslett JN, Kunkel LM (2002) Microarray analysis of normal and dystrophic skeletal muscle. Int J Dev Neurosci 20:359–365 [DOI] [PubMed] [Google Scholar]
- 5.Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ (2002) Gene expression profiling of 12633 genes in Alzheimer hippo-campal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res 70:462–473 [DOI] [PubMed] [Google Scholar]
- 6.Haroutunian V, Katsel P, Schmeidler J (2009) Transcriptional vulnerability of brain regions in Alzheimer’s disease and dementia. Neurobiol Aging 30:561–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thornton S, Sowders D, Aronow B, Witte DP, Brunner HI, Giannini EH, Hirsch R (2002) DNA microarray analysis reveals novel gene expression profiles in collagen-induced arthritis. Clin Immunol 105:155–168 [DOI] [PubMed] [Google Scholar]
- 8.van der Pouw Kraan TC, van Baarsen LG, Rustenburg F, Baltus B, Fero M, Verweij CL (2007) Gene expression profiling in rheumatology. Methods Mol Med 136:305–327 [DOI] [PubMed] [Google Scholar]
- 9.Burke W (2003) Genomics as a probe for disease biology. N Engl J Med 349:969–974 [DOI] [PubMed] [Google Scholar]
- 10.Izuhara K, Saito H (2006) Microarray-based identification of novel biomarkers in asthma. Allergol Int 55:361–367 [DOI] [PubMed] [Google Scholar]
- 11.Demmer RT, Behle JH, Wolf DL, Handfield M, Kebschull M, Celenti R, Pavlidis P, Papapanou PN (2008) Transcriptomes in healthy and diseased gingival tissues. J Periodontol 79:2112–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jonsson D, Ramberg P, Demmer RT, Kebschull M, Dahlen G, Papapanou PN (2011) Gingival tissue transcriptomes in experimental gingivitis. J Clin Periodontol 38:599–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kebschull M, Guarnieri P, Demmer RT, Boulesteix AL, Pavlidis P, Papapanou PN (2013) Molecular differences between chronic and aggressive periodontitis. J Dent Res 92:1081–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papapanou PN, Behle JH, Kebschull M, Celenti R, Wolf DL, Handfield M, Pavlidis P, Demmer RT (2009) Subgingival bacterial colonization profiles correlate with gingival tissue gene expression. BMC Microbiol 9:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kebschull M, Papapanou PN (2011) Periodontal microbial complexes associated with specific cell and tissue responses. J Clin Periodontol 38:17–27 [DOI] [PubMed] [Google Scholar]
- 16.Kramer B, Kebschull M, Nowak M, Demmer RT, Haupt M, Korner C, Perner S, Jepsen S, Nattermann J, Papapanou PN (2013) Role of the NK cell-activating receptor CRACC in periodontitis. Infect Immun 81:690–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nowak M, Kramer B, Haupt M, Papapanou PN, Kebschull J, Hoffmann P, Schmidt-Wolf IG, Jepsen S, Brossart P, Perner S, Kebschull M (2013) Activation of invariant NK T cells in periodontitis lesions. J Immunol 190:2282–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kebschull M, Demmer RT, Grun B, Guarnieri P, Pavlidis P, Papapanou PN (2014) Gingival tissue transcriptomes identify distinct periodontitis phenotypes. J Dent Res 93:459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoecklin-Wasmer C, Guarnieri P, Celenti R, Demmer RT, Kebschull M, Papapanou PN (2012) MicroRNAs and their target genes in gingival tissues. J Dent Res 91:934–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papapanou PN, Sedaghatfar MH, Demmer RT, Wolf DL, Yang J, Roth GA, Celenti R, Belusko PB, Lalla E, Pavlidis P (2007) Periodontal therapy alters gene expression of peripheral blood monocytes. J Clin Periodontol 34:736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hummon AB, Lim SR, Difilippantonio MJ, Ried T (2007) Isolation and solubilization of proteins after TRIzol extraction of RNA and DNA from patient material following prolonged storage. Biotechniques 42(467–470):472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spiess AN, Mueller N, Ivell R (2003) Amplified RNA degradation in T7-amplification methods results in biased microarray hybridizations. BMC Genomics 4:44. [DOI] [PMC free article] [PubMed] [Google Scholar]