

Abstract
Fom3, a cobalamin-dependent radical S-adenosylmethionine (SAM) methylase, has recently been shown to catalyze the methylation of carbon 2” of cytidylyl-2-hydroxyethylphosphonate (HEP-CMP) to form cytidylyl-2-hydroxypropylphosphonate (HPP-CMP) during the biosynthesis of fosfomycin, a broad-spectrum antibiotic. It has been hypothesized that a 5’-deoxyadenosyl 5’-radical (5’-dA•) generated from the reductive cleavage of SAM abstracts a hydrogen atom from HEP-CMP to prime the substrate for addition of a methyl group from methylcobalamin (MeCbl); however, the mechanistic details of this reaction remain elusive. Moreover, it has been reported that Fom3 catalyzes the methylation of HEP-CMP to give a mixture of the (S)-HPP and (R)-HPP stereoisomers, which is rare for an enzyme catalyzed reaction. Herein, we describe a detailed biochemical investigation of a Fom3 that is purified with one equivalent of its cobalamin cofactor bound, which is almost exclusively in the form of MeCbl. Electron paramagnetic resonance and Mössbauer spectroscopies confirm that Fom3 contains one [4Fe-4S] cluster. Using deuterated enantiomers of HEP-CMP, we demonstrate that the 5’-dA• generated by Fom3 abstracts the C2”-pro-R hydrogen of HEP-CMP and that methyl addition takes place with inversion of configuration to yield solely (S)-HPP-CMP. Fom3 also sluggishly converts cytidylyl-ethylphosphonate to the corresponding methylated product, but more readily acts on cytidylyl-2-fluoroethylphosphonate, which exhibits a lower C2” homolytic bond-dissociation energy. Our studies suggest a mechanism in which the substrate C2”-radical, generated upon hydrogen atom abstraction by the 5’-dA•, directly attacks MeCbl to transfer a methyl radical (CH3•) rather than a methyl cation (CH3+), directly forming cob(II)alamin in the process.
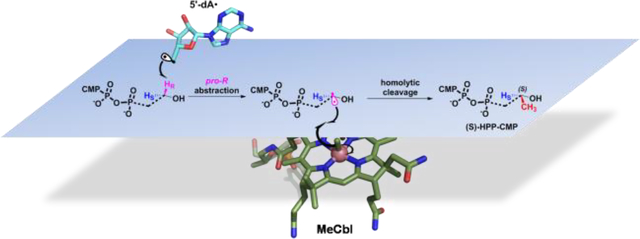
Graphical Abstract
Fosfomycin is a clinically-approved broad-spectrum antibiotic that displays bactericidal activity against both Gram-positive and Gram-negative bacteria. It has been approved by the Food and Drug Administration for the treatment of uncomplicated urinary tract infections as well as some gastrointestinal infections.1, 2 Fosfomycin, whose chemical structure contains a reactive epoxide ring and a carbon phosphorus bond, both of which are essential for its activity, functions by blocking peptidoglycan biosynthesis through the covalent inhibition of MurA, which catalyzes the first step in the bacterial cell wall biogenesis pathway. Although fosfomycin was first discovered in 1969,3 the recent world-wide rise in antibiotic resistant bacteria has generated a resurgence in interest in the drug,4 because its unique mechanism of action makes cross-resistance uncommon, allowing it to be used simultaneously or synergistically with other classes of antibiotics.
Fosfomycin is produced both by streptomyces and by pseudomonas, which, interestingly, employ different biosynthetic pathways to generate the molecule. Genetic and biochemical studies of fosfomycin biosynthesis in Streptomyces fradiae and Streptomyces wedmorensis indicated that at least five gene products are directly involved in the biosynthesis of fosfomycin from phosphoenolpyruvate (PEP) (Figure 1).5–7 Fom1, a PEP mutase that is conserved in both Streptomyces and Pseudomonas, catalyzes the conversion of PEP to phosphonopyruvate (PnPy) in the first committed step of the fosfomycin biosynthetic pathway.8 Next, Fom2 catalyzes the decarboxylation of PnPy to form phosphonoacetaldehyde (PnAA), which is subsequently converted to 2-hydroxyethylphosphonate (HEP) by FomC, an alcohol dehydrogenase. Recent studies have shown that HEP is converted to cytidylyl-2-hydroxyethylphosphonate (HEP-CMP) by Fom1, which, in addition to its PEP mutase domain, contains a cytidyltransferase domain.9 HEP-CMP is then converted to cytidylyl-2-hydroxypropylphosphonate (HPP-CMP) by Fom3, a cobalamin-dependent radical S-adenosylmethionine (SAM) methylase.10 HPP-CMP is then hydrolyzed to 2-hydroxypropylphosphonate (HPP) by FomD, as described in the work herein and in recent works.10–13 In the final step of fosfomycin biosynthesis in Streptomyces, (S)-HPP is converted to fosfomycin by the nonheme iron peroxidase Fom4, which is also referred to as HppE.14–16
Figure 1.

Fosfomycin biosynthetic pathway in streptomyces.
Fom3 is one of the first cobalamin-dependent RS methylases to be studied, both in vivo and in vitro.1, 10, 17 Its initial in vitro characterization involved its purification from inclusion bodies, which was followed by refolding the protein and reconstituting its iron-sulfur (FeS) and cobalamin cofactors. Using a single time-point NMR assay, the authors claimed that Fom3 methylates HEP to afford HPP; however, whether 5’-deoxyadenosine (5’-dA) and S-adenosylhomocysteine (SAH) (the expected co-products of this reaction) were formed was not reported.17 In two recent studies, however, which were conducted with S. wedmorensis and S. fradiae Fom3s that were purified from the soluble fraction of Escherichia coli cell extract upon heterologous expression, neither HPP, SAH, nor 5’-dA was detected when HEP was used as the substrate. By contrast, when HEP was replaced with HEP-CMP in the same reaction, which contained SAM, Fom3, and methyl viologen (MV) and NADPH (to satisfy the requirement for a low-potential reductant), the generation of HPP-CMP, 5’-dA, and SAH was observed both by Sato et al. and by Lanz et al.10, 18 These findings, along with the newly discovered bifunctional role of Fom1,9 provided strong evidence that HEP-CMP is indeed the true substrate for Fom3.
It was thought that the inherent insolubility of Fom3 and many other cobalamin-dependent RS methylases is due to a lack of readily available cobalamin during their overproduction in E. coli, which might promote misfolding. Recently, a plasmid (pBAD42-BtuCEDFB) was engineered that harbors the genes responsible for cobalamin uptake and trafficking in E. coli. Studies showed that this plasmid markedly increases the intracellular cobalamin concentration, resulting in a significant increase in yield of four class B RS methylases: TsrM, Fom3, PhpK, and ThnK.18 Herein, we take advantage of this new strategy for overproducing class B RS methylases and characterize S. fradaie Fom3 purified from E. coli soluble crude lysate with both its [4Fe-4S] cluster and cobalamin cofactors present. We use UV-vis, electron paramagnetic resonance (EPR), and Mössbauer spectroscopies to show that as-isolated Fom3 contains almost one complete [4Fe–4S] cluster and almost one full equivalent of bound cobalamin, which is found almost exclusively in its MeCbl form. Through the use of deuterated enantiomers of HEP-CMP, we show that the 5’-deoxyadenosyl 5’-radical (5’-dA•) abstracts the C2”-pro-R hydrogen atom of HEP-CMP and that methyl addition takes place with inversion of configuration to afford 2”-(S)-HPP-CMP. While Fom3 will also convert 2”-thioethylphosphonate-CMP to its methylated product, its ability to convert ethylphosphonate-CMP and 2-fluoroethylphosphonate-CMP to the corresponding methylated product argues for a mechanism involving a direct attack of a C-2” carbon radical onto MeCbl to yield a cob(II)alamin species.
Materials and Methods
Materials
Ferric chloride was obtained from EMD Biosciences (Gibbstown, NJ). Bradford reagent for protein concentration determination as well as the bovine serum albumin (BSA) standard was purchased from Pierce, Thermo Fisher Scientific (Rockford, IL). Dithiothreitol (DTT), L-(+)-arabinose, isopropyl β-D-1-thiogalactopyranoside (IPTG), ampicillin, spectinomycin, and kanamycin were purchased from Gold Biotechnology (St. Louis, MO). SUMO express protease was purchased from Lucigen (Middleton, WI). SAM was synthesized from ATP and L-methionine and purified as described previously.19 E. coli flavodoxin (flv) and flavodoxin reductase (flr) were purified as described previously.20 All reagents for chemical syntheses were obtained from Sigma–Aldrich unless specified further. All other materials and reagents have been previously reported,21, 22 or were of the highest available quality.
General Methods
UV-vis spectra were recorded on a Cary 50 spectrometer from Varian using the WinUV software package to control the instrument. Sonic disruption of E. coli was conducted with a 550 sonic dismembrator (Fisher Scientific, Waltham, MA) in an anaerobic chamber (Coy, Grass Lakes, MI). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was conducted as described previously.23 Ultra-high-performance liquid chromatography with detection by tandem mass spectrometry (LC-MS/MS) was conducted on an Agilent Technologies 1200 system coupled to an Agilent Technologies 6470 QQQ mass spectrometer. The system was operated with the associated Mass Hunter software package, which was also used for data collection and analysis.
Overexpression and Purification of Fom3
Overexpression and purification of Fom3 was conducted as previously described.18 Briefly, genes on plasmid pSUMO-Fom3 were co-expressed with those on pBAD42-BtuCEDFB—which encodes the genes responsible for cobalamin uptake in E. coli, and pDB1282—which encodes the isc operon to allow for expression in the presence of Fe/S cluster assembly proteins. The plasmids were used to transform E. coli BL21 (DE3) cells, which were cultured in ethanolamine media supplemented with hydroxocobalamin. The resulting SUMO-Fom3 fusion protein was purified by immobilized metal-affinity chromatography (IMAC), and native Fom3 was produced after incubating SUMO-Fom3 with Ulp1, a SUMO protease. Using methods previously described,21, 24 native Fom3 was isolated by reapplying the protein-Ulp1 mixture to Ni-NTA resin and collecting the flow through. The concentration of Fom3 was determined by the Bradford assay using BSA (fraction V) as a standard25 and using the correction factor of 1.18.18
Quantification of Iron, Sulfide, and Cobalamin
The amount of cobalamin bound to Fom3 was determined by boiling the enzyme for 5 min in 0.1 M potassium cyanide to generate the dicyanocobalamin form of the cofactor. The concentration of the released dicyanocobalamin was determined from its UV-vis spectrum (ɛ367 = 30,800 M−1 cm−1).26 The methods of Beinert et al. were used to determine the amount of iron and sulfide present in Fom3.27, 28 Quantification of AdoCbl, MeCbl, and OHCbl was done by LC-MS/MS as previously described.22 These reactions were all performed in dim lighting and in amber tubes to prevent the photolysis of alkylcobalamins.
Determination of Fom3 Activity
The activity of Fom3 was determined using previously described methods.18 Reactions were conducted under anaerobic conditions and contained the following: Fom3 (1 – 20 μM), SAM (1.5 mM), HEP-CMP (1.5 mM) or HEP-CMP analog (1.5 mM), MV (1 mM) and NADPH (2 mM). When the flavodoxin reducing system was used as the requisite low-potential reductant, MV and NADPH were replaced with flavodoxin (30 μM), flavodoxin reductase (15 μM) and 1 mM NADPH. In some reactions, MV and NADPH were replaced with Ti(III)citrate (1 mM).29 Reactions were initiated with reductant, and aliquots were removed at designated times from 0 to 60 min and added to 400 mM H2SO4 (final concentration) to quench the reactions. Standard curves for product quantification by LC-MS/MS contained 100 μM tyrosine (internal standard) and varying concentrations of SAH and 5’-dA or HEP-CMP and HPP-CMP.
Quantification of Product and Cobalamin Concentrations by LC-MS/MS
The quantification of cobalamin and products of the reaction was performed on an Agilent Technologies 6470 QQQ mass spectrometer using methods previously described.18, 22 Quenched reactions monitoring cobalamin concentration were separated on an Agilent Zorbax Extend-C18 RRHD column (2.1 mm × 50 mm, 1.8 μm particle size), while quenched reactions measuring SAH and 5’-dA or HEP-CMP and HPP-CMP were separated on Agilent Zorbax Eclipse-C18 RRHD (2.1 mm × 50 mm, 1.8 μm particle size) or Agilent SB-Aq RRHD (2.1 mm × 50 mm, 1.8 μm particle size) columns, respectively.
Electron Paramagnetic Resonance (EPR) Spectroscopy
Fom3 was diluted to a final concentration of 220 μM for each EPR sample, and all samples were flash-frozen in cryogenic liquid isopentane in an anaerobic chamber. EPR samples were prepared by adding dithionite to a final concentration of 3 mM and SAM or HEP-CMP to a final concentration of 1.5 mM when appropriate. Each sample was incubated for 2 min at room temperature before freezing. Continuous-wave EPR (CW-EPR) spectra were acquired on an ESP300 Bruker X-band spectrometer equipped with an ER 410ST resonator. Temperature was controlled by an ER 4112-HV Oxford Instruments variable-temperature helium-flow cryostat. Acquisition and control of experimental parameters was performed using EWIN 2012 software on an external PC via a GPIB interface.30 The conversion time and the time constant were 40.96 ms, the modulation amplitude was 5 G, and the microwave power was 20 mW.
Mössbauer spectroscopy
57FeCl3 was generated by incubating 57Fe2O3 overnight at 70 °C with HCl (6 molar equivalents HCl/57Fe2O3) in an anaerobic chamber. The concentration of 57FeCl3 was determined by iron analysis as previously described.27 57 Fe-labeled Fom3 was purified from E. coli cells cultured in 57Fe-enriched (50 μM) media. All samples were made with 350 μM Fom3 and, when applicable, 1 mM SAM or 1 mM HEP-CMP. All samples were frozen and stored in liquid nitrogen prior to use. Mössbauer spectra were recorded using a spectrometer from SEE Co (Edina, MN) and a Janis SVT-400 variable-temperature cryostat (Woburn, MA), where the external magnetic field of 53 mT was applied parallel to the γ radiation. Quoted isomer shifts are referenced relative to the centroid of the spectrum of α-Fe recorded at room temperature. Analysis of the resultant spectra was conducted using the program WMOSS (www.wmoss.org, SEE Co., Edina, MN).
Synthesis of HPP-CMP, HEP-CMP and Substrate Analogs
The syntheses of HPP-CMP, HEP-CMP and other substrate analogs were performed by coupling various C-phosphonate triethylammonium salts with the P-imidazolide of cytidine monophosphate. The crude product was initially purified by chromatography on a Sephadex DEAE A-25 column using a linear gradient of 10–1000 mM aqueous ammonium bicarbonate. The product was then further purified by high-performance liquid chromatography (HPLC) on an Agilent 1100 Series system coupled with an Agilent 1100 Series variable wavelength detector and quaternary pump. The NMR spectra of all compounds were collected on a Bruker AV-3-HD-500 instrument. High-resolution ESI mass spectra were collected on a Waters LCT Premier instrument.
Cloning of the Streptomyces fradiae fomD Gene
The fomD gene from Streptomyces fradiae, codon-optimized for expression in E. coli and synthesized by GeneArt (Thermofisher Scientific), was amplified from a plasmid encoding FomD using the polymerase chain reaction (PCR). The forward primer, 5’-GTC CAG AGC TCG GTC TCC AGG TAT GAC CGA AGC AGC-3’, contains 11 bases of the plasmid, a Bsa1 recognition site (bold), a four-base 5’ spacer preceding the Bsa1 cut site (underlined), and the first 14 bases of the fomD gene. The reverse primer is the reverse compliment of the forward primer. The fomD gene was purchased with the Xho1 cut site at the C-terminus of the gene. The PCR product was treated with Dpn1 for two hours and then used to transform E. coli DH5α cells. The DNA was isolated, and the correct DNA sequence was verified at the Penn State Genomics Core Facility (University Park), resulting in a construct with a Bsa1cut site at the N-terminus of the gene. The construct and the plasmid pSUMO were treated with Bsa1 and Xho1, and the fomD gene was then ligated into pSUMO to generate a construct that encodes a FomD fusion to the C-terminus of SUMO, which contains an N-terminal hexahistidine (His6) tag. The correct DNA sequence was verified and designated as pSUMO-FomD.
Overexpression of the fomD Gene
The plasmid pSUMO-FomD was used to transform E. coli BL21 (DE3) cells. A starter culture was inoculated from a single colony and then incubated for 24 h at 37 °C while shaking at 250 rpm. Bacterial growth was carried out at 37 °C in 16 L of lysogeny broth (LB) while cultures were agitated at 180 rpm. At an optical density at 600 nm (OD600) of ~0.55, the flasks were placed on ice for 1 h prior to induction of gene expression with IPTG at a final concentration of 200 μM. Cultures were incubated for 16 h at 18 °C before cells were collected by centrifugation at 7000×g for 12 min. Approximately 30 g of cells was obtained.
Purification of FomD
All purification steps and subsequent manipulations of FomD were performed in a Coy anaerobic chamber. Cell paste (30 g) was resuspended in 100 mL of lysis buffer (50 mM HEPES, pH 7.5, 300 mM KCl, 10 mM imidazole, 10 mM β-mercaptoethanol) with lysozyme (1 mg/mL) and DNase (0.1 mg/mL) for 30 min at room temperature and then cooled to 4 °C before lysing. Cells were lysed by sonication and centrifuged at 50 000×g for 1 h to separate insoluble material. The resulting supernatant was loaded onto a pre-equilibrated column of Ni-NTA resin and purified by IMAC. The resin was washed with 200 mL of wash buffer (50 mM HEPES, pH 7.5, 300 mM KCl, 25 mM imidazole, 10 mM β-mercaptoethanol, 10% glycerol) before eluting with elution buffer (50 mM HEPES, pH 7.5, 300 mM KCl, 500 mM imidazole, 10 mM β-mercaptoethanol, 10% glycerol). The protein was collected and concentrated by ultracentrifugation using an Amicon centrifugal filter device (EMD Millipore; Billerica, MA) with a 10 kDa cutoff. The FomD-SUMO fusion protein was then exchanged into cleavage buffer (50 mM HEPES, pH 7.5, 300 mM KCl, 10 mM β-mercaptoethanol, 15% glycerol) using a PD-10 column and treated with the SUMO protease Ulp1 overnight on ice (0 °C) to generate native FomD. The cleavage reaction was then reapplied to the Ni-NTA resin to capture the SUMO protein and Ulp1 protease along with other proteins that bind nonspecifically to the resin, allowing the untagged FomD to be collected in the flow-through fraction. FomD was concentrated and exchanged into gel-filtration buffer (50 mM HEPES, pH 7.5, 300 mM KCl, 15% glycerol, 5 mM DTT) as described in previous steps.
Determination of FomD Activity
Reactions contained FomD (100 μM) and synthetic racemic HPP-CMP (6 mM) in a 5 mm NMR tube that was equipped with an insert containing D2O for locking the field. The data were collected as proton decoupled 31P-NMR. The earliest time point was at 5 min after initiating the reaction with FomD. Time points at 10, 15, 30, and 60 min were collected manually, with each time point correlating to 25 scans. The remaining time points were collected automatically at 1 h intervals using 1425 dummy scans (~59 min total) followed by 25 scans (~1 min total) of data collection. The concentrations of substrate and product were calculated by integrating the 31P peak areas.
Determination of the Stereochemistry of the Fom3 Product
Large-scale (10 mL) Fom3 reactions were conducted under anaerobic conditions and contained the following: Fom3 (5μM), SAM (1 mM), HEP-CMP (0.5 mM) and Ti(III)citrate (1 mM). Four identical reactions were performed in parallel. Reactions were initiated with Fom3 and incubated for 2 h before removing them from the anaerobic chamber and quenching them in acid. The HPP-CMP produced from the Fom3 reaction was purified using the same method as that described for synthetic HPP-CMP. After acid hydrolysis, the product was incubated with Fom4 as described in supplementary information. Authentic (R)-HPP and (S)-HPP were used as controls. 31P-NMR spectra were collected on a Bruker AV-3-HD-500 instrument with phosphoric acid as an internal standard. Fom4 was overproduced and purified as previously described.13
Results
Overexpression and Purification of Fom3
Studies on Fom3 have been hindered by its inherent insolubility upon overproduction in E. coli. In the first in vitro characterization of Fom3—which was from Streptomyces wedmorenis—the enzyme was purified by resolubilizing inclusion bodies, which was followed by in vitro refolding of the protein and reconstitution of its MeCbl and iron-sulfur (Fe/S) cluster cofactors.31 More recently, Sato et al. were able to purify Fom3 from a soluble fraction of E. coli cell extract. Their yields were not reported, but their protein was overwhelmingly produced as insoluble inclusion bodies—as judged from their SDS–PAGE analysis—and did not have cobalamin bound.10 Recently, we developed a strategy that makes use of an accessory plasmid (pBAD42-BtuCEDFB) to enhance the soluble fraction of cobalamin-dependent RS enzymes overproduced in E. coli. This plasmid encodes genes that are responsible for cobalamin uptake and transport in E. coli. It was generated with the thought that enhanced accessibility to cobalamin might allow for better in vivo folding, thereby helping to further solubilize some cobalamin-dependent enzymes. As has recently been described, we overexpressed the fom3 gene from Streptomyces fradiae in E. coli BL21 (DE3) along with pBAD42-BtuCEDFB and pDB1282, which encodes genes involved in Fe/S cluster biosynthesis. Purification of Fom3 was performed using methods previously described,21 which involved isolation of a hexahistidine- (His6)-tagged SUMO-Fom3 fusion protein by IMAC. Native Fom3 was generated by removing the SUMO tag using the SUMO protease Ulp1 and was purified by reapplying the cleavage mixture to a nickel-NTA column while collecting the flow-through rather than the eluate.
The UV-vis spectrum of as-isolated Fom3 displays a broad absorption between 350 and 600 nm, which is consistent both with the presence of an Fe/S cluster and with the presence of cobalamin (Figure 2a). The stoichiometry of bound cobalamin was determined by treating Fom3 with potassium cyanide to yield the dicyano adduct of cobalamin, which has a strong absorption feature at 367 nm (ɛ367 = 30,800 M−1 cm−1).26 Using the absorption spectrum shown in Figure 2b along with the molar absorptivity of dicyanocobalamin, Fom3 is determined to bind 0.85 cobalamins per polypeptide. Analysis for iron and sulfide show as-isolated Fom3 to contain 3.6 mol of the former and 3.4 mol of the latter per polypeptide.
Figure 2.

UV-vis spectrum of as-isolated Fom3 (11.1 μM). (b) UV-vis spectrum of Fom3 (10 μM) treated with potassium cyanide.
Fom3 Reactions
Until recently, it was believed that Fom3 catalyzes the methylation of HEP to form HPP. However, studies by Sato et al. showed that HEP-CMP is the true substrate for S. wedmorensis Fom3, affording HPP-CMP.10 In their studies, HPP-CMP formation required the addition of MeCbl to their reactions, and only MV/NADPH served as the low-potential reductant. More recent studies by Lanz et al., however, indicate that Fom3 from S. fradiae can catalyze multiple turnovers in the absence of added MeCbl. These recent observations suggest that MeCbl is a true cofactor in Fom3 and that MeCbl does not re-bind to the enzyme after each turnover, as has been previously suggested.10 Furthermore, S. fradiae Fom3 converts HEP-CMP to HPP-CMP when using an in vivo reducing system composed of E. coli flavodoxin (flv), flavodoxin reductase (flr), and NADPH. In fact, the protein is even more active in the presence of the in vivo reducing system, exhibiting a kcat value of 5.4 h−1, while kcat in the presence of MV/NADPH is 2.1 h−1 (Figure 3). Although these turnover numbers are low, they are consistent with those of other cobalamin-dependent RS methylases that cleave SAM to generate a 5’-dA•, such as ThnK, GenK, PoyC, and CysS.32–35 By contrast, TsrM, a cobalamin-dependent RS methylase that does not generate a 5’-dA•, displays a significantly higher turnover number (~20 min−1).18
Figure 3.

Time-dependent formation of HPP-CMP by Fom3 (10 μM) in the presence of SAM (1.5 mM), HEP-CMP (1.5 mM), and NADPH (2 mM) and MV (1 mM) (black) or flavodoxin (30 μM), flavodoxin reductase (15 μM), and NADPH (1 mM) (red).
Ti(III)citrate, which exhibits a reduction potential for the Ti(IV/III) couple that is less than −800 mV, was also used as a reductant.36 This reductant has been used routinely in reactions of other corrinoid iron-sulfur enzymes.37 When we repeated the Fom3 reaction using 10 μM Fom3 and Ti(III)citrate as the low-potential reductant, a linear increase of product over 60 min is not observed, and the rate of product formation rapidly decreases after the 5 min time point (Figure 4a). We ascribe this decrease in rate to product inhibition by HPP-CMP. Given that the addition of SAH nucleosidase does not relieve this inhibition, it is unlikely due to a build-up of SAH or 5’-dA, both of which are acted upon by this enzyme. Therefore, to determine the activity of Fom3 in the presence of Ti(III)citrate more accurately, we monitored the time-dependent formation of HPP-CMP over 30 min using only 1 μM Fom3 (Figure 4b). As shown in Figure 4c, a steady-state kinetic analysis of the Fom3 reaction in the presence of Ti(III)citrate shows that the enzyme exhibits a kcat value of ~0.78 min−1, and a Km value for HEP-CMP of ~74 μM.
Figure 4.

Time-dependent formation of HPP-CMP by Fom3 in the presence of SAM, HEP-CMP, and Ti(III)citrate with (a) 10 μM Fom3 and (b) 1 μM Fom3. (c) Steady-state kinetic analysis of the Fom3 reaction using Ti(III)citrate and 1 μM Fom3.
Analysis of cobalamin
Unlike TsrM, which is isolated with many different forms of bound cobalamin (i.e. hydroxocobalamin, adenosylcobalamin, cob(I)alamin, and cob(II)alamin), Fom3 is isolated almost exclusively (~90%) with bound MeCbl, which is the form of the cofactor that acts as the direct methyl donor (Figure S1). We believe that Fom3 is purified with bound MeCbl, while as-isolated TsrM is void of MeCbl, because Trp, the substrate for TsrM, is present in E. coli. By contrast, HEP-CMP is not found in E. coli. Therefore, the presence in E. coli of SAM and the flavodoxin reducing system, which promotes reduction of hydroxocobalamin to cob(I)alamin, supports formation of bound MeCbl with no pathway for its decay.
To shed light on the rate-limiting step during HPP-CMP formation, an analysis of the bound forms of cobalamin in Fom3 during turnover was conducted by an established LC-MS/MS method. Similar to what was observed with TsrM,22 Fom3 exists almost exclusively with bound MeCbl under turnover conditions (Figure S2), suggesting that methyl transfer from MeCbl to HEP-CMP is slower than methyl transfer from SAM to cob(I)alamin.
Analysis of the Fom3 Fe/S Cluster by CW-EPR Spectroscopy
Although Fom3 was examined by EPR spectroscopy in a previous study,17 the protein’s spectral envelope was poorly defined. Herein, EPR was used to study the Fe/S cluster of Fom3 in the presence and in the absence of substrates. Figure 5a displays the EPR spectrum of the dithionite-reduced [4Fe-4S]+ cluster of as-isolated Fom3 at 10 K. The spectrum is similar to that of most other RS enzymes and consistent with the presence of a [4Fe–4S]+ cluster. To assess whether SAM or HEP-CMP has an effect on the electronic properties of the Fe/S cluster, dithionite-reduced Fom3 was analyzed in the presence of each substrate. Analysis of dithionite-reduced Fom3 in the presence of SAM (Figure 5b) or HEP-CMP (Figure 5c) results in only slight changes to the spectrum in Figure 5a.
Figure 5.

X-band EPR spectra of (a) Fom3 (220 μM) at 10 K in the presence of 3 mM dithionite with added (b) 1.5 mM SAM or (c) 1.5 mM HEP-CMP with microwave frequency of 9.621 GHz.
Analysis of the Fe/S Cluster by Mössbauer Spectroscopy
Mössbauer spectroscopy was used to determine the type and stoichiometry of Fe/S cluster(s) in Fom3. The Mössbauer spectrum of as-isolated Fom3 (Figure 6, top, vertical bars) exhibits a quadrupole doublet with isomer shift and quadrupole splitting parameters (δ = 0.45 mm/s, ΔEQ = 1.05 mm/s, 98% of total intensity, black line) that are typical of valence-delocalized [4Fe-4S]2+ clusters.38 Upon incubating as-isolated Fom3 with SAM (Figure 6, bottom, vertical bars), a new weak feature at ~1.75 mm/s is observed (see red arrow). This feature most likely arises from a small amount of a site-differentiated [4Fe-4S]2+ cluster and has been observed for other [4Fe-4S]2+-containing enzymes.39, 40 Addition of HEP-CMP to Fom3 does not significantly perturb the Mössbauer spectrum whether or not SAM is also present (Figure S3).
Figure 6.

Mössbauer spectra of Fom3 (black bars, with simulation as a gray line) and Fom3 in the presence of SAM (red bars), recorded at 4.2 K in an externally applied field of 53 mT oriented parallel to the gamma beam.
Methylation of HEP-CMP with d3-SAM
To demonstrate definitively that the methyl group of SAM is transferred to cobalamin and then subsequently to HEP-CMP, S-adenosyl-[methyl-d3]-methionine (d3-SAM) was incubated with Fom3, HEP-CMP, unlabeled MeCbl, and MV/NADPH under turnover conditions, and the isotopic composition of the products of the reaction was analyzed by LC-MS/MS. As shown in Figure 7a, MeCbl containing three deuteriums (d3-MeCbl) is observed (m/z = 674.5), indicating that SAM transfers its methyl group to cobalamin with all of its original three hydrogens intact. The intact methyl group of MeCbl is then transferred to HEP-CMP to generate d3-HPP-CMP (m/z = 449) (Figure 7b). The observation of d3-HPP-CMP and almost no unlabeled HPP-CMP indicates that unlabeled MeCbl does not exchange on and off the enzyme on the time scale of the reaction, as has previously been suggested.10
Figure 7.

MS analysis of (a) cobalamin and (b) HPP-CMP formed in a reaction containing Fom3 (10 μM), d3-SAM (1.5 mM), HEP-CMP (1.5 mM), NADPH (2 mM), and methyl viologen (1 mM).
Determination of the Site of Hydrogen Atom Abstraction
It has been proposed that 5’-dA generated during the Fom3 reaction is the result of a 5’-dA• abstracting a hydrogen atom from C2” of HEP-CMP. However, an alternative mechanism can be proposed, whereby the 5’-dA• abstracts a hydrogen atom from the hydroxyl group, which would subsequently lower the pKa of C2” to allow for subsequent deprotonation followed by a nucleophilic attack of a C2” carbanion onto MeCbl (Figure S4). To determine the site of hydrogen atom abstraction, 2”-d2-HEP-CMP (Figure 8a) was synthesized and used in a Fom3 reaction under turnover conditions, and LC-MS/MS was used to assess the mass of the 5’-dA formed after 1 h of incubation. As can be seen in Figure 8b, nearly all of the 5’-dA produced exhibits an m/z of 253.1 (MRM transition: 253.1 to 136), which is consistent with the presence of one deuterium atom (5’-dAD). Only a small fraction (~3.1%) of 5’-dA detected is at natural abundance (MRM transition: 252.1 to 136). The 5’-dA product is also observed by LC-MS (Figure 8c) in scan mode, in which m/z 253.1 (5’-dAD) is overwhelmingly the predominant ion formed, further confirming that C2” is the site of hydrogen atom abstraction.
Figure 8.

(a) Structure of 2”-d2-HEP-CMP. (b) MS/MS analysis of 5’-dAH (black) and 5’-dAD (red) and (c) MS analysis of 5’-dA product in a reaction with Fom3, 2”-d2-HEP-CMP, SAM, NADPH, and MV.
Stereoselectivity of Hydrogen Atom Abstraction
Two deuterated substrate isotopomers, [2”-R-2H]-HEP-CMP (pro-R-HEP-CMP) and [2”-S-2H]-HEP-CMP (pro-S-HEP-CMP), were synthesized as probes to determine the stereoselectivity of hydrogen atom abstraction in the Fom3 reaction by assessing deuterium transfer to 5’-dA or deuterium remaining in HEP-CMP in reactions containing either of the isotopomers. In reactions containing pro-R-HEP-CMP, formation of 5’-dAD is observed (Figure 9a), as is formation of unlabeled HPP-CMP. By contrast, in reactions containing pro-S-HEP-CMP, formation of 5’-dA at natural abundance is observed, as is HPP-CMP containing a single deuterium (Figure 9a). These results indicate that the 5’-dA• abstracts the pro-R hydrogen atom from C2” of HEP-CMP and are consistent with past feeding studies with labeled forms of HEP, which suggested that the pro-R hydrogen is removed during fosfomycin biosynthesis.41, 42 However, at the time of those studies, the step at which the pro-R hydrogen is removed was unknown. To determine if hydrogen atom abstraction is rate-limiting in the reaction, the rate of 5’-dA formation in reactions with pro-R-HEP-CMP and pro-S-HEP-CMP was monitored. As can be seen in Figure 9b, Fom3 catalyzes the formation of 5’-dAD with pro-R-HEP-CMP and 5’-dAH with pro-S-HEP-CMP with turnover numbers of 5.3 h−1 and 5.6 h−1, respectively, suggesting that this step is not rate-limiting under the conditions of the reaction. It is therefore likely that the reductive cleavage of SAM limits the overall rate of the reaction.
Figure 9.

(a) LC-MS analysis of 5’-dA in Fom3 reaction with pro-R-HEP-CMP (red) and pro-S-HEP-CMP (blue). (b) Time-dependent formation of 5’-dAH (blue) and 5’-dAD (red) in a reaction with Fom3 (10 μM), SAM (1.5 mM), pro-S-HEP-CMP (1.5 mM) (blue) or pro-R-HEP-CMP (1.5 mM), and flavodoxin (30 μM), flavodoxin reductase (15 μM), and NADPH (1 mM).
Stereoselectivity of Fom3 Methylation
Recently, Sato et al. concluded that the methylation of HEP-CMP by Fom3 is not stereoselective, affording a 1:1 mixture of (S)-HPP-CMP and (R)-HPP-CMP. This conclusion was based on the observation of two sets of doublet methyl signals after the isolated product of the reaction was analyzed by 1H NMR spectroscopy. Given that enzymes rarely catalyze the production of racemic products, as well as the differences between the behavior of our enzyme preparation of Fom3 and that of Sato et al., we re-investigated the stereoselectivity of Fom3 methylation. When we performed a similar NMR analysis on HPP-CMP isolated from the Fom3 reaction, we did not observe two sets of doublets, as reported by Sato et al. However, we also did not observe two sets of doublets for racemic HPP-CMP that we synthesized chemically (Figure S5), indicating that a different method of analysis would be needed to assess the stereochemical outcome of the Fom3 reaction. To distinguish between the two enantiomers, we used the iron-dependent epoxidase HppE (i.e. Fom4), which is known to convert (S)-HPP to fosfomycin and (R)-HPP to (2-oxopropyl)phosphonate (the ketone product). These two products of the Fom4 reaction can be differentiated easily by 31P-NMR.
Our initial efforts to cleave the pyrophosphate bond of HPP-CMP to generate HPP focused on FomD (Figure 1), a putative pyrophosphatase whose function had not been verified in in vitro reactions. FomD from S. fradiae was overproduced and purified and then used in a reaction containing racemic HPP-CMP that was chemically synthesized. As shown in Figure S6, the progress curve of the reaction is biphasic, with a quick burst of HPP formation followed by a slower phase of HPP formation. We believe that FomD hydrolyzes one of the two enantiomers much more rapidly than the other (most likely (S)-HPP-CMP, vide infra); however, even after 15 h of incubation, HPP-CMP could still be detected (Figure S7). Because we wanted to observe both potential products of the Fom4 reaction easily, we turned to acid hydrolysis of HPP-CMP, which would not bias the ratio of (S)-HPP and (R)-HPP produced.
The Fom3 reaction was scaled up to accumulate sufficient HPP-CMP for analysis, and then was treated with 6 M HCl at 110 °C for 12 h to cleave the pyrophosphate bond to afford HPP and CMP. To ensure that these harsh cleavage conditions do not racemize or invert the newly generated stereocenter in HPP-CMP, we synthesized diethyl (S)-(2-hydroxypropyl)phosphonate (40) and diethyl (R)-(2-hydroxypropyl)phosphonate (43) and hydrolyzed the two authentic compounds under the same conditions (6M HCl, 110 °C, 12 h) (Figure S8). The resulting HPPs, as well as racemic HPP, were given to Fom4 and analyzed by 31P-NMR. As shown in Figure 10A, racemic HPP is cleanly converted into fosfomycin and the ketone product in a 1:1 ratio. When 40 and 43 are hydrolyzed by hydrochloric acid and given to Fom4, the (R)-precursor is converted into the ketone analog as shown in Figure 10B, while the (S)-precursor is converted into fosfomycin as shown in Figure 10C. Our results agree with a previous report by Wang et al.,16 indicating that the conditions of acid hydrolysis do not affect the stereochemistry of the HPP product. It should be noted that the 31P-NMR spectra are proton-coupled; therefore, the phosphorous peak in fosfomycin is split into a doublet of doublets by the two adjacent hydrogens. In the ketone analog, the phosphorous peak is split by two equivalent hydrogens into a triplet. As shown in Figure 10D, when HPP-CMP produced by Fom3 is hydrolyzed and given to Fom4, a fosfomycin-identical peak is observed, with no trace of the ketone analog. Therefore, we can conclude that Fom3 methylates HEP-CMP in an enantioselective manner, giving (S)-HPP-CMP as the overwhelmingly predominant, if not the exclusive, product.
Figure 10.

31P-NMR of Fom4 products using a) racemic HPP; b) (R)-HPP; c) (S)-HPP; d) HPP derived from methylated product of Fom3. The peaks at 4.5 ppm were sodium phosphate as internal standard. The peak in d at 3.5 ppm refers to cytidine monophosphate, the co-product of acid hydrolysis. Full spectra are provided in supplementary information.
Use of HEP-CMP Analogs to Probe the Mechanism of Fom3
In the prevailing mechanism for cobalamin-dependent RS methylases that methylate sp3-hybridized carbon centers, the radical formed upon hydrogen-atom abstraction from the substrate is presumed to perform a nucleophilic attack onto the methyl group of MeCbl, initiating a homolytic cleavage of the cobalt–carbon bond to give the methylated product and cob(II)alamin. Upon reduction of cob(II)alamin, the resulting cob(I)alamin attacks SAM to regenerate MeCbl (Figure 11). In the Fom3 reaction, however, an alternate mechanism can be envisaged, whereby MeCbl undergoes a heterolytic cleavage. In this mechanism, the substrate radical undergoes deprotonation of the hydroxyl group to form a ketyl radical anion. Subsequently, the C2” carbanion can attack MeCbl nucleophilically, which would undergo heterolytic cleavage to generate the cob(I)alamin form of the cofactor. The resulting alkoxy radical could then be reduced and reprotonated to generate the HPP-CMP product (Figure 11).
Figure 11.

Possible mechanisms for Fom3 catalyzed methylation of HEP-CMP.
To distinguish between these two mechanisms, we synthesized and used the following HEP-CMP analogs in Fom3 reactions, each of which lacks a hydroxyl group adjacent to C2”: cytidylyl-ethylphosphonate (EP-CMP), cytidylyl-propylphosphonate (PP-CMP), cytidylyl-2-methoxyethylphosphonate(MEP-CMP), cytidylyl-thioethylphosphonate (TEP-CMP) and cytidylyl-fluoroethylphosphonate (FEP-CMP) (Figure 13A). If Fom3 can use any of the analogs as a substrate, save (TEP-CMP), then a heterolytic cleavage mechanism could be ruled out, because the proton on the hydroxyl group that is removed during catalysis is no longer present. The five analogs, together with HEP-CMP as positive control, were incubated with Fom3 under turnover conditions using either SAM or d3-SAM and Ti(III)citrate as the required low-potential reductant, and LC-MS was used to analyze product formation. In reactions using HEP-CMP, EP-CMP, FEP-CMP and TEP-CMP, m/z values of 444.1, 428.1, 446.1 and 460.1 are observed after 60 min of incubation, corresponding to the methylated product of those substrates, respectively (Black traces in Figure 12A, B, C and D, respectively). In parallel reactions using d3-SAM, product peaks shift to m/z values of 447.1, 431.1, 449.1 and 463.1 respectively (Red traces in Figure 12A, B, C and D, respectively), indicating that three SAM-derived deuterium atoms are incorporated into each product. The PP-HEP and MEP-CMP analogs did not support detectable formation of the corresponding methylated product.
Figure 13.

a) Chemical structures of HEP-CMP analogs used to probe the mechanism of Fom3; b) Progress curves for SAH formation in Fom3 reactions; d) Progress curves for 5’-dA formation in Fom3 reactions. The Fom3 reactions contained 20 μM Fom3, 1 mM SAM and 1 mM Ti(III)citrate. The red, blue, black and green dots represent HEP-CMP, TEP-CMP, FEP-CMP and EP-CMP. No product formation was observed using PP-CMP and MEP-CMP as substrates.
Figure 12.

MS analysis of Fom3 reaction using HEP-CMP (a), EP-CMP (b), FEP-CMP (c) and TEP-CMP(d). The assay was performed with 20 μM Fom3 and 1 mM Ti(III)Citrate for 2 h with SAM (black traces) or d3-SAM (red traces).
The formation of SAH and 5-dA was also quantified in each reaction to gain insight into the rates of product formation, given that SAH and the methylated product are typically produced in equivalent amounts. As shown in Figures 13B and 13C, HEP-CMP is turned over with a kcat of ~1 min−1, while, TEP-CMP is turned over with a slightly diminished kcat of ~0.5 min 1. EP-CMP and FEP-CMP were indeed turned over, but at very low rates. The higher turnover rate with FEP-CMP than with EP-CMP is most likely due to the stabilizing effect of the fluorine atom adjacent to the alkyl radical produced upon hydrogen atom abstraction.43–45 Therefore, it appears that the ability of Fom3 to act upon various substrate analogs depends both on steric and or electronic effects. Nevertheless, given that both FEP-CMP and EP-CMP are viable substrates for Fom3, the heterolytic pathway is unlikely, suggesting that the substrate radical produced upon hydrogen atom abstraction most likely directly attacks the methyl group of MeCbl, initiating a homolytic cleavage of the cobalt bond to give the methylated product and cob(II)alamin.
DISCUSSION
In this work, we performed spectroscopic and detailed mechanistic studies on a robust preparation of Fom3. EPR and Mössbauer spectroscopies were used to investigate the enzyme’s Fe/S cluster, confirming its [4Fe-4S] configuration. Interestingly, we note that the unusual partial resolution of the low-energy line of the quadrupole doublets associated with the [4Fe-4S] cluster of the cobalamin-dependent RS enzyme TsrM is not observed in the spectrum of Fom3. The addition of SAM results in a change to the Mössbauer spectrum that is consistent with a site-differentiated [4Fe-4S] cluster. This behavior is similar to that observed for most RS enzymes, but distinctly different from that of TsrM, which is the only RS enzyme found to date that does not catalyze a reductive cleavage of SAM. Our results suggest that the unique properties of the [4Fe-4S] cluster of TsrM are not representative of all cobalamin-dependent RS enzymes and may reflect the fact that TsrM is an outlier in the RS superfamily. Consistent with our observations of SAM binding in Fom3, when HEP-CMP is added to the enzyme, no change to the Mössbauer spectrum is observed, suggesting that SAM, rather than HEP-CMP, binds to the Fe/S cluster.
We also achieved considerable progress with respect to the catalytic activity of Fom3. In the groundbreaking study by Sato et al., it was reported that Fom3 from S. wedmorensis is only active in the presence of MV/NADPH and that no detectable amount of product is observed when the in vivo flv/flr/NADPH reducing system is used as the required low-potential reductant. Herein, we report that Fom3 from S. fradiae, overproduced and isolated as described in Materials and Methods, is more than two-fold more active in the presence of flv/flr/NADPH as compared to in the presence of MV/NADPH, which is ~five-fold more active than the preparation by Sato et al.10 Interestingly, when we use Ti(III)citrate as the low-potential reductant, the activity of Fom3 increases dramatically, affording a turnover number as high as 2.3 min−1, which is the highest of any cobalamin-dependent RS enzyme that catalyzes a reductive cleavage of SAM.
One of the most stunning conclusions from the study by Sato et al. was that Fom3 methylated HEP-CMP with a lack of stereoselectivity, giving rise to a racemic mixture of HPP-CMP. Given that enzymes rarely catalyze nonstereoselective reactions, we developed synthetic and biochemical methods to reinvestigate this aspect of the Fom3 reaction. Using HEP-CMP that is stereoselectively deuterated at C2′′, we find that the 5’-dA• abstracts the C2′′-proR hydrogen atom of the substrate. The methylated product, however, exhibits a 1H NMR spectrum containing only a single doublet signal. Surprisingly, a chemically generated racemic mixture of HPP-CMP also exhibits the same 1H NMR spectrum. To determine the stereoselectivity of methylation, the HPP-CMP product was cleaved by treatment with acid, and the resulting HPP was incubated with Fom4, which had been shown previously to catalyze the production of fosfomycin from (S)-HPP, but the production of (2-oxopropyl)phosphonate from (R)-HPP. Our data clearly show that Fom3 only produces (S)-HPP-CMP; (R)-HPP-CMP is not observed within our limits of detection.
The stereochemical course of the cobalamin-dependent RS enzymes GenD1 and MoeK5 can be directly deduced from the structures of their substrates and products, because methylation occurs at stereogenic centers.46, 47 In both cases, the stereochemical outcomes are inversion of configuration. Moreover, the stereochemistry of methylation in the biosynthesis of bottromycin and gentamicin has been experimentally determined. In those systems, inversion of configuration was observed after hydrogen abstraction. Our investigation of the stereochemical course of the Fom3 reaction, as well as the recent work by Schweifer and Hammerschmidt,48 agree with these findings, suggesting that in the structures of these enzymes, MeCbl and the RS machinery are placed on the opposite sides of the carbon to be methylated. The only known crystal structure of a cobalamin-dependent RS enzyme, OxsB, though not an RS methylase, is consistent with this hypothesis.49
During our studies, we also functionally annotated FomD, which had been suggested to hydrolyze HPP-CMP to HPP and CMP (Figure 1).10 FomD from S. fradiae catalyzed a relatively rapid hydrolysis of about 50% of a racemic mixture of (S)- and (R)-HPP-CMP, with a much slower rate of hydrolysis of the remaining 50%. This behavior suggests that FomD prefers one stereoisomer over the other, which is most likely (S)-HPP-CMP, given that only this stereoisomer gives rise to fosfomycin.
Throughout the course of this study, we developed a number of methods for the chemical syntheses of stereoselectively labeled substrates and products, which we leveraged to generate substrate analogs that might allow us to delve deeper into the Fom3 mechanism. The simplest mechanism for cobalamin-dependent RS methylases involves the abstraction of a hydrogen atom from the carbon center undergoing methylation to afford a carbon-centered radical that attacks the methyl group of MeCbl to generate the methylated product and cob(II)alamin. This mechanism is very likely operative in the reactions of cobalamin-dependent RS methylases like CysS, PoyC, and ThnK, which are all capable of methylating carbons that have no heteroatom substituents. However, alternative mechanisms can be proposed for Fom3 and GenK, which operate on substrates containing α-hydroxy groups. For example, hydrogen-atom abstraction could take place from the hydroxyl group rather than from the carbon center undergoing methylation. This step could yield a sufficiently acidic carbon species that could undergo deprotonation to yield a ketyl radical anion (Figure 11), in which the carbanion resonance form attacks the methyl group of MeCbl to generate cob(I)alamin. Reduction of the remaining alkoxy radical would then yield the final product. A second mechanism involves abstraction of a hydrogen atom from the carbon center undergoing methylation with subsequent loss of a proton from the hydroxyl group to yield the same ketyl radical anion (Figure 11). Our use of site-specifically deuterated HEP-CMP as a substrate clearly rules out hydrogen atom abstraction from the hydroxyl group of the substrate; however, a ketyl radical anion species originating from hydrogen atom abstraction from the target carbon center is still a viable possibility. We therefore generated a series of substrate analogs (cytidylyl-ethylphosphonate, cytidylyl-propylphosphonate, cytidylyl-2-methoxyethylphosphonate, cytidylyl-2-fluoroethylphosphonate), all of which lack an α-hydroxy group. The ethyl and fluoroethyl analogs can be methylated on their C-2” position by Fom3, but the rates at which they are turned over are much lower than that of the natural substrate. We attribute these lower rates to substrate orientation in the active site (i.e., loss of hydrogen bonding by removal of the –OH group) as well as to a significantly higher HBDE associated with cleaving the target C–H bond. It is noteworthy that the fluoroethyl-containing analog supports a significantly greater rate of turnover than the ethyl-containing analog, because the HBDE associated with generating the α-fluoroalkyl radical is lower by several kcal/mol. Product formation was not observed with the methoxy- or propyl-containing analogs, which we attribute to steric issues. The similar activities of Fom3 with HEP-CMP and TEP-CMP support the premise of hydrogen bonding in the active site and radical stabilization for efficient generation of the substrate radical in the Fom3 reaction. In summary, our results suggest that Fom3 catalyzes its reaction by abstracting the C2”-pro-R hydrogen atom of HEP-CMP with a subsequent direct substrate radical attack on MeCbl (homolytic mechanism in Figure 11) to afford 2”-(S)-HPP-CMP. In closing, we add that two papers were recently published on the stereochemical course of the reaction of Fom3 from Streptomyces wedmorensis while this paper was in review. In both instances, the C2”-pro-R hydrogen atom of HEP-CMP was found to be abstracted, and the methyl group of MeCbl was found to be transferred to afford only 2”-(S)-HPP-CMP, supporting our conclusions with S. fradiae Fom3.11, 12
Supplementary Material
ACKNOWLEDGMENT
The authors thank the Huck Institute for Life Sciences Genomics Core for DNA sequencing.
Funding Sources
This work was supported by NIH grants GM-122595 and AI-111419 to S. J. B and GM127079 to C.K. S.J.B. is an investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS
- 2-HEP
2-hydroxyethylphosphonate\
- 2-HPP
2-hydroxypropylphosphonate
- 5’-dA
5’-deoxyadenosine
- 5-dA•
5’-deoxyadenosyl 5’-radical
- AdoCbl
adenosylcobalamin
- DTT
dithiothreitol
- EPR
electron paramagnetic resonance
- Fe/S
iron sulfur
- HEP-CMP
cytidylyl-2-hydroxyethylphosphonate
- HPP-CMP
cytidyl-2-hydroxypropylphosphonate
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- MeCbl
methylcobalamin
- MV
methylviologen
- OHCbl
hydroxocobalamin
- RS
radical SAM
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- Trp
tryptophan
Footnotes
ASSOCIATED CONTENT
Supporting Information
Figures S1 – Figures S8, sequence of codon-optimized fomD gene, and synthetic procedures are available free of charge on the ACS Publications website at DOI: http://pubs.acs.org.
REFERENCES
- [1].Falagas ME, Vouloumanou EK, Samonis G, and Vardakas KZ (2016) Fosfomycin, Clin. Microbiol. Rev 29, 321–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Falagas ME, Giannopoulou KP, Kokolakis GN, and Rafailidis PI (2008) Fosfomycin: use beyond urinary tract and gastrointestinal infections, Clin. Infect. Dis 46, 1069–1077. [DOI] [PubMed] [Google Scholar]
- [3].Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, Wolf FJ, Miller TW, Chaiet L, Kahan FM, Foltz EL, Woodruff HB, Mata JM, Hernandez S, and Mochales S (1969) Phosphonomycin, a new antibiotic produced by strains of streptomyces, Science 166, 122–123. [DOI] [PubMed] [Google Scholar]
- [4].Institute of Medicine Forum on Microbial, T. (2010) The National Academies Collection: Reports funded by National Institutes of Health, In Antibiotic Resistance: Implications for Global Health and Novel Intervention Strategies: Workshop Summary, National Academies Press (US) National Academy of Sciences., Washington (DC). [PubMed] [Google Scholar]
- [5].Hidaka T, Goda M, Kuzuyama T, Takei N, Hidaka M, and Seto H (1995) Cloning and nucleotide sequence of fosfomycin biosynthetic genes of Streptomyces wedmorensis, Mol. Gen. Genet 249, 274–280. [DOI] [PubMed] [Google Scholar]
- [6].Kuzuyama T, Hidaka T, Imai S, and Seto H (1993) Studies on the biosynthesis of fosfomycin. V. Cloning of genes for fosfomycin biosynthesis, J. Antibiot 46, 1478–1480. [DOI] [PubMed] [Google Scholar]
- [7].Woodyer RD, Shao Z, Thomas PM, Kelleher NL, Blodgett JA, Metcalf WW, van der Donk WA, and Zhao H (2006) Heterologous production of fosfomycin and identification of the minimal biosynthetic gene cluster, Chem. Biol 13, 1171–1182. [DOI] [PubMed] [Google Scholar]
- [8].Kim SY, Ju KS, Metcalf WW, Evans BS, Kuzuyama T, and van der Donk WA (2012) Different biosynthetic pathways to fosfomycin in Pseudomonas syringae and Streptomyces species, Antimicrob. Agents Chemother 56, 4175–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cho S-H, Kim S-Y, Tomita T, Shiraishi T, Park J-S, Sato S, Kudo F, Eguchi T, Funa N, Nishiyama M, and Kuzuyama T (2017) Fosfomycin biosynthesis via transient cytidylylation of 2-hydroxyethylphosphonate by the bifunctional Fom1 enzyme, ACS Chem. Biol 12, 2209–2215. [DOI] [PubMed] [Google Scholar]
- [10].Sato S, Kudo F, Kim S-Y, Kuzuyama T, and Eguchi T (2017) Methylcobalamin-dependent radical SAM C-methyltransferase Fom3 recognizes cytidylyl-2-hydroxyethylphosphonate and catalyzes the nonstereoselective C-methylation in fosfomycin biosynthesis, Biochemistry 56, 3519–3522. [DOI] [PubMed] [Google Scholar]
- [11].McLaughlin MI, and van der Donk WA (2018) Stereospecific radical-mediated B12-dependent methyl transfer by the fosfomycin biosynthesis enzyme Fom3, Biochemistry, 10.1021/acs.biochem.8b00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sato S, Kudo F, Kuzuyama T, Hammerschmidt F, and Eguchi T (2018) C-Methylation catalyzed by Fom3, a cobalamin-dependent radical S-adenosyl-L-methionine enzyme in fosfomycin biosynthesis, proceeds with inversion of configuration, Biochemistry, doi: 10.1021/acs.biochem.8b00614. [DOI] [PubMed] [Google Scholar]
- [13].Sato S, Miyanaga A, Kim S-Y, Kuzuyama T, Kudo F, and Eguchi T (2018) Biochemical and structural analysis of FomD that catalyzes the hydrolysis of cytidylyl (S)-2-hydroxypropylphosphonate in fosfomycin biosynthesis, Biochemistry, 10.1021/acs.biochem.8b00690. [DOI] [PubMed] [Google Scholar]
- [14].Olivares P, Ulrich EC, Chekan JR, van der Donk WA, and Nair SK (2017) Characterization of two late-stage enzymes involved in fosfomycin biosynthesis in Pseudomonads, ACS Chem. Biol 12, 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].van der Donk WA (2006) Rings, Radicals, and regeneration: the early years of a bioorganic laboratory, J. Org. Chem 71, 9561–9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang C, Chang WC, Guo Y, Huang H, Peck SC, Pandelia ME, Lin GM, Liu HW, Krebs C, and Bollinger JM Jr. (2013) Evidence that the fosfomycin-producing epoxidase, HppE, is a non-heme-iron peroxidase, Science 342, 991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Allen KD, and Wang SC (2014) Initial characterization of Fom3 from Streptomyces wedmorensis: the methyltransferase in fosfomycin biosynthesis, Arch. Biochem. Biophys 543, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lanz N, Blaszczyk AJ, McCarthy E, Wang B, Wang R, Jones B, and Booker SJ (2018) Enhanced solubilization of class B radical S-adenosylmethionine methylases by improved cobalamin uptake in Escherichia coli, Biochemistry 57, 1475–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Iwig DF, and Booker SJ (2004) Insight into the polar reactivity of the onium chalcogen analogues of S-adenosyl-L-methionine, Biochemistry 43, 13496–13509. [DOI] [PubMed] [Google Scholar]
- [20].Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu-Gogonea C, Souder MG, Tu L, and Booker SJ (2004) Lipoyl synthase requires two equivalents of S-adenosyl-L-methionine to synthesize one equivalent of lipoic acid, Biochemistry 43, 6378–6386. [DOI] [PubMed] [Google Scholar]
- [21].Blaszczyk AJ, Silakov A, Zhang B, Maiocco SJ, Lanz ND, Kelly WL, Elliott SJ, Krebs C, and Booker SJ (2016) Spectroscopic and electrochemical characterization of the iron-sulfur and cobalamin cofactors of TsrM, an unusual radical S-adenosylmethionine methylase, J. Am. Chem. Soc 138, 3416–3426. [DOI] [PubMed] [Google Scholar]
- [22].Blaszczyk AJ, Wang B, Silakov A, Ho JV, and Booker SJ (2017) Efficient methylation of C2 in L-tryptophan by the cobalamin-dependent radical S-adenosylmethionine methylase TsrM requires an unmodified N1 amine, J. Biol. Chem 292, 15456–15467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4, Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- [24].Blaszczyk AJ, Wang RX, and Booker SJ (2017) TsrM as a model for purifying and characterizing cobalamin-dependent radical S-adenosylmethionine Methylases, Methods Enzymol. 595, 303–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- [26].Ljungdahl LG, LeGall J, and Lee JP (1973) Isolation of a protein containing tightly bound 5-methoxybenzimidazolylcobamide (factor 3m) from Clostridium thermoaceticum, Biochemistry 12, 1802–1808. [DOI] [PubMed] [Google Scholar]
- [27].Beinert H (1978) Micro methods for the quantitative determination of iron and copper in biological material, Methods Enzymol. 54, 435–445. [DOI] [PubMed] [Google Scholar]
- [28].Beinert H (1983) Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins, Anal. Biochem 131, 373–378. [DOI] [PubMed] [Google Scholar]
- [29].Zehnder AJ, and Wuhrmann K (1976) Titanium (III) citrate as a nontoxic oxidation-reduction buffering system for the culture of obligate anaerobes, Science 194, 1165–1166. [DOI] [PubMed] [Google Scholar]
- [30].EWWIN. Software Package, S. S. S. 2012.
- [31].Allen KD, and Wang SC (2014) Initial characterization of Fom3 from Streptomyces wedmorensis: The methyltransferase in fosfomycin biosynthesis, Arch. Biochem. and Biophys. 543, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Marous DR, Lloyd EP, Buller AR, Moshos KA, Grove TL, Blaszczyk AJ, Booker SJ, and Townsend CA (2015) Consecutive radical S-adenosylmethionine methylations form the ethyl side chain in thienamycin biosynthesis, Proc. Natl. Acad. Sci 112, 10354–10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Parent A, Guillot A, Benjdia A, Chartier G, Leprince J, and Berteau O (2016) The B12-Radical SAM enzyme PoyC catalyzes valine Cβ-methylation during polytheonamide biosynthesis, J. Am. Chem. Soc 138, 15515–15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim HJ, Liu Y. n., McCarty RM, and Liu H. w. (2017) Reaction catalyzed by GenK, a cobalamin-dependent radical S-adenosyl-L-methionine methyltransferase in the biosynthetic pathway of gentamicin, proceeds with retention of configuration, J. Am. Chem. Soc 139, 16084–16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang Y, Schnell B, Baumann S, Müller R, and Begley TP (2017) Biosynthesis of branched alkoxy groups: iterative methyl group alkylation by a cobalamin-dependent radical SAM enzyme, J. Am. Chem. Soc 139, 1742–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Guo M, Sulc F, Ribbe MW, Farmer PJ, and Burgess BK (2002) Direct assessment of the reduction potential of the [4Fe−4S]1+/0 couple of the Fe protein from Azotobacter vinelandii, J. Am. Chem. Soc 124, 12100–12101. [DOI] [PubMed] [Google Scholar]
- [37].Parthasarathy A, Stich TA, Lohner ST, Lesnefsky A, Britt RD, and Spormann AM (2015) Biochemical and EPR-spectroscopic investigation into heterologously expressed vinyl chloride reductive dehalogenase (VcrA) from Dehalococcoides mccartyi Strain VS, J. Am. Chem. Soc 137, 3525–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pandelia ME, Lanz ND, Booker SJ, and Krebs C (2015) Mossbauer spectroscopy of Fe/S proteins, Biochim. Biophys. Acta 1853, 1395–1405. [DOI] [PubMed] [Google Scholar]
- [39].Kent TA, Emptage MH, Merkle H, Kennedy MC, Beinert H, and Munck E (1985) Mossbauer studies of aconitase. Substrate and inhibitor binding, reaction intermediates, and hyperfine interactions of reduced 3Fe and 4Fe clusters, J. Biol. Chem 260, 6871–6881. [PubMed] [Google Scholar]
- [40].Molle T, Clemancey M, Latour JM, Kathirvelu V, Sicoli G, Forouhar F, Mulliez E, Gambarelli S, and Atta M (2016) Unanticipated coordination of tris buffer to the radical SAM cluster of the RimO methylthiotransferase, J. Biol. Inorg. Chem. 21, 549–557. [DOI] [PubMed] [Google Scholar]
- [41].Hammerschmidt F (1988) Biosynthese von Naturstoffen mit einer PC-Bindung. III. Synthese der (R)- und (S)-(2-Amino[1-D1]ethyl)phosphonsäure und deren Hydroxylierung zu (2-Amino-1-hydroxyethyl)phosphonsäure in Acanthamoeba castellanii (Neff), Vol. 1988. [Google Scholar]
- [42].Hammerschmidt F, and Kaehlig H (1991) Biosynthesis of natural products with a phosphorus-carbon bond. 7. Synthesis of [1,1–2H2]-, [2,2–2H2]-, (R)- and (S)-[1–2H1](2-hydroxyethyl)phosphonic acid and (R,S)-[1–2H1](1,2-dihydroxyethyl)phosphonic acid and incorporation studies into fosfomycin in Streptomyces fradiae, J. Org. Chem 56, 2364–2370. [Google Scholar]
- [43].Dolbier WR (1996) Structure, reactivity, and chemistry of fluoroalkyl radicals, Chem. Rev 96, 1557–1584. [DOI] [PubMed] [Google Scholar]
- [44].Ni C, and Hu J (2016) The unique fluorine effects in organic reactions: recent facts and insights into fluoroalkylations, Chem. Soc. Rev. 45, 5441–5454. [DOI] [PubMed] [Google Scholar]
- [45].Zhang X-M (1998) Radical substituent effects of α-fluorine and α-trifluoromethyl groups, J. Org. Chem 63, 3590–3594. [Google Scholar]
- [46].Huang C, Huang F, Moison E, Guo J, Jian X, Duan X, Deng Z, Leadlay PF, and Sun Y (2015) Delineating the biosynthesis of gentamicin x2, the common precursor of the gentamicin C antibiotic complex, Chem. Biol 22, 251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ostash B, Doud EH, Lin C, Ostash I, Perlstein DL, Fuse S, Wolpert M, Kahne D, and Walker S (2009) Complete characterization of the seventeen step moenomycin biosynthetic pathway, Biochemistry 48, 8830–8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schweifer A, and Hammerschmidt F (2018) Stereochemical course of methyl transfer by cobalamin-dependent radical SAM methyltransferase in fosfomycin biosynthesis, Biochemistry 57, 2069–2073. [DOI] [PubMed] [Google Scholar]
- [49].Bridwell-Rabb J, Zhong A, Sun HG, Drennan CL, and Liu HW (2017) A B12-dependent radical SAM enzyme involved in oxetanocin A biosynthesis, Nature 544, 322–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.