

Abstract
Despite the relevance for understanding structure-function relationships, robust prediction of proton donors and nucleophiles in enzyme active sites remains challenging. Here we tested three types of state-of-the-art computational methods to calculate the pKa’s of the buried and hydrogen bonded catalytic dyads in five enzymes. We asked the question what determines the pKa order, i.e., what makes a residue proton donor vs. nucleophile. The continuous constant pH molecular dynamics simulations captured the experimental pKa orders and revealed that the negative nucleophile is stabilized by increased hydrogen bonding and solvent exposure as compared to the proton donor. Surprisingly, this simple trend is not apparent from crystal structures and the static structure-based calculations. While the generality of the findings awaits further testing via a larger set of data, they underscores the role of dynamics in bridging enzyme structures and functions.
Catalytic functions of enzymes are often carried out by titratable groups that can act as proton donors (acids) and nucleophiles (bases).1 Thus, accurate prediction of the relative order of active-site pKa’s helps establish the structure-function relationships.1–3 Over the past two decades, tremendous progress has been made in the development of computational methods for pKa calculations;4–7 however, reliable identification of proton donor and nucleophile remains challenging. This is because most enzyme active sites are deeply buried, giving rise to large pKa shifts relative to the solution (or model) pKa’s, and site-specific protonation/deprotonation can be highly coupled to each other due to hydrogen bonding and electrostatics. Accurate calculation of these balancing forces is non-trivial.6,8,9 Structure-based calculations are further complicated by the ionization induced conformational reorganization and water penetration which are not readily accounted for.10 Current empirical methods lack of rules for establishing the pKa order for coupled sites.
Commonly used computational approaches for pKa predictions can be grouped into three classes:6 methods based on empirical functions, macroscopic methods based on solving the Poisson-Boltzmann (PB) equation, and microscopic methods based on constant pH molecular dynamics (MD). A popular empirical method PropKa estimates the contributions to pKa shifts using structure-based energy calculations for desolvation, hydrogen bonding and Coulomb interactions.11,12 In the PB-based methods, electrostatic potential is computed using the continuum model, in which the protein is considered as a low dielectric cavity embedded in a high dielectric medium. Popular tools include the MCCE program13 and DelPhiPKa which employs a smooth Gaussian dielectric function.14 Several web servers are also available, for example, APBS-PDB2PQR,15,16 H++,17 and DelPhiPKa.18 Unlike the empirical and PB-based macroscopic methods, constant pH MD determines pKa’s using the Henderson-Hasselbalch equation, where the ratio of the protonated and deprotonated states is estimated by simulations at different pH.19–25 Thus, in addition to pKa values, constant pH MD also reports on the details of pH-dependent conformational dynamics.
Recently, we became interested in the pH-dependent catalysis and inhibition of the aspartic protease BACE1 β-secretase 1 (BACE1), which is a major drug target for Alzheimer’s disease.26 Correct assignment of the proton donor (higher pKa) and nucleophile (lower pKa) is a pre-requisite for studying pH-dependent enzyme dynamics and its relationship to substrate and inhibitor binding.26,27 In this work, we asked the question, what is the molecular determinant of the microscopic pKa order of the aspartyl dyad in BACE1 and two close homologs, β-secretase 2 (BACE2) and cathepsin D (CatD)? In all three structures, the catalytic dyad is completely buried and hydrogen bonded to each other. As comparison and control, we also examined two classic enzymes with abundant experimental data, hen egg white lysozyme (HEWL) and staphylococcal nuclease (SNase). In these enzymes the catalytic residues are partially buried, and in one of them (HEWL), the dyad carboxylates are spatially separated (titration is not coupled). We tested the empirical PropKa,11,12 the PB solver APBS,15,16 and the hybrid-solvent22 as well as the all-atom23 continuous constant pH MD (CpHMD) methods. CpHMD simulations reproduced the experimental pKa orders, and revealed that the negative nucleophile is more stabilized by hydrogen bonding and solvent exposure as compared to the proton donor. Surprisingly, this simple trend is not apparent from the crystal structures (and therefore the static structure-based calculations).
Five enzymes are considered here (Fig. 1), BACE1, BACE2, CatD, HEWL and SNase. BACE1 is an aspartyl protease which catalyzes the β-site cleavage of the amyloid precursor protein.28 Kinetic experiments gave the pKa’s of 5.2 and 3.5 for Asp32 and Asp228, which act as the general acid and base, respectively.29 BACE2 and CatD are two close homologs of BACE1 with the respective catalytic dyads Asp48/Asp241 and Asp33/Asp231. HEWL is a glycoside hydrolase that catalyzes the hydrolysis of the glycosidic bond in polysaccharides comprising the cell wall.30 The catalytic dyad is comprised of Glu35 (proton donor) and Asp52 (nucleophile), which have NMR-derived pKa of 6.1 and 3.6, respectively.31 SNase is a phosphodiesterase which catalyzes the cleavage of the phosphodiester bond in DNA and RNA hydrolysis.32 The active site of SNase involves Asp19, Asp21, Asp40 and Glu43.32 Here we focus on the hydrogen bonded residues Asp19 and Asp21, with the respective NMR-derived pKa’s of 2.2 and 6.5.33 For simplicity in discussion, we will refer to Asp19 with the lower pKa as the nucleophile component of the “dyad”.
Figure 1.
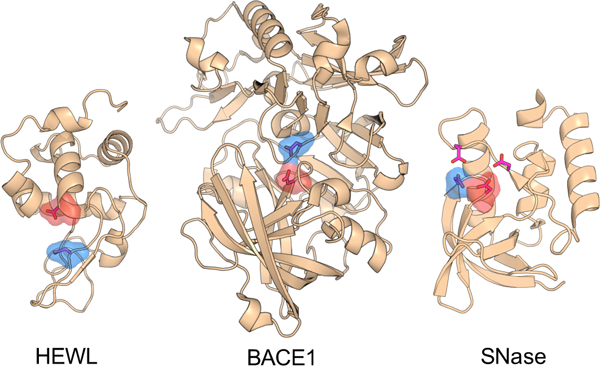
Crystal structures of HEWL (PDB ID 2LZT), BACE1 (PDB ID 1SGZ) and SNase (PDB ID 3BDC). The structures of BACE2 and CatD are very similar to BACE1 and not shown here. The active-site residues are represented by the stick model. The catalytic dyads in HEWL and BACE1 as well as the two hydrogen bonded catalytic residues in SNase are highlighted by surface rendering. The proton donor and nucleophile are colored blue and red, respectively.
To calculate the catalytic pKa’s, we employed the empirical method PropKa,11,12 the PB solver APBS,15,16 and the hybrid-solvent22 as well as all-atom CpHMD.23 The hybrid-solvent CpHMD is the most validated CpHMD method,7 and makes use of explicit solvent for accurate conformational sampling and generalized Born (GB) model for rapid calculation of solvation forces affecting protonation states. The recently developed all-atom CpHMD with particle mesh Ewald (PME) for long-range electrostatics23 and titratable water for charge neutrality36,37 is most rigorous but requires much longer simulation time23 (see later discussion). Thus, it was not applied to BACE2 and CatD.
Table 1 lists the calculated pKa’s of the five enzymes using the empirical, PB and CpHMD methods in comparison to experimental data. The catalytic pKa’s of BACE1/BACE2/CatD are most challenging to predict, as the dyads are completely buried and hydrogen bonded to each other (i.e. coupled). Since PropKa cannot determine the order of coupled residues, it offers two alternative sets of pKa’s. As to the PB results, the two pKa’s are too similar to be assigned to the proton donor and nucleophile. In contrast, both CpHMD methods reproduce the experimental pKa orders. Note, in the absence of experimental data for BACE2, the pKa order is expected to be the same as the homologous BACE1. We also note that, the CpHMD titration data was fit to the Hill equation (Eq. 1) and the two-proton model (Eq. 2). The former yields the microscopic site-specific pKa’s, while the latter yields the macroscopic stepwise pKa’s, which can be directly compared to experiment. If the titration of two residues is highly coupled, i.e., involving shared protons, the two site-specific pKa’s are similar, while the stepwise pKa’s are split (see an example in our recent study of a salt-bridge triad38). This is however not the case for the enzymes studied here. The site-specific and stepwise pKa’s are nearly identical for HEWL and SNase, and are only slightly different for BACE1/BACE2/CatD (up to 0.3 pH units). This suggests that, despite hydrogen bonding between the dyad, titration can be assigned to a specific residue, consistent with the NMR experiments.31,33
Table 1.
Experimental and calculated pKa’s of the catalytic residues in BACE1/BACE2/CatD, HEWL and SNasea
| Expt | PropKa | APBS | Hybr | All-atom | |
|---|---|---|---|---|---|
| BACE1 | |||||
| Asp32 | 5.2 | 8.0/4.4 | 5.5/5.4 | 4.1/4.2 | 3.7/4.1 |
| Asp228 | 3.5 | 4.3/7.9 | 5.2/5.1 | 1.9/1.8 | −0.4/−0.6 |
| BACE2 | |||||
| Asp48 | n/d | 6.9/5.2 | 6.3/5.6 | 3.5/3.7 | n/d |
| Asp241 | n/d | 5.2/6.9 | 6.2/5.7 | 2.2/1.9 | n/d |
| CatD | |||||
| Asp33 | >5 | 8.4/4.6 | 10.5/6.2 | 4.4/4.7 | n/d |
| Asp231 | 4.1 | 3.1/8.3 | 11.9/6.6 | 3.2/2.9 | n/d |
| HEWL | |||||
| Glu35 | 6.1 | 6.5 | 4.9/3.5 | 5.9/5.9 | 7.7/7.8 |
| Asp52 | 3.6 | 3.9 | −0.3/2.3 | 3.7/3.7 | 5.7/5.6 |
| SNase | |||||
| Asp19 | 2.2 | 4.2/2.9 | 7.5/5.0 | 1.6/1.5 | 3.5/3.3 |
| Asp21 | 6.5 | 2.3/3.7 | 7.1/4.7 | 4.3/4.4 | 5.8/6.0 |
The nucleophile (lower experimental pKa or by homology to BACE1) is highlighted in italics. The experimental pKa’s of HEWL and SNase are taken from NMR data,31,33 while those of BACE1 and CatD are inferred from kinetic experiments.29,34 The same structures with missing residues added were used in all calculations. The empirical calculations (PropKa 3.111,12 ) returned two sets of pKa’s for the coupled dyads in BACE1/BACE2/CatD and SNase. For the APBS calculations (version 1.4.115,16), two pKa’s are listed corresponding to the use of an effective protein dielectric constant of 4 or 20. The van der Waals surface based on the CHARMM radii was used. For the CpHMD simulations, the first pKa corresponds to the microscopic residue-specific pKa, while the second one corresponds to the macroscopic stepwise pKa. The hybrid-solvent CpHMD data for BACE1 and CatD were taken from our previous work.26,35
Although the correct pKa orders are predicted by the CpHMD methods, there are deviations between the calculated and experimental pKa’s. For the hybrid-solvent method, both dyad pKa’s in BACE1 series and SNase are too low by up to 1.5 pH units, as the major source of error is the underestimation of desolvation penalty 22,26 by the GBSW implicit-solvent model.39 For the PME-based allatom CpHMD, however, the pKa error can be either positive or negative due to the balance of two opposing factors. The desolvation penalty is too high, which overly upshifts the pKa’s of buried carboxylic groups, while the attractive electrostatics (e.g., hydrogen bonding) in the interior is too strong, which overly downshifts the pKa’s of these groups. Both factors are related to the lack of polarizability in the protein interior represented by the additive force field (dielectric constant is too low40). These errors are also exacerbated by limited sampling; estimation of free energies in explicit solvent converges much slower than in the GB solvent. Asp228 of BACE1 shows the largest pKa error, as it is subject to the strongest hydrogen bonding in the largest protein among all.
We turn to HEWL, which is the “easiest” case among the test set of enzymes, as the two carboxyl groups are spatially well separated (by about 7 Å in the crystal structure) and only partially buried. All four methods correctly predicted the experimental pKa order of the catalytic dyad. In fact, a correct pKa order was also predicted by other PB methods such as MCCE13 and DelPhiPKa.14 As to the hydrogen bonded and partially buried active-site residues in SNase, the four methods perform similarly as for BACE1 homologs. PropKa cannot assign the pKa order; PB method give very similar pKa’s; and the two CpHMD methods correctly predict the pKa order. Taken together, these data suggests that CpHMD can reliably differentiate the proton donor and nucleophile components of coupled carboxyl dyads.
The distinct performance of CpHMD prompted us to investigate the physical origin of the pKa order. We considered hydrogen bonding and desolvation, which are the major contributors to the pKa shifts of the catalytic residues relative to the model values. Coulomb interactions with other residues are not considered here, as they are negligible for the carboxyl dyads in BACE1 series,26,35 which is typical for buried and coupled catalytic dyads. Since catalytic dyads are subject to a similar degree of hydrophobic burial, we first examined the hydrogen bond patterns of dyad residues in the five enzymes. Surprisingly, in all five enzymes, the calculated occupancy of hydrogen bonds formed by the nucleophile carboxylate in the titration pH range is always larger than the proton donor, and its pH dependence is well correlated with the pH profile of deprotonation (Fig. 2, Fig. S8 and S9). The hybrid-solvent CpHMD results (Fig. 2) are in agreement with the all-atom CpHMD results (Fig. S6). Interestingly, the carboxyl sidechains are always hydrogen bond acceptors, which suggests that hydrogen bonding stabilizes the deprotonated form and explains the match between the pH profiles of carboxylate hydrogen bonding and deprotonation (Fig. 2, compare curves in the left and right panels).
Figure 2. Deprotonated state of the nucleophile is stabilized by hydrogen bonding.
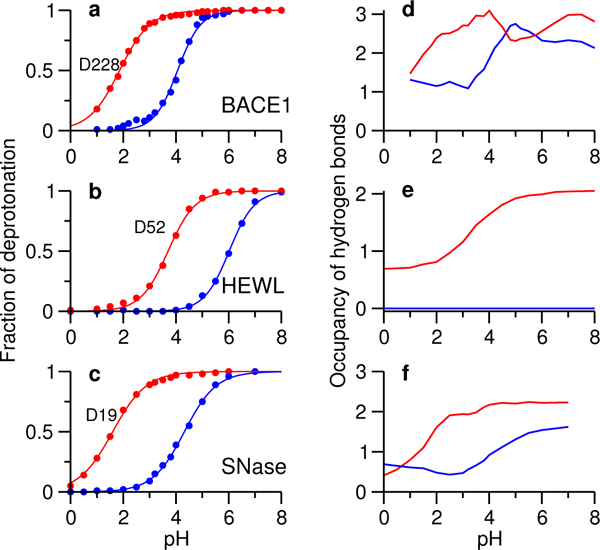
The fraction of deprotonation at different pH for Asp32/Asp228 in BACE1 (a), Glu35/Asp52 in HEWL (b), and Asp19/Asp21 in SNase (c) in the hybrid-solvent CpHMD simulations. Total occupancy of hydrogen bonds formed by the proton donor (blue) and nucleophile (red) carboxylates in BACE1 (d), HEWL (e) and SNase (f) at different simulation pH.
We were curious as to whether the difference in hydrogen bonding between the two carboxyl groups is present in the crystal structures. In BACE1 and its homologs BACE2 and CatD, the aspartyl dyad are hydrogen bonded to each other. Additionally, the BACE1 proton donor Asp32 forms hydrogen bonds with Gly34 and Ser35, while the nucleophile Asp228 forms hydrogen bonds with Gly230 and Thr231. PropKa calculation showed that these hydrogen bonds (two for each carboxyl group) make nearly identical contributions to the pKa down shifts: −1.58 for Asp32 and −1.53 for Asp228. The hydrogen bond patterns in the homologous BACE2 and CatD are very similar to BACE1. The proton donor forms hydrogen bonds with a serine and a glycine, while the nucleophile forms hydrogen bonds with a threonine and a glycine. PropKa calculation gave identical contributions (about −1.4 units) to the pKa down shifts. The hydrogen bond environment of the carboxyl dyads in the CpHMD simulations is similar to the crystal structures, except that the catalytic nucleophile can form (accept) a hydrogen bond with not only the sidechain but also the backbone of the aforementioned threonine: Thr231 in BACE1 (Fig. 3d), Thr244 in BACE2 (Fig. S8), and Thr234 in CatD (Fig. S9). Interestingly, the occupancies of the carboxylate–threonine hydrogen bonds increase with pH (Fig. 3a and Fig. S8 and S9), consistent the pH profiles of the total occupancy of hydrogen bonds. The all-atom CpHMD simulations confirmed these trends (Fig. S6).
Figure 3. pH-dependent hydrogen bonding of the nucleophiles in HEWL, BACE1 and SNase.
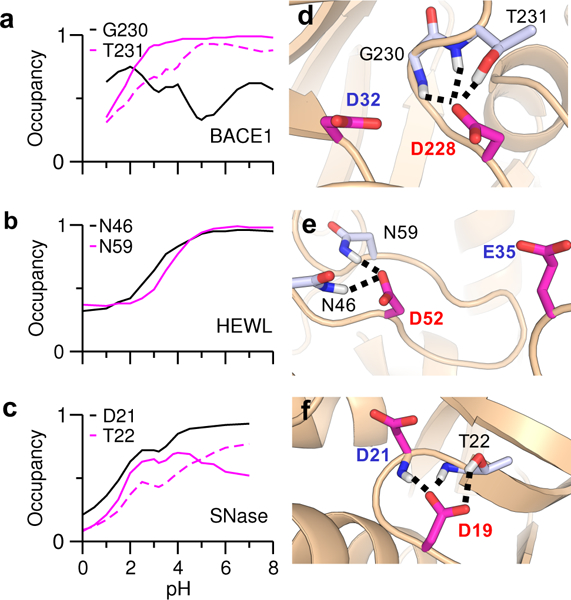
Occupancy of individual hydrogen bonds at different simulation pH for the dyad nucleophiles, Asp228 in BACE1 (a), Asp52 in HEWL (b), and Asp 19 in SNase (c) in the hybrid-solvent CpHMD simulations. The carboxylate–backbone hydrogen bond not present in the crystal structure is indicated by the dashed lines. The residues forming hydrogen bonds with the dyad nucleophile are indicated. Representative snapshots showing the hydrogen bonds formed by the nucleophile in BACE1 (d), HEWL (e), and SNase (f). The proton donor and nucleophiles are labeled in blue and red, respectively.
In the crystal structure of HEWL, the proton donor Glu35 forms a hydrogen bond with Ala110, while the nucleophile Asp52 forms hydrogen bonds with Asn44, Asn46 and Asn59. Based on these hydrogen bonds, PropKa calculation gave the pKa shifts of −0.6 for Glu35 and −1.92 for Asp52. These results are largely in agreement with the CpHMD simulations, although in the simulations, Glu35 does not form any hydrogen bond, while Asp52 forms hydrogen bonds with Asn46 and Asn59 only (Fig. 3b and e, Fig. S7). The occupancies of the two hydrogen bonds increase with pH, matching the profile of Asp52 deprotonation. Consistently, hydrogen bonding is the major energetic factor that lowers the pKa of Asp52 in the PropKa calculation. This agreement is likely the reason for the same pKa order calculated by PropKa and CpHMD simulations.
In the crystal structure of SNase, Asp19 and Asp21 are hydrogen bonded to each other. The nucleophile Asp19 forms a hydrogen bond with the sidechain of Thr22, while Asp21 forms hydrogen bond with both the sidechain and backbone of Thr41. These hydrogen bonds result in similar pKa shifts for the dyad residues (−1.33 for Asp19 and −1.4 for Asp21). These hydrogen bonds are present in the CpHMD simulations; however, Asp19 can form an additional hydrogen bond with the backbone of Thr22 in the simulations (Fig. 3c and f, Fig. S7). Considering the five enzymes together, we can see that for a coupled carboxyl dyad, crystal structure shows no hydrogen bonding difference, while CpHMD simulation reveals one additional hydrogen bond that stabilizes the charged nucleophile. For a non-coupled carboxyl dyad, both crystal structure and simulation show more hydrogen bonding for the charged nucleophile. Thus, we hypothesize that hydrogen bonding is the molecular determinant for the pKa order of the carboxyl dyads.
Desolvation is another major contributor to the pKa shifts of catalytic dyads. In contrast to hydrogen bonding, which stabilizes the charged form and hence shifts the carboxylate pKa down, desolvation favors the neutral state and shifts the carboxylate pKa up. We previously mentioned that it is expected that the dyad residues are buried to a similar degree. We test whether it is indeed the case for the five enzymes. For BACE1, BACE2 and CatD, the PropKa calculations gave 100% burial for the carboxyl dyads, which is consistent with the CpHMD simulations showing below 10% fractional solvent accessible surface area. (SASA). Interestingly, in the titration pH range of the nucleophile, it is slightly more exposed to solvent than the proton donor for all three enzymes (Fig. 4a and Fig. S10). For HEWL, the PropKa calculation showed that Glu35 and Asp52 are 66%) and 50%) buried, respectively. Consistently, CpHMD simulations showed about 20% fractional SASA values for the two carboxylates, and in the titration pH range of the nucleophile Asp52, its solvent exposure is slightly higher than the proton donor Glu35 (Fig. 4b). For SNase, the PropKa. calculation gave 60%) and 86%) burial for the coupled carboxylates Aspl9 and Asp21, respectively, which is in agreement with the CpHMD simulations which showed 20–25%) and 10%) fractional SASA values for the two residues, respectively (Fig. 4c). The same trends are seen in the all-atom CpHMD simulations (Fig. S11). Taken together, the above analysis suggests that the catalytic dyads have similar solvent exposure based on both CpHMD simulations and crystal structures; however, the nucleophile is always slightly more exposed than the proton donor during dynamics even if they are both 100%) buried in the crystal structure.
Figure 4. Solvent exposure of the carboxyl dyads in HEWL, BACE1 and SNase.
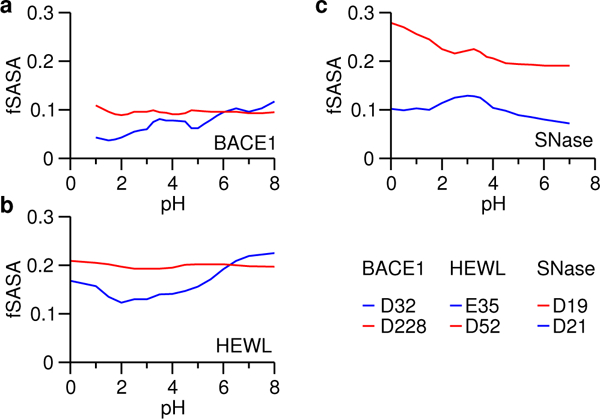
Fractional SASA (fSASA) refers to the solvent accessible surface area of the carboxy late oxygens in the protein environment relative to that in solution. Data for the nucleophiles are shown in red.
In summary, CpHMD simulations recapitulated the experimental pKa orders of coupled and buried carboxyl dyads in enzyme active sites, which presents a challenge for the commonly used empirical calculations and PB electrostatic calculations. Intriguingly, CpHMD data revealed that the negative nucleophile is stabilized by a higher occupancy of hydrogen bonds than the proton donor in both spatially separated and hydrogen bonded (coupled) carboxyl dyads. For the latter, the higher occupancy is captured in the CpHMD simulations but not PropKa calculations based on the crystal structures. Additionally, simulations showed a consistently, slightly higher solvent exposure for the negative nucleophile than the proton donor even if the dyad is 100%) buried according to the crystal structure. Although these findings are initially surprising, increased hydrogen bonding and solvent exposure stabilize the charged state, consistent with the lower pKa of the nucleophile. Increased solvent exposure also makes it easier for the general base (one form of nucleophile) to engage in the deprotonation of a nearby water, which initiates the catalytic process. While the generality of the findings emerged from the study awaits further testing using a larger set of data, they demonstrate how adding dynamics helps advance our understanding of the structure-function relationships. As such, molecular dynamics based pKa prediction approaches hold a unique potential.
Methods and Protocols.
Five enzymes, BACE1 (PDB ID 1SGZ), BACE2 (PDB ID 3ZKQ), CatD (PDB ID ILYA), HEWL (PDB ID 2LZT) and the hyperstable Δ+PHS SNase (PDB ID 3BDC) were studied in this work. While all five proteins were subject to the hybrid-solvent CpHMD titration simulations,22 three of them, BACE1, HEWL and SNase were subject to the PME-based a.ll-atom CpHMD simulations.23 The carboxyl dyad pKa’s were also calculated by the empirical PropKa method (version 3.1)11,12 and the PB method APBS (version 1.4.1)15,16 based on the same crystal structures with missing residues added.
All CpHMD simulations were carried out using the CHAR.MM package (version c36a.6).41 CHAR.MM22/CMAP force field was used to represent proteins.42,43 and the CHAR.MM modified TIP3P model41 was used to represent water. pH replica-exchange protocol was used in all CpHMD simulations.22 In the hybrid-solvent CpHMD titration of BACE1, BACE2 and CatD, the simulations lasted 20, 20 and 30 ns per replica, corresponding to an aggregate sampling time of 480 ns, 400 ns, 720 ns, respectively. For the smaller HEWL and SNase, 5 ns per replica was used, corresponding to an aggregate sampling time of 80 ns. In the all-atom PME CpHMD titration of BACE1, HEWL and SNase, 30 ns per replica was used, corresponding to an aggregate sampling time of 720 ns, 700 ns, and 660 ns, respectively.
To verify structural stability at different pH, the backbone root-mean-square deviation (RMSD) from the starting structure was calculated (Fig. S1–2 for HEWL and SNase; pH-dependent conformational dynamics of BACE1 was discussed previously26). As expected, the backbone RMSD for the hyper-stable SNase was about 1 Å with a small fluctuation throughout the pH range, while for HEWL, the RMSD remained below 2 Å with larger fluctuation at pH below 2. The latter is likely a result of the acid-induced structural change, consistent with the simulations using Monte-Carlo based constant pH method.44 To verify sampling convergence, time series of the pH-dependent unprotonated fractions of the dyads and the pKa values were calculated (Fig. S3–S5). We note that pH replica exchange enhances the sampling of both protonation and conformational states, allowing a significant reduction of the convergence time per replica (by at least one order of magnitude in our experience) relative to the single pH constant pH simulations. 22
The fraction of unprotonated states was calculated using the following definition: λ < 0.1 for the protonated state and λ > 0.9 for the deprotonated state. The microscopic site-specific pKa’s were calculated by fitting the fraction of unprotonated states S at different pH to the modified Hill equation,
| (1) |
where n is the Hill coefficient. We also calculated the macroscopic stepwise pKa’s by fitting the total number of bound protons to the dyad, Nprot, to the following model based on statistical mechanics,33,36,45,46
| (2) |
where pK1 and pK2 are the two macroscopic pKa’s, and the denominator represents the partition function.
Supplementary Material
Acknowledgement
Financial support is provided by National Institutes of Health (GM098818 and GM118772).
Footnotes
Supporting Information Available
Supporting Information contains detailed computational protocols and supplementary figures.
References
- (1).Harris TK; Turner GJ Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life 2002, 53, 85–98. [DOI] [PubMed] [Google Scholar]
- (2).Ondrechen MJ; Clifton JG; Ringe D THEMATICS: A simple computational predictor of enzyme function from structure. Proc. Natl. Acad. Sci. USA 2001, 98, 12473–12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nielsen JE; Mccammon JA Calculating pKa values in enzyme active sites. Protein Sci. 2003, 12, 1894–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Khandogin J In Multi-scale quantum models for biocatalysis; York DM, Lee T-S, Eds.; Springer: New York, 2009; Chapter 10. Modeling protonation equilibria in biological macro-molecules, pp 261–284. [Google Scholar]
- (5).Wallace JA; Shen JK Predicting pKa values with continuous constant pH molecular dynamics. Methods Enzymol. 2009, 466, 455–475. [DOI] [PubMed] [Google Scholar]
- (6).Alexov E; Mehler EL; Baker N; Baptista AM; Huang Y; Milletti F; Nielsen JE; Farrell D; Carstensen T; Olsson MHM; Shen JK; Warwicker J; Williams S; Word JM Progress in the prediction of pKa values in proteins. Proteins 2011, 79, 3260–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chen W; Morrow BH; Shi C; Shen JK Recent development and application of constant pH molecular dynamics. Mol. Simul. 2014, 40, 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Schutz CN; Warshel A What are the dielectric constants of proteins and how to validate electrostatic models? Proteins 2001, 44, 400–417. [DOI] [PubMed] [Google Scholar]
- (9).Gunner MR; Zhu X; Klein MC MCCE analysis of the pKas of introduced buried acids and bases in staphylococcal nuclease. Proteins 2011, 79, 3306–3319. [DOI] [PubMed] [Google Scholar]
- (10).Harms MJ; Castañeda CA; Schlessman JL; Sue GR; Isom DG; Cannon BR; García-Moreno E, The B pKa values of acidic and basic residues buried at the same internal location in a protein are governed by different factors. J. Mol. Biol. 2009, 389, 34–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Li H; Robertson AD; Jensen JH Very fast empirical prediction and rationalization of protein pKa values. Proteins 2005, 61, 704–721. [DOI] [PubMed] [Google Scholar]
- (12).Søndergaard CR; Mats HM Olsson MR; Jensen JH Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- (13).Georgescu RE; Alexov EG; Gunner MR Combining conformational flexibility and continuum electrostatics for calculating pKas in proteins. Biophys. J. 2002, 83, 1731–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang L; Li L; Alexov E pKa predictions for proteins, RNAs, and DNAs with the Gaussian dielectric function using DelPhi pKa. Proteins 2015, 86, 2186–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Baker NA; Sept D; Joseph S; Holst MJ; McCammon JA Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dolinsky TJ; Nielsen JE; McCammon JA; Baker NA PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Anandakrishnan R; Aguilar B; Onufriev AV H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang L; Zhang M; Alexov E DelPhiPKa web server: predicting pKa of proteins, RNAs and DNAs. Bioinformatics 2016, 32, 614–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Baptista AM; Martel PJ; Petersen SB Simulation of protein conformational freedom as a function of pH: constant-pH molecular dynamics using implicit titration. Proteins 1997, 27, 523–544. [PubMed] [Google Scholar]
- (20).Mongan J; Case DA; McCammon JA Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem. 2004, 25, 2038–2048. [DOI] [PubMed] [Google Scholar]
- (21).Khandogin J; Brooks III, Constant CL pH molecular dynamics with proton tautomerism. Biophys. J. 2005, 89, 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wallace JA; Shen JK Continuous constant pH molecular dynamics in explicit solvent with pH-based replica exchange. J. Chem. Theory Comput. 2011, 7, 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Huang Y; Chen W; Wallace JA; Shen J All-Atom Continuous Constant pH Molecular Dynamics With Particle Mesh Ewald and Titratable Water. J. Chem. Theory Comput. 2016, 12, 5411–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Swails JM; York DM; Roitberg AE Constant pH Replica Exchange Molecular Dynamics in Explicit Solvent Using Discrete Protonation States: Implementation, Testing, and Validation. J. Chem. Theory Comput. 2014, 10, 1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Goh GB; Hulbert BS; Zhou H; Brooks III, Constant CL pH molecular dynamics of proteins in explicit solvent with proton tautomerism. Proteins 2014, 82, 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ellis CR; Shen J pH-Dependent Population Shift Regulates BACE1 Activity and Inhibition. J. Am. Chem. Soc. 2015, 137, 9543–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ellis CR; Tsai C-C; Hou X; Shen J Constant pH Molecular Dynamics Reveals pH-Modulated Binding of Two Small-Molecule BACE1 Inhibitors. J. Phys. Chem. Lett. 2016, 7, 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shimizu H; Tosaki A; Kaneko K; Hisano T; Sakurai T; Nukina N Crystal structure of an active form of BACE1, an enzyme responsible for amyloid β protein production. Mol. Cell. Biol. 2008, 28, 3663–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Toulokhonova L; Metzler WJ; Witmer MR; Copeland RA; Marcinkeviciene J Kinetic studies on β-site amyloid precursor protein-cleaving enzyme (BACE). J. Biol. Chem. 2003, 278, 4582–4589. [DOI] [PubMed] [Google Scholar]
- (30).Vocadlo DJ; Davies GJ; Laine R; Withers SG Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature 2001, 412, 835–838. [DOI] [PubMed] [Google Scholar]
- (31).Webb H; Tynan-Connolly BM; Lee GM; Farrell D; O’Meara F; Søndergaard CR; Teilum K; Hewage C; McIn-tosh LP; Nielsen JE Remeasuring HEWL pKa values by NMR spectroscopy: methods, analysis, accuracy, and implications for theoretical pKa calculations. Proteins 2011, 79, 685–702. [DOI] [PubMed] [Google Scholar]
- (32).Cotton FA; Hazen EE Jr.; Legg MJ Staphylococcal nuclease: Proposed mechanism of action based on structure of enzyme–thymidine 3’,5’-bisphosphate–calcium ion complex at 1.5-Å resolution. Proc. Natl. Acad. Sci. USA 1979, 76, 2551–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Castañeda CA; Fitch CA; Majumdar A; Khangulov V; Schlessman JL; García-Moreno E, Molecular B determinants of the pKa values of Asp and Glu residues in staphylococcal nuclease. Proteins 2009, 77, 570–588. [DOI] [PubMed] [Google Scholar]
- (34).Goldfarb NE; Lam MT; Bose AK; Patel AM; Duck-worth AJ; Dunn BM Electrostatic Switches That Mediate the pH-Dependent Conformational Change of “Short” Re-combinant Human Pseudocathepsin D. Biochemistry 2005, 44, 15725–15733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ellis CR; Tsai C-C; Lin F-Y; Shen J Conformational dy-namics of cathepsin D and binding to a small-molecule BACE1 inhibitor. J. Comput. Chem. 2017, 38, 1260–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wallace JA; Shen JK Charge-leveling and proper treatment of long-range electrostatics in all-atom molecular dynamics at constant pH. J. Chem. Phys. 2012, 137, 184105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chen W; Wallace J; Yue Z; Shen J Introducing titratable water to all-atom molecular dynamics at constant pH. Biophys. J. 2013, 105, L15–L17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yue Z; Chen W; Zgurskaya HI; Shen J Constant pH Molecular Dynamics Reveals How Proton Release Drives the Conformational Transition of a Transmembrane Efflux Pump. J. Chem. Theory Comput. 2017, 13, 6405–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Im W; Lee MS; Brooks III, Generalized CL Born model with a simple smoothing function. J. Comput. Chem. 2003, 24, 1691–1702. [DOI] [PubMed] [Google Scholar]
- (40).Lemkul JA; Huang J; Roux B; MacKerell AD Jr. An Empirical Polarizable Force Field Based on the Classical Drude Oscillator Model: Development History and Recent Applications. Chem. Rev. 2016, 116, 4983–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Brooks BR et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).MacKerell AD Jr. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (43).MacKerell AD Jr.; Feig M; Brooks CL III Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- (44).Machuqueiro M; Baptista AM Is the prediction of pKa values by constant-pH molecular dynamics being hindered by inherited problems? Proteins 2011, 79, 3437–3447. [DOI] [PubMed] [Google Scholar]
- (45).Baptista AM; Teixeira VH; Soares CM Constant-pH molecular dynamics using stochastic titration. J. Chem. Phys. 2002, 117, 4184–4200. [Google Scholar]
- (46).Ullmann GM Relations between protonation constants and titration curves in polyprotic acids: a critical view. J. Phys. Chem. B 2003, 107, 1263–1271. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.