

Abstract
Systemic lupus erythematosus (SLE) is a complex disease characterized by numerous autoantibodies and clinical involvement in multiple organ systems. The immunological events triggering the onset and progression of clinical manifestations have not yet been fully defined, but a central role for B cells in the pathogenesis has been brought to the fore in the last several years. The breakdown of B cell tolerance is likely a defining and early event in the disease process and may occur by multiple pathways, including alterations in factors that affect B cell activation thresholds, B cell longevity, and apoptotic cell processing. Both antibody dependent and independent mechanisms of B cells are important in SLE. Thus, autoantibodies contribute to autoimmunity by multiple mechanisms including immune-complex mediated Type III hypersensitivity reactions, type II antibody-dependent cytotoxicity, and by instructing innate immune cells to produce pathogenic cytokines including IFNα, TNF and IL-1. Recent data have highlighted the critical role of toll-like receptors as a link between the innate and adaptive immune system in SLE immunopathogenesis. Given the large body of evidence implicating abnormalities in the B cell compartment in SLE, there has been a therapeutic focus on developing interventions that target the B cell compartment. A number of different approaches to targeting B cells have been utilized including B cell depletion with monoclonal antibodies against B cell-specific molecules, induction of negative signaling in B cells, and blocking B cell survival and activation factors. Overall, therapies targeting B cells are beginning to show promise in the treatment of SLE and continue to elucidate the diverse roles of B cells in this complex disease.
Keywords: B lymphocytes, systemic lupus erythematosus, anti-CD20, rituximab, belimumab, atacicept
Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune disease with considerable heterogeneity in clinical manifestations and disease course, characterized by pathogenic autoantibody formation, immune complex deposition, and end organ damage. The mortality and morbidity of patients with SLE has significantly improved during the last few decades. In the 1950s, the four-year survival rate for lupus was approximately 50%, while more recent series estimate 5 year and 10 year survival rates of 96 and 85% respectively (1). Despite these improvements, SLE continues to be associated with significant morbidity and a three- to five-fold increased mortality compared to the general population. Moreover, there are a group of SLE patients who continue to suffer from aggressive disease that does not respond to conventional treatments.
The continuing need for more effective therapies with less toxic side effects has led to the interest in targeted biologic therapies for severely affected patients who are refractory to or can not tolerate traditional therapies. However, major obstacles in finding efficacious therapies for SLE include the challenges of clinical trial design given the low prevalence of disease, great clinical heterogeneity, relapsing-remitting course, and lack of well-established endpoints (2–4). These challenges have contributed to the fact that there have been no new drugs approved for the treatment of SLE in over 50 years. Despite these difficulties there is reason for optimism, including concerted efforts toward improving lupus clinical trial methodology (4) that have recently led to a successful outcome in two clinical trials of a B cell targeted biologic in SLE. Moreover, our understanding about the pathogenesis of SLE has grown substantially in the past decade, leading to an explosion of promising biologic therapies. Here we will review the state-of-the-art pathophysiology of SLE providing the rationale for B cell targeted therapies, followed by a critical evaluation of the efficacy and safety of B cell depletion with anti-CD20 monoclonal antibody therapy in the management of the disease, and other promising B cell targeted approaches.
Role of B cells in systemic lupus erythematosus
Although multiple immunologic abnormalities are important for the development and clinical expression of SLE, a large body of evidence strongly points to the B cell as a critical player in the pathogenesis of this disease (5).
B cell tolerance loss
As SLE is characterized by the generation of large amounts of autoantibodies directed against chromatin and a variety of other self-antigens the loss of B cell tolerance is believed to play a key role in the disease. Evidence that the breakdown of B cell tolerance likely occurs very early in SLE and may precede or trigger other immune abnormalities is provided by the demonstration that SLE patients express ANAs several years before the onset of clinical disease. The lag time observed between the appearance of ANAs and clinical expression of SLE may be explained by the need for epitope spreading and generation of increasingly pathogenic autoantibodies (6).
It is notable that numerous single gene defects affecting the B cell compartment can lead to lupus-like disease in murine models, and many of these defects share a common endpoint, the loss of B cell tolerance. At least three broad categories of defects that can lead to a lupus-like phenotype have been defined in the mouse and are instructive for thinking about B cell abnormalities in SLE. These defects may affect (1) B cell activation thresholds (e.g. FcR), (2) B cell longevity (e.g. BAFF transgenics), or (3) apoptotic cell/autoantigen processing (e.g. mer knock-out). Although many alterations in B cell signaling or co-stimulatory molecules that may alter (1) and/or (2) have been shown to lead to a lupus-like phenotype in the mouse, their relevance for human SLE and even spontaneous murine models of SLE is not well defined. Mohan and colleagues have recently elucidated the mechanism by which the Sle1 susceptibility locus derived from lupus-prone New Zealand Mixed (NZM2410) mice contributes to the development of autoimmunity. A gene within this locus encoding a member of the SLAM (signaling lymphocyte activation molecule) family was found to be highly expressed in immature B cells and altered in these lupus-prone mice in such a way as to impair signaling and impede antigen driven negative selection (B cell deletion, receptor revision, and anergy induction) (7). Interestingly, this suggests that some of the genes that contribute to lupus may function by down-regulating BCR signaling at the immature stage and impairing B cell tolerance.
In contrast, other B cell signaling defects may cause up-regulated signaling, as exemplified by loss of inhibitory Fc receptor function. Thus, FcγRIIb deficiency leads to a lupus-like phenotype in mice, although with different penetrance depending on mouse strain (8). Moreover, deficiency of the inhibitory FcγRIIb reduces the threshold for autoreactive B cell activation (9), and restoration of proper FcγRIIb expression on B cells in lupus prone mice prevents the expansion and accumulation of autoantibody producing plasma cells (10). These findings are even more meaningful given that polymorphisms in FcγRIIb are associated with human SLE and have a direct functional consequence on B cell signaling (11, 12). Thus, a FcγRIIb membrane spanning Ile232 to Thr232 substitution associated with lupus in Asian populations caused decreased FcR lipid raft association and greater, more sustained calcium mobilization and downstream biochemical signaling events upon B cell receptor engagement (11). Moreover, other defects may lead to decreased expression of FcgRIIb as recently elegantly demonstrated in human lupus memory B cells (12). Such alterations in B cell signaling proteins could explain earlier observations of increased calcium responses to BCR ligation in SLE B cells (13). Crow and colleagues recently took an interesting approach to this question in human lupus by comparing the gene expression signatures specifically in memory B cells and found the under-expression of a number of inhibitory receptors including CD22 and CD72, as well as the over-expression of type I interferon inducible genes (14). Overall, such defects in B cell signaling pathways are important because they may contribute to the loss of peripheral B cell tolerance in SLE.
Alterations in B cell longevity also can lead to lupus like phenotypes, as exemplified by transgenic expression of BAFF (B cell activator of the TNF family), a key cytokine that promotes B cell survival. These mice develop a lupus-like phenotype with excessive numbers of mature B cells, spontaneous germinal center (GC) reactions, autoantibodies, high plasma cell (PC) numbers, and immunoglobulin (Ig) deposition in the kidney (15). Moreover, lupus-prone mice have elevated levels of circulating BAFF, and administration of soluble BAFF receptor ameliorates disease progression and improves survival (16). The importance of BAFF in human SLE has been demonstrated by the finding of elevated serum levels in SLE patients and the correlation with serum IgG and autoantibody levels (17). Excessive BAFF may be another factor that promotes the survival of autoreactive B cells in the periphery (18, 19). Thus, two recent reports have shown that BAFF is a key limiting resource for early (transitional) B cell survival, with autoreactive B cells surviving selection into the mature pool only when BAFF was in excess (18, 19). Cancro and colleague have put forth an appealing model of B cell selection and tolerance induction that postulates that the number of cells entering the transitional B cell pool must exceed the available space if appropriately stringent negative selection is to occur. Events that compromise this rule, including a decrease in incoming cell numbers or increases in BAFF, may circumvent negative selection of autoreactivity (20) (Figure 1).
Figure 1. A model for B cell development, selection, and function after B cell depletion therapy.
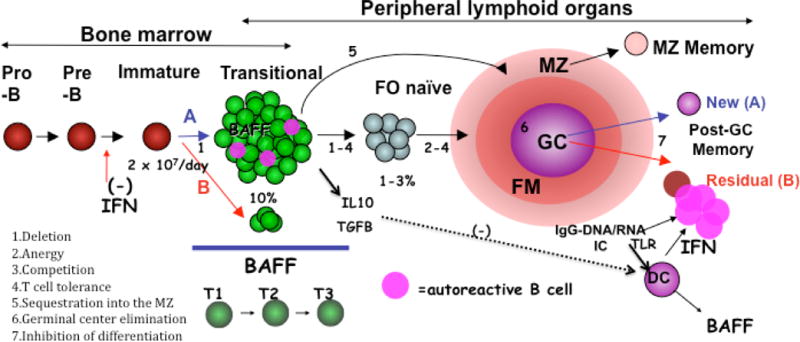
The outcome of BCD is going to depend on how well established autoimmunity is eradicated and how the immune system reconstitutes. B cells are continually generated in the bone marrow, and once rituximab is cleared can develop through well recognized stages (T=transitional, FO=follicular naïve, FM=follicular mantle, MZ=marginal zone, GC=germinal center) with defined tolerance check points as shown. Autoreactive B cells (depicted in pink) are deleted in the BM immature subsets, and cells in the transitional subsets undergo further selection, the stringency of which is determined in part by available BAFF (BAFF excess due to overproduction, peripheral lymphopenia, or reduced BM output reduces selective stringency). A favorable reconstitution profile (denoted A) will be characterized by an abundance of newly emerging transitional B cells in an environment that favors stringent negative selection of autoreactivity (high numbers of transitional B cells relative to BAFF). A non-favorable reconstitution profile (B in red) will be characterized by a higher fraction of residual memory B cells induced by an environment of TLR activation (via DNA and RNA containing immune complex activation of plasmacytoid dendritic cells), yielding large quantities of interferon and inhibition of new BM B cell lymphopoiesis. The outcome of BCD will also depend on the balance between protective (regulatory, anti-inflammatory) B cells and pathogenic (effector, pro-inflammatory) B cells and their corresponding cytokines. We postulate that physiologically, transitional cells predominantly produce IL10 (or TGFB) which in a normal environment exerts anti-inflammatory actions. This situation would be altered in autoimmune disease. Finally, BCDT may restore the physiological balance between protective and pathogenic B cell functions by creating an environment dominated by transitional cells with anti-inflammatory and tolerogenic functions and Treg inducing activity.
The impaired clearance of apoptotic debris may also lead to SLE and may do so in part by providing large amounts of self antigen and immune complexes that deliver stimulatory signals to autoreactive B cells. Several publications in recent years indicate that such apoptotic blebs and immune complexes contain ligands for Toll-like receptors (TLR), including RNA or DNA, which can provide costimulatory signals for autoreactive B cells (21–24), as further described below.
Abnormalities in the B cell compartment in human SLE
Recent work has demonstrated the role of the breakdown of peripheral B cell tolerance mechanisms in human SLE (25, 26). Thus, Sanz and colleagues have shown that an important tolerance checkpoint operates in healthy subjects to censor autoreactive (9G4) B cells in the mature naïve compartment thereby preventing the expansion of these cells into the memory compartment (27), a checkpoint recently corroborated by others (28) and further shown by us to be faulty in SLE (26). Other work by Nussenzweig and colleagues has shown that 50–75% of newly produced human B cells are autoreactive and must be silenced by tolerance mechanisms (29). Key checkpoints to censor autoreactive B cell clones occur at the immature B cell stage in the bone marrow (BM) and between new transitional emigrants and mature B cells in the periphery. Notably, in this system up to 20% of mature naïve B cells were self-reactive, indicating the need for additional censoring mechanisms at this stage as suggested by Sanz and colleagues and recently shown to be the case in the transition to the IgM memory compartment (28). Nussenzweig has also studied a small number of SLE patients (n=3) and found the transitional B cell checkpoint to be defective although with some heterogeneity, with 41–50% of antibodies from the mature naïve compartment retaining autoreactivity with HEp-2 cell lysates. This data is intriguing, although the precise point of tolerance breakdown, the variability in this finding in different patient subsets, and the cause of this tolerance checkpoint defect remain to be defined.
A large body of additional evidence indicates that B cells are abnormal in human SLE, with defects ranging from increased calcium flux on signaling through the BCR to high or aberrant expression of costimulatory molecules such as CD80, CD86, and CD40 ligand (30). B cell alterations in SLE peripheral blood appear to be more prominent and fluid than in other autoimmune diseases. A range of abnormalities in B cell homeostasis in SLE have been observed, including a naïve B cell lymphopenia, expansion of peripheral blood plasma cells, increased transitional B cells, and expansion of activated memory B cells subsets (31–34). We surmise that this reflects the systemic nature of the autoimmune process with frequent cycles of activation, differentiation, and traffic between secondary lymphoid organs and target tissues. Of note, the frequency and absolute number of plasma cells in peripheral blood has correlated with disease activity and anti-dsDNA titers (35). More recent studies have begun to refine subsets of plasma cells, including a HLADR high fraction, that may better delineate plasmablasts enriched for autoreactivity and correlating with disease activity (36). Similarly, our group has elucidated the heterogeneity that exists in human B cell memory and find that active SLE patients have an expansion of effector memory B cell populations positive for markers of homing to sites of systemic inflammation (CXCR3+) and negative for lymphoid homing markers (CD62L−) (33, 37). Other groups have found a subset of CD27-IgD-CD95+ memory B cells with an activated phenotype that correlated with disease activity and serologic abnormality (34). Abnormal expansion of peripheral blood B cells with a pre-GC phenotype has also been reported and proposed to indicate exuberant or abnormally regulated GG reactions in SLE (38). However, in many cases these cells may actually represent immature transitional B cells as opposed to pre-GC cells (39).
Overall, the frequency of these diverse B cell subset abnormalities, their association with each other and other immunologic abnormalities, and relationship with disease activity and disease subsets remain to be better elucidated in larger longitudinal cohorts of SLE patients. It is noteworthy, however, that a number of the B cell targeted therapies have been shown to reverse at least some of these peripheral B cell abnormalities. For example, we have shown that CD20 targeted B cell depletion normalizes naïve lymphopenia, effector memory B cell expansion (CD27-IgD- double negative B cells), the presence of plasma cell precursors, and expansion of autoreactive memory B cell populations (32). In other studies, BAFF blockade has also been shown to alter peripheral B cell homeostasis in SLE with decreases in transitional B cells, naïve B cells, and plasmablasts, but with less significant effects on the memory B cell compartment at least at early time points (40).
The diverse role of B cells in the initiation and propagation of autoimmunity
B cells may participate in the immune dysregulation of SLE at multiple levels by: (1) serving as the precursors of antibody-secreting cells, (2) taking up and presenting autoantigens to T cells, (3) helping to regulate and organize inflammatory responses through cytokine and chemokine secretion (such as interleukin-10, interleukin-6, interferon-γ, and lymphotoxin-α), and (4) regulating other immune cells (5) (Figure 2). The importance of these latter functions has been demonstrated in murine SLE, where B cells have been found to be critical to the development of disease even when they are unable to secrete autoantibodies. Thus, genetically B cell deficient JH knockout MRL/lpr lupus prone mice have strikingly attenuated disease, with the expected absence of autoantibodies but also the surprising lack of T cell activation (41). Shlomchik and colleagues used a novel approach to further elucidate the role of B cells in SLE by generating MRL/lpr mice that express a mutant transgene encoding surface Ig that cannot be secreted. These mice have B cells, but no circulating Ig, yet develop T cell activation and nephritis (5). This landmark study was the first to highlight that B cells can play a pathogenic role in lupus independent of serum autoantibody and has been recently supported by our observations that B cell depletion with anti-CD20 has robust effects in the treatment of murine lupus without changes in autoantibodies (42). This notion has also been corroborated in humans by our own and others findings that clinical improvement in SLE patients treated with rituximab correlates with B cell depletion and precedes by several months any decline in serum levels of relevant autoantibodies (43).
Figure 2. The diverse role of B cells in SLE.
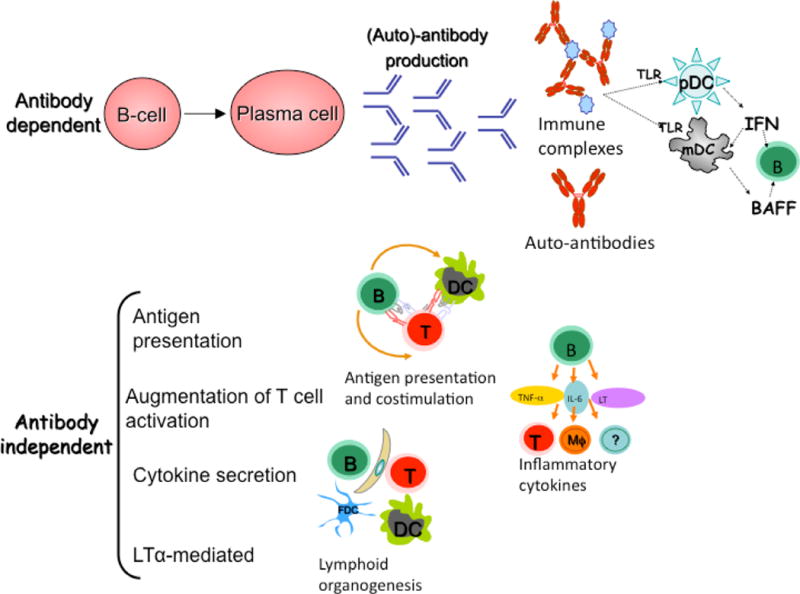
B cells contribute to SLE disease pathogenesis by both antibody dependent and independent mechanisms. In addition to direct binding of autoantibodies to target antigens on cells which can lead to cellular cytotoxicity and complement activation, immune complexes can also activate complement and cause toll-like receptor (TLR) activation. Antibody independent functions for B cells include antigen presentation and co-stimulation to T cells, such that B cells can affect T cell function in diverse ways, contributing to T cell activation, polarization, and even recruitment of follicular T helper cells to the GC. Other antibody independent functions for B cells include pro-inflammatory and anti-inflammatory cytokine secretion and chemokine secretion- including LTα which affects lymphoid organogenesis.
Autoantibody independent functions for B cells in SLE
There has been much speculation about what the key autoantibody independent functions of B cells are in SLE (Figure 2). A particularly novel function may be direct effects on lymphoid neogenesis through the production of LTα. The formation of tertiary or ectopic lymphoid tissue formation is a process that may lead to dysregulated B/T-cell interactions and local amplification of autoimmune responses in multiple autoimmune diseases, including RA, Sjogren’s syndrome, type I diabetes, multiple sclerosis, inflammatory bowel disease, Hashimoto’s thyroditis, and SLE (44, 45). For example, recent reports suggest the importance of this process in human lupus nephritis (46).
Moreover, B cells can produce numerous other cytokines and may do so in a polarized fashion, mimicking Th1/Th2 cells. So called effector B cells (Be1 and Be2) can participate in feedback regulation of T helper cells (47), although the relevance of these mechanisms for SLE have yet to be demonstrated. Along similar lines, B cells play a key role in the recruitment of CXCR5+ follicular T helper (TFH) cells to the germinal center (48). TFH cells provide critical assistance for follicular and germinal center B cells, inducing activation, differentiation, and antibody production. Recent data indicates that TFH cells and a newly defined population of extrafollicular helper T cells are dependent on B cells for development (49). The influence of B cells on TFH cells via ICOSL and OX40L costimulation may be important in SLE as excessive activity of TFH cells induces hyperactive GC, breakdown of B cell tolerance, autoantibody production and a lupus-like phenoytype in sanroque mice (50).
From an autoimmunity standpoint, it is important to remember that B cells may either stimulate or inhibit pathogenic responses (Table 1). Thus, evidence is accumulating for regulatory B cells capable of preventing or suppressing autoimmunity in different mouse models (51, 52). This protective role can be mediated by inducing T cell anergy during antigen presentation or inducing Treg expansion or activity (52). B cells may also directly suppress Th1 and Th17-mediated diseases (53). These activities are mediated, at least in part, by the production of IL-10 or TGFβ and may control a variety of auto-inflammatory diseases including: inflammatory arthritis, inflammatory bowel disease, autoimmune diabetes, experimental autoimmune encephalitis and contact hypersensitivity (54–57). Understanding the imbalance between these opposing B cell functions in disease and how this imbalance may be restored after targeted biologic therapy is an important area of ongoing research.
Table 1.
The balance between pro-inflammatory and protective B cell functions.
| Induce autoimmunity | Suppress autoimmunity | |
|---|---|---|
| •T cell activation | •T cell anergy | |
| •Th1 cytokines (Be 1) | •Th2 cytokines (Be2) | |
| •Treg inhibition | •Treg priming/expansion | |
| •DC recruitment | •DC inhibition (IL-10) | |
| •Pathogenic Autoab | •Protective Ab | |
| •Pro-inflammatory cytokines | •Anti-inflammatory cytokines | |
| TNF, IFNγ, IL-12p40, IL-6 | •Formation of ectopic lymphoid tissue | IL-10, TGFβ |
The interface between innate and adaptive immunity: Autoantibodies, IFN, and Toll like receptor activation
Once B cell tolerance is broken, autoantibodies can contribute to disease pathogenesis in SLE through a number of classical effector roles, including the formation of damaging immune complexes (immune-complex mediated Type III hypersensitivity reactions) and direct pathogenesis (type II antibody-dependent cytotoxicity) (Figure 2) (58). However, as alluded to previously, intriguing evidence has emerged that autoantibodies can play an active role in propagating the autoimmune process in SLE, through immune cell activation involving RNA- or DNA-containing autoantigens and toll like receptors (TLR) (21–24, 59, 60). Notably, plasmacytoid dendritic cells (DC) may be activated by costimulation of TLRs (TLR-7, -8, or-9) and FcRs via immune complex binding, stimulating the secretion of large quantities of IFN-α (61), a cytokine with important immunomodulatory functions that include activation and maturation of DCs and stimulation of both T and B cells. In combination with TLR-7 and -9 activation of myeloid DCs to produce BAFF, a feedback loop is generated that triggers more B cell activation (Figure 1). Moreover, by binding to cell surface autoantibodies and subsequently to TLR-7 or -9 within B cells, RNA- or DNA-containing autoantigens or immune complexes can directly trigger activation and proliferation of autoantibody-producing B cells (21, 62, 63). In support of the importance of TLR signaling to lupus disease pathogenesis and B cell activation, deficiency of TLR-7 or -9 can prevent autoantibody production in a variety of mouse models (62, 64). Of addition interest, it was recently shown that the murine autoimmune Yaa locus represents a TLR-7 duplication which increases the responsiveness of B cells to TLR-7 ligands, thus contributing to a break in B cell tolerance (65). Notably, one of the mechanisms by which antimalarials likely exert their effect in SLE is via inhibition of TLR signaling.
As reviewed elsewhere, recent evidence suggests a prominent role for IFN α activation in SLE. It is important to note that IFN α may contribute to B cell abnormalities in SLE, promoting the differentiation of activated B cells into plasmablasts (66) and in conjunction with TLR stimulation triggering B cell expansion (67) and a lowered activation threshold for autoreactive B cells (68). We have also recently reported a novel role for IFN α activation in the bone marrow of SLE patients by decreasing B cell lymphopoeisis, contributing to B cell lymphopenia, and theoretically decreasing the stringency of B cell negative selection (69).
Although the relative importance of autoantibody dependent vs. independent roles of B cells in human SLE pathogenesis remains to be defined, given the multiple pathogenic roles of autoantibodies in SLE enumerated above, one would postulate that the most effective therapeutic interventions might decrease autoantibody levels. This may require targeting of the plasma cell compartment directly or indirectly. In order to effectively do so, it is important to understand the nuances of plasma cell biology. In particular, the premise that autoreactive plasma cells are short-lived and continually replenished from ongoing immune responses has recently been called into question. Indeed, there is accumulating evidence that some autoreactive plasma cell populations may be long-lived and resistant to conventional therapies (70).
Therapeutic targeting of the B cell compartment
A number of different approaches to targeting B cells have been utilized: 1) B cell depletion with monoclonal antibodies against B cell-specific molecules (e.g., anti-CD20), 2) induction of negative signaling in B cells (e.g. anti-CD22); 3) blocking B cell survival and activation factors (e.g., anti-BAFF), and 4) blocking costimulatory interactions between B and T cells. Many of these agents are currently undergoing formal testing in clinical trials or are under development (Table 2).
Table 2.
Approaches to target B cells
| Compound | Description | Stage of development |
|---|---|---|
|
| ||
| •B cell depletion | ||
| •Rituximab | chimeric anti-CD20 monoclonal antibody | Phase III SLE |
| •Ocrellzumab | humanized anti-CD20 monoclonal antibody | Phase III SLE |
| •TRU-015 | anti-CD20 SMIP-small modular Immunopharmaceutical | Phase III RA |
| •Ofatumumab | fully human anti-CD20 monoclonal antibody | Phase III RA |
| •Inhibitory signaling | ||
| •Epratuzumab | humanized anti-CD22 monoclonal antibody | Phase II in SLE |
| •Co-stimuletory blockade | ||
| •Anti-CD40L | monoclonal antibody against CD40L | no longer In development |
| •CTLA4-lg | fusion protein of CTLA4: blocks B7-CD28 costimulation | Phase II SLE |
| •Cytokine blockade | ||
| •Belimumab/Benlyata fully human anti-BAFF monoclonal antibody | advanced development in SLE | |
| •BR3-Fc | fusion protein of the BR3 BAFF receptor | Phase I RA |
| •Anti-BR3 | fully human antibody against BR3 | Phase I RA |
| •Atacicept | fusion protein of the TACI receptor blocks BAFF/APRIL | Phase I, II RA, SLE |
B cell depletion
Anti-CD20 monoclonal antibody
The largest body of clinical data regarding B cell depletion involves anti-CD20 targeted therapy with the monoclonal antibody rituximab. CD20 is a member of the tetraspan family of integral membrane proteins (71) and is specifically expressed on immature, naïve, memory, and germinal center (GC) B cells, but not on early pre B cells or plasma cells. In vitro rituximab can kill B cells by complement mediated cytotoxicity (CMC), antibody dependent cell mediated cytotoxicity (ADCC), and induction of apoptosis (72). Elegant studies in a murine model of human CD20 expression have demonstrated that different B cell subsets may be more dependent on certain mechanisms of depletion than others because of tissue microenvironment effects (73). Of note, the kinetics of B cell depletion in tissues is slower than that in the peripheral blood, and certain tissue bound subsets may be incompletely depleted. The latter point may be particularly relevant for human autoimmune diseases where complete depletion of autoreactive B cell clones may be critical for full therapeutic potential. Although anti-CD20 is usually effective in depleting B cells from peripheral blood, success in depleting B cells from other sites such as lymph nodes or tertiary lymphoid tissues may be highly variable (74, 75). Failure to deplete in these tissue sites may lead to non-response or early relapse.
Clinical data on B cell depletion in SLE
FDA-approved for lymphoma in 1997 and rheumatoid arthritis refractory to anti-tumor necrosis factor in 2006, B cell depletion with rituximab has also shown benefit in open label studies and case series for many other autoimmune diseases (76). Initial evidence for the potential benefit of B cell depletion in SLE has been gathered from the combination of the original dose escalation trials of rituximab performed by our own group at the University of Rochester (77) and by Albert and colleagues at the University of Pennsylvania (78) as well as from several open label series and numerous published and unpublished case reports (79–81).
Recently, Isenberg and colleagues published an update of their large single center experience with rituximab use in 50 SLE patients with severe refractory disease to conventional immunosuppressives, the longest follow-up to date. Six months after the initial treatment, 42 and 47% of patients showed complete and partial remissions, respectively. Among the patients followed for 7.5 years, one remained B cell depleted for over 7 years and 53% had experienced a disease flare. Three quarters of flares occurred within a year of initial treatment, and 80% of these patients were retreated with good effect. Similarly, in an article reviewing the world-wide experience, Sfikakis et al. reported that rituximab was well tolerated in a total of 100 patients with severe, refractory SLE (82). Ramos et al. also recently reported favorable results through a systematic review of off-label use in 188 cases from the literature between 2002 and 2007 (83). Interestingly, in this latter review 52% of patients received concomitant cyclophosphamide, and a higher rate of therapeutic response was observed in those patients (98% vs. 82%, p<0.001). The UCLA group recently reported their experience with rituximab in 35 Hispanic and African American patients with refractory SLE (84). They observed significant clinical and immunologic responses, with particular benefit in arthritis and nephritis. Additionally, in data from the Autoimmunity and Rituximab (AIR) French registry, 104 SLE patients were treated with rituximab (largely without cyclophosphamide) for a variety of indications (including cutaneous/articular involvement in 41%, renal involvement in 30%, and autoimmune cytopenia in 23%) and had an overall clinician observed efficacy of 73% (85).
These results support the conclusion that BCDT may be effective for patients with active SLE that is refractory to standard immunosuppression. Moreover, the combination of rituximab and cyclophosphamide may offer synergy since both agents can target B cells (note too that even if not used in direct combination, refractory patients have often received cyclophosphamide in the recent past). Although most studies have found adequate peripheral blood B cell depletion, as noted previously depletion may be less complete in tissue sites. In SLE this is suggested by the finding of residual circulating B cells by high sensitivity flow cytometry, predominantly of a memory phenotype, and a reconstitution in poor responders that is dominated by memory B cells (32, 86). The problem of incomplete B cell depletion is highlighted by our original open-label dose escalation phase I/II trial of rituximab in 17 moderately active SLE patients (77). In the majority of patients with effective B cell depletion (11/17), the SLAM score was significantly improved at 2 and 3 months, respectively (p=0.0016 and p=0.0022), an improvement that persisted for at least 12 months. Six patients in this study had incomplete B cell depletion (including 1 of 4 in the high dose group) and no clinical improvement. These results highlight that B cell depletion can be effective in a non-refractory group of SLE patients as monotherapy when high dose steroids and other treatments do not confound the results. Incomplete B cell depletion was associated with certain Fc receptor genotypes (21), African American ancestry, lower serum rituximab levels (22), and the development of human anti-chimeric antibodies (HACAs). Another phase I/II prospective open-label study also found that a third of patients developed HACAs, which tended to correlate with less complete B cell depletion, as measured by early B cell return and/or ability to respond to vaccinations (78). The results raise the concern that rituximab may be more immunogenic in active SLE.
Despite considerable enthusiasm for rituximab in SLE based on this open-label experience, two recent placebo-controlled trials in non-renal lupus (EXPLORER) and renal lupus (LUNAR) failed to meet primary endpoints. At the 2008 American College of Rheumatology meeting. Merrill et al. (87) reported the EXPLORER trial, a randomized, double-blind, placebo-controlled phase ll/lll multicenter trial comparing rituximab to placebo in a 2:1 randomization in 257 active SLE patients. Both groups had significant and sustained decreases in global BILAG and Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) scores over time, but with no differences in primary and secondary endpoints. In a pre-specified subgroup analysis of black and Hispanic subjects, who overall had lower placebo responses (15.7% PCR including MCR), there was a statistically significant difference with the addition of rituximab (33.8% PCR including MCR).
Although the majority of lupus manifestations have been reported to respond to B cell depletion in case series and open-label studies, there have been no organ-specific clinical trials with the exception of the recently completed LUNAR study in lupus nephritis. Organ-specific trials may be advantageous due to less disease heterogeneity and possibly better outcome measures. Moreover, we can theoretically select patients based on a pathophysiology that might be particularly responsive to B cell depletion. For example, rituximab treatment may be particularly efficacious for disease manifestations predominantly mediated by B cells (directly or indirectly) and/or short-lived plasma cells (for example, cytopenias) but not those mediated by antibodies produced by long-lived plasma cells.
Recent data from small open label studies have supported the benefit of rituximab in lupus nephritis. Von Vollenhoven and colleagues evaluated a combination of cyclophosphamide and rituximab in 18 patients with active nephritis (10 class III/IV, 7 class V, 1 unknown). On repeat renal biopsy, improvement in the histopathologic class of nephritis and a decrease in the renal activity index occurred in a majority of patients. Moreover, a reduction in the number of CD3, CD4, and CD20 cells in the renal interstitium was noted in 50% of the patients on repeat biopsy (88) (updated results at the 2008 ACR meeting). In a similarly small study of 10 patients with LN treated with rituximab and oral prednisolone (at 0.5 mg/kg/day for 10 weeks then tapered by 4 mg every 2 weeks), Sfikakis and colleagues found a complete renal response in 5 and a partial response in another 3. Notably, T cell activation as measured by CD40 ligand on CD4 cells decreased significantly. The effect on T cell activation was greatest when complete response was attained and remained low in patients who maintained a complete response (89). Other retrospective studies (90), prospective cohorts (91), and open-label studies (92) have suggested similar benefit.
Thus, there was great hope for the phase lll, randomized, double-blind, placebo-controlled, multicenter study (LUNAR) evaluating the efficacy and safety of rituximab plus mycophenolate in SLE patients with active proliferative nephritis (types lll/lV) (n=144) that was recently reported at the 2009 ACR meeting (93). Rituximab was administered as in EXPLORER using RA dosing at baseline and 6 months, with the primary endpoint the fraction of patients achieving a complete or partial renal response at 52 weeks. Although there were numerically more responders in the rituximab group (57% vs. 46%), the study did not show a statistically significant difference in primary or clinical secondary endpoints. Blacks and Hispanics randomized to rituximab had greater responses compared to placebo than Whites, but statistical significance was not achieved. Rituximab did have a greater effect on levels of anti-dsDNA (p=0.007) and complement (p=0.025) at Wk 52. Adverse events were similar between groups with no new or unexpected safety signals. Overall, this study failed to indicate that the addition of rituximab to mycophenolate and steroids provides additional benefit, at least at 52 weeks, which some may argue is short follow-up in a nephritis study.
Conclusions and perspectives: Does BCD work in SLE?
Given that there have been notable examples of therapeutics that appeared effective in open label studies until well-controlled studies provided contrary evidence, the results of the two recent randomized trials of rituximab in SLE should give us pause. Two alternative explanations are possible, either B cell depletion is not effective in SLE or trial design was suboptimal for demonstrating clinical efficacy. Due to the large body of pre-clinical and open-label data in refractory disease suggesting efficacy, we favor the latter explanation. Several recent papers have addressed the formidable challenges of trial design in SLE (3, 94–96). As different manifestations of SLE may result from different mechanisms, one targeted therapy is unlikely to be effective for all patients. It is also important to note that the most impressive open-label results with rituximab for SLE have been observed in (1) refractory patients (often previously receiving cyclophosphamide) and (2) those treated with one or two low doses of cyclophosphamide in combination with rituximab, an approach not used in either EXPLORER or LUNAR.
How does B cell depletion work in SLE?
B cell depletion has the potential to induce disease amelioration by inhibiting autoantibody production and/or by interfering with other B cell pathogenic functions. In some diseases at least (RA, ANCA vasculitis, and IgM antibody-associated polyneuropathy) autoantibody decline seems to be associated with clinical improvement (97, 98). The autoantibody decline suggests that the autoreactive plasma cells are relatively short-lived and thus disappear if their B cell precursors are eliminated. Conversely, subsequent increases in autoantibody titers (RF, ANCA) seem to be closely associated with B cell repopulation and sometimes may even precede the expansion of B cells (97). Collectively, the data suggest that at least in some patients repopulation may be dominated by the preferential expansion of residual autoreactive B cells perhaps due to a competitive advantage (or lack of competition) in a depleted peripheral compartment (32, 86, 99). The picture appears to be more complicated in SLE where total IgM and IgG antibody levels as well as disease-specific autoantibodies remain relatively stable for at least 1 year, particularly when cyclophosphamide is not co-administered. This implicates the importance of the autoantibody independent role of B cells in ongoing disease. Indeed, limited studies of the effects of B cell depletion on other immune cells have demonstrated decreases in T cell activation (100) and increases in T regulatory cell numbers and function (101).
We have found that upon longer follow-up, however, SLE patients with good B cell depletion could be split into those whose anti-ds DNA levels progressively declined after 1 year and eventually normalized and those in whom the levels failed to diminish (32). As a group, serological responders experienced dramatic and sustained clinical responses. Interestingly, patients in this subset have a robust B cell reconstitution with up to 80% of all peripheral blood B cells displaying a transitional phenotype (B cells intermediate between immature and mature naïve) (102, 103). We speculate that the reconstitution phenotype observed in long-term responders is the consequence of profound B cell depletion induced by rituximab and the ensuing bone marrow derived repopulation and suggests the potential emergence of a protective, regulatory B cell population (Figure 1). In addition, it seems likely that tissue depletion of autoreactive B cells that may continue to generate plasma cells of heterogeneous life span may have been less successful in patients with good blood depletion but poor serological response. Of significant interest, expansion of transitional cells also correlates with prevention of diabetes after B cell depletion in NOD mice (104). Overall, these studies raise critical questions regarding the factors that influence depletion, the kinetics and quality of repopulation, and the mechanisms responsible for long-term remission in a subset of patients.
Potential complications of B cell depletion therapy
Given that primary and secondary immune responses may be compromised even a year after B cell depletion therapy (105) (106) (107), it is recommended that immunizations be given 1 month prior to starting rituximab, and yearly influenza vaccine given as late as possible after the last dose. More careful studies of humoral immune responses after rituximab and the correlation with B cell subset recovery are necessary, however. We would predict that primary immune responses may be impaired post-rituximab even after recovery of normal absolute B cell counts if the B cells are predominantly immature transitional cells, given the hypo-responsiveness of transitional B cells (39).
Because of the lack of expression of CD20 on plasma cells (the major antibody producing cells) it has been long appreciated that immunoglobulin levels are not significantly affected in patients who are treated with rituximab, at least not in the short term after a single course of therapy. Pre-existing anti-tetanus toxoid and anti-pneumococcal polysaccharide antibodies remain stable for prolonged periods of time in both SLE and RA (108). However, hypogammaglobulinemia with low IgG levels has been observed in children treated with rituximab (109), and IVIG has been advocated in this group as well as prophylactically in infants (110). With repeated B cell depletion, development of low immunoglobulin levels is more common, although not clearly associated with increased infectious complications. A recent meta-analysis supports prior literature regarding the safety of B cell depletion in that treatment of RA patients with rituximab was not associated with an increased incidence of serious infections (111), even after repeated courses (112) or with TNF blockade after B cell depletion (113). The caveat here is that the database is relatively small for RA and even smaller for SLE. Moreover, there have been reports of fulminant hepatitis B reactivation and rare cases of PML (105). Thus, patients should be screened for hepatitis B prior to the use of rituximab and viral prophylaxis considered in high-risk patients.
Unfortunately, there are no screening methods to identify patients at risk for development of PML. A recent paper reported on 57 cases of PML occurring in the setting of rituximab, 52 in the patients with lymphoproliferative disease, 2 for SLE, and 1 for RA, with a mortality rate of 89% (114). Many of these patients were heavily immunosuppressed with medications in addition to rituximab currently or historically. Although the frequency of PML in RA patients receiving rituximab is rare (3/100,000), the incidence may be higher in SLE, which is independently associated with PML (105). Overall, the data suggests that patients who receive rituximab have an increased risk of PML. Callabrese and Molloy (115) recently provided a cogent discussion of this topic and made several important points: (1) PML is a risk associated with all immunosuppressive therapies, (2) As PML has been observed in SLE in the absence of exposure to rituximab, it is unclear how much additional risk is imparted by B cell depletion, and (3) Given that PML is a devastating complication and the risk of it occurring is not zero, a risk-benefit discussion should take place with patients.
Other agents that target B cells
Other B cell depleting antibodies
Other monoclonal antibodies that target CD20 are in various phases of development, including ocrelizumab (humanized anti-CD20) (Table 2). Theoretically, a fully human antibody may be better tolerated during infusions due to less immunogenicity. This could translate into more complete B cell depletion especially in SLE where HACAs are more common after rituximab. Interestingly, the trial of ocrelizumab in lupus nephritis may answer the question of synergy with cyclophosphamide given that a subset of patients will receive B cell depletion in combination.
Epratuzumab is a humanized anti-CD22 that also induces B cell depletion although less pronounced than with anti-CD20 and preferentially of naïve and transitional B cells. However, anti-CD22 also blocks the activation and proliferation of B cells, possibly acting as a negative regulator of B cell function. A recent press release regarding a phase IIb trial of epratuzumab in SLE reported superior response rates compared to placebo at week 12. Other approaches to B cell depletion under development include antibodies against CD19, which is expressed on virtually all B cells and is lost at a later stage than CD20 as B cells differentiate into plasma cells. In animal models of autoimmunity anti-CD19 depletes a wider spectrum of B cells than anti-CD20, e.g. peritoneal B cells including B1 B cells are depleted with anti-CD19 but spared with anti-CD20 (116). Moreover, treatment with anti-CD19 in these models was associated with a decrease in total immunoglobulins indicating an effect on plasma cells.
Inhibition of co-stimulation
As an alternative to selective B cell depletion, there has been interest in targeting co-stimulatory signaling pathways. Direct inhibition of B-T cell collaboration through inhibition of the CD40-CD40L pathway has been demonstrated to be effective in mouse models of lupus (117). Two studies of anti-CD40L antibodies in SLE have been reported (118, 119), but either failed to show clinical efficacy over placebo or were complicated by unexpected thromboembolic events (119). Interestingly, two small mechanistic studies demonstrated beneficial immune effects (n=5 for each), including a marked reduction in autoreactive anti-dsDNA producing B cells (120) and substantial reductions in abnormal B cell populations (30). These reports suggest that anti-CD40L can interfere with aberrant germinal center reactions in SLE and translate into clinical benefit, if administered with the proper pharmacokinetics for adequate costimulatory blockade.
An alternative co-stimulatory target in SLE includes the CD28 and CTLA4 receptors and their B cell co-ligands B7-1 and B7-2. Blockade of B7 stimulation on B cells with a fusion protein of the extracellular domain of CTLA and immunoglobulin constant regions has yielded promising results in murine SLE (121). A double-blind, placebo-controlled phase II trial evaluated the efficacy of abatacept (CTLA4Ig) in reducing BILAG flares in 180 non-renal SLE patients with active polyarthritis, serositis, or discoid lesions. Although primary and secondary endpoints were not met, post hoc analyses suggested that the outcome measures used may have been suboptimal and also that there may be greater efficacy of abatacept in the polyarthritis subset of disease.
Inhibition of cytokines with B cell effects
There has been active interest in developing antagonists of BAFF in the treatment of SLE. Belimumab (fully human monoclonal antibody against BAFF) for SLE has now been evaluated in multiple double-blind, placebo-controlled trials. Although the original Phase II study did not meet its primary efficacy endpoint, when only the serologically active subjects were analyzed post hoc there was a statistically significant improvement in disease activity using a novel responder index. During long-term open treatment, the frequency of SELENA-SLEDAI flares declined to 7% at 3 years, suggestive of sustained improvement of disease activity with long-term therapy. After these initial encouraging results in phase II trials, two large phase III trials were initiated and have recently reported favorable clinical responses compared to placebo. The first of these studies BLISS-52 was presented at the 2009 ACR meeting, and the second BLISS-72 was reported in November 2009 to have also met its primary efficacy endpoint. Given these initial reports, it is important to consider whether BAFF blockade would be expected to yield different results compared to anti-CD20. Indeed, this would be surprising given that both agents deplete B cells, with anti-CD20 causing even more pronounced B cell depletion and likely better depletion of memory B cell subsets. On the other hand, BAFF blockade may have additional effects on other immune cells important in SLE including T cells and dendritic cells (122). Moreover, rituximab causes a compensatory increase in BAFF, which may theoretically have adverse effects on B cell selection. Despite these speculations, it is likely that the distinct trial results with BAFF blockade vs. anti-CD20 in SLE are more related to trial design than biologic differences between the two therapies.
Additional agents neutralizing BAFF are in various active stages of development, including fusion proteins of immunoglobulin Fc and the BAFF receptors, either BAFF-R (BR3) or transmembrane activation and calcium modulating cyclophilin ligand interactor (TACI) (Atacicept). Atacicept blocks both BAFF and its related cytokine APRIL and was well tolerated in a SLE phase I study, with reductions in total IgG, IgM, and autoantibodies (123). However, a phase II study of atacicept in combination with mycophenolate mofetil for lupus nephritis was terminated because of infections. A phase II/III trial of atacicept for non-renal lupus is ongoing and still recruiting. Because of its effects on plasma cells TACI-Ig would appear to have great potential in autoantibody mediated diseases. Indeed, another potential explanation for the disappointing results of B cell depletion in SLE is the lack of effect on autoantibodies and long-lived plasma cells (124). However, a challenge will be to eliminate autoantibody producing plasma cells without increasing the risk of infection due to adverse effects on protective antibodies.
Conclusions
In summary, recent controlled clinical trials of B cell targeting agents in SLE have had variable benefit but have contributed to our understanding of how to conduct trials in lupus and have recently shown promise. New agents capable of affecting long-lived plasma cells are now being developed (125). These agents should allow eradication of autoantibodies, but will need to be used carefully to prevent infectious complication and to ensure that autoimmune plasma cells do not re-populate the long-lived plasma cell compartment. The impact of these emerging targeted therapies on patient survival is likely to be dramatic, and studies on the immune system of patients in ongoing clinical trials should continue to provide invaluable insight into the pathogenesis of human SLE.
Acknowledgments
Dr. Anolik has been supported by several grants including U19 Autoimmunity Center of Excellence AI56390, R01 AI077674-01A1, the Lupus Foundation of American, and the Lupus Research Institute. Dr. Anolik has received grants from Amgen Pharmaceuticals, Genentech, Proteolix, and Vaccinex. She has served as a consultant for Genentech and Roche. We thank Andreea Coca for expert review of the manuscript and Inaki Sanz and Gregg Silverman for input on figures. The collaborations and thoughtful discussions with colleagues, especially Inaki Sanz and John Looney, are gratefully noted.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bongu A, Chang E, Ramsey-Goldman R. Can morbidity and mortality of SLE be improved? Best Practice and Research Clinical Rheumatology. 2002;16:313–32. doi: 10.1053/berh.2001.0228. [DOI] [PubMed] [Google Scholar]
- 2.Gordon C, Bertsias GK, Ioannidis JP, et al. EULAR recommendations for points to consider in conducting clinical trials in systemic lupus erythematosus (SLE) Ann Rheum Dis. 2008 Apr 3; doi: 10.1136/ard.2007.083022. [DOI] [PubMed] [Google Scholar]
- 3.Dall’Era M, Wofsy D. Clinical trial design in systemic lupus erythematosus. Curr Opin Rheumatol. 2006 Sep;18(5):476–80. doi: 10.1097/01.bor.0000240357.22680.63. [DOI] [PubMed] [Google Scholar]
- 4.Bertsias G, Gordon C, Boumpas DT. Clinical trials in systemic lupus erythematosus (SLE): lessons from the past as we proceed to the future–the EULAR recommendations for the management of SLE and the use of endpoints in clinical trials. Lupus. 2008;17(5):437–42. doi: 10.1177/0961203308090031. [DOI] [PubMed] [Google Scholar]
- 5.Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189(10):1639–48. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arbuckle M, McClain M, Rubertone M, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–33. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 7.Kumar KR, Li L, Yan M, et al. Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science. 2006 Jun 16;312(5780):1665–9. doi: 10.1126/science.1125893. [DOI] [PubMed] [Google Scholar]
- 8.Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. 2000 Aug;13(2):277–85. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 9.Fukuyama H, Nimmerjahn F, Ravetch JV. The inhibitory Fcgamma receptor modulates autoimmunity by limiting the accumulation of immunoglobulin G+ anti-DNA plasma cells. Nat Immunol. 2005 Jan;6(1):99–106. doi: 10.1038/ni1151. [DOI] [PubMed] [Google Scholar]
- 10.McGaha TL, Sorrentino B, Ravetch JV. Restoration of tolerance in lupus by targeted inhibitory receptor expression. Science. 2005 Jan 28;307(5709):590–3. doi: 10.1126/science.1105160. [DOI] [PubMed] [Google Scholar]
- 11.Floto RA, Clatworthy MR, Heilbronn KR, et al. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat Med. 2005 Oct;11(10):1056–8. doi: 10.1038/nm1288. [DOI] [PubMed] [Google Scholar]
- 12.Mackay M, Stanevsky A, Wang T, et al. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J Exp Med. 2006 Sep 4;203(9):2157–64. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liossis SN, Kovacs B, Dennis G, Kammer GM, Tsokos GC. B cells from patients with systemic lupus erythematosus display abnormal antigen receptor-mediated early signal transduction events. J Clin Invest. 1996;98(11):2549–57. doi: 10.1172/JCI119073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olferiev M, Crow M. Activation of Interferon and Ubiquitin Pathways in Lupus Memory B Cells. Arthritis & Rheumatism. 2009;60(10 supplement):S744. [Google Scholar]
- 15.Mackay F, Woodcock S, Lawton P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190:1697–710. doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gross J, Johnston J, Mudri S. TACI and BCMA are receptors for a TNF homologue implicated in B cell autoimmune disease. Nature. 2000;404:995–9. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 17.Stohl W, Metyas S, Tan SM, et al. B lymphocyte stimulator overexpression in patients with systemic lupus erythematosus: longitudinal observations. Arthritis & Rheumatism. 2003;48(12):3475–86. doi: 10.1002/art.11354. [DOI] [PubMed] [Google Scholar]
- 18.Lesley R, Xu Y, Kalled SL, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF.[see comment] Immunity. 2004;20(4):441–53. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 19.Thien M, Phan TG, Gardam S, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004 Jun;20(6):785–98. doi: 10.1016/j.immuni.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 20.Miller J, Stadanlick JE, Cancro MP. Space, selection, and surveillance: setting boundaries with BLyS. J of Immunology. 2006;176:6405–10. doi: 10.4049/jimmunol.176.11.6405. [DOI] [PubMed] [Google Scholar]
- 21.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors.[see comment] Nature. 2002;416(6881):603–7. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 22.Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19(6):837–47. doi: 10.1016/s1074-7613(03)00323-6. [DOI] [PubMed] [Google Scholar]
- 23.Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. Journal of Experimental Medicine. 2004;199(12):1631–40. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005 Feb;115(2):407–17. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yurasov S, Wardemann H, Hammersen J, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005 Mar 7;201(5):703–11. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cappione A, 3rd, Anolik JH, Pugh-Bernard A, et al. Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J Clin Invest. 2005 Nov;115(11):3205–16. doi: 10.1172/JCI24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pugh-Bernard AE, Silverman GJ, Cappione AJ, et al. Regulation of inherently autoreactive VH4-34 B cells in the maintenance of human B cell tolerance. J Clin Invest. 2001;108(7):1061–70. doi: 10.1172/JCI12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsuiji M, Yurasov S, Velinzon K, Thomas S, Nussenzweig MC, Wardemann H. A checkpoint for autoreactivity in human IgM+ memory B cell development. J Exp Med. 2006 Feb 20;203(2):393–400. doi: 10.1084/jem.20052033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wardemann H, Yurasov S, Schaefer A, Young J, Meffre E, Nussenzweig M. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–7. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 30.Grammer A, Lipsky PE. B cell abnormalities in systemic lupus erythematosus. Arthritis Res Ther. 2003;5:S22–7. doi: 10.1186/ar1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Odendahl M, Jacobi A, Hansen A, et al. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunology. 2000;165:5970–9. doi: 10.4049/jimmunol.165.10.5970. [DOI] [PubMed] [Google Scholar]
- 32.Anolik J, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis & Rheumatism. 2004;50:3580–90. doi: 10.1002/art.20592. [DOI] [PubMed] [Google Scholar]
- 33.Wei C, Anolik J, Cappione A, et al. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol. 2007 May 15;178(10):6624–33. doi: 10.4049/jimmunol.178.10.6624. [DOI] [PubMed] [Google Scholar]
- 34.Jacobi AM, Reiter K, Mackay M, et al. Activated memory B cell subsets correlate with disease activity in systemic lupus erythematosus: Delineation by expression of CD27, IgD, and CD95. Arthritis & Rheumatism. 2008;58(6):1762–73. doi: 10.1002/art.23498. [DOI] [PubMed] [Google Scholar]
- 35.Jacobi AM, Odendahl M, Reiter K, et al. Correlation between circulating CD27high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis & Rheumatism. 2003;48(5):1332–42. doi: 10.1002/art.10949. [DOI] [PubMed] [Google Scholar]
- 36.Jacobi AM, Mei H, Hoyer BF, et al. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with SLE. Ann Rheum Dis. 2009 Feb 5; doi: 10.1136/ard.2008.096495. [DOI] [PubMed] [Google Scholar]
- 37.Wei C, Palanichamy A, Jenks S, et al. B Cell Signatures as Biomarkers in SLE. Arthritis & Rheumatism. 2009;60(11 supplement) [Google Scholar]
- 38.Arce E, Jackson DG, Gill MA, Bennett LB, Banchereau J, Pascual V. Increased frequency of pre-germinal center b cells and plasma cell precursors in the blood of children with systemic lupus erythematosus. J Immunol. 2001;167(4):2361–9. doi: 10.4049/jimmunol.167.4.2361. [DOI] [PubMed] [Google Scholar]
- 39.Palanichamy A, Barnard J, Zheng B, et al. Novel human transitional B cell populations revealed by B cell depletion therapy. Journal of Immunology. 2009 May 15;182(10):5982–93. doi: 10.4049/jimmunol.0801859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sabahi R, Owen T, Barnard J, et al. Immunologic effects of BAFF antagonism in the treatment of human SLE. Arthritis & Rheumatism. 2007;9(11) [Google Scholar]
- 41.Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. 1994;180(4):1295–306. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bekar K, Owen T, Dunn R, et al. Prolonged effects of short-term anti-CD20 B cell depletion therapy in murine systemic lupus erythematosus Arthritis & Rheumatism. 2009 doi: 10.1002/art.27515. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Looney RJ, A J, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: A phase I/II dose-escalation trial of rituximab. Arthritis & Rheumatism. 2004;50(8):2580–9. doi: 10.1002/art.20430. [DOI] [PubMed] [Google Scholar]
- 44.Lorenz RG, Chaplin DD, McDonald KG, McDonough JS, Newberry RD. Isolated lymphoid follicle formation is inducible and dependent upon lymphotoxin-sufficient B lymphocytes, lymphotoxin beta receptor, and TNF receptor I function. J Immunol. 2003 Jun 1;170(11):5475–82. doi: 10.4049/jimmunol.170.11.5475. [DOI] [PubMed] [Google Scholar]
- 45.Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 46.Guttikonda R, Chang A, Liu N, Clark M. Lymphocytes and plasma cells in human lupus nephritis. Arthritis and Rheumatism. 2006;54:S. [Google Scholar]
- 47.Harris D, Haynes L, Sayles P, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nature Immunology. 2000;1:475–81. doi: 10.1038/82717. [DOI] [PubMed] [Google Scholar]
- 48.Ebert LM, Horn MP, Lang AB, Moser B. B cells alter the phenotype and function of follicular-homing CXCR5+ T cells. Eur J Immunol. 2004 Dec;34(12):3562–71. doi: 10.1002/eji.200425478. [DOI] [PubMed] [Google Scholar]
- 49.Odegard JM, Marks BR, DiPlacido LD, et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008 Nov 24;205(12):2873–86. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vinuesa CG, Cook MC, Angelucci C, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005 May 26;435(7041):452–8. doi: 10.1038/nature03555. [DOI] [PubMed] [Google Scholar]
- 51.Lund FE. Cytokine-producing B lymphocytes – key regulators of immunity. Current Opinion in Immunology. 2008;20(3):332–8. doi: 10.1016/j.coi.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fillatreau S, Gray D, Anderton SM. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol. 2008;8(5):391–7. doi: 10.1038/nri2315. [DOI] [PubMed] [Google Scholar]
- 53.Lampropoulou V, Hoehlig K, Roch T, et al. TLR-Activated B Cells Suppress T Cell-Mediated Autoimmunity. J Immunol. 2008 Apr 1;180(7):4763–73. doi: 10.4049/jimmunol.180.7.4763. 2008. [DOI] [PubMed] [Google Scholar]
- 54.Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3(10):944–50. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 55.Mauri C, Gray D, Mushtaq N, Londei M. Prevention of Arthritis by Interleukin 10-producing B Cells. J Exp Med. 2003 Feb 17;197(4):489–501. doi: 10.1084/jem.20021293. 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X, Jensen PE. Cutting Edge: Primary B Lymphocytes Preferentially Expand Allogeneic FoxP3+ CD4 T Cells. J Immunol. 2007 Aug 15;179(4):2046–50. doi: 10.4049/jimmunol.179.4.2046. 2007. [DOI] [PubMed] [Google Scholar]
- 57.Mann MK, Maresz K, Shriver LP, Tan Y, Dittel BN. B Cell Regulation of CD4+CD25+ T Regulatory Cells and IL-10 Via B7 is Essential for Recovery From Experimental Autoimmune Encephalomyelitis. J Immunol. 2007 Mar 15;178(6):3447–56. doi: 10.4049/jimmunol.178.6.3447. 2007. [DOI] [PubMed] [Google Scholar]
- 58.Martin F, Chan AC. B cell immunobiology in disease: evolving concepts from the clinic. Ann Rev Immunol. 2006;24:467–96. doi: 10.1146/annurev.immunol.24.021605.090517. [DOI] [PubMed] [Google Scholar]
- 59.Bave U, Alm GV, Ronnblom L. The combination of apoptotic U937 cells and lupus IgG is a potent IFN-alpha inducer. Journal of Immunology. 2000;165(6):3519–26. doi: 10.4049/jimmunol.165.6.3519. [DOI] [PubMed] [Google Scholar]
- 60.Bave U, Magnusson M, Eloranta M, Perers A, Alm G, Ronnblom L. Fc gamma RIIa is expressed on natural IFN-alpha producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J Immunology. 2003;171:3296–302. doi: 10.4049/jimmunol.171.6.3296. [DOI] [PubMed] [Google Scholar]
- 61.Ronnblom L, Alm GV. A pivotal role for the natural interferon alpha-producing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus.[comment] Journal of Experimental Medicine. 2001;194(12):F59–63. doi: 10.1084/jem.194.12.f59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus.[see comment] Immunity. 2006 Sep;25(3):417–28. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 63.Herlands RA, Christensen SR, Sweet RA, Hershberg U, Shlomchik MJ. T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity. 2008 Aug;29(2):249–60. doi: 10.1016/j.immuni.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med. 2006 Mar 20;203(3):553–61. doi: 10.1084/jem.20052438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006 Jun 16;312(5780):1669–72. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 66.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003 Aug;19(2):225–34. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 67.Bekeredjian-Ding IB, Wagner M, Hornung V, et al. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J Immunol. 2005 Apr 1;174(7):4043–50. doi: 10.4049/jimmunol.174.7.4043. [DOI] [PubMed] [Google Scholar]
- 68.Uccellini MB, Busconi L, Green NM, et al. Autoreactive B cells discriminate CpG-rich and CpG-poor DNA and this response is modulated by IFN-alpha. J Immunol. 2008 Nov 1;181(9):5875–84. doi: 10.4049/jimmunol.181.9.5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barnard J, Palanichamy A, Bauer J, Koeuth T, Baechler E, Anolik J. Interferon activation in human SLE bone marrow inhibits B cell lymphopoeisis Arthritis & Rheumatism. 2008;10(11) [Google Scholar]
- 70.Hoyer B, Moser K, Hauser A, et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med. 2004;199:1577. doi: 10.1084/jem.20040168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tedder TF, Engel P. CD20: a regulator of cell-cycle progression of B lymphocytes. Immunol Today. 1994;15(9):450–4. doi: 10.1016/0167-5699(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 72.Maloney DG, Smith B, Rose A. Rituximab: mechanism of action and resistance. Seminars in Oncology. 2002;29(1 Suppl 2):2–9. doi: 10.1053/sonc.2002.30156. [DOI] [PubMed] [Google Scholar]
- 73.Gong Q, Ou Q, Ye S, et al. Importance of celllular microenvironment and circulatory dynamics in B cell immunotherapy. J of Immunology. 2005;174:817–26. doi: 10.4049/jimmunol.174.2.817. [DOI] [PubMed] [Google Scholar]
- 74.Mamani-Matsuda M, Cosma A, Weller S, et al. The human spleen is a major reservoir for long-lived vaccinia virus-specific memory B cells. Blood. 2008 May 1;111(9):4653–9. doi: 10.1182/blood-2007-11-123844. [DOI] [PubMed] [Google Scholar]
- 75.Vos K, Thurlings RM, Wijbrandts CA, van Schaardenburg D, Gerlag DM, Tak PP. Early effects of rituximab on the synovial cell infiltrate in patients with rheumatoid arthritis. Arthritis Rheum. 2007 Mar;56(3):772–8. doi: 10.1002/art.22400. [DOI] [PubMed] [Google Scholar]
- 76.Levesque MC. Translational Mini-Review Series on B Cell-Directed Therapies: Recent advances in B cell-directed biological therapies for autoimmune disorders. Clin Exp Immunol. 2009 Aug;157(2):198–208. doi: 10.1111/j.1365-2249.2009.03979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Looney RJ, Anolik JH, Campbell D, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: A phase I/II dose-escalation trial of rituximab. Arthritis & Rheumatism. 2004;50(8):2580–9. doi: 10.1002/art.20430. [DOI] [PubMed] [Google Scholar]
- 78.Albert D, Dunham J, Khan S, et al. Variability in the biological response to anti-CD20 B cell depletion in systemic lupus erythaematosus. Ann Rheum Dis. 2008 Dec;67(12):1724–31. doi: 10.1136/ard.2007.083162. [DOI] [PubMed] [Google Scholar]
- 79.Lindholm C, Borjesson-Asp K, Zendjanchi K, Sundqvist AC, Tarkowski A, Bokarewa M. Longterm clinical and immunological effects of anti-CD20 treatment in patients with refractory systemic lupus erythematosus. J Rheumatol. 2008 May;35(5):826–33. [PubMed] [Google Scholar]
- 80.Jonsdottir T, Gunnarsson I, Risselada A, Welin Henriksson E, Klareskog L, van Vollenhoven RF. Treatment of refractory SLE with rituximab plus cyclophosphamide: clinical effects, serological changes, and predictors of response. Ann Rheum Dis. 2007 Sep 7;:ard.2007.079095. doi: 10.1136/ard.2007.079095. [DOI] [PubMed] [Google Scholar]
- 81.Amoura Z, Mazodier K, Michel M, et al. Efficacy of Rituximab in Systemic Lupus Erythematosus: A Series of 22 Cases. Arthritis & Rheumatism. 2007;56(9):S458. [Google Scholar]
- 82.Sfikakis PP, Boletis JN, Tsokos GC. Rituximab anti-B-cell therapy in systemic lupus erythematosus: pointing to the future. Curr Opin Rheumatol. 2005 Sep;17(5):550–7. doi: 10.1097/01.bor.0000172798.26249.fc. [DOI] [PubMed] [Google Scholar]
- 83.Ramos-Casals M, Soto MJ, Cuadrado MJ, Khamashta MA. Rituximab in systemic lupus erythematosus: A systematic review of off-label use in 188 cases. Lupus. 2009 Aug;18(9):767–76. doi: 10.1177/0961203309106174. [DOI] [PubMed] [Google Scholar]
- 84.Karpouzas G, Gogia M, Moran R, Hahn B. Rituximab Therapy Induces Durable Remissions in Hispanic and African American Patients with Refractory Systemic Lupus Erythematosus (SLE) Arthritis & Rheumatism. 2009 [Google Scholar]
- 85.Terrier Bea. Tolerance and Efficacy of Rituximab (RTX) in Systemic Lupus Erythematosus (SLE): Data of 104 Patients From the AIR (« Auto-immunity and Rituximab ») Registry. Arthritis & Rheumatism. 2009 [Google Scholar]
- 86.Anolik JH, Barnard J, Owen T, et al. Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis & Rheumatism. 2007 Sep;56(9):3044–56. doi: 10.1002/art.22810. [DOI] [PubMed] [Google Scholar]
- 87.Merrill J, Neuwelt C, Wallace D, et al. Efficacy and Safety of rituximab in patients with moderately to severely active systemic lupus erythematosus: results from the randomized, double-blind phase II/III study EXPLORER. American College of Rheumatology Annual Meeting. 2008 www.abstractsonline.com.
- 88.Gunnarsson I, Sundelin B, Jonsdottir T, Jacobson S, Henriksson E, van Vollenhoven R. Histopathologic and clinical outcome of rituximab treatment in patients with cyclophosphamide-resistant proliferative lupus nephritis. Arthritis & Rheumatism. 2007;56(4):1263–72. doi: 10.1002/art.22505. [DOI] [PubMed] [Google Scholar]
- 89.Sfikakis PP, Boletis JN, Lionaki S, et al. Remission of proliferative lupus nephritis following B cell depletion therapy is preceded by down-regulation of the T cell costimulatory molecule CD40 ligand: an open-label trial. Arthritis Rheum. 2005 Feb;52(2):501–13. doi: 10.1002/art.20858. [DOI] [PubMed] [Google Scholar]
- 90.Melander C, Sallee M, Trolliet P, et al. Rituximab in severe lupus nephritis: early B-cell depletion affects long-term renal outcome. Clin J Am Soc Nephrol. 2009 Mar;4(3):579–87. doi: 10.2215/CJN.04030808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pepper R, Griffith M, Kirwan C, et al. Rituximab is an effective treatment for lupus nephritis and allows a reduction in maintenance steroids. Nephrol Dial Transplant. 2009 Jul 17; doi: 10.1093/ndt/gfp336. [DOI] [PubMed] [Google Scholar]
- 92.Li EK, Tam LS, Zhu TY, et al. Is combination rituximab with cyclophosphamide better than rituximab alone in the treatment of lupus nephritis? Rheumatology (Oxford) 2009 Aug;48(8):892–8. doi: 10.1093/rheumatology/kep124. [DOI] [PubMed] [Google Scholar]
- 93.Furie R, Looney J, Rovin B, et al. Efficacy and Safety of Rituximab in Subjects with Active Proliferative Lupus Nephritis (LN): Results From the Randomized, Double-Blind Phase III LUNAR Study. Arthritis Rheum. 2009 [Google Scholar]
- 94.Isenberg D, Gordon C, Merrill J, Urowitz M. New therapies in systemic lupus erythematosus - trials, troubles and tribulations…. working towards a solution. Lupus. 2008 Nov;17(11):967–70. doi: 10.1177/0961203308095139. [DOI] [PubMed] [Google Scholar]
- 95.Looney J, Anolik J, Sanz I. A perspective on B-cell targeting therapy for SLE. Modern Rheumatology. 2009 Aug 8; doi: 10.1007/s10165-009-0213-x. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Coca A, Anolik J. Two negative randomized controlled trials in lupus: now what? Faculty of 1000. 2009 doi: 10.3410/M1-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cambridge G, Stohl W, Leandro M, Migone T, Hilbert D, Edwards J. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: Relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis & Rheumatism. 2006;54(3):723–32. doi: 10.1002/art.21650. [DOI] [PubMed] [Google Scholar]
- 98.Keogh KA, Wylam ME, Stone JH, Specks U. Induction of remission by B lymphocyte depletion in eleven patients with refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2005 Jan;52(1):262–8. doi: 10.1002/art.20718. [DOI] [PubMed] [Google Scholar]
- 99.Rouziere A-S, Kneitz C, Palanichamy A, Dorner T, Tony H-P. Regeneration of the immunoglobulin heavy-chain repertoire after transient B-cell depletion with an anti-CD20 antibody. Arthritis Research & Therapy. 2005;7(4):R714–R24. doi: 10.1186/ar1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sfikakis P, Boletis J, Lionaki S, et al. Remission of proliferative lupus nephritis following anti-B cell therapy is preceded by downregulation of the T cell costimulatory molecule CD40 Ligand. Arthritis & Rheumatism. 2004;50:S227. doi: 10.1002/art.20858. [DOI] [PubMed] [Google Scholar]
- 101.Vigna-Perez M, Hernandez-Castro B, Paredes-Saharopulos O, et al. Clinical and immunologic effects of rituximab in patients with lupus nephritis refractory to conventional therapy: a pilot study. Arthritis Res Ther. 2006;8:R83. doi: 10.1186/ar1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Anolik JH, Barnard J, Cappione A, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis & Rheumatism. 2004;50(11):3580–90. doi: 10.1002/art.20592. [DOI] [PubMed] [Google Scholar]
- 103.Anolik J, Barnard J, Owen T, et al. Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis & Rheumatism. 2007;56(9):3044–56. doi: 10.1002/art.22810. [DOI] [PubMed] [Google Scholar]
- 104.Hu C-y, Rodriguez-Pinto D, Du W, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007 Dec 3;117(12):3857–67. doi: 10.1172/JCI32405. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Looney RJ, Srinivasan R, Calabrese LH. The effects of rituximab on immunocompetency in patients with autoimmune disease. Arthritis Rheum. 2008 Jan;58(1):5–14. doi: 10.1002/art.23171. [DOI] [PubMed] [Google Scholar]
- 106.van der Kolk L, Baars J, Prins M, van Oers M. Rituximab treatment results in impaired secondary humoral immune responsiveness. Blood. 2002;100:2257–9. [PubMed] [Google Scholar]
- 107.Bearden CM, Agarwal A, Book BK, et al. Rituximab inhibits the in vivo primary and secondary antibody reresponse to a neoantigen, bacteriophage phiX174. American Journal of Transplantation. 2005;5(1):50–7. doi: 10.1111/j.1600-6143.2003.00646.x. [DOI] [PubMed] [Google Scholar]
- 108.Cambridge G, Leandro MJ, Edwards JC, et al. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis & Rheumatism. 2003;48(8):2146–54. doi: 10.1002/art.11181. [DOI] [PubMed] [Google Scholar]
- 109.Willems M, Haddad E, Niaudet P, et al. Rituximab therapy for childhood-onset systemic lupus erythematosus. J Pediatr. 2006 May;148(5):623–7. doi: 10.1016/j.jpeds.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 110.Zecca M, Nobili B, Ramenghi U, et al. Rituximab for the treatment of refractory autoimmune hemolytic anemia in children. Blood. 2003 May 15;101(10):3857–61. doi: 10.1182/blood-2002-11-3547. 2003. [DOI] [PubMed] [Google Scholar]
- 111.Salliot C, Dougados M, Gossec L. Risk of serious infections during rituximab, abatacept and anakinra treatments for rheumatoid arthritis: meta-analyses of randomised placebo-controlled trials. Ann Rheum Dis. 2009 Jan;68(1):25–32. doi: 10.1136/ard.2007.083188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Keystone E, Fleischmann R, Emery P, et al. Safety and efficacy of additional courses of rituximab in patients with active rheumatoid arthritis: an open-label extension analysis. Arthritis Rheum. 2007 Dec;56(12):3896–908. doi: 10.1002/art.23059. [DOI] [PubMed] [Google Scholar]
- 113.Genovese MC, Breedveld FC, Emery P, et al. Safety of biologic therapies following rituximab treatment in rheumatoid arthritis patients. Ann Rheum Dis. 2009 Jan 20; doi: 10.1136/ard.2008.101675. [DOI] [PubMed] [Google Scholar]
- 114.Carson KR, Focosi D, Major EO, et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a Review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol. 2009 Aug;10(8):816–24. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 115.Calabrese LH, Molloy ES. Therapy: rituximab and PML risk-informed decisions needed! Nat Rev Rheumatol. 2009 Oct;5(10):528–9. doi: 10.1038/nrrheum.2009.193. [DOI] [PubMed] [Google Scholar]
- 116.Yazawa N, Hamaguchi Y, Poe JC, Tedder TF. Immunotherapy using unconjugated CD19 monoclonal antibodies in animal models for B lymphocyte malignancies and autoimmune disease. Proceedings of the National Academy of Sciences of the United States of America. 2005 Oct 18;102(42):15178–83. doi: 10.1073/pnas.0505539102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang X, Huang W, Schiffer LE, et al. Effects of anti-CD154 treatment on B cells in murine systemic lupus erythematosus. Arthritis & Rheumatism. 2003;48(2):495–506. doi: 10.1002/art.10929. [DOI] [PubMed] [Google Scholar]
- 118.Kalunian KC, Davis JC, Jr, Merrill JT, Totoritis MC, Wofsy D, Group I-LS Treatment of systemic lupus erythematosus by inhibition of T cell costimulation with anti-CD154: a randomized, double-blind, placebo-controlled trial. Arthritis & Rheumatism. 2002;46(12):3251–8. doi: 10.1002/art.10681. [DOI] [PubMed] [Google Scholar]
- 119.Boumpas DT, Furie R, Manzi S, et al. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis & Rheumatism. 2003;48(3):719–27. doi: 10.1002/art.10856. [DOI] [PubMed] [Google Scholar]
- 120.Huang W, Sinha J, Newman J, et al. The effect of anti-CD40 ligand antibody on B cells in human systemic lupus erythematosus. Arthritis and Rheumatism. 2002;46:1554–62. doi: 10.1002/art.10273. [DOI] [PubMed] [Google Scholar]
- 121.Daikh DI, Wofsy D. Cutting edge: reversal of murine lupus nephritis with CTLA4Ig and cyclophosphamide. Journal of Immunology. 2001;166(5):2913–6. doi: 10.4049/jimmunol.166.5.2913. [DOI] [PubMed] [Google Scholar]
- 122.Chang SK, Mihalcik SA, Jelinek DF. B lymphocyte stimulator regulates adaptive immune responses by directly promoting dendritic cell maturation. J Immunol. 2008 Jun 1;180(11):7394–403. doi: 10.4049/jimmunol.180.11.7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dall’Era M, Chakravarty E, Wallace D, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis & Rheumatism. 2007 Dec;56(12):4142–50. doi: 10.1002/art.23047. [DOI] [PubMed] [Google Scholar]
- 124.Munafo A, Priestley A, Nestorov I, Visich J, Rogge M. Safety, pharmacokinetics and pharmacodynamics of atacicept in healthy volunteers. European Journal of Clinical Pharmacology. 2007 Jul;63(7):647–56. doi: 10.1007/s00228-007-0311-7. [DOI] [PubMed] [Google Scholar]
- 125.Neubert K, Meister S, Moser K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nature Medicine. 2008 Jul;14(7):748–55. doi: 10.1038/nm1763. [DOI] [PubMed] [Google Scholar]