

SUMMARY
Loss of function in the Scn1a gene leads to a severe epileptic encephalopathy called Dravet syndrome (DS). Reduced excitability in cortical inhibitory neurons is thought to be the major cause of DS seizures. Here, in contrast, we show enhanced excitability in thalamic inhibitory neurons that promotes the non-convulsive seizures that are a prominent yet poorly understood feature of DS. In a mouse model of DS with a loss of function in Scn1a, reticular thalamic cells exhibited abnormally long bursts of firing caused by the downregulation of calcium-activated potassium SK channels. Our study supports a mechanism in which loss of SK activity causes the reticular thalamic neurons to become hyperexcitable and promote non-convulsive seizures in DS. We propose that reduced excitability of inhibitory neurons is not global in DS and that non-GABAergic mechanisms such as SK channels may be important targets for treatment.
Graphical Abstract
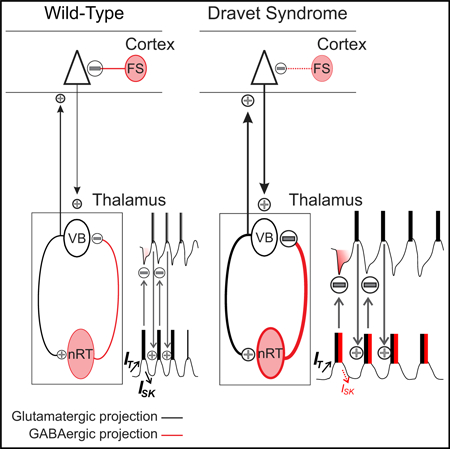
In Brief
In a mouse model of Dravet syndrome (DS) resulting from voltage-gated sodium channel deficiency, Ritter-Makinson et al. find that inhibitory neurons of the reticular thalamic nucleus are paradoxically hyperexcitable due to compensatory reductions in a potassium SK current. Boosting this SK current treats nonconvulsive seizures in DS mice.
INTRODUCTION
SCN1A encodes the alpha subunit of the voltage-gated sodium channel Nav1.1, which is widely expressed in the brain and heart. As regulators of cardiac (Auerbach et al., 2013; Kalume et al., 2013) and brain rhythms, mutations in SCN1A dramatically affect human health. SCN1A is one of the most commonly mutated genes associated with epilepsy, including Dravet syndrome (DS) (Claes et al., 2001, 2009). This age-dependent, early-onset epileptic encephalopathy is characterized by non-convulsive and convulsive seizures, developmental delay, intellectual and motor disabilities, autistic features, sleep impairment, and a high risk of sudden death. Non-convulsive seizures are often intractable and may account for the cognitive decline, behavioral disorders, and reduction in social life associated with DS (Guzzetta, 2011). The absence of non-convulsive seizures is typical of borderline forms of DS, in which cognitive ability is at least partially preserved (Dalla Bernardina et al., 1986).
In mouse models of DS, reduced Scn1a function in GABAergic cortical and hippocampal inhibitory neurons reduces action potential (AP) firing and network inhibition, which is thought to lead to runaway excitation (altered excitatory-inhibitory balance) and convulsive seizures (Cheah et al., 2012; Dutton et al., 2013; Ogiwara et al., 2007; Tai et al., 2014; Yu et al., 2006). Knockout of Scn1a in parvalbumin and somatostatin neurons increases the susceptibility to thermally induced convulsive seizures, whereas selective knockout in either neuron subtype phenocopies distinct DS neuropsychiatric behavioral phenotypes (Rubinstein et al., 2015). Identifying the molecular and brain circuit mechanisms by which reduced SCN1A causes non-convulsive seizures could lead to novel therapeutic strategies.
Although less studied, another hub of Scn1a-expressing GABAergic neurons exists in the nucleus reticularis thalami (nRT) (Papale et al., 2013), which dynamically modulates interactions between the thalamus and cerebral cortex (Crick, 1984; Halassa and Acsády, 2016). The nRT neurons are intrinsic oscillators that inhibit the excitatory relay thalamic nuclei (Gentet and Ulrich, 2003; Houser et al., 1980), positioning them to regulate thalamocortical oscillations, including seizures. Given the abundant expression of Scn1a in nRT (Papale et al., 2013) and that disruptions in nRT correlate with neurological (Makinson et al., 2017; Paz et al., 2010; Slaght et al., 2002) and psychiatric disorders (Ferrarelli and Tononi, 2011; Zhang et al., 2010), we asked whether Scn1a deficiency results in nRT pathology and is involved in non-convulsive seizures using a well-established mouse model containing a human Scn1a mutation (DS mice; Ogiwara et al., 2007).
RESULTS
Scn1a Deficiency Alters the Electric Membrane Excitability of nRT Neurons
We first investigated the effect of Scn1a deficiency on GABAergic parvalbumin-positive nRT neurons. The input resistance (Rin) of nRT neurons was increased in DS mice (Figure S1A), while the membrane capacitance was not significantly reduced (Figure S1A), indicating a decrease in the total membrane conductance. Moreover, the AP threshold was depolarized and the AP width was increased in DS mice, as expected from Scn1a reduction (Cheah et al., 2012) (Figure S1B). However, nRT neurons from DS mice fired more APs in response to depolarization (Figure S1C), consistent with an increased Rin.
Given that post-hyperpolarization rebound bursting plays a critical role in thalamic function (Beenhakker and Huguenard, 2009; Paz and Huguenard, 2015), we investigated whether Scn1a deficiency altered rebound bursting in nRT cells. In response to similar hyperpolarizations, the number of APs within the rebound burst was higher in nRT neurons from DS mice (Figures 1A and 1B), indicating that GABAergic nRT neurons exhibit enhanced intrinsic excitability in DS mice.
Figure 1. SK Deficiency in nRT Neurons Underlies Enhanced Cellular Bursting in DS Mice.

(A) Representative traces show that Dravet syndrome (DS) neurons (red) exhibit enhanced post-inhibitory rebound burst firing upon hyperpolarization induced by −60 pA current pulses, compared to wild-type (WT) (black). The number of APs in the rebound burst are marked above.
(B) Number of APs in the rebound burst (WT, n = 30 cells, 4 mice; DS, n = 43 cells, 6 mice). Data are represented as means ± SEMs, compared with parametric t test with Welch’s correction and α = 0.05, **p < 0.001.
(C–G) T-type calcium current (IT) is similar in nRT neurons from DS mice and WT littermates.
(C) Representative traces showing IT from nRT cells of DS animals and WT littermates (C–F: DS, n = 26 neurons, 6 mice; WT, n = 14 neurons, 4 mice).
(D) IT density, maximal amplitude divided by individual cell capacitance.
(E) IT decay time constant.
(F) IT half-maximal voltage (V50%) taken from Boltzmann function.
(G) Normalized IT amplitude plotted as a function of the pre-pulse membrane potential best fitted with a Boltzmann function (R2 = 0.99 for both fits) (DS, n = 16 cells, 6 mice; WT littermates, n = 13 cells, 4 mice).
(H) ISK traces obtained by digital subtraction in nRT neurons from a WT (black) mouse and a DS (red) mouse (also see Figures S1G and S1H and Method Details).
(I) ISK density, maximal current divided by individual cell capacitance. Data are represented as means ± SEMs, compared with the Mann-Whitney U test. **p = 0.0085. (WT, n = 8 cells, 3 mice; DS, n = 10 cells, 3 mice).
(J and K) SK2 relative mRNA expression levels (encoded Kcnn2, short, J, and long, K, transcript variants) normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA, in nRT. Expression levels were calculated using the ΔΔCT method and expressed as arbitrary units. Mann-Whitney U test, **p < 0.01.
(L) Number of APs on the rebound burst, plotted against hyperpolarization step (mV) with vehicle or 100 μM EBIO treatment. WT, n = 6 neurons, 3 mice; DS, n = 6 neurons, 3 mice. Data are represented as means ± SEMs. Data points compared by individual unpaired t test, not corrected for multiple comparisons, *p < 0.03.
(M) Schematic of the minimal thalamic circuit used for computational modeling, including TCVB and nRT neurons, TCVB to nRT a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-d-aspartate (NMDA) excitatory synapses, and nRT to TCVB GABA-A and GABA-B inhibitory synapses. Only the nRT neuron (black) is simulated (synaptic currents are absent) to obtain the results shown in Figures 1N and 1O.
(N) Voltage traces of an nRT neuron in the WT parameter domain (black trace; and ) and of a neuron in the DS domain with decreased SK maximum conductance (red trace; and ), in response to a hyperpolarizing injected current for 500 ms.
(O) Map of the average number of APs in the rebound burst in the nRT neuron model as a function of maximal conductance parameters. APs numbers are computed over 30 simulations with different realizations of noise in the model. The means of these average numbers of APs are indicated in the WT (white) and DS (red) domains.
We next examined the effects of Scn1a deficiency on thalamocortical (TC) neurons in the somatosensory ventrobasal thalamus (VB), which has also been reported to express Scn1a (Ogiwara et al., 2013). We observed no differences in the passive membrane, tonic, or rebound firing properties of TCVB neurons in DS mice (Figures S2A–S2C). Finally, spontaneous excitatory synaptic currents were not altered in nRT and TCVB neurons from DS mice (Figures S1D, S1E, and 2D–2F, respectively) and were similar to those reported in our previous studies (Paz et al., 2011).
SK Current Deficit Underlies Enhanced Cellular Burst Firing in nRT Neurons
Low-threshold T-type calcium and calcium-activated small potassium (SK) conductances play a major role in nRT post-inhibitory rebound bursts (Jahnsen and Llinás, 1984; Clemente-Perez et al., 2017; Cueni et al., 2008). In DS mice, the peak T-current (IT) density, voltage dependence, and biophysical properties of the steady-state inactivation (SSI) IT were not altered in nRT neurons (Figure 1C–1G) and were comparable in size and kinetics to IT previously measured in the nRT neurons of normal rodents (Paz et al., 2010; Clemente-Perez et al., 2017). These results suggest that enhanced rebound burst firing in DS nRT neurons is not due to a stronger IT.
We next asked whether alterations in SK channels (Cueni et al., 2008) underlie the enhanced burst firing. Whole-cell patch-clamp recordings showed a reduced density of SK currents (ISK) in DS nRT neurons (Figures 1H, 1I, S1G, and S1H). qPCR of nRT isolated from DS mice showed reduced levels of Kcnn2S (Figures 1J and 1K), which is the predominant SK2 sub-type in nRT (Wimmer et al., 2012). In a rescue experiment, the SK agonist 1-ethyl-2-benzimidazolinone (EBIO) (Cueni et al., 2008) was sufficient to normalize the rebound bursting properties in DS nRT neurons (Figure 1L), but it did not affect the firing of TCVB neurons (Figures S2G and S2H).
In a biophysical model (Adelman et al., 2012; Destexhe et al., 1994; Paz et al., 2013) (Figure 1M), reducing SK conductance phenocopied the changes observed in DS nRT neurons, namely the enhanced number of APs within the rebound burst (Figure 1N) and the enhanced Rin (Figure S1F). The increased Rin (Figure S1F) and mean AP number (Figure 1O) were robust across the parameter domains tested. Notably, reducing the transient sodium conductance altered neither the number of APs in the rebound burst nor the Rin (Figures S3J and S3K), suggesting that SK reduction is sufficient to cause enhanced Rin and AP firing in the nRT, independent of changes in transient sodium conductance.
Scn1a Deficiency Leads to Thalamic Microcircuit Hyperexcitability
Rebound burst firing in nRT neurons is important for generating and maintaining oscillatory activity in the intra-thalamic microcircuit (Beenhakker and Huguenard, 2009; Jahnsen and Llinás, 1984; Bazhenov et al., 2000; Deschênes et al., 1984; Steriade et al., 1987). The rebound burst in nRT neurons provides phasic GABAergic inhibition onto excitatory thalamic relay neurons, which leads to rebound burst firing in relay cells and subsequent re-excitation of the nRT, with this sequence reiterating to maintain thalamic network oscillation (Beenhakker and Huguenard, 2009; Paz and Huguenard, 2015). Therefore, we asked whether the intra-thalamic microcircuit between the nRT and relay nuclei is hyperexcitable in Scn1a-deficient mice.
To assess this, we used horizontal thalamic slice preparations that conserve the connectivity between nRT and VB (Huntsman et al., 1999). In wild-type (WT) mice, stimulation of the internal capsule consistently evoked rhythmic oscillatory bursting activity in VB (Figure 2A) in the spindle frequency range (~7–10 Hz), similar to previous reports (Paz et al., 2011; Huntsman et al., 1999; Clemente-Perez et al., 2017; Barthó et al., 2014; Sorokin et al., 2017; Halassa et al., 2011). Evoked repetitive bursting in VB necessitates “ping-pong”-type interactions between nRT and VB; when these connections are severed, no circuit oscillations are observed in VB (von Krosigk et al., 1993). Similar stimulations in DS mice induced longer circuit oscillations in the thalamus (Figure 2B), with more bursts (Figure 2C) occurring at a lower frequency (Figure 2D) in DS mice. These metrics support the intrathalamic nRT-VB microcircuit being hyperexcitable in DS mice.
Figure 2. SK Deficiency in nRT Neurons Underlies Intra-thalamic Circuit Hyperexcit-ability in DS Mice.

(A) Representative evoked intra-thalamic circuit oscillations in thalamic slices from WT and DS mice. Black circles indicate stimulation artifact. Left and right boxes indicate sections of the recording that were expanded, bottom left and right, respectively. Double-sided arrow indicates the length of the evoked oscillation in DS recording. Top right inset: schematic of the intrathalamic oscillatory microcircuit between VB and nRT. Stimulation of the internal capsule (black circle and lightning bolt) leads to an interplay of activity between nRT and TCVB cells that resembles a ping-pong game causing a circuit oscillation whose strength can be assessed by VB recordings in horizontal thalamic slice preparations (see also Figure S6F).
(B–D) Data analyzed by the Mann-Whitney U test,WT, n = 9 mice; DS, n = 12 mice. Data are represented as means ± SEMs.
(B) Duration of evoked oscillation, **p < 0.01.
(C) Number of bursts within the evoked oscillation, *p < 0.05.
(D) Burst frequency within the evoked oscillation, **p < 0.01.
(E) Schematic of the full nRT-TCVB circuit used for the computational modeling of thalamic oscillations in WT and DS conditions.
(F) Top: representative nRT and TCVB voltage traces triggered by a stimulus (*) mimicking the electrical stimulation of the internal capsule, in the WT and DS conditions. Bottom: probability distributions of the duration of oscillations in the circuit over 1,000 simulations with different realizations of the stochastic current. The mean oscillation duration in the nRT-TCVB circuit model in the DS condition is ~5 times that in the WT condition. The maximal conductances are as in Figure 1N.
(G, I, and K) Maps of the duration (G), burst number(I), and frequency (K) in the nRT-TCVB circuit model, as a function of CaT and SK maximal conductances, computed over 30 simulations with different realizations of noise. The mean of these averages in the WT and DS domains (white and red boundaries, respectively) are indicated.(H, J, and L) Plots of means ± SEMs of the duration(H), burst number (J), and frequency (L) using 9 samples with randomly maximal conductances drawn from the WT domain and 12 samples with randomly maximal conductances drawn from the DS domain, with groups compared using the Mann-Whitney U test (H, **p = 0.0062; J, *p = 0.0208; L, **p = 0.0002).
We then assessed the impact of reduced SK conductance at the circuit level in our computational model, including TCVB and nRT neurons (Figure 2E). Reduced SK elicited enhanced network oscillations (Figure 2F), displaying longer oscillations (Figures 2G and 2H), more bursts within each oscillation (Figures 2I and 2J), and lower oscillation frequency (Figures 2K and 2L) in the DS domain. Longer nRT bursts and the consequent TCVB hyperpolarization account for the counterintuitive decrease in the frequency of bursting in DS mice (Figures 2K and 2L).
In the WT domain (at normal SK conductance levels), a 10% reduction in the sodium maximal conductance did not affect burst AP number and Rin in the nRT neuron model (Figures S1J and S1K) or oscillation properties in the nRT-TCVB circuit model (Figures S1L–S1N) (i.e., it did not account for experimental observations). Moreover, reduced sodium conductance left unchanged the effects of reduced SK conductance in the DS domain in both the nRT and nRT-TCVB models (Figures S1J–S1N) (i.e., it did not preclude the mechanistic effect unraveled between reduced SK and the nRT-TCVB microcircuit hyperexcitability). Even a 50% decrease in the sodium maximal conductance did not affect these conclusions (data not shown). The model thus suggests that in nRT neurons, reduced SK, but not reduced sodium conductance, renders the thalamus “epileptic like” by enhancing thalamic rhythmogenesis.
“Silent” Non-convulsive Seizures in Human and Mouse DS
Given that the nRT is a powerful modulator of cortical rhythms (Steriade et al., 1993), we next asked whether enhanced bursting in the nRT-VB circuit could underlie seizures in DS mice. DS presents with a complex seizure phenotype that includes non-convulsive seizures (e.g., typical and atypical absence seizures) (Dravet, 2011). The cellular mechanisms underlying these seizures remain unknown (Dravet, 2011), and non-convulsive seizures have not been described in DS mouse models.
Thus, we set out to compare the seizure phenotype in DS mice to human DS patients (Figure 3). Non-convulsive seizures occurred in mice on average 15 ± 8 times per hour, lasting0.5–6 s (n = 9), and 7 ± 4 times per hour, lasting on average 10 s in DS patients (n = 3). Notably, non-convulsive seizures in DS mice and DS patients exhibited similar spectral electroencephalogram (EEG) signatures with fundamental frequencies of 4–7 Hz that varied, depending on the cortical region (Figures 3B, 3C, 3E, 3F, S3, and S4), characterized by poly-spike-and-wave components (Figure 3, note the similarities between 3C and 3G), which were different from the spike-and-wave discharges of typical absence seizures.
Figure 3. DS Mice Exhibit Non-convulsive Seizures Similar to Human Patients with DS.

(A) Diagram of the mouse electrocorticography (ECoG) and thalamic depth electrode recording montage. ECoG was recorded from somatosensory (S1) and visual (V1) cortices, with depth electrodes implanted in the thalamus.
(B) Top: example of an ECoG signature of a representative non-convulsive seizure in DS mice recorded from S1 (black), along with simultaneous multiunit (MU) recordings in the thalamus—in this case, the ventrobasal thalamus (orange). Inset: magnification of ECoG poly-spikes overlaid on a slower oscillation. Bottom: spectrogram showing frequency components of the recorded seizure.
(C) MU recordings in the somatosensory thalamus simultaneously with ECoG show in both nRT and VB clusters of APs (“population bursts” of AP firing) phase locked with ECoG poly-spikes.
(D) Power spectral density of the seizure shown in (B). Peak fundamental frequency is 6–7 Hz.
(E) Diagram of human EEG recording montage.
(F) Top: EEG signature of a non-convulsive seizure in a patient with DS recorded in two different locations on the scalp. Inset: spikes are not typical spike-and-wave discharges and have a higher frequency component in addition to the slower oscillation. Bottom: spectrogram showing frequency components of the recorded seizure.
(G) Expanded EEG shows epileptic activity characterized by a sequence of poly-spikes overlaid on a slower oscillation (compare with C).
(H) Power spectral density of the seizure shown in (E). Peak fundamental frequency of the depicted seizure is ~5 Hz, but it can vary across the different cortical regions for the same seizure (see Figures S3 and S4).
SK Enhancement Treats Non-convulsive Seizures in DS Mice
Recordings in nRT and VB showed high-frequency burst firing, phase locked to cortical poly-spikes during spontaneously occurring non-convulsive seizures (Figures 3B, 3C, and S5A–S5D) and during the convulsive hyperthermia-triggered seizures (Figures S5E and S5F) in freely behaving mice. Pharmacological boosting of SK channels in the nRT by acute infusion of EBIO reduced the number of spontaneous non-convulsive seizures (Figures 4A and 4B). Given the close apposition of nRT and VB, we cannot exclude the potential that intra-nRT infusions of EBIO also affected the VB. However, the lack of EBIO effects on the firing of TCVB neurons in vitro (Figures S2G and S2H) suggests that the abrogation of seizures mainly resulted from the effects of EBIO on the nRT. Next, we investigated more clinically tractable systemic injections of EBIO and found that these also reduced the number of seizures (Figures 4C–4F). Thus, pharmacological enhancers of ISK could be a treatment for non-convulsive seizures in DS.
Figure 4. Local or Systemic EBIO Treatment Reduces Non-convulsive Seizure Frequency in DS Mice.

(A) Diagram of targeting of the thalamus with guide cannulas and tungsten electrodes for unit recordings. Inset: depth electrodes were positioned in nRT.
(B) Quantification of effects of intra-nRT administration of EBIO (800 nL of 0.4 mM) on non-convulsive seizure number within 1 hr of infusion. Data are from n = 4 DS mice. *p = 0.0189, two-tailed t test.
(C) Diagram of systemic administration of EBIO (25 mg/kg, subcutaneously [s.c.]).
(D) Quantification of effects of systemic administration of EBIO (25 mg/kg, s.c.) on non-convulsive seizure number within 30 min of EBIO infusion. Data are from n = 9 DS mice. **p = 0.0039 Wilcoxon signed-rank test.
(E) Representative ECoG and multiunit activity (MUA) traces from one DS mouse after vehicle and EBIO treatments.
(F) Magnification of the recordings indicated with red line in (E). Note the lack of seizures in ECoG and switch from bursting to single spike (arrows) firing mode after EBIO injection.
Optogenetic Toggling of Thalamic Bursting Modulates Seizures in Scn1a-Deficient Mice
The nRT exerts its effects on cortical rhythms through its TC relay targets (Pinault, 2004), and non-convulsive seizures are prominent in the S1 cortex. Therefore, we hypothesized that if nRT → VB → S1 was causally involved in seizures, interrupting the bursting in VB should disrupt the seizures (Figure 5A). To test this, we perturbed VB firing using stable step function opsin (SSFO), a bistable opsin (Yizhar et al., 2011) that can switch the firing of TCVB cells from bursting to tonic mode (Sorokin et al., 2017). Unilateral SSFO activation in VB after seizure detection (Figure 5B) rapidly eliminated VB bursts, toggled tonic firing, and stopped ongoing seizures (Figures 5C–5E). Thus, VB bursting output may maintain non-convulsive seizures in DS. The light itself did not disrupt seizures (Figure S6), demonstrating that optical disruption of seizures requires SSFO activation.
Figure 5. Optogenetic Disruption of Thalamic Bursting Aborts Non-convulsive Seizures in DS Mice.

(A) Diagram showing that nRT modulates cortical rhythms through the relay thalamus.
(B) Diagram of optogenetic targeting of the thalamus. Inset: depth electrodes were positioned in VB thalamus.
(C) Top: example of optogenetic seizure interruption of an ongoing non-convulsive seizure in a DS mouse. The blue bar corresponds to a brief (50 ms) pulse of blue light to activate SSFO-expressing thalamocortical neurons. Unilateral activation of SSFO in the thalamus immediately interrupts the seizure. Bottom: spectrogram showing frequency components of the recorded seizure and decrease in power after SSFO activation.
(D) Magnification of the seizure interruption shown in (C). Activation of SSFO switches burst firing in the thalamus to tonic firing.
(E) Quantification of 1–25 Hz broadband power 2 s before and after optogenetic manipulation. Data are from 32 trials across 4 mice. ****p < 0.0001 Mann-Whitney test.
DISCUSSION
Here, we discovered that in DS, Scn1a deficiency and subsequent reduction of ISK in the nRT drives thalamic hyperexcitability to promote non-convulsive cortical seizures.
Less Is More: How Does Scn1a Deficiency in nRT Neurons Lead to Thalamic Network Hyperexcitability?
Our finding that GABAergic nRT neurons in DS mice have a more depolarized AP threshold is expected, and confirms previous studies (Kalume et al., 2015; Hedrich et al., 2014). However, we also found that nRT neurons exhibit an overall increase in excitability, displaying stronger spiking during depolarizing current pulses and stronger post-hyperpolarization rebound bursts of APs. This is at least in part due to a compensatory decrease in SK channel expression and consequent shift of the current balance toward inward currents and increased Rin. These changes were sufficient to maintain abnormal oscillations in the intrathalamic network formed by reciprocal connections between nRT and relay TC neurons in the somatosensory thalamus. Although many other compensations likely occur in DS, the reduction in ISK is particularly powerful because of its known role in thalamic rhythmogenesis. Our findings are in agreement with previous studies showing that blockade of ISK with apamin causes cellular and circuit hyperexcitability in thalamic slices (Kleiman-Weiner et al., 2009). The thalamic hypersynchrony due to blockade of ISK is not as strong as that obtained with GABA blockers (Kleiman-Weiner et al., 2009), which may explain how SK deficiency in nRT results in atypical spike-and-wave discharge absence seizures.
The identification of SK2 as a modifier of DS non-convulsive seizures adds to the list of genetic modifiers of SCN1A epilepsies (Calhoun et al., 2017; Hawkins and Kearney, 2016; Hawkins et al., 2016; Martin et al., 2007). Notably, we not only found that Kcnn2 is reduced in the nRT but also that pharmacological enhancement of ISK in nRT can treat non-convulsive seizures in our DS mouse model. Intra-cerebellar or systemic injections of the SK channel booster EBIO have previously been used to reduce burst firing in the cerebellum, to enhance motor performance, and to reduce dyskinesia in tottering mice (Walter et al., 2006); systemic administration of chlorzoxazone—a Food and Drug Administration (FDA)-approved activator of SK channels—has been shown to improve motor performance in mice (Gao et al., 2012) and in human patients (Feil et al., 2013). Although systemic EBIO treatment likely affects structures other than the nRT, our observation that local intra-nRT injections of EBIO are sufficient to treat seizures suggests that the thalamus could be a target to treat non-convulsive seizures in DS. Furthermore, TCVB neurons, known not to express SK2, exhibited normal firing properties in DS mice and were not affected by EBIO.
We propose the following working model of thalamic involvement in DS pathology: Scn1a deficiency leads to a compensatory reduction of ISK in nRT neurons, which contributes to enhanced Rin and a shift toward inward currents, which enhance the intrinsic excitability of nRT neurons in response to both hyperpolarization (rebound bursts) and depolarizing stimuli (Figure S6F). The increased excitability of nRT neurons results in enhanced bursts of APs following activation from either excitatory TCVB neurons or cortex. Enhanced output from nRT neurons leads to longer-lasting hyperpolarizations in the target TCVB neurons, resulting in stronger rebound bursting that re-excites nRT neurons. This cycle iterates to maintain non-convulsive seizures.
Our model fits within the framework of the dysfunctional cortex in DS. A hyperactive cortex could recruit the nRT via glutamatergic projections to amplify abnormal cortical inputs due to the intrinsic hyperexcitability of nRT cells (enhanced Rin) and via extensive projections from nRT onto sensory and limbic TC nuclei. It may be that the seizures initiate in the cortex, which in turn recruits the nRT and thalamus to maintain the seizure (Figure S5D). While further investigation is needed, it is clear that the thalamus, both nRT and VB, is required for seizure maintenance and is thus a promising target.
Treatment Strategies for DS: What Are the Implications of Enhanced Excitability of GABAergic nRT in Light of Reduced GABAergic Function in the Cortex?
Previous work on DS showed that sodium channel deficits in GABAergic neurons result in reduced firing, leading to the hypothesis that reduced network inhibition in the neocortex and hippocampus promotes convulsive seizures (Cheah et al., 2012; Dutton et al., 2013; Ogiwara et al., 2007; Tai et al., 2014; Yu et al., 2006). This model suggests that the optimal treatment of seizures would be to enhance firing of GABAergic neurons. However, our study shows that in contrast to the cortex, thalamic GABAergic neurons of the nRT show enhanced intrinsic excitability and rebound burst firing that result in thalamic network hyperexcitability. If cortical hyperexcitability is due to reduced firing of GABAergic cells, but thalamic hyperexcitability is due to enhanced firing of GABAergic cells, then drug treatments that enhance firing of GABAergic cells in both regions will not yield optimal results or may even worsen seizures. Moreover, given that thalamic modulation treats non-convulsive seizures and that the thalamus is implicated in sleep, cognition, and motor planning (Beenhakker and Huguenard, 2009; Saalmann and Kastner, 2015; Karussis et al., 2000; Guo et al., 2017), we propose that the thalamus could be an important target for treating non-convulsive seizures and perhaps associated comorbidities such as sleep impairments and cognitive dysfunction in DS. When designing new therapeutic strategies to enhance GABAergic neuron function, it will be important to keep in mind that these should be combined with drugs that reduce nRT bursting. Our results align with the recent finding that although reduced firing in GABAergic neurons occurs during development in DS, this change is temporary and does not contribute to the chronic epilepsy in DS mice (Favero et al., 2018).
What Are the Implications for Understanding DS Pathophysiology Beyond Seizures?
Mice lacking SK2 show fragmented sleep patterns and reduced sleep spindles (Cueni et al., 2008), similar to DS mice (Kalume et al., 2015), supporting the idea that reductions in SK2 underlie thalamic circuit dysfunction and sleep impairments in DS. Over-expressing SK2 increases sleep spindles and reduces sleep fragmentation (Wimmer et al., 2012), indicating that enhancement of SK function in DS may restore sleep impairments, in addition to treating seizures.
Here, we describe a cellular and circuit mechanism underlying non-convulsive seizures in DS and compare this phenomenon with human patients. Non-convulsive seizures are debilitating, account for the cognitive decline in human patients with DS (Guzzetta, 2011; Dalla Bernardina et al., 1986), and are comorbid with autism spectrum disorders in multiple mouse models (Holder and Quach, 2016; Jung et al., 2013; Gheyara et al., 2014). It remains to be elucidated which subnetworks of nRT, in addition to the somatosensory subnetwork, are affected in their firing pattern, and if so, how these pathological firing patterns may spread to the different thalamic nuclei. Unraveling the cognitive impact of the DS mutations requires cell-specific and inducible mouse models. Given the recently uncovered framework of nRT organization and function (Clemente-Perez et al., 2017) and the role of nRT SK channels in attention deficit disorder (Wells et al., 2016), we speculate that the nRT could be a key brain substrate and thus therapeutic target for the comorbid neurological (seizure) and psychiatric (autism) components of DS.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeanne Paz (jeanne.paz@gladstone.ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
DS mice
We performed all experiments per protocols approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco and Gladstone Institutes. Precautions were taken to minimize stress and the number of animals used in each set of experiments. Mice were separately housed after surgical implants. Mice modeling DS, Scn1aR1407X+/− mice (Ogiwara et al., 2007), hereafter referred to as DS mice, and WT littermates were used in this study and maintained on a mixed background (80%–88% C57BL6J (Jax 000664)/12%–20% C3HeB/FeJ (Jax 000658). DS mice that are homozygous for the Scn1a mutation die by age post-natal 15 (P15) and therefore were not studied here. Heterozygous DS mice have a high mortality rate between post-natal days 20 and 71 and a 31-fold higher risk of death than WT littermates (Gheyara et al., 2014). By 73 days, only 18% of Scn1aRX/+ mice remain alive (Gheyara et al., 2014) and could be recorded with chronic multioptrode devices. Both male and female mice were used for these experiments and ages ranged between postnatal days 18–80.
Human Subjects
All procedures were approved by the Regional and Institutional Committee of Science and Research Ethics of Scientific Council of Health. Per institutional guideline, this clinical case study was exempted from IRB approval. The total number of human subjects cited in this study (n = 3) are also reported in the text of the results. The subjects ages ranged from 5–7 years and included 1 boy and 2 girls.
METHOD DETAILS
Viral injections
Stereotaxic viral injections were carried out as described (Clemente-Perez et al., 2017, Paz et al., 2013, Paxinos and Franklin, 2012). Briefly, in WT or DS mice, adeno-associated virus carrying genes for eYFP alone (rAAV5/CamkIIa-eYFP) (7.4 ×1012 genome copies per ml)) or Stable Step Function Opsin (SSFO; rAAV5/CamkIIa-hChR2(C128S;D156A)-eYFP (5.2×1012 genome copies per ml)) was injected into the ventrobasal nucleus of the thalamus (VB) using the following coordinates: 1.6 mm posterior to Bregma, 1.6 lateral relative to midline, and two separate injections of 400 nL of virus at 3.2 and 3.5 mm ventral to the cortical surface (total injected volume, 800 nl). Viruses were acquired from (UNC Vector Core SCR_002448). We allowed the viruses to express for anywhere between 3 weeks and 6 months, depending on the nature of the experiment.
Immunostaining, microscopy, and image analysis
Mice were anesthetized with a lethal dose of ketamine (300 mg/kg) and xylazine (30 mg/kg) and perfused with 4% paraformaldehyde in 1× PBS. Serial coronal sections (50 μm thick) were cut on a Leica SM2000R Sliding Microtome. Sections were mounted in an antifade medium (Vectashield) and imaged using a Biorevo BZ-9000 Keyence microscope at 10×–20×. The expression of the viral constructs in different brain regions was confirmed using the mouse brain atlas (Paxinos and Franklin, 2012).
Slice preparation
Mice were euthanized with 4% isoflurane, perfused with ice-cold sucrose cutting solution containing 234 mM sucrose, 2.5 mM KCl, 1.25 mM NaH2PO4, 10 mM MgSO4, 0.5 mM CaCl2, 26 mM NaHCO3, and 11 mM glucose, equilibrated with 95% O2 and 5% CO2, pH 7.4, and decapitated. We prepared 250-μm (for patch-clamp electrophysiology) or 400-μm (for thalamic microcircuit studies)-thick horizontal thalamic slices containing VB thalamus and nRT with a Leica VT1200 microtome (Leica Microsystems). We incubated the slices, initially at 32°C for 1 h and then at 24–26°C, in artificial cerebro-spinal fluid (aCSF) containing 126 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2, 26 mM NaHCO3, and 10 mM glucose, equilibrated with 95% O2 and 5% CO2, pH 7.4. The thalamic slice preparation was performed as described (Paz et al., 2011, Clemente-Perez et al., 2017, Paz et al., 2013).
Patch-clamp electrophysiology from thalamic slices
Recordings were performed as described (Paz et al., 2011, Clemente-Perez et al., 2017, Paz et al., 2013). We visually identified nRT and TC neurons by differential contrast optics with a Zeiss (Oberkochen) Axioskop microscope and an infrared video camera. Recording electrodes made of borosilicate glass had a resistance of 2.5–4 MΩ when filled with intracellular solution. Access resistance was monitored in all the recordings, and cells were included for analysis only if the access resistance was < 25 MΩ. We corrected offline the potentials for −15 mV liquid junction potential. Intrinsic and bursting properties were recorded in the presence of kynurenic acid (2 mM, Sigma), the internal solution contained 67 mM potassium gluconate, 67 mM KCl, 1 mM MgCl2, 0.1 mM CaCl2, 10 mM HEPES, and 1.1 mM EGTA, 4mM ATP, 6mM phosphocreatine, pH adjusted to 7.4 with KOH (290 mOsm). For EPSCs and voltage-clamp recordings, the internal solution contained 120 mM potassium gluconate, 11 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, and 1 mM EGTA, pH adjusted to 7.4 with KOH (290 mOsm) recorded in the presence of picrotoxin (50 μM, Tocris). For EBIO recordings, 0.4 M EBIO was initially dissolved in DMSO and then diluted to 100 μM in aCSF containing kynurenic acid (2 mM) and picrotoxin (50 μM, Tocris). We only focused on studying the subset of cells in nRT that expresses IT and exhibits rebound burst firing which is the majority cell type in nRT (Clemente-Perez et al., 2017). Recordings with EBIO were done 5–10 minutes after drug application. IT measurements were conducted as described with steady-state inactivation protocols (Clemente-Perez et al., 2017). This protocol, which was established by the Huguenard lab, characterizes steady-state inactivation of IT by using depolarizing commands to activate near threshold (−65mV), well-clamped currents. While a reasonable voltage clamp is possible in at least some nRT cells in slices from young animals, the yield is much too low to allow us to assess the currents in our adult DS and WT slices. Instead, we show well-controlled voltage clamp of modestly sized IT, demonstrate that they have the prototypical slower decay that distinguishes them from relay cells. Using this protocol, we obtain an admittedly incomplete picture of IT properties. Nevertheless, we can reliably obtain inactivation slope and half-inactivation voltage, as well as amplitude of IT at our fixed test voltage (see (Paz et al., 2010) and (Clemente-Perez et al., 2017)).
For ISK recordings, apamin (100 nm final concentration) was diluted in aCSF containing the synaptic blockers kynurenic acid (2 mM, Sigma) and picrotoxin (50 μM, Tocris). Recordings for ISK measurements were done 5–10 minutes after drug application. ISK was calculated by digitally subtracting IT using measured in aCSF with and without apamin as described (Cueni et al., 2008, Wells et al., 2016). Currents were subtracted using Clampfit 10.5 (Molecular Devices, SCR_011323).
Extracellular thalamic oscillations
We placed horizontal slices (400 μm) containing somatosensory thalamus in an interface chamber at 34°C and superfused them at a rate of 2 mL min−1 with oxygenated aCSF (same as recipe used for patch-clamp electrophysiology, except supplemented with 0.3 mM glutamine for cellular metabolic support). Extracellular multiunit activity (MUA) recordings were obtained with a linear 16-channel multi-electrode array (Neuronexus) that spanned the nRT and VB. MUA signals were amplified 10,000 times and band-pass filtered between 100 Hz and 6 kHz using the RZ5 from Tucker-Davis Technologies (TDT, SCR_006495). Position of recording array was visually checked for each recording to confirm position of electrodes in nRT and VB. We delivered electrical stimuli to the internal capsule with a pair of tungsten microelectrodes (50–100 kΩ, FHC). The stimuli were 100 μs in duration, 50 V in amplitude, and delivered once every 30 s.
We then used custom MATLAB software to analyze thalamic MUA recordings.
Detection of Kcnn2 mRNA in brain tissue
DS mice and WT littermate control mice (age 3–5 months) were euthanized with Fatal Plus, and nRT were microdissected in ice-cold sucrose cutting solution. Tissue was immediately flash-frozen and then RNA was isolated using QIAGEN RNeasy kits with on-column DNase digest. Superscript First-Strand cDNA synthesis kits were used to convert RNA into cDNA. qRT-PCR experiments were performed using the following primers for mouse Kcnn2S (short transcript variant, Genbank: NM_080465.2): F- 5′ AGT GGT CTG GAG GAA GAG GA 3′ and R- 5′ CAT TTG CAC GTT CTC CCG AA 3′ and mouse Kccn2L (long transcript variant, Genbank: NM_001312905.1): F-5′ TGG TTA CAG ACT GAG ACT CTT GT 3′ and R- 5′ TCC TCT TCC TCC AGA CCA CT 3′ and the mouse house-keeping gene Gapdh (Genbank: NM_001289726.1 and Genbank: NM_008084.3): F- 5′ GGT CGG TGT GAA CGG ATT TG 3′ and R- 5′ GCA ACA ATC TCC ACT TTG CC 3′. Reactions were performed using Sybr Green Master Mix (Applied Biosystems) on a 7900HT Fast Thermal Cycler (Applied Biosystems) and levels of Kcnn2 mRNA were determined using the ΔΔCT method.
Surgical implantation of devices for simultaneous recording of seizure activity and manipulation of brain regions
The devices for simultaneous ECoG, MUA recordings and optical or pharmacological manipulations in freely behaving mice were all custom made in the Paz lab. In general, recordings were optimized for assessment of somatosensory subnetworks (S1 cortex and somatosensory nRT) and thus do not necessarily generalize to other nRT domains, which should be investigated in the future. For optogenetic studies, we designed devices containing multiple screws for acquisition of ECoG signal, along with local field potential (LFP) and MUA signal recoded from tungsten electrode wires that were positioned approximately 250 and 500 mm from the tip of an optic fiber (200 μm core, ThorLabs) (Paz et al., 2013, Clemente-Perez et al., 2017).
Cortical screws were implanted bilaterally in S1 (−0.5 mm posterior from Bregma, ± 3.25 mm lateral), and bilaterally in V1 (−2.9 mm posterior from Bregma, ± 3.25 mm lateral). For optogenetic manipulation of VB, optrodes were implanted at −1.5 posterior from Bregma, 1.5 mm lateral, with the tips of the optical fiber and electrodes at 3.0 mm, 3.25 and 3.5 mm ventral to the cortical surface, respectively.
Mice were also instrumented for local infusion of drug into nRT using cannulas (Plastics One). Briefly, we made custom-built devices in which a guide cannula was attached to our devices for recording ECoG. Tungsten wires were glued to the guide cannula, so that they protruded 250 and 400 mm from the tip of the insert cannula (500 μM protrusion from tip of guide cannula). To target nRT, guide cannulas were implanted at −1.3 mm posterior from Bregma, 2 mm lateral, and 2.5 mm deep.
Mice were allowed to recover for at least 1 week before recording. ECoG and thalamic LFP signals were recorded using RZ5 (TDT) and sampled at 1221 Hz, with thalamic MUA signals sampled at 24 kHz. A video camera that was synchronized to the signal acquisition was used to continuously monitor the animals. Animals were briefly anesthetized with ~2% isoflurane at the start of each recording to connect for recording. Each recording trial lasted 30–180 min. To control for circadian rhythms, we housed our animals using a regular light/dark cycle, and performed recordings between roughly 11:00 AM and 6:00 PM. All the recordings were performed during wakefulness. The location of the optrodes or cannulas was validated by histology after euthanasia in mice that did not experience sudden death and whose brains we were able to recover and process.
ECoG spectral analysis
We used the continuous wavelet transformation for spectral analysis to decompose signals into both time and frequency (Sorokin et al., 2016, 2017). We used a basis of Morlet wavelets from 1–128 Hz with seven octaves and 10 wavelets per octave(Sorokin et al., 2017). Video monitoring was used to distinguish non-convulsive and convulsive seizures.
In vivo optogenetics during free behavior
We simultaneously passed a fiber optic with an inline rotating joint (Doric) through a concentric channel in the electrical commutator, and connected it to the 200-μm core fiber optic in each animal’s headpiece while recording ECoG/MU. The fiber optic was connected to 450- and 532-nm wavelength laser control boxes, which were triggered externally using the RZ5 (TDT). The tip of the optical fiber rested 100–500 mm from the most ventral tungsten electrode on each optrode to allow maximal activation of the desired brain region. We used 5–35 mW of laser power, measured at the end of the optical fiber before connecting to the animals. Light was turned on by the scientist upon visual seizure detection.
Pharmacological manipulations during free behavior
All animals were habituated to the recording chamber prior to drug studies. To determine the effects of systemic EBIO administration on non-convulsive seizure frequency in DS mice, we used a randomized-block design to record half of mice on test Day 1 with vehicle (s.c. 250 μl of 10% DMSO/90% saline) and half of the mice with EBIO (s.c. 25 mg/kg, dissolved in 10% DMSO and 90% saline) (Wells et al., 2016). We then reversed the drug condition for test day 2 (which was 48 h later). Non-convulsive seizures were manually scored offline by an experienced user during the first 30 min of vehicle or EBIO application. Note, as EBIO also changed the brain state (more time spent in stage II sleep), the user could not be blinded to the experimental condition.
To determine the effects of local infusion of EBIO into the nRT, we implanted mice with cannulas (Plastics One) targeting the right nRT. After recovery from surgery, 800 nL of vehicle (0.25% DMSO in saline) was infused into the right nRT at a rate of 200 nL per minute using a syringe pump. ECoG and thalamic LFP/MUA were then recorded for 1 h post drug administration. EBIO (400 μM, 0.0015% DMSO in saline) was then infused into the right nRT and the ECoG, and thalamic LFP/MUA was recorded for an additional hour. Non-convulsive seizures were manually scored offline by an experienced user during the first 60 min of vehicle or EBIO application.
Human Seizure Recordings
All procedures were approved by the Regional and Institutional Committee of Science and Research Ethics of Scientific Council of Health. Per institutional guideline, this clinical case study was exempted from IRB approval. The original video-EEG recording of three patients with DS associated with SCN1A pathogenic variant was reviewed by an experienced pediatric neurophysiologist (MRC). The international 10–20 system was used for electrode placement. From our cohort of patients with DS, we selected patients in whom long-term video-EEG recording was performed (> 19 hours) and non-convulsive seizures were recorded. Atypical absences where defined as discharge of irregular high voltage spikes and slow waves accompanied by decrease awareness and at time eyelids and/or upper limb slight myoclonic jerks.
Computational modeling
A computational model of the negative feedback loop between TC neurons and thalamic reticular nucleus (nRT) neurons was built to assess the oscillatory response of thalamic slices in WT and DS mice. Excitability in TC and nRT neurons was described using the Hodgkin-Huxley formalism, based on previous modeling (Paz et al., 2013, Destexhe et al., 1994), with some parameters fitted from present intracellular recordings. The nRT model included the leak, fast sodium, delayed rectifier potassium, low-threshold calcium (CaT), calcium-activated potassium (SK) and calcium-activated non-specific cationic (CAN) currents, and the TC model incorporated the leak, fast sodium, delayed rectifier potassium, low-threshold calcium (CaT) and hyperpolarization-activated (H) currents. Both neuron models included first-order calcium dynamics. The negative feedback loop between TC and nRT neurons was implemented with AMPA and NMDA currents at synapses from TC to nRT neurons and GABA–A and GABA–B currents at synapses from nRT to TC neurons. A noise current with zero mean was introduced in both nRT and TC neurons to account for the stochasticity observed experimentally in the thalamic circuit. The WT domain was defined, in the space of CaT and SK maximal conductances of the nRT neuron, as the parameter region yielding realistic nRT action potential firing frequencies and circuit oscillations with burst numbers and durations consistent with those obtained in slices. The DS domain was defined as the region with a similar CaT maximal conductance range but with lower SK maximal conductance, consistent with the smaller ISK found in intracellular nRT recordings in DS mice.
Thalamic reticular nucleus neuron
The membrane potential of the nRT neuron model followed , where the leak current was . The action potential currents and were derived from [(Traub et al., 1991)], with doubled kinetics and a leftward shift of sodium inactivation and potassium activation voltage–dependences (−10 mV and −20 mV, respectively) to qualitatively reproduce the action potentials’ properties observed experimentally in mice nRT neurons. The T–type calcium current was derived from (Destexhe et al., 1994), with activation and inactivation kinetics respectively slowed by factors 10 and 2, to account for voltage–clamp data observed (~200 ms inactivation at −75 mV) and reversal potential computed as , with the extracellular calcium concentration, R the gas constant, T the absolute temperature and F the Faraday constant. The SK–type calcium–activated potassium current was , with second order activation(Destexhe et al., 1994),(Adelman et al., 2012) following with and . The non–specific cationic CAN current followed: , with first-order activation , with and . Calcium dynamics followed first-order dynamics , where the surface to volume ratio is with the soma radius and the width of a sub–membrane shell where calcium freely diffuses, is the basal intracellular calcium concentration and the intracellular calcium time constant. Parameters were: CnRT = 1μFcm−2, , , , , , , , and as detailed in legends, , , , , , , , , , , .
TC neuron
The model of TC neurons was as described(Paz et al., 2013), with: and parameters corresponding to the physiological condition.
Synaptic architecture
The negative feedback loop between TC and nRT neurons was implemented as the interaction of one RTN and one TC neuron. The TC→nRT connexion comprised AMPA and NMDA components. The AMPA current was with glutamatergic activation kinetics following first order deactivation with time constant τAMPA and soft–bound increment at every TC action potential: , where δ() is the Dirac function and represents TC spike arrival times. The NMDA current was with a glutamatergic activation scheme similar to that of the current and where the voltage–dependence was taken from (Lacey et al., 2012). The nRT→TC connexion comprised GABA–A and GABA–B components with formalisms similar to that of AMPA currents. To model synaptic interactions between TC and nRT thalamic populations, every spike of each model neuron evoked ten spikes in the other neuron of the model, with delays uniformly distributed within 1–3 ms. Synaptic parameters were , VAMPA = 0mV, τAMPA = 1ms, ΔmAMPA = 0.25, , VNMDA = 0mV, τNMDA = 75ms, ΔmNMDA = 0.1, , VGABA−A = −95mV, τGABA−A = 1ms, ΔmGABA−A = 0.25, , VGABA−B = −95mV, τGABA−B = 75ms, ΔmGABA−B = 0.1.
Source of noise in nRT and TC neurons
We introduced a source of noise in nRT and TC neurons. Multiple sources of noise exist in real neurons, notably due to the stochasticity in the gating dynamics of membrane and synaptic channels. Introducing stochasticity allows network oscillations to end at one point – because of membrane voltage fluctuations – as observed experimentally (without noise, oscillations are endless in the model, once triggered). To do so, we added to nRT and TC neurons a small stochastic current mimicking noisy fluctuations in intrinsic and synaptic currents, , where time–varying conductances were built as standard normally–distributed random processes, filtered with time constant τNoise and normalized to admit a given standard deviation σNoise. Note that these conductance had zero mean, so they had no net depolarizing/hyperpolarizing effect on the membrane potential of nRT and TC neurons per se and their sole influence was to introduce stochasticity. The time–varying conductances affecting nRT and TC neurons, and , were independent. Stochastic parameters were σNoise = 10−3mScm−2, VNoise = 0mV, τNoise = 20ms.
Simulation protocols
In simulations mimicking intracellular recordings in nRT neurons, all synaptic conductances and the noise conductance were set to zero, the TC model was not computed and the nRT neuron was stimulated with for 500 ms, as applied experimentally. In network simulations, the optogenetic stimulation applied to TC neurons was mimicked with for 25 ms.
Definition of WT and DS domains in the parameter space
To define WT and DS domains, we first selected the range in which the nRT neuron model produced realistic action potential firing frequencies in the range 50–230 Hz, as found experimentally. This domain corresponded to . In this range of realistic nRT spiking frequencies, the WT domain was operationally defined as values for which the nRT–TC network model produced oscillations ranging within 5–35 bursts and lasting within 0.5–6 s, as found experimentally. These criteria defined a domain corresponding to large values (~1 – 2.5mScm−2), with large AHP repolarizations, as found observed in intracellular nRT recordings (Figure 1). The DS domain was then defined – within the range of realistic nRT spiking frequencies – as models with values smaller than those found in the WT domain, consistent with the smaller size of the ISK in intracellular nRT recordings of DS mice (Figure 1H,I).
Numerical simulations
Ordinary differential equations were solved using the forward Euler scheme with 0.01 ms time step. Simulated time was 25 s maximum and was interrupted after 300 ms in the absence of discharge. The maps for circuit simulations were obtained taking the mean of 30 repetitions of the network simulations with different noise realizations. The specific quantitative details of the results obtained obviously depended on parameter values but the behaviors observed were qualitatively very robust with regard to variations in intrinsic and synaptic parameter.
QUANTIFICATION AND STATISTICAL ANALYSIS
All numerical values are given as means and error bars are standard error of the mean (SEM) unless stated otherwise. Data analysis was performed with MATLAB (SCR_001622), Origin 9.0 (Microcal Software, SCR_002815), GraphPad Prism 6 (SCR_002798), and SigmaPlot (SCR_003210).
Comparison of thalamic circuit oscillations in thalamic slices
We used custom MATLAB software to detect extracellular spikes based on taking the first derivative of the MU signal and thresholding over background. Spikes were excluded if their waveform lasted 2 ms longer. An experienced user confirmed parameter settings to optimize for all recordings. Bursts were defined as three or more spikes per burst, with the max interburst interval as 1.2 s. We quantified the number of total evoked bursts and oscillation duration recorded in the most active VB channels using on average 10–20 sweeps per recording. All data were compared using a Mann-Whitney test, with alpha = 0.05. (*p < 0.05, **p < 0.01, ***p < 0.001).
Analysis of electrophysiological properties
The input resistance (Rin) and membrane time constant (τm)
were measured from the membrane hyperpolarizations in response to low intensity current steps (−20 to −60 pA) in linear portion of the V–I plots. Rin and τm were compared with Mann-Whitney test, with α = 0.05, (ns, non-significant, p > 0.05) using GraphPad Prism 6 (SCR_002798).
T currents (IT)
Data for IT half-maximal voltage (V50%) were taken from the Boltzmann function calculated using Origin 9.0 (Microcal Software, SCR_002815). Most data were compared using a Mann-Whitney test, with alpha = 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001). Number of APs on first burst measured against hyperpolarization step was compared through individual unpaired t tests not corrected for multiple comparisons, with alpha = 0.05, (*p < 0.05, **p < 0.01, ***p < 0.001). Mann-Whitney tests and individual unpaired t tests were done using GraphPad Prism 6 (SCR_002798).
SK currents (ISK)
were recorded at a constant holding voltage following a hyperpolarizing step to gate IT as described (Cueni et al., 2008). Apamin subtraction was carried out by digitally subtracting averaged traces (typically 2 – 5 sweeps) obtained following bath application of 100 nM apamin. This subtraction protocol to measure ISK was adapted from (Cueni et al., 2008). We assessed the quality and stability of voltage–clamp control in these recordings as described (Cueni et al., 2008). First, we tested whether apamin–sensitive currents, obtained by digital subtraction, remained stable for the period required to perform application of pharmacological substances via the bath or through the recording pipette (~10 min). Second, we carried out the subtraction when all recording conditions, such as input resistance, series resistance and capacitive currents remained unaltered before and after 100 nM apamin application. See also Figures S1G and S1H.
What is the advantage of using a Steady-state inactivation (SSIT) protocol for quantifying IT and ISK?
SSIT used to measure IT allows us to have a well-controlled voltage-clamp as shown by the fact that the decay time constants (orange lines show decay exponential fit) of the IT do not vary depending on the voltage step used to deinactivate the IT (see Figure S1G). The voltage-clamp was verified before and after apamin application, and cells that showed more than 10% of variation in voltage clamp were excluded from the study. Although IT is small with this SSIT protocol, it has a major advantage of offering a well-controlled voltage-clamp which allows a reliable quantification of IT, which is well-established for measuring IT in brain slices from adult animals (see (Paz et al., 2010)). Note that the subtraction by this optimized SSIT method, with small amplitude IT, does not require clipping of the subtraction artifact as has been used in previous studies. Thus, the kinetics of the ISK activation can be obtained without artifact using the SSIT protocol.
Analysis of cortical rhythm modulation by selective optical activation of thalamic neurons during free behavior
ECoG spectral analysis
Spectrogram images in Figures 3 and 5 were generated for frequencies between 0 and 25 Hz using the short-time Fourier transform with 0.5 s Hamming windows and 98% overlap between segments. The power spectral density estimates were plotted as the mean values of the spectrogram at each point at 0–25 Hz during the seizure event.
Optogenetic seizure interruption analysis
To compare the effects of light on EcoG power, we calculated relative changes in averaged band power between pre-stimulus (PreStim, 2 s before light pulse) and post-stimulus (PostStim, 2 s after end of light pulse) periods across multiple recording trials (Figure 5). Average power at 1–25 Hz was calculated using a modified periodogram. The difference between groups was analyzed using a Mann-Whitney rank sum test (p < 0.005).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| rAAV5/Camklla-eYFP | UNC Vector Core | N/A |
| rAAV5/Camklla-hChR2(C128S;D156A)-eYFP | UNC Vector Core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1 -EBIO (1 -Ethyl-2-benzimidazolinone) | Tocris | Cat. # 1041 |
| Apamin | Sigma | Cat. # A1289 |
| Kynurenic Acid | Sigma | Cat. # K3375 |
| Picrotoxin | Tocris | Cat. #1128/1G |
| Experimental Models: Organisms/Strains | ||
| Mouse: Scn1aR1407+/− C57BL6J:C3HeB/FeJ | (Ogiwara et al., 2007) | N/A |
| Oligonucleotides | ||
| Forward Primer for Kcnn2S: F- 5′ AGT GGT CTG GAG GAA GAG GA 3′ | This paper | Custom synthesis |
| Reverse Primer for Kcnn2S: R- 5′ CAT TTG CAC GTT CTC CCG AA 3′ | This paper | Custom synthesis |
| Forward Primer for Kcnn2L: F-5′ TGG TTA CAG ACT GAG ACT CTT GT 3′ | This paper | Custom synthesis |
| Reverse Primer for Kcnn2L: R- 5′ TCC TCT TCC AGA CCA CT 3′ | This paper | Custom synthesis |
| Forward Primer for Gapdh: F- 5′ GGT CGG TGT GAA CGG ATT TG 3′ | This paper | Custom synthesis |
| Reverse Primer for Gapdh: R- 5′ GCA ACA ATC TCC ACT TTG CC 3′ | This paper | Custom synthesis |
| Software and Algorithms | ||
| Clampfit 10.5 | Molecular Devices | SCR_011323 |
| MATLAB | Mathworks | SCR_001622 |
| Origin 9.0 | OriginLab | SCR_002815 |
| Graphpad Prism 6 | Graphpad | SCR_002798 |
| SigmaPlot | SigmaPlot | SCR_003210 |
Highlights.
Reticular thalamic neurons (nRT) are hyperexcitable in Dravet syndrome (DS)
This results from a potassium SK channel deficiency
Boosting SK in nRT neurons treats DS non-convulsive seizures
Disrupting bursting of thalamocortical neurons stops DS nonconvulsive seizures
ACKNOWLEDGMENTS
S.R.-M. is supported by the American Epilepsy Society and the Dravet Syndrome Foundation Postdoctoral Research Fellowship. B.H. is supported by the American Epilepsy Society Postdoctoral Research Fellowship. J.T.P. is supported by NIH/NINDS R01NS096369, the DoD, the NSF, Gladstone Institutes, the Kavli Institute for Fundamental Neuroscience, the Michael Prize, and the Jewett Foundation. A.C.-P. and F.S.C. are supported by NSF Graduate Research Fellowships. S.S.H. is supported by the ARCS Foundation and Weill Institute Foundation scholarships. O.H.J.E.R. and F.E.H. are supported by Dutch NWO-ALW VIDI 016.121.346 and ZON-MW TOPGO.L.10.066 and the Erasmus MC MRace. M.-C.C. is supported by the Belgian American Education Foundation. We thank Scott Brovarney and Juan Alcauter for help with animal husbandry and technical help with histology. We thank Jordan Sorokin and John Huguenard for sharing their MATLAB code for the analysis of evoked thalamic oscillations. We thank Meredith Calvert from the Gladstone Histology & Light Microscopy Core for experimental and technical support. We thank Drs. Daniel Lowenstein and Adam Numis for their feedback on the interpretation of the human EEG and Brett Mensh for critical feedback on the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.018.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adelman JP, Maylie J, and Sah P (2012). Small-conductance Ca2+-activated K+ channels: form and function. Annu. Rev. Physiol 74, 245–269. [DOI] [PubMed] [Google Scholar]
- Auerbach DS, Jones J, Clawson BC, Offord J, Lenk GM, Ogiwara I, Yamakawa K, Meisler MH, Parent JM, and Isom LL (2013). Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One 8, e77843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthó P, Slézia A, Mátyás F, Faradzs-Zade L, Ulbert I, Harris KD, and Acsády L (2014). Ongoing network state controls the length of sleep spindles via inhibitory activity. Neuron 82, 1367–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhenov M, Timofeev I, Steriade M, and Sejnowski T (2000). Spiking-bursting activity in the thalamic reticular nucleus initiates sequences of spindle oscillations in thalamic networks. J. Neurophysiol 84, 1076–1087. [DOI] [PubMed] [Google Scholar]
- Beenhakker MP, and Huguenard JR (2009). Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron 62, 612–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun JD, Hawkins NA, Zachwieja NJ, and Kearney JA (2017). Cacna1g is a genetic modifier of epilepsy in a mouse model of Dravet syndrome. Epilepsia 58, e111–e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, Rubenstein JL, and Catterall WA (2012). Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 109, 14646–14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, and De Jonghe P (2001). De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet 68, 1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes LR, Deprez L, Suls A, Baets J, Smets K, Van Dyck T, Deconinck T, Jordanova A, and De Jonghe P (2009). The SCN1A variant database: a novel research and diagnostic tool. Hum. Mutat 30, E904–E920. [DOI] [PubMed] [Google Scholar]
- Clemente-Perez A, Makinson SR, Higashikubo B, Brovarney S, Cho FS, Urry A, Holden SS, Wimer M, Dávid C, Fenno LE, et al. (2017). Distinct Thalamic Reticular Cell Types Differentially Modulate Normal and Pathological Cortical Rhythms. Cell Rep 19, 2130–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F (1984). Function of the thalamic reticular complex: the searchlight hypothesis. Proc. Natl. Acad. Sci. USA 81, 4586–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueni L, Canepari M, Luján R, Emmenegger Y, Watanabe M, Bond CT, Franken P, Adelman JP, and Lüthi A (2008). T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat. Neurosci 11, 683–692. [DOI] [PubMed] [Google Scholar]
- Dalla Bernardina B, Capovilla G, and Chiamenti C (1986). Myoclonic epilepsies of infancy and early childhood: nosological and prognostic approach In Advances in Epileptology, Wada JA and Penry JK, eds. (Raven Press; ), pp. 175–179. [Google Scholar]
- Deschênes M, Paradis M, Roy JP, and Steriade M (1984). Electrophysiology of neurons of lateral thalamic nuclei in cat: resting properties and burst discharges. J. Neurophysiol 51, 1196–1219. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Sejnowski TJ, and Steriade M (1994). A model of spindle rhythmicity in the isolated thalamic reticular nucleus.J. Neurophysiol 72, 803–818. [DOI] [PubMed] [Google Scholar]
- Dravet C (2011). The core Dravet syndrome phenotype. Epilepsia 52 (Suppl 2), 3–9. [DOI] [PubMed] [Google Scholar]
- Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, and Escayg A (2013). Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol. Dis 49, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero M, Sotuyo NP, Lopez E, Kearney JA, and Goldberg EM (2018). A Transient Developmental Window of Fast-Spiking Interneuron Dysfunction in a Mouse Model of Dravet Syndrome. J. Neurosci 38, 7912–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil K, Claaßen J, Bardins S, Teufel J, Krafczyk S, Schneider E, Schniepp R, Jahn K, Kalla R, and Strupp M (2013). Effect of chlorzoxazone in patients with downbeat nystagmus: a pilot trial. Neurology 81, 1152–1158. [DOI] [PubMed] [Google Scholar]
- Ferrarelli F, and Tononi G (2011). The thalamic reticular nucleus and schizophrenia. Schizophr. Bull 37, 306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Todorov B, Barrett CF, van Dorp S, Ferrari MD, van den Maagdenberg AMJM, De Zeeuw CI, and Hoebeek FE (2012). Cerebellar ataxia by enhanced Ca(V)2.1 currents is alleviated by Ca2+-dependent K+-channel activators in Cacna1a(S218L) mutant mice. J. Neurosci 32, 15533–15546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentet LJ, and Ulrich D (2003). Strong, reliable and precise synaptic connections between thalamic relay cells and neurones of the nucleus reticularis in juvenile rats. J. Physiol 546, 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheyara AL, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W, Finucane MM, Sanchez PE, and Mucke L (2014). Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol 76, 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ZV, Inagaki HK, Daie K, Druckmann S, Gerfen CR, and Svoboda K (2017). Maintenance of persistent activity in a frontal thalamocortical loop. Nature 545, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzzetta F (2011). Cognitive and behavioral characteristics of children with Dravet syndrome: an overview. Epilepsia 52 (Suppl 2), 35–38. [DOI] [PubMed] [Google Scholar]
- Halassa MM, and Acsády L (2016). Thalamic Inhibition: Diverse Sources, Diverse Scales. Trends Neurosci 39, 680–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Siegle JH, Ritt JT, Ting JT, Feng G, and Moore CI (2011). Selective optical drive of thalamic reticular nucleus generates thalamic bursts and cortical spindles. Nat. Neurosci 14, 1118–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NA, and Kearney JA (2016). Hlf is a genetic modifier of epilepsy caused by voltage-gated sodium channel mutations. Epilepsy Res 119, 20–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NA, Zachwieja NJ, Miller AR, Anderson LL, and Kearney JA (2016). Fine Mapping of a Dravet Syndrome Modifier Locus on Mouse Chromosome 5 and Candidate Gene Analysis by RNA-Seq. PLoS Genet 12, e1006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich UBS, Liautard C, Kirschenbaum D, Pofahl M, Lavigne J, Liu Y, Theiss S, Slotta J, Escayg A, Dihné M, et al. (2014). Impaired action potential initiation in GABAergic interneurons causes hyperexcitable networks in an epileptic mouse model carrying a human Na(V)1.1 mutation. J. Neurosci 34, 14874–14889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder JL Jr., and Quach MM (2016). The spectrum of epilepsy and electroencephalographic abnormalities due to SHANK3 loss-of-function mutations. Epilepsia 57, 1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser CR, Vaughn JE, Barber RP, and Roberts E (1980). GABA neurons are the major cell type of the nucleus reticularis thalami. Brain Res 200, 341–354. [DOI] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, and Huguenard JR (1999). Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science 283, 541–543. [DOI] [PubMed] [Google Scholar]
- Jahnsen H, and Llinás R (1984). Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. J. Physiol 349, 205–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Seo JS, Kim BS, Lee D, Jung K-H, Chu K, Lee SK, and Jeon D (2013). Social deficits in the AY-9944 mouse model of atypical absence epilepsy. Behav. Brain Res 236, 23–29. [DOI] [PubMed] [Google Scholar]
- Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, and Catterall WA (2013). Sudden unexpected death in a mouse model of Dravet syndrome. J. Clin. Invest 123, 1798–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F, Oakley JC, Westenbroek RE, Gile J, de la Iglesia HO, Scheuer T, and Catterall WA (2015). Sleep impairment and reduced interneuron excitability in a mouse model of Dravet syndrome. Neurobiol. Dis 77, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karussis D, Leker RR, and Abramsky O (2000). Cognitive dysfunction following thalamic stroke: a study of 16 cases and review of the literature.J. Neurol. Sci 172, 25–29. [DOI] [PubMed] [Google Scholar]
- Kleiman-Weiner M, Beenhakker MP, Segal WA, and Huguenard JR (2009). Synergistic roles of GABAA receptors and SK channels in regulating thalamocortical oscillations. J. Neurophysiol 102, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey CJ, Bryant A, Brill J, and Huguenard JR (2012). Enhanced NMDA receptor-dependent thalamic excitation and network oscillations in stargazer mice. J. Neurosci 32, 11067–11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinson CD, Tanaka BS, Sorokin JM, Wong JC, Christian CA, Goldin AL, Escayg A, and Huguenard JR (2017). Regulation of Thalamic and Cortical Network Synchrony by Scn8a. Neuron 93, 1165–1179.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, and Escayg A (2007). The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum. Mol. Genet 16, 2892–2899. [DOI] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Take-uchi T, Itohara S, Yanagawa Y, Obata K, et al. (2007). Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci 27, 5903–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, Yanagawa Y, Tamamaki N, Hensch TK, Itohara S, and Yamakawa K (2013). Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum. Mol. Genet 22, 4784–4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, Makinson CD, Christopher Ehlen J, Tufik S, Decker MJ, Paul KN, and Escayg A (2013). Altered sleep regulation in a mouse model of SCN1A-derived genetic epilepsy with febrile seizures plus (GEFS+). Epilepsia 54, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, and Franklin K (2012). Mouse Brain in Stereotaxic Coordinates (Elsevier Academic Press; ). [Google Scholar]
- Paz JT, and Huguenard JR (2015). Microcircuits and their interactions in epilepsy: is the focus out of focus? Nat. Neurosci 18, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Christian CA, Parada I, Prince DA, and Huguenard JR (2010). Focal cortical infarcts alter intrinsic excitability and synaptic excitation in the reticular thalamic nucleus. J. Neurosci 30, 5465–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Bryant AS, Peng K, Fenno L, Yizhar O, Frankel WN, Deisseroth K, and Huguenard JR (2011). A new mode of corticothalamic transmission revealed in the Gria4(−/−) model of absence epilepsy. Nat. Neurosci 14, 1167–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Davidson TJ, Frechette ES, Delord B, Parada I, Peng K, Deisseroth K, and Huguenard JR (2013). Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat. Neurosci 16, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D (2004). The thalamic reticular nucleus: structure, function and concept. Brain Res. Rev 46, 1–31. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, and Catterall WA (2015). Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain 138, 2219–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saalmann YB, and Kastner S (2015). The cognitive thalamus. Front. Syst. Neurosci 9, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaght SJ, Leresche N, Deniau J-M, Crunelli V, and Charpier S (2002). Activity of thalamic reticular neurons during spontaneous genetically determined spike and wave discharges. J. Neurosci 22, 2323–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin JM, Paz JT, and Huguenard JR (2016). Absence seizure susceptibility correlates with pre-ictal b oscillations. J. Physiol. Paris 110 (4 Pt A), 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin JM, Davidson TJ, Frechette E, Abramian AM, Deisseroth K, Huguenard JR, and Paz JT (2017). Bidirectional Control of Generalized Epilepsy Networks via Rapid Real-Time Switching of Firing Mode. Neuron 93, 194–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Domich L, Oakson G, and Deschênes M (1987). The deafferented reticular thalamic nucleus generates spindle rhythmicity. J. Neurophysiol 57, 260–273. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, and Sejnowski TJ (1993). Thalamocortical oscillations in the sleeping and aroused brain. Science 262, 679–685. [DOI] [PubMed] [Google Scholar]
- Tai C, Abe Y, Westenbroek RE, Scheuer T, and Catterall WA (2014). Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 111, E3139–E3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Wong RK, Miles R, and Michelson H (1991). A model of a CA3 hippocampal pyramidal neuron incorporating voltage-clamp data on intrinsic conductances. J. Neurophysiol 66, 635–650. [DOI] [PubMed] [Google Scholar]
- von Krosigk M, Bal T, and McCormick DA (1993). Cellular mechanisms of a synchronized oscillation in the thalamus. Science 261, 361–364. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alviña K, Womack MD, Chevez C, and Khodakhah K (2006). Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat. Neurosci 9, 389–397. [DOI] [PubMed] [Google Scholar]
- Wells MF, Wimmer RD, Schmitt LI, Feng G, and Halassa MM (2016). Thalamic reticular impairment underlies attention deficit in Ptchd1(Y/-) mice. Nature 532, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer RD, Astori S, Bond CT, Rovó Z, Chatton J-Y, Adelman JP, Franken P, and Lüthi A (2012). Sustaining sleep spindles through enhanced SK2-channel activity consolidates sleep and elevates arousal threshold.J. Neurosci 32, 13917–13928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, et al. (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, and Catterall WA (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci 9, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Zhang Z-W, Zak JD, and Liu H (2010). MeCP2 is required for normal development of GABAergic circuits in the thalamus. J. Neurophysiol 103, 2470–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.