

Abstract
The neuronal ceroid lipofuscinoses (NCLs) are a group of inherited neurodegenerative disorders characterized by progressive declines in neurological functions following normal development. The NCLs are distinguished from similar disorders by the accumulation of autofluorescent lysosomal storage bodies in neurons and many other cell types, and are classified as lysosomal storage diseases. At least 13 genes contain pathogenic sequence variants that underlie different forms of NCL. Naturally occurring canine NCLs can serve as models to develop better understanding of the disease pathologies and for preclinical evaluation of therapeutic interventions for these disorders. To date 14 sequence variants in 8 canine orthologs of human NCL genes have been found to cause progressive neurological disorders similar to human NCLs in 12 different dog breeds. A mixed breed dog with Labrador Retriever and Beagle parents developed progressive neurological signs consistent with NCL starting at approximately 11 to 12 months of age, and when evaluated with magnetic resonance imaging at 21 months of age exhibited diffuse brain atrophy. Due to the severity of neurological decline the dog was euthanized at 23 months of age. Cerebellar and cerebral cortical neurons contained massive accumulations of autofluorescent storage bodies the contents of which had the appearance of tightly packed membranes. A whole genome sequence, generated with DNA from the affected dog contained a homozygous C-to-T transition at position 30,574,637 on chromosome 22 which is reflected in the mature CLN5 transcript (CLN5:c.619C>T) and converts a glutamine codon to a termination codon (p.Gln207Ter). The identical nonsense mutation has been previously associated with NCL in Border Collies, Australian Cattle Dogs, and a German Shepherd-Australian Cattle Dog mix. The current whole genome sequence and a previously generated whole genome sequence for an Australian Cattle Dog with NCL share a rare homozygous haplotype that extends for 87 kb surrounding 22:30,574,637 and includes 21 polymorphic sites. When genotyped at 7 of these polymorphic sites, DNA samples from the German Shepherd-Australian Cattle Dog mix and from 5 Border Collies with NCL that were homozygous for the CLN5:c.619 T allele also shared this homozygous haplotype, suggesting that the NCL in all of these dogs stems from the same founding mutation event that may have predated the establishment of the modern dog breeds. If so, the CLN5 nonsence allele is probably segregating in other, as yet unidentified, breeds. Thus, dogs exhibiting similar NCL-like signs should be screened for this CLN5 nonsense allele regardless of breed.
Keywords: dog, neuronal ceroid lipofuscinosis, CLN5, lysosomal storage disease, neurodegeneration, whole genome sequencing
1. Introduction
The neuronal ceroid lipofuscinoses (NCLs) are rare inherited progressive neurodegenerative diseases that in people are characterized by apparently normal development followed by progressive declines in cognitive and motor functions, loss of vision, seizures, respiratory impairment, and in most cases premature death [1]. The neurological signs are usually accompanied by generalized brain atrophy, retinal degeneration, and massive accumulation of autofluorescent lysosomal storage bodies in the central nervous system as well as other tissues and organs [1]. Prior to the advent of modern molecular genetics, NCL was classified into different types based primarily on the age of onset, pattern of clinical signs, and rate of disease progression. Currently, however, the mutations responsible for most cases of human NCL are known, and the different types are now classified as CLN1 through CLN14 based on the gene in which the disease-causing mutation resides [2]. In people, NCL-causing sequence variants occur in at least 13 genes [2]. The disease phenotype, including age of onset, spectrum of clinical signs, and rate of disease progression can vary not only according to the gene in which the causative mutation occurs, but also influenced by the specific mutation within that gene. The onset of disease signs can occur as early as infancy to as late as middle age, with a similar variability in the severity of disease signs.
Progress in developing treatments for the NCLs has been hampered by the lack of adequate animal models. Transgenic mouse models have been made for most of the NCLs, but many of these models fail to recapitulate the human disease phenotype and therapies tested in these models have not translated to human application [3-32]. Large animal models with nervous systems more similar to those of humans in size and complexity are likely to be more suitable for therapy development. Indeed, an enzyme replacement therapy for treating CLN2 disease that was successful in delaying disease onset and slowing the progression of clinical signs in a dog model is now being used successfully to treat children with this disorder [33-37]. The availability of dog models for the other NCLs would likely accelerate therapy development for these disorders as well.
Diseases with clinical signs similar to the human NCLs have been described in over 20 dog breeds and in several mixed breed dogs [38, 39]. NCL has been associated with sequence variants in 8 canine orthologs of human NCL genes [38, 39]. Among these canine disorders the onset of clinical signs ranges from a few months to 7 years of age [38].
With one exception, the parents of mixed breed dogs with NCL were purebred dogs of breeds in which the same causative mutation had been found to occur [38]. The exception was an offspring of a cross between an Australian Cattle Dog and a German Shepherd Dog. The affected dog was homozygous for a CLN5:c.619C > T variant previously associated with NCL in purebred Australian Cattle Dogs and Border Collies [38]. To our knowledge, neither an NCL-like disease phenotype nor the CLN5 sequence variant have been identified in German Shepherd Dogs. A mixed breed dog exhibiting NCL-like signs reported to be the offspring of a Beagle and a Labrador Retriever was brought to our attention. A late-adult onset NCL has been reported in the latter breed, but no causative mutation was identified [40]. There have been no published reports of NCL-like disease in Beagles. Studies were therefore undertaken to determine whether the affected dog did indeed suffer from NCL, and if so, to identify the causative mutation(s).
2. Materials and methods
2.1. Subject dog
These studies were conducted with approval of the University of Missouri Institutional Animal Care and Use Committee and with the informed consent of the dog’s owner. The affected dog (Fig. 1) was one of a litter of four produced by a mating between a female Beagle and a Labrador Retriever. The affected dog first began to exhibit obvious neurological signs at approximately 10 to 12 months of age, and these signs became progressively worse over time. However, abnormalities in the dog’s stance were observed as early as 6 months of age. The neurological signs included anxiety, loss of housetraining, almost complete loss of responsiveness to previously learned commands and verbal prompting, compulsive circling, disruption of previously normal sleep patterns, loss of the ability to vocalize, impaired ability to prehend and swallow food, severe loss of coordination and ability to climb up or down stairs, development of tremors and seizures that were not responsive to phenobarbital, exhibition of trance-like behavior, and severe visual impairment in both dim and bright lighting conditions. Based on behavioral observations, visual impairment began with a loss of visual acuity, as the dog could visually track large but not small objects until at least 18 months of age. Due to the severity of the neurological signs, particularly the difficulty in eating, the owner elected to have the dog euthanized at approximately 23 months of age and the body was submitted for necropsy.
Fig. 1.

Photograph of the affected dog at approximately 20 months of age.
The affected dog’ owner was informed by the owner of one of the littermates that the littermate had died after having exhibited similar signs. No DNA or tissues samples were collected from the littermate, and no disease diagnosis was made. Definitive health information on the two remaining littermates could not be obtained. We were unable to obtain information about the health histories of the parents or to obtain samples for DNA isolation from these dogs. Based on physical appearance, the mother of the affected dogs appeared to be of Beagle lineage and the father appeared to be of Labrador Retriever lineage, but we were unable to obtain pedigree information on these dogs. The breeding that produced the affected puppies was apparently unplanned since all four littermates were donated to a dog rescue service.
2.2. Clinical evaluations and necropsy
The affected dog was examined by a veterinary neurologist (JRM) at approximately 21.5 months of age. The examination included full general physical examination and neurologic examination, as well as magnetic resonance imaging of the brain performed using a GE 1.5 Tesla Optima MR360 instrument. Cerebrospinal fluid cytology and fluid analyses as well as complete blood count and blood chemistry analyses were also performed at this time.
After euthanasia a complete necropsy was performed by a veterinary pathologist in the Pathology Department of Angell Animal Medical Center. Whole blood and parts of the formalin-fixed brain were shipped to the University of Missouri for further analyses. Fixed tissue samples were examined with fluorescence and electron microscopy [41]. For electron microscopy, small pieces of the cerebellar cortex, the deep cerebellar nuclei, and the parietal cerebral cortex were washed in 0.17 M sodium cacodylate buffer and then transferred to electron microscopy fixative (2% glutaraldehyde, 1.12% paraformaldehyde, 0.13 M sodium cacodylate, 1 mM CaCl2, pH 7.4) and incubated in the latter fixative at room temperature with gentle agitation for at least 48 hours prior to being further processed for electron microscopic examination.
2.3. DNA analysis
DNA from the affected dog was purified from EDTA anti-coagulated blood as previously described [42] and submitted to the University of Missouri DNA Core Facility for PCR-free library preparation and sequencing on the Illumina NextSeq 500 platform. The resulting paired-end sequence data was analyzed using a modified best-practices GATK pipeline to align to the reference genome (CanFam3.1) and call variants. VCF files were generated from the affected dog’s whole genome sequence and from the whole genome sequences of 141 other dogs with a variety of diseases that served as controls. Based on owner reports, 135 of the control dogs were purebred. The breeds represented in the control cohort are listed in the supplementary material. The 142 VCF files were loaded into commercial software (Golden Helix SVS) for sorting the variants by Alternative Allele Counts and by Gene Name. A previously described, a TaqMan allelic discrimination assay [43] was used to genotype archived DNA samples from randomly selected Beagles, Labrador Retrievers, German Shepherd Dogs for a C-to-T transition at position 30,574,637 on chromosome 22. The direct sequencing of PCR amplicons was used to determine the genotype at 7 polymorphic sites near 22:30,574,637, as detailed in the supplementary material. In an attempt to more definitively determine the breed ancestry of the affected dog we shipped 50 μg of purified DNA from the affected dog to each of four different commercial testing companies to perform DNA tests that are advertised to identify the breed backgounds of mixed breed dogs. To avoid potential legal issues, we identify the companies here only by the code letters A,B, C and D. However companies offering this type of testing can easily be found by performing an internet search.
3. Results
3.1. Physical, clinical, and neurological findings
The affected dog was evaluated by a veterinary neurologist (JRM) when the dog was 22 months of age due to intermittent facial spasms and focal seizures of up to 45 minutes duration for each episode during which the dog remained alert. These episodes began when the dogs was approximately 21 months of age and occurred on an almost daily basis, usually multiple times each day, and were not prevented with phenobarbital. Some episodes included shaking of one hind limb. The dog was extremenly anxious and would pace and bump into objects. Mild proprioceptive ataxia was observed and the dog would cross over her front legs when walking or standing. Crainial nerves, postural reactions, spinal reflexes and touch sensation were assessed as normal.
Magnetic resonance imaging of the brain at this time (Precontrast: T2WFSE, T2 FLAIR, MERGE (T2*GRE), T1WFSE; DWI; postcontrast (2.8 mL Omniscan IV): T1WFSE; 3D FSPGR) revealed diffuse brain atrophy characterized by widening of the cerebral and cerebellar sulci, generalized ventriculomegaly, and decreased size of the interthalamic adhesion (Fig. 2). In addition, there was diminished grey-white matter distinction. No abnormal contrast enhancement or intracranial mass effects detected. Cerebrospinal fluid evaluation and blood count and chemistries did not indicate any central nervous system inflammation.
Fig 2.

T2 weighted magnetic resonance (MR) mid-sagittal (A) and axial (B) images at the level of the interthalamic adhesion from an approximately 2 year old normal dog. T2 weighted MR mid-sagittal (C) and axial (D) images at the level of the interthalamic adhesion from the affected dog. Diffuse brain atrophy is noted in the affected dog (C, D) as evidenced by enlargement of the lateral (5-pointed star) and third (arrowhead) ventricles, widening of the cerebral (solid arrow) and cerebellar (dotted arrow) sulci, and narrowing of the interthalamic adhesion (dotted line). Comparable structures in the age-matched, control dog (A, B) are marked with identical symbols to highlight normal structure.
3.2. Pathology findings
Gross examination of the affected dog at necropsy confirmed the diffuse brain atrophy observed with MR imaging. The brain was diffusely reduced in size, evidenced by increased space separating the brain from the cranial vault. There was mild dilation of the lateral ventricle. Edema of the lungs was the only other gross finding.
Multiple tissues were examined microscopically including cerebrum, thalamus, cerebellum, brainstem, pituitary gland, liver, spleen, kidney, adrenal gland, heart, lung, thyroid gland, parathyroid gland, stomach, small intestine, pancreas, and eye globe. Neuronal cell bodies in the brain and periphery (retina, autonomic ganglia, myenteric/submucosal plexuses) exhibited accumulation of eosinophilic large granules to globules, often with an eccentric or perinuclear distribution. The cytoplasmic inclusions are PAS-positive (Fig. 3). Scattered degenerate neurons were observed in the cerebral cortex. Numerous reactive glial cells identified by GFAP immunostaining were present in layer 6 of the cerebral cortex (Fig. 4) and the granular layer and deep cerebellar nuclei of the cerebellum (Fig. 5). Intense GFAP immunostaining was also present in the nerve fiber layer of the retina (Fig. 6). Purkinje cell numbers were significantly decreased in the cerebellar cortex, and the granular layer appeared thinned. The optic nerve head was vacuolated with increased glial cells. Hepatocytes and renal tubular epithelial cells had finely globular/granular eosinophilic cytoplasm, with no PAS reactivity. Lung sections are congested with mildly increased macrophages in alveoli.
Fig. 3.

Light micrograph of PAS-stained section of a brainstem nucleus from the affected dog depicting massive accumulation of PAS-stained cytoplasmic inclusions in large neurons (arrow).
Fig. 4.

Immunohistochemical demonstration of reactive astrocytes in the parietal cerbral cortex of the affected dog. GFAP immunostaining of sections of the cerebral cortex demonstrated numerous reactive astrocytes (arrows) in cortical layers 6A (polymorphic cell layer) (A) and 6B (B). Reactive astrocytes were sparse elsewhere in the cerebral cortex.
Fig. 5.

Immunohistochemical demonstration of reactive astrocytes in the cerebellum of the affected dog. GFAP immunostained reactive astrocytes (arrows) were abundant in the in granulary layer of the cerebellar cortex arrows in (A) and in the deep cerebellar nuclei (arrows in B).
Fig. 6.
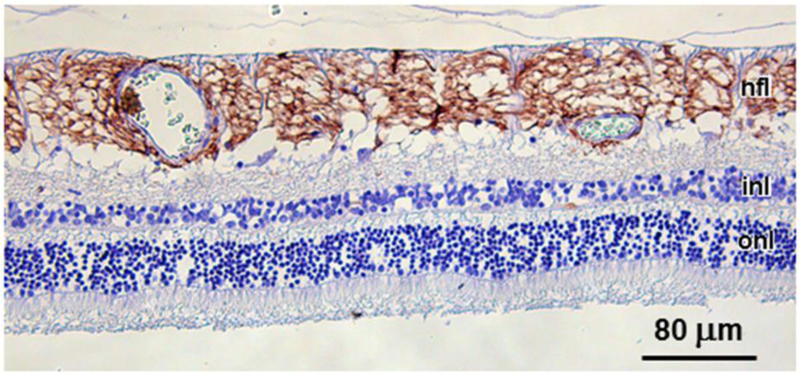
GFAP immunostaining was intense in the nerve fiber layer (nfl) of the affected dog, but was not present in any other retinal cell layers, including the inner nuclera layer (inl) or outer nuclear layer (onl).
The disease-related inclusion bodies that were present in brain and retina exhibited autofluorescence typical of lipofuscin and ceroid [44-48] (Fig. 7). In the cerebellar cortex, the autofluorescent inclusions were present primarily in the Purkinje cell bodies, with few scattered autofluorescent bodies in the molecular or granule cell layers (Fig. 7A, arrows). In the deep cerebellar nuclei, large neurons contained abundant autofluorescent inclusions that were aggregated at one pole of the cell body (Fig. 7B, arrows). In the cerebral cortex, almost all neurons contained the disease-related autofluorescent inclusions (Fig. 7C, arrows). In the retina, these inclusions were present only in ganglion cells (Fig. 7D, arrow). The ganglion cells were much more sparse and smaller in size than ganglion cells from normal dog retinas.
Fig. 7.

Fluorescence micrographs of sections of the cerebellar cortex (A), a deep cerebellar nucleus (B), the parietal cerebral cortex (C), and the retina (D) from the affected dog. Disease-specific autofluorescent cytoplasmic inclusions are indicated by arrows. In (A) m:molecular layer, p:Purkinkje layer, g:granule cell layer. In (D) gcl:ganglion cell layer; onl:outer nuclerar layer. Bar in (B) indicates the magnification of all four micrographs.
Electron microscopic examination of the disease-specific storage bodies in cerebral cortical neurons and cerebellar Purkinje cells revealed that the stored material consisted primarily of whorls of densely-packed membrane-like structures (Figs. 8 and 9). The storage body contents were very similar in appearance in both the Purkinje cells and cortical neurons. In the cerebral cortex, however, a minority of the storage bodies contained scattered electron-dense amorphous inclusions (Fig. 8) that were not observed in the cerebellum. The material in the storage bodies was enclosed by surrounding membranes, but due to the initial fixation in formalin, these outer membranes were only partially preserved (Figs. 8 and 9).
Fig. 8.

Electron micrographs of storage bodies in cerebral cortical neurons of the affected dog. The contents of most storage bodies consisted of tightly packed clusters of membrane-like structures (A). In some storage bodies, electron dense amorphous patches of material were embedded within the membrane-like aggregates (arrows in B). Membranes enclosing the storage material (arrowheads) were only partially preserved due to the initial fixation in formalin.
Fig. 9.

Electron micrographs of storage bodies in cerebellar Purkinje cells of the affected dogaffected dog. As with the cerebral cortical neurons, the contents of most storage bodies consisted of tightly packed clusters of membrane-like structures, but none of the electron dense amorphous patches of material were observed in the Purkinje cell storage bodies. As with the cerebral cortex, membranes enclosing the storage material (arrows) were only partially preserved due to the initial fixation in formalin.
3.3. Molecular genetic analyses
The affected dog’s genome contained 8 variants that were predicted to alter the primary structure of an encoded polypeptide, absent from 141 control canine whole genome sequences, and homozygous in the affected dog. None of these variants were in genes predicted to cause lysosomal storage diseases. Therefore, the exclusion criteria were relaxed so that variants that occurred in two or fewer of the 282 reference alleles were retained for consideration. These relaxed exclusion criteria identified 7 additional rare variants that were homozygous in the affected dog, including a C-to-T transition at position 30,574,637 on chromosome 22 (CLN5:c.619C>T). As previously reported, the 22:30,574,637T (CLN5:c.619T) allele was homozygous in the whole genome of an Australian Cattle Dog with NCL [43]. The variant occurs in exon 4 of CLN5 where it converts codon 207 from CAA (Gln) to TAA (Stop). DNA samples from 68 Beagles, 65 Labrador Retrievers, and 70 German Shepherd Dogs were randomly selected from those in the University of Missouri canine DNA Repository and genotyped at the chromosome 22:30,574,637 locus. All of the randomly selected samples were homozygous for the 22:30,574,637C allele.
We compared the genomic region surrounding chromosome 22:30,574,637 in the whole genome sequence of the affected dog and in the previously reported whole genome sequence of an Australian Cattle Dog with that was homozygous for the 22:30,574,637T allele [43]. Both dogs were homozygous for an identical 21-variant haplotype that extends for 87 kb from 22:30,523,216 to 22:30,610,464. Of the 141 whole genome sequences from control dogs, 140 were homozygous for at least one allele that was not part of the 21-variant haplotype. Thus, this full-length haplotype could have occurred in only one of the whole genome sequences, an epileptic Standard Schnauzer that was potentially heterozygous for the haplotype but homozygous for 22:30,574,637C allele (Table 1). In addition, we genotyped 6 individual DNA samples at 7 of the 21 polymorphic sites in the haplotype region. These 6 samples were all from dogs previously diagnosed with NCL due to homozygosity for the 22:30,574,637T nonsense allele. Five of the genotyped DNA samples were from Border Collies from Japan and the other genotyped sample was from the previously described mixed breed dog of Australian Cattle dog and German Shepherd ancestry. As indicated in Table 1, all of these samples were homozygous for the same alleles that were found in the whole genome sequences from the affected dog and the Australian Cattle Dog with NCL.
Table 1:
Genotypes of dogs with NCL at polymorphic sites near the CLN5 missense mutation at 22:30574637
| Genotypes | ||||||
|---|---|---|---|---|---|---|
| Chromosome 22 Position |
Reference Allele |
Variant Allele |
Variant Allele Frequencies |
WGS for NCL Dogs† |
WGS for Epileptic Std. Schnauzer‡ |
DNA from NCL Dogs# |
| 30523216 | G | T€ | 0.152 | T€/T | T/T | T/T |
| 30524512 | AAAAAAAAA | - | 0.192 | −/− | −/− | N.D.* |
| 30535524 | T | TT | 0.408 | TT/TT | TT/TT | N.D. |
| 30538412 | - | CT | 0.461 | CT/CT | CT/CT | N.D. |
| 30542756 | C | A | 0.755 | A/A | A/A | N.D. |
| 30546129 | AC | - | 0.069 | −/− | AC/− | N.D. |
| 30550783 | - | G | 0.755 | G/G | G/G | N.D. |
| 30553284 | AAA | - | 0.111 | −/− | AAA/− | N.D. |
| 30553293 | AAATTA | - | 0.367 | −/− | AAATTA/− | −/− |
| 30567376 | C | T | 0.193 | T/T | T/T | T/T |
| 30569433 | TTT | - | 0.536 | −/− | −/− | N.D. |
| 30569456 | A | G | 0.768 | G/G | G/G | N.D. |
| 30574637 | C | T | 0.013 | T/T | C/C | T/T |
| 30578990 | A | c | 0.178 | C/C | C/C | C/C |
| 30589554 | A | - | 0.565 | −/− | A/− | N.D. |
| 30594474 | C | A | 0.938 | A/A | A/A | N.D. |
| 30598322 | G | C | 0.383 | C/C | C/C | C/C |
| 30600625 | - | ATA | 0.925 | ATA/ATA | ATA/ATA | N.D. |
| 30601475 | T | - | 0.732 | −/− | −/− | N.D. |
| 30603161 | T | C | 0.925 | C/C | C/C | N.D. |
| 30608518 | T | A | 0.183 | A/A | A/T | A/A |
| 30610464 | c | T | 0.105 | T/T | C/T | T/T |
Genotypes in whole genome sequences for the affected dog and an Australian Cattle Dog with NCL that was homozygous for the 22:30,574,637T allele
Genotypes in the whole genome sequence for an epileptic Standard Schnauzer that did not suffer from NCL
Genotypes in genomic DNA samples from dogs with NCL including 6 Border Collies and a dog with a German Shepherd Dog sire and an Australian Cattle Dog dam (all of these dogs were homozygous for the 22:30,574,637T allele)
Red alleles are in the NCL-associated haplotype
N.D. = not determined
3.4. Breed background analysis
Since breed background determination based on physical appearance can be unreliable, DNA samples from the affected dog were submitted to four different companies that analyze the breed backgrounds of mixed breed dogs based on patterns of polymorphic markers in the genome of the dog being evaluated. The results provided by the companies are summarized in Table 2. Although there were significant differences in the results provided by the different companies, analyses from 3 of 4 companies were in agreement on significant contributions from the following breeds: Treeing Walker Coonhound, Beagle, Shetland Sheepdog, and Siberian Husky. To our knowledge, NCL has not been reported in any of these breeds. On the other hand, only Company C indicated a contribution from a breed (Border Collie) in which NCL associated with the CLN5:c.619T variant has been reported [49].
Table 2.
Breed contributions to the genome of the affected dog as determined by 4 companies.
| Breed Contribution to Affected Dog (%) | ||||
|---|---|---|---|---|
| Breed | Company A | Company B | Company C | Company D |
| Treeing Walker Coonhound | 38.2 | 25 | -- | 25 |
| Beagle | -- | 12.5 | 25 | 12.5 |
| Harrier | -- | -- | 25 | -- |
| Mountain Cur | 19.4 | -- | -- | -- |
| Miniature Poodle | -- | 12.5 | -- | 12.5 |
| Shetland Sheepdog | 12.5 | 12.5 | -- | 12.5 |
| Siberian Husky | 9.4 | -- | 12.5 | 12.5 |
| Border Collie | Trace | -- | 12.5 | -- |
| Fox Terrier | -- | -- | 12.5 | -- |
| Labrador Retriever | -- | -- | 12.5 | -- |
| Rat Terrier | -- | -- | 12.5 | -- |
| German Shepherd Dog | 5.3 | -- | -- | -- |
| Chow Chow | trace | -- | -- | -- |
| Dalmatian | trace | -- | -- | -- |
| Herding breeds | -- | trace | -- | -- |
| Guard breeds | -- | trace | -- | -- |
| Sporting breeds | -- | trace | -- | -- |
| Terrier breeds | -- | trace | -- | -- |
| Unknown mix | -- | -- | -- | 25 |
4. Discussion and Conclusions
In 2005, Australian investigators reported that a young-adult onset NCL in Border Collies was caused by a homozygous nonsense sequence variant in CLN5 [49]. Eleven years later, whole genome sequence analysis identified the identical homozygous CLN5 variant in Australian Cattle Dogs with NCL [43]. More recently, the same homozygous CLN5 variant was found in a dog with an Australian Cattle Dog dam and a German Shepherd Dog sire [38]. In the present study whole genome sequence analysis identified the same CLN5 nonsense mutation in a mixed breed dog in which Broder Collie, Australian Cattle Dog, or German Shepherd Dog ancestry appears to be minimal or non-existent (Table 2).
A homozygous 2 bp deletion and frameshift variant in CLN5 was identified in Golden Retriever siblings with NCL [50]. Although the precise cellular function of the CLN5 protein remains to be determined, both the latter deletion and the CLN5 22:30,574,637C>T nonsense variant are predicted to encode truncated polypeptides with little or no residual biological activity [50]. As might be expected with homozygous nullifying mutations, similar disease phenotypes have been reported for the affected Golden Retrievers and the dogs of various breed backgrounds with the homozygous CLN5 nonsense mutation. In all these cases, clinical signs have typically become apparent at 12 to 20 months of age. The affected dogs exhibited seizures and progressive declines in vision, cognition and coordination of movement which culminated in euthanasia due to reduced quality of life before the third birthday. Also similar to the affected dog, previous descriptions of dogs with CLN5-related NCL described MRIs indicative of diffuse brain atrophy, and autofluorescent storage bodies with varied ultrastructure that included aggregates of membrane-like structures [38, 41, 50].
Most cases of genetically defined canine NCL have been found only in the breed from which the variant was discovered [38]. One exception was a homozygous single base pair deletion and frame shift in MFSD8 that was first identified as the likely cause of NCL in a Chinese Crested dog [51] but subsequently also identified in Chihuahuas with NCL [52-54]. Since the members of both breeds are very small and share other physical attributes, a founding event may have occurred in one breed and the variant may have been introduced into the other breed by an interbreed mating. We proposed that interbreed mating may also explain the occurrence of the same homozygous CLN5 nonsense variant in both Border Collies and Australian Cattle Dogs with NCL [43]. When the CLN5 nonsense variant was found to be homozygous in a cross between a German Shepherd Dog and an Australian Cattle Dog, we suggested that the German Shepherd Dog parent may have had an Australian Cattle Dog ancestor with the disease allele [38]. With the new finding of the identical NCL-causing CLN5 sequence variant in a mixed breed dog with little or no apparent Australian Cattle Dog or Border Collie ancestry, recent transfer of the disease allele from the breed in which it originated to other breeds seems less plausible. An alternative explanation for the distribution of the CLN5 nonsense variant among breeds is that the CLN5 nonsense variant arose in a common ancestor of multiple breeds. The breed background data in Table 1 and our previous work [38] suggest that the CLN5 22:30,574,637T variant is particularly likely to be present in Treeing Walker Coonhounds, Beagles, Shetland Sheepdogs, Siberian Huskies and German Shepherd Dogs. Therefore dogs from these breeds in particular that are exhibiting NCL-like neurological signs should be genotyped for the presence of this variant. To our knowledge, NCL has not been reported in any of these breeds.
A rare, homozygous, 21-variant, 87 kb haplotype surrounds the CLN5 nonsense variant in whole genome sequences from both the affected dog and a previously described Australian Cattle Dog with NCL [43], indicating that the mutation in both dogs stems from a common foundation event. Demonstration that 5 Border Collies and a previously reported mixed breed dog with Australian Cattle Dog dam and a German Shepherd Dog sire were homozygous for at least 7 of the haplotype alleles suggests that the same founding event was responsible for their NCL and supports the possibility that the founding event occurred before the establishment of the modern dog breeds. The PRCD:c.5A transition that causes progressive retinal atrophy [55] and the ADAMTS17:c.1473+1 G>A transition that causes lens luxation [56] are examples of other sequence variants that are responsible for recessive diseases in many breeds [57, 58] and probably have an ancient origins. If, in fact, the founding CLN5 nonsense mutation event predated breed establishment, it is likely that CLN5 nonsense allele is segregating in additional as yet unrecognized breeds. The fact that CLN5-associated NCL has not been reported to date in more breeds indicates that the nonsense allele is almost certainly rare in most breeds in which it occurs. However, the lack of reports of NCL in more breeds is also likely to be due in part of the failure to publicize most cases of NCL-like disorders in dogs. Indeed, after the first report that NCL in Border Collies is associated with the 22:30,574,637T nonsense variant in CLN5, a survey of Border Collies from four kennels in Japan found the frequency T allele to be almost 35% [59], indicating that dogs from lines that certainly produced affected offspring continued to be used for breeding. In addition, by the time we described a form of NCL in American Bulldogs and subsequently reported the causative sequence variant in CTSD [60, 61], the frequency of the mutant allele among American Bulldogs had become quite high [60]. Based on these two examples, it is quite possible that cases of NCL associated with homozygosity for the CLN5 22:30,574,637T variant have occurred in Treeing Walker Coonhounds, Beagles, Shetland Sheepdogs, Siberian Huskies, German Shepherd Dogs and other breeds but gone unreported. It is not unusual for owners of dogs with progressive neurological diseases to have their dogs euthanized without seeking to identify the cause of the disease and without the attending veterinarian publishing a description of the disease phenotype.
Supplementary Material
Acknowledgements
This work was funded in part by grant EY023968 from the U.S. National Institutes of Health and AKC Canine Health Foundation grant 02257. Our thanks to Cheryl Jensen for expert technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Mole SE, Williams RE, Goebel HH, The Neuronal Ceroid Lipofuscinoses (Batten Disease), Oxford University Press, Oxford, Great Britain, 2011. [Google Scholar]
- [2].Warrier V, Vieira M, Mole SE, Genetic basis and phenotypic correlations of the neuronal ceroid lipofusinoses Biochim Biophys Acta 1832 (2013) 1827–1830. [DOI] [PubMed] [Google Scholar]
- [3].Aldrich A, Bosch ME, Fallet R, Odvody J, Burkovetskaya M, Rama Rao KV, Cooper JD, Drack AV, Kielian T, Efficacy of phosphodiesterase-4 inhibitors in juvenile Batten disease (CLN3) Ann Neurol 80 (2016) 909–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bertamini M, Marzani B, Guarneri R, Guarneri P, Bigini P, Mennini T, Curti D, Mitochondrial oxidative metabolism in motor neuron degeneration (mnd) mouse central nervous system Eur J Neurosci 16 (2002) 2291–2296. [DOI] [PubMed] [Google Scholar]
- [5].Cooper JD, Moving towards therapies for juvenile Batten disease? Exp Neurol 211 (2008) 329–331. [DOI] [PubMed] [Google Scholar]
- [6].Cooper JD, Messer A, Feng AK, Chua-Couzens J, Mobley WC, Apparent loss and hypertrophy of interneurons in a mouse model of neuronal ceroid lipofuscinosis: evidence for partial response to insulin-like growth factor-1 treatment J Neurosci 19 (1999) 2556–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Damme M, Brandenstein L, Fehr S, Jankowiak W, Bartsch U, Schweizer M, Hermans-Borgmeyer I, Storch S, Gene disruption of Mfsd8 in mice provides the first animal model for CLN7 disease Neurobiology of Disease 65 (2014) 12–24. [DOI] [PubMed] [Google Scholar]
- [8].Drack AV, Mullins RF, Pfeifer WL, Augustine EF, Stasheff SF, Hong SD, Immunosuppressive Treatment for Retinal Degeneration in Juvenile Neuronal Ceroid Lipofuscinosis (Juvenile Batten Disease) Ophthalmic Genet 36 (2015) 359–364. [DOI] [PubMed] [Google Scholar]
- [9].Elger B, Schneider M, Winter E, Carvelli L, Bonomi M, Fracasso C, Guiso G, Colovic M, Caccia S, Mennini T, Optimized synthesis of AMPA receptor antagonist ZK 187638 and neurobehavioral activity in a mouse model of neuronal ceroid lipofuscinosis ChemMedChem 1 (2006) 1142–1148. [DOI] [PubMed] [Google Scholar]
- [10].Finn R, Kovacs AD, Pearce DA, Treatment of the Ppt1(−/−) mouse model of infantile neuronal ceroid lipofuscinosis with the N-methyl-D-aspartate (NMDA) receptor antagonist memantine J Child Neurol 28 (2013) 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Geraets RD, Langin LM, Cain JT, Parker CM, Beraldi R, Kovacs AD, Weimer JM, Pearce DA, A tailored mouse model of CLN2 disease: A nonsense mutant for testing personalized therapies PLoS ONE 12 (2017) e0176526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hawkins-Salsbury JA, Cooper JD, Sands MS, Pathogenesis and therapies for infantile neuronal ceroid lipofuscinosis (infantile CLN1 disease) Biochim Biophys Acta 1832 (2013) 1906–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Katz ML, Johnson GS, Tullis GE, Lei B, Phenotypic characterization of a mouse model of juvenile neuronal ceroid lipofuscinosis Neurobiology of Disease 29 (2008) 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Katz ML, Rice LM, Gao CL, Dietary carnitine supplements slow disease progression in a putative mouse model for hereditary ceroid-lipofuscinosis Journal of Neuroscience Research 50 (1997) 123–132. [DOI] [PubMed] [Google Scholar]
- [15].Kohlschutter A, Schulz A, CLN2 Disease (Classic Late Infantile Neuronal Ceroid Lipofuscinosis) Pediatr Endocrinol Rev 13 Suppl 1 (2016) 682–688. [PubMed] [Google Scholar]
- [16].Kovacs AD, Pearce DA, Attenuation of AMPA receptor activity improves motor skills in a mouse model of juvenile Batten disease Exp Neurol 209 (2008) 288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kovacs AD, Pearce DA, Finding the most appropriate mouse model of juvenile CLN3 (Batten) disease for therapeutic studies: the importance of genetic background and gender Dis 8 (2015) 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Perentos N, Martins AQ, Watson TC, Bartsch U, Mitchell NL, Palmer DN, Jones MW, Morton AJ, Translational neurophysiology in sheep: measuring sleep and neurological dysfunction in CLN5 Batten disease affected sheep Brain 138 (2015) 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Petkau TL, Blanco J, Leavitt BR, Conditional loss of progranulin in neurons is not sufficient to cause neuronal ceroid lipofuscinosis-like neuropathology in mice Neurobiology of Disease 106 (2017) 14–22. [DOI] [PubMed] [Google Scholar]
- [20].Ruther K, [NCL in animal models] Ophthalmologe 107 (2010) 621–627. [DOI] [PubMed] [Google Scholar]
- [21].Seehafer SS, Ramirez-Montealegre D, Wong AM, Chan CH, Castaneda J, Horak M, Ahmadi SM, Lim MJ, Cooper JD, Pearce DA, Immunosuppression alters disease severity in juvenile Batten disease mice J Neuroimmunol 230 (2011) 169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Seigel GM, Lotery A, Kummer A, Bernard DJ, Greene ND, Turmaine M, Derksen T, Nussbaum RL, Davidson B, Wagner J, Mitchison HM, Retinal pathology and function in a Cln3 knockout mouse model of juvenile Neuronal Ceroid Lipofuscinosis (batten disease) Mol Cell Neurosci 19 (2002) 515–527. [DOI] [PubMed] [Google Scholar]
- [23].Selden NR, Al-Uzri A, Huhn SL, Koch TK, Sikora DM, Nguyen-Driver MD, Guillaume DJ, Koh JL, Gultekin SH, Anderson JC, Vogel H, Sutcliffe TL, Jacobs Y, Steiner RD, Central nervous system stem cell transplantation for children with neuronal ceroid lipofuscinosis J Neurosurg Pediatrics 11 (2013) 643–652. [DOI] [PubMed] [Google Scholar]
- [24].Tamaki SJ, Jacobs Y, Dohse M, Capela A, Cooper JD, Reitsma M, He D, Tushinski R, Belichenko PV, Salehi A, Mobley W, Gage FH, Huhn S, Tsukamoto AS, Weissman IL, Uchida N, Neuroprotection of host cells by human central nervous system stem cells in a mouse model of infantile neuronal ceroid lipofuscinosis Cell Stem Cell 5 (2009) 310–319. [DOI] [PubMed] [Google Scholar]
- [25].Tang CH, Lee JW, Galvez MG, Robillard L, Mole SE, Chapman HA, Murine cathepsin F deficiency causes neuronal lipofuscinosis and late-onset neurological disease Mol Cell Biol 26 (2006) 2309–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zeman RJ, Peng H, Etlinger JD, Clenbuterol retards loss of motor function in motor neuron degeneration mice Exp Neurol 187 (2004) 460–467. [DOI] [PubMed] [Google Scholar]
- [27].Augustine EF, Mink JW, Juvenile NCL (CLN3 Disease): Emerging Disease-Modifying Therapeutic Strategies Pediatr Endocrinol Rev 13 Suppl 1 (2016) 655–662. [PubMed] [Google Scholar]
- [28].Ghosh A, Rangasamy SB, Modi KK, Pahan K, Gemfibrozil, food and drug administration-approved lipid-lowering drug, increases longevity in mouse model of late infantile neuronal ceroid lipofuscinosis J Neurochem 141 (2017) 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Groh J, Berve K, Martini R, Fingolimod and Teriflunomide Attenuate Neurodegeneration in Mouse Models of Neuronal Ceroid Lipofuscinosis Mol Ther 25 (2017) 1889–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kinarivala N, Trippier PC, Progress in the Development of Small Molecule Therapeutics for the Treatment of Neuronal Ceroid Lipofuscinoses (NCLs) J Med Chem 59 (2016) 4415–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mirza M, Volz C, Karlstetter M, Langiu M, Somogyi A, Ruonala MO, Tamm ER, Jagle H, Langmann T, Progressive retinal degeneration and glial activation in the CLN6 (nclf) mouse model of neuronal ceroid lipofuscinosis: a beneficial effect of DHA and curcumin supplementation PLoS ONE 8 (2013) e75963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Roberts MS, Macauley SL, Wong AM, Yilmas D, Hohm S, Cooper JD, Sands MS, Combination small molecule PPT1 mimetic and CNS-directed gene therapy as a treatment for infantile neuronal ceroid lipofuscinosis J Inherit Metab Dis 35 (2012) 847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Katz ML, Coates JR, Sibigtroth CM, Taylor JD, Carpentier M, Young WM, Wininger FA, Kennedy D, Vuillemenot BR, O'Neill CA, Enzyme replacement therapy attenuates disease progression in a canine model of late-infantile neuronal ceroid lipofuscinosis (CLN2 disease) Journal of Neuroscience Research 92 (2014) 1591–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Vuillemenot BR, Kennedy D, Cooper JD, Wong AM, Sri S, Doeleman T, Katz ML, Coates JR, Johnson GC, Reed RP, Adams EL, Butt MT, Musson DG, Henshaw J, Keve S, Cahayag R, Tsuruda LS, O'Neill CA, Nonclinical evaluation of CNS-administered TPP1 enzyme replacement in canine CLN2 neuronal ceroid lipofuscinosis Molecular Genetics & Metabolism 114 (2015) 281–293. [DOI] [PubMed] [Google Scholar]
- [35].Whiting RE, Jensen CA, Pearce JW, Gillespie LE, Bristow DE, Katz ML, Intracerebroventricular gene therapy that delays neurological disease progression is associated with selective preservation of retinal ganglion cells in a canine model of CLN2 disease Experimental Eye Research 146 (2016) 276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Whiting RE, Narfstrom K, Yao G, Pearce JW, Coates JR, Castaner LJ, Jensen CA, Dougherty BN, Vuillemenot BR, Kennedy D, O'Neill CA, Katz ML, Enzyme replacement therapy delays pupillary light reflex deficits in a canine model of late infantile neuronal ceroid lipofuscinosis Experimental Eye Research 125 (2014) 164–172. [DOI] [PubMed] [Google Scholar]
- [37].Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, Dyke JP, Cahan H, Slasor P, Jacoby D, Kohlschutter A, Group CLNS, Study of Intraventricular Cerliponase Alfa for CLN2 Disease New England Journal of Medicine 378 (2018) 1898–1907. [DOI] [PubMed] [Google Scholar]
- [38].Katz ML, Rustad E, Robinson GO, Whiting REH, Student JT, Coates JR, Narfstrom K, Canine neuronal ceroid lipofuscinoses: Promising models for preclinical testing of therapeutic interventions Neurobiology of Disease 108 (2017) 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lingaas F, Guttersrud OA, Arnet E, Espenes A, Neuronal ceroid lipofuscinosis in Salukis is caused by a single base pair insertion in CLN8 Animal Genetics 49 (2018) 52–58. [DOI] [PubMed] [Google Scholar]
- [40].Rossmeisl JH Jr., Duncan R, Fox J, Herring ES, Inzana KD, Neuronal ceroid-lipofuscinosis in a Labrador Retriever Journal of Veterinary Diagnostic Investigation 15 (2003) 457–460. [DOI] [PubMed] [Google Scholar]
- [41].Kolicheski A, Barnes Heller HL, Arnold S, Schnabel RD, Taylor JF, Knox CA, Mhlanga-Mutangadura T, O'Brien DP, Johnson GS, Dreyfus J, Katz ML, Homozygous PPT1 Splice Donor Mutation in a Cane Corso Dog With Neuronal Ceroid Lipofuscinosis Journal of Veterinary Internal Medicine 31 (2017) 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Katz ML, Khan S, Awano T, Shahid SA, Siakotos AN, Johnson GS, A mutation in the CLN8 gene in English Setter dogs with neuronal ceroid-lipofuscinosis Biochemical & Biophysical Research Communications 327 (2005) 541–547. [DOI] [PubMed] [Google Scholar]
- [43].Kolicheski A, Johnson GS, O'Brien DP, Mhlanga-Mutangadura T, Gilliam D, Guo J, Anderson-Sieg TD, Schnabel RD, Taylor JF, Lebowitz A, Swanson B, Hicks D, Niman ZE, Wininger FA, Carpentier MC, Katz ML, Australian Cattle Dogs with Neuronal Ceroid Lipofuscinosis are Homozygous for a CLN5 Nonsense Mutation Previously Identified in Border Collies Journal of Veterinary Internal Medicine 30 (2016) 1149–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Eldred GE, Katz ML, The autofluorescent products of lipid peroxidation may not be lipofuscin-like Free Radical Biology & Medicine 7 (1989) 157–163. [DOI] [PubMed] [Google Scholar]
- [45].Katz ML, Gao CL, Rice LM, Formation of lipofuscin-like fluorophores by reaction of retinal with photoreceptor outer segments and liposomes Mechanisms of Ageing & Development 92 (1996) 159–174. [DOI] [PubMed] [Google Scholar]
- [46].Katz ML, Robison WG Jr., Herrmann RK, Groome AB, Bieri JG, Lipofuscin accumulation resulting from senescence and vitamin E deficiency: spectral properties and tissue distribution Mechanisms of Ageing & Development 25 (1984) 149–159. [DOI] [PubMed] [Google Scholar]
- [47].Katz ML, Shanker MJ, Development of lipofuscin-like fluorescence in the retinal pigment epithelium in response to protease inhibitor treatment Mechanisms of Ageing & Development 49 (1989) 23–40. [DOI] [PubMed] [Google Scholar]
- [48].Katz ML, Stientjes HJ, Gao CL, Christianson JS, Iron-induced accumulation of lipofuscin-like fluorescent pigment in the retinal pigment epithelium Investigative Ophthalmology & Visual Science 34 (1993) 3161–3171. [PubMed] [Google Scholar]
- [49].Melville SA, Wilson CL, Chiang CS, Studdert VP, Lingaas F, Wilton AN, A mutation in canine CLN5 causes neuronal ceroid lipofuscinosis in Border collie dogs Genomics 86 (2005) 287–294. [DOI] [PubMed] [Google Scholar]
- [50].Gilliam D, Kolicheski A, Johnson GS, Mhlanga-Mutangadura T, Taylor JF, Schnabel RD, Katz ML, Golden Retriever dogs with neuronal ceroid lipofuscinosis have a two-base-pair deletion and frameshift in CLN5 Molecular Genetics & Metabolism 115 (2015) 101–109. [DOI] [PubMed] [Google Scholar]
- [51].Guo J, O'Brien DP, Mhlanga-Mutangadura T, Olby NJ, Taylor JF, Schnabel RD, Katz ML, Johnson GS, A rare homozygous MFSD8 single-base-pair deletion and frameshift in the whole genome sequence of a Chinese Crested dog with neuronal ceroid lipofuscinosis BMC Veterinary Research [Electronic Resource] 10 (2015) 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ashwini A, D'Angelo A, Yamato O, Giordano C, Cagnotti G, Harcourt-Brown T, Mhlanga-Mutangadura T, Guo J, Johnson GS, Katz ML, Neuronal ceroid lipofuscinosis associated with an MFSD8 mutation in Chihuahuas Molecular Genetics & Metabolism 118 (2016) 326–332. [DOI] [PubMed] [Google Scholar]
- [53].Faller KM, Bras J, Sharpe SJ, Anderson GW, Darwent L, Kun-Rodrigues C, Alroy J, Penderis J, Mole SE, Gutierrez-Quintana R, Guerreiro RJ, The Chihuahua dog: A new animal model for neuronal ceroid lipofuscinosis CLN7 disease? Journal of Neuroscience Research 94 (2016) 339–347. [DOI] [PubMed] [Google Scholar]
- [54].Karli P, Oevermann A, Bauer A, Jagannathan V, Leeb T, MFSD8 single-base pair deletion in a Chihuahua with neuronal ceroid lipofuscinosis Animal Genetics 47 (2016) 631. [DOI] [PubMed] [Google Scholar]
- [55].Kohyama M, Tada N, Mitsui H, Tomioka H, Tsutsui T, Yabuki A, Rahman MM, Kushida K, Mizukami K, Yamato O, Real-time PCR genotyping assay for canine progressive rod-cone degeneration and mutant allele frequency in Toy Poodles, Chihuahuas and Miniature Dachshunds in Japan J Vet Med Sci 78 (2016) 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Farias FH, Johnson GS, Taylor JF, Giuliano E, Katz ML, Sanders DN, Schnabel RD, McKay SD, Khan S, Gharahkhani P, O'Leary CA, Pettitt L, Forman OP, Boursnell M, McLaughlin B, Ahonen S, Lohi H, Hernandez-Merino E, Gould DJ, Sargan DR, Mellersh C, An ADAMTS17 splice donor site mutation in dogs with primary lens luxation Investigative Ophthalmology & Visual Science 51 (2010) 4716–4721. [DOI] [PubMed] [Google Scholar]
- [57].Donner J, Anderson H, Davison S, Hughes AM, Bouirmane J, Lindqvist J, Lytle KM, Ganesan B, Ottka C, Ruotanen P, Kaukonen M, Forman OP, Fretwell N, Cole CA, Lohi H, Frequency and distribution of 152 genetic disease variants in over 100,000 mixed breed and purebred dogs PLoS Genet 14 (2018) e1007361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gould D, Pettitt L, McLaughlin B, Holmes N, Forman O, Thomas A, Ahonen S, Lohi H, O'Leary C, Sargan D, Mellersh C, ADAMTS17 mutation associated with primary lens luxation is widespread among breeds Vet ophthalmol 14 (2011) 378–384. [DOI] [PubMed] [Google Scholar]
- [59].Mizukami K, Kawamichi T, Koie H, Tamura S, Matsunaga S, Imamoto S, Saito M, Hasegawa D, Matsuki N, Tamahara S, Sato S, Yabuki A, Chang HS, Yamato O, Neuronal ceroid lipofuscinosis in Border Collie dogs in Japan: clinical and molecular epidemiological study (2000-2011) Thescientificworldjournal 2012 (2012) 383174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Awano T, Katz ML, O'Brien DP, Taylor JF, Evans J, Khan S, Sohar I, Lobel P, Johnson GS, A mutation in the cathepsin D gene (CTSD) in American Bulldogs with neuronal ceroid lipofuscinosis Molecular Genetics & Metabolism 87 (2006) 341–348. [DOI] [PubMed] [Google Scholar]
- [61].Evans J, Katz ML, Levesque D, Shelton GD, de Lahunta A, O'Brien D, A variant form of neuronal ceroid lipofuscinosis in American bulldogs Journal of Veterinary Internal Medicine 19 (2005) 44–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.