

Abstract
Objective:
To examine the prospective associations between exposure to perfluoroalkyl substances (PFASs) and longitudinal measurements of glucose metabolism in high-risk overweight and obese Hispanic children.
Methods:
Forty overweight and obese Hispanic children (8–14 years) from urban Los Angeles underwent clinical measures and 2-hour oral glucose tolerance tests (OGTT) at baseline and a follow-up visit (range: 1–3 years after enrollment). Baseline plasma perfluorooctanoic acid (PFOA), perfluorooctane sulfonate (PFOS), perfluorohexane sulfonic acid (PFHxS), and the plasma metabolome were measured by liquid-chromatography with high-resolution mass spectrometry. Multiple linear regression models were used to assess the association between baseline PFASs and changes in glucose homeostasis over follow-up. A metabolome-wide association study coupled with pathway enrichment analysis was performed to evaluate metabolic dysregulation associated with plasma PFASs concentrations. We performed a structural integrated analysis aiming to characterize the joint impact of all factors and to identify latent clusters of children with alterations in glucose homeostasis, based on their exposure and metabolomics profile.
Results:
Each ln (ng/ml) increase in PFOA and PFHxS concentrations was associated with a 30.6 mg/dL (95% CI: 8.8–52.4) and 10.2 mg/dL (95% CI: 2.7–17.7) increase in 2-hour glucose levels, respectively. A ln (ng/ml) increase in PFHxS concentrations was also associated with 17.8 mg/dL increase in the glucose area under the curve (95% CI: 1.5–34.1). Pathway enrichment analysis showed significant alterations of lipids (e.g., glycosphingolipids, linoleic acid, and de novo lipogenesis), and amino acids (e.g., aspartate and asparagine, tyrosine, arginine and proline) in association to PFASs exposure. The integrated analysis identified a cluster of children with increased 2-h glucose levels over follow up, characterized by increased PFAS levels and altered metabolite patterns.
Conclusions:
This proof-of-concept analysis shows that higher PFAS exposure was associated with dysregulation of several lipid and amino acid pathways and longitudinal alterations in glucose homeostasis in Hispanic youth. Larger studies are needed to confirm these findings and fully elucidate the underlying biological mechanisms.
Keywords: Perfluoroalkyl substances, Type 2 diabetes, Glucose metabolism, Metabolomics, Children
1. Introduction
Type 2 diabetes is a silent epidemic in youth and its incidence has continuously increased over the past 2 decades, with the fastest rise observed among Hispanics compared to non-Hispanic whites (Lascar et al., 2018; Mayer-Davis et al., 2017). Young-onset type 2 diabetes has a more aggressive disease phenotype that can lead to premature development of complications and long-term adverse health effects with direct impacts on quality of life (Lascar et al., 2018). A growing body of evidence indicates that early life environmental exposures can result in metabolic abnormalities and increased type 2 diabetes risk in later life (Alderete et al., 2017; Alonso-Magdalena et al., 2011; Heindel et al., 2017), yet there has been little study of the environmental contributions to diabetes risk in minority ethnic groups.
Perfluoroalkyl substances (PFASs) include chemicals that have been used for decades as industrial surfactants and in textile coatings, firefighting foams and consumer products (e.g., cookware, food containers, and clothing) (Grandjean and Clapp, 2014). Rodent studies suggest that perinatal and/or early postnatal exposure to PFOA, PFOS and PFHxS induces increased insulin levels and impaired glucose tolerance in off-spring (Hines et al., 2009; Wan et al., 2014; Lv et al., 2013; Zhao et al., 2011). Cross-sectional epidemiological studies in adults from the National Health and Nutrition Examination Survey (NHANES) support observations from animal studies, showing that increased serum PFOA and PFOS levels were associated with increased fasting glucose (Liu et al., 2018), increased insulin levels, and insulin resistance (indicated by increased homeostatic model assessment for insulin resistance, HOMA-IR) (Lin et al., 2009). Further, PFOA serum levels was positively associated with diabetes prevalence in US men in the NHANES study (He et al., 2017), and a higher incidence of type 2 diabetes in women in the Nurses’ Health Study II (He et al., 2017). Few previous studies assessed associations between PFASs exposures with diabetes risk in children. The National Health and Nutrition Examination Survey (NHANES) study suggested that increased serum perfluorononanoic acid (PFNA) concentrations were associated with decreased β-cell function and increased risk for hyperglycemia in US adolescents (aged 12–20 years) (Lin et al., 2009), while the European Youth Heart Study showed that childhood PFOA exposure at 9 years of age was associated with decreased β-cell function at 15 years of age (Domazet et al., 2016). However, a pregnancy cohort in the US showed negative associations between plasma PFAS concentrations and insulin resistance at the age of 8 years (Fleisch et al., 2017).
The mechanisms underlying the effects of PFASs exposure on dysregulated glucose metabolism and diabetes risk remain unknown. In a high-throughput targeted metabolomics study, PFOA exposure has been associated with metabolic disruption in liver tissue of mice, such as alterations in lipid metabolism (e.g., glycerophospholipids, linoleic acid, and arachidonic acid) and amino acids (e.g., tyrosine, tryptophan, arginine and proline) (Yu et al., 2016). To our knowledge, only one human study has examined PFASs exposure and the plasma metabolome showing that circulating levels of PFASs in elderly adults was associated with dysregulated glycerophosphocholines and fatty acids metabolism (Salihovic et al., 2019). Dysregulation of lipid and amino acid pathways have been well-characterized and found to be strongly associated with the risk of developing type 2 diabetes in human studies (Guasch-Ferre et al., 2016; Padberg et al., 2014). Collectively, results from these studies indicate that PFASs exposure might cause metabolic perturbations in lipid and amino acid pathways, thereby contributing to increased risk for type 2 diabetes.
The objective of this study was to examine the associations between PFASs exposure and longitudinal measurements of glucose metabolism in overweight and obese Hispanic children. We also aimed to understand the underlying metabolic disturbances due to PFASs exposure by performing a high-resolution metabolomics analysis. We hypothesized that higher PFASs exposures would be associated with dysregulated glucose homeostasis and alterations in key metabolic pathways implicated in type 2 diabetes pathophysiology.
2. Methods
2.1. Study population
We examined participants from the Study of Latino Adolescents at Risk of Type 2 Diabetes (SOLAR) project, which is a longitudinal cohort that recruited participants in two waves from 2001 to 2012 (Goran et al., 2004; Weigensberg et al., 2003). Participants lived in Los Angeles, California, and were recruited predominantly from metabolic clinics as well as word of mouth, health fairs, and advertisements in the local communities. Inclusion criteria were Hispanic/Latino ethnicity (defined as self-reported race/ethnicity for all participants, parents, and grandparents), age- and sex-specific BMI equal or above the sex- and age-specific 85th percentile of the Centers for Disease Control and Prevention (CDC) growth charts, and absence of type 1 or type 2 diabetes. Participants were also excluded if they were using a medication or diagnosed with a condition known to influence insulin and/or glucose metabolism or body composition. Participants underwent repeated, detailed phenotyping for clinical risk factors of type 2 diabetes using as well as body composition testing. The original sample size from the full cohort was 258 children with the necessary 2-hour OGTT outcome measures, covariate data, and plasma samples. Of these 258 children, we chose a random sample of 40 for this proof-of-concept study. These 40 participants did not significantly differ from the 258 based on important characteristics such as age, body fat percent, or measures from the 2-hour OGTT (Supplementary Table 1). These participants had their baseline visit between the 2001–2011 and were followed for an average of 1.3 years (standard deviation: 0.5). Study protocol was approved by the University of Southern California (USC) Institutional Review Board (IRB) and informed written consent (parental consent for participants < 18 years) and assent (when applicable) were obtained for each participant before initiation of the study.
2.2. Clinical assessments
Participants attended annual clinical visits at the Los Angeles County Hospital or the USC University Hospital. At the visits, participants received a comprehensive medical history and physical examination and pubertal staging was determined using the Tanner scale (Marshall and Tanner, 1969; Marshall and Tanner, 1970). Height (m) and weight (kg) were measured to determine body mass index (BMI) and the age- and sex-specific CDC growth charts were used to classify BMI status (Kuczmarski et al., 2000). A DEXA scan was also performed to determine body fat percent using a Hologic QDR 4500 W (Hologic, Bedford, MA). A 2-hour OGTT was performed at each clinical visit using a glucose load of 1.75 g per kg body weight, to a maximum of 75 g, of anhydrous glucose dissolved in water. Baseline and post-challenge samples were assayed for glucose and insulin at fasting as well as 30, 60, and 120 min after glucose intake and were used to calculate glucose and insulin areas under the curve (AUC).The homeostatic model assessment (HOMA-IR), an estimate of insulin resistance, was calculated using fasting glucose and insulin values from the 2-hour OGTT by using the formula: HOMA-IR = fasting glucose (mg/dL) × fasting insulin (μU/mL) / 405 (Matthews et al., 1985). Glucose was assayed using a Yellow Springs Instruments analyzer (YSI INC. Yellow Springs, OH, USA) that uses a membrane bound glucose oxidase technique. Lastly, insulin was assayed using an automated enzyme immunoassay (Tosoh AIA 600 II analyzer, Tosoh Bioscience, Inc., South San Francisco, CA, USA).
2.3. High-resolution metabolomics
High resolution metabolomics (HRM) profiling was completed using standardized methods (Soltow et al., 2013). Samples were prepared and analyzed in a single batch and included six analyses of pooled human plasma (CHEAR-Ref) for quality control purposes and reference standardization. Plasma aliquots were removed from storage at −80 °C, thawed and 50 μL was treated with 100 μL of ice-cold LC-MS grade acetonitrile. Plasma was then equilibrated for 30 min on ice, centrifuged (16.1 × g at 4 °C) for 10 min to remove precipitated proteins, transferred to a 200 μL autosampler vial and maintained at 4 °C until analysis (< 22 h). Sample extracts were analyzed using liquid chromatography and Fourier transform high-resolution mass spectrometry (Dionex Ultimate 3000, Q-Exactive HF, Thermo Scientific). The chromatography system was operated in a dual pump configuration that enabled parallel analyte separation and column flushing. For each sample, 10 μL aliquots were analyzed in triplicate using hydrophilic interaction liquid chromatography (HILIC) with electrospray ionization (ESI) source operated in positive mode for metabolomic profiling and reverse phase chromatography with ESI operated in negative mode for quantification of PFASs. Analyte separation for HILIC was accomplished by a 2.1 mm × 50 mm × 2.5 μm Waters XBridge BEH Amide XP HILIC and an eluent gradient (A = 2% formic acid, B = water, C = acetonitrile) consisting of an initial 1.5 min period of 2.5% A, 22.5% B, 75% C followed by a linear increase to 2.5% A, 77.5% B, 20% C at 4 min and a final hold of 1 min. RPC separation was by 2.1 mm × 50 mm × 3 μm endcapped C18 column (Higgins) using an eluent gradient (A = 2% 5 mM ammonium acetate, B = water, C = acetonitrile) consisting of an initial 2 min period of 5%A, 90%B, 5%C, followed by a linear increase to 5%A, 0%B, 95%C at 6 min and held for the remaining 4 min. For both methods, mobile phase flow rate was held at 0.35 mL/min for the first 1.5 min, increased to 0.5 mL/min and held for the final 4 min. The high-resolution mass spectrometer was operated at 120,000 resolution and mass-to-charge ratio (m/z) range 85–1275. Probe temperature, capillary temperature, sweep gas and S-Lens RF levels were maintained at 200 °C, 300 °C, 1 arbitrary units (AU), and 45, respectively, for both polarities. Additional source tune settings were optimized for sensitivity using a standard mixture, positive tune settings for sheath gas, auxiliary gas, sweep gas and spray voltage setting were 45 AU, 25 AU and 3.5 kV, respectively; negative settings were 30 AU, 5AU and −3.0 kV. Maximum C-trap injection times of 100 milliseconds and automatic gain control target of 1 × 106 for both polarities. During untargeted data acquisition, no exclusion or inclusion masses were selected, and data was acquired in MSI mode only. Raw data files were then extracted using apLCMS (Yu et al., 2009) with modifications by xMSanalyzer (Uppal et al., 2013). Uniquely detected ions consisted of m/z, retention time and ion abundance, referred to as m/z features. Prior to data analysis, m/z features were batch corrected using ComBat (Johnson et al., 2007) and filtered to remove those with coefficient of variation (CV) ≥ 100% and > 10% non-detected values.
2.4. Quantification of plasma levels of PFASs
Concentrations of PFOA, PFOS and PFHxS were quantified by reference standardization using the LC-HRMS method described in Section 2.3 with reverse phase chromatography for analyte separation and negative mode ESI (Go et al., 2015). Analyte identification was confirmed by matching MS2 ion dissociation patterns, precursor m/z and retention time to authentic reference standards, and concentrations in the Children’s Health Exposure Analysis Resource (CHEAR) pooled plasma reference sample were quantified by methods of addition and comparison against NIST standard reference material 1950 (Metabolites in Frozen Human Plasma) (Simon-Manso et al., 2013). Using the CHEAR reference samples, the response factor for each analyte was determined using the M-H adduct, and plasma concentrations were calculated in study samples by single point calibration via response factors (calculated as the ratio between the known concentration of the compound being quantified and ion intensity in CHEAR reference samples. Calculated limit of detection (LOD) for PFOA, PFOS and PFHxS was 0.02, 0.1 and 0.03ng/mL, respectively. PFOA, PFOS, and PFHxS were detected in 97.5%, 97.5%, and 100% of participants. Due to the moderate to high correlation of PFAS (r from 0.4 to 0.7, Table 2), we also performed a principal component (PC) analysis for the 3 PFAS (PFOA, PFOS, PFHxS), and we selected the first component (“PC1”), as a composite variable representing PFAS burden, which explained 96.7% of the variance. This variable was used as the primary exposure variable in the integrated analysis with the metabolites and the outcomes of interest.
Table 2.
Distribution of polyfluoroalkyl substances (PFASs) concentrations and Spearman correlation coefficients for PFASs measured at baseline among overweight and obese Hispanic participants living in urban Los Angeles, CA who had their baseline visits between 2001 and 2011.
| Exposure | PFHxS | PFOS | PFOA |
|---|---|---|---|
| Plasma concentration of PFASs (ng/ml) | |||
| Geometric mean | 1.65 | 12.22 | 2.78 |
| Geometric mean SD | 2 | 1.91 | 1.29 |
| Minimum | 0.47 | 1.95 | 1.88 |
| Maximum | 12.81 | 65.3 | 5.37 |
| Below LOD (%) | 2.5 | 0 | 2.5 |
| Spearman correlation coefficients | |||
| PFHxS | 1.00 | ||
| PFOS | 0.58** | 1.00 | |
| PFOA | 0.44* | 0.71** | 1.00 |
p-Value < 0.005.
p-Value < 0.0001.
2.5. Statistical analysis
Geometric means (GMs), and interquartile ranges were calculated for plasma concentrations of all PFASs. Additionally, Spearman correlation coefficients among PFAS concentrations were calculated. Since the distribution of PFASs were right-skewed, exposures were natural log transformed for statistical analyses. Changes in metabolic outcomes were calculated as the respective follow-up measure subtracted from the baseline measure (e.g., fasting glucose at follow-up – baseline fasting glucose). We fitted generalized additive models (GAM) with penalized spline smooth terms and visually assessed plotted splines to determine linearity of exposure–outcome associations. We found no evidence of nonlinear associations of PFAS concentrations with type 2 diabetes outcomes except for the association of PFHxS concentrations with changes in the glucose AUC (pGAM < 0.05). We therefore fitted multiple linear regression models to estimate the relationships between change in metabolic outcomes (i.e., fasting glucose and insulin, glucose and insulin AUC, HOMA-IR) in relation to each PFAS exposure, as continuous natural log–transformed plasma concentrations at baseline. For glucose AUC and PFHxS, cubic spline models were also fitted to investigate the non-linear association. Baseline pubertal status was classified as pre-puberty (Tanner Stage 1), puberty (Tanner Stage 2–4), and post puberty (Tanner Stage 5). A modified version of the Hollingshead Four-Factor Index of Social Status was used to assess socioeconomic status in participants where information was available (n = 33). This index takes into account the occupation and education of each parent/guardian residing in the child’s home in order to generate a single measure of a family’s social status. Social position was then categorized as ≤25th percentile (n = 10), > 25th percentile and < 75th percentile (n = 19), ≥75th percentile (n = 4), and missing (n = 7). Multiple linear models adjusted for the baseline outcome, baseline social position, age, sex, and change in age at follow-up (Model 1). Models were further adjusted for baseline pubertal status as well as baseline and change in body fat percent at follow-up as adiposity and puberty are strong predictors of the outcome of interest (Model 2). Selections of covariates retained in the models were based on directed acyclic graphs (Supplementary Fig. 1) (Textor et al., 2011). Prior studies have shown more pronounced associations between PFASs exposure and insulin resistance in females (Halldorsson et al., 2012), therefore effect modification by sex was examined via an interaction term and stratification. As an exploratory analysis, effect modification of obesity (≥98th versus < 98th CDC percentile) and puberty (Tanner 1 versus Tanner 2–5) were also examined.
The metabolome-wide association study was performed in order to identify metabolites and global metabolic changes associated with PFASs exposure, including PFOA, PFOS and PFHxS. This untargeted analysis fitted multiple linear models that were used to examine the associations between plasma PFASs concentrations (independent variables) and the log2 transformed m/z features (dependent variables) after controlling for baseline age, sex, and social position. The log-2 transformation of metabolite data was used to meet the assumptions of linear regression. The m/z features with a Benjamini-Hochberg false discovery rate (FDR) of ≤20% (Benjamini and Hochberg, 1995) were then selected for visualization by Manhattan plots and metabolic pathway enrichment analysis using Mummichog (Li et al., 2013) with 10,000 permutations. Using this pathway enrichment analysis, we then identified significantly dysregulated pathways associated with PFASs exposure (p ≤ 0.05).
We performed an integrated latent variable analysis to characterize the joint impact of all factors and identify latent clusters of children with alterations in glucose homeostasis, based on their PFAS exposure and metabolomics profile by LUCIDus R package (https://CRAN.R-project.org/package=LUCIDus). For the estimation of the number of latent clusters, we used Bayesian Information Criteria. We used the PFASs composite variable as the primary exposure variable instead of the three individual PFASs since highly correlated exposure parameters invalidate the presumption in this analysis. Metabolites were annotated using adduct of protonated ion (M + H) (Supplementary Table 2) and were selected from the most significantly altered metabolic pathways (based on p value and the numbers of significant metabolite features) associated with the plasma PFAS in the Mummichog analysis. The outcome variable was determined as the change in 2-hour post-load glucose levels from baseline to follow-up visit (binary: increase vs. decrease). The LUCIDus package provides effect estimates for the association of estimated latent clusters with changes in 2 h glucose levels. It also provides the distribution of exposure and metabolites in each identified cluster (assigned based on the posterior probability using cutoff point of 0.05) (Supplementary Table 3).
All statistical analyses were performed using SAS, version 9.4 (SAS, Institute, Cary, NC) and the R statistical environment version 3.1.2.
3. Results
3.1. Characteristics of the study population
Overweight and obese Hispanic children (BMI percentile, mean ± SD: 96.8 ± 3.5) included in this study were between 8 and 14 years of age at baseline (Table 1). Half of children were female, and most were in the early to mid-stages of puberty. On average, children had normal fasting (< 100 mg/dL) and 2-hour glucose levels (< 140 mg/dL) at baseline. Mean fasting insulin levels were 16.1 mg/dL (SD 17.1) and mean HOMA-IR was 3.5 (SD 2.5). Fasting glucose levels were significantly higher in the follow up clinical visit compared to baseline (mean: 89.4 mg/dL vs. 97.7 mg/dL, p < 0.05, Table 1).
Table 1.
General baseline and follow-up characteristics of overweight and obese Hispanics participants living in urban Los Angeles, CA who had their baseline visits between 2001 and 2011.
| Baseline mean (SD) | Follow-up mean (SD) | |
|---|---|---|
| Age (years) | 11.4 ± 2.0 | 12.6 ± 2.2**n |
| Sex (N; male, female) | 19/21 | 19/21 |
| Puberty status (%)* | 33/55/12 | 22/53/25 |
| BMI percentile (%) | 96.8 ± 3.5 | 96.5 ± 4.5 |
| Body fat percent (%) | 38.6 ± 5.6 | 37.7 ± 6.3 |
| Clinical fasting glucose (mg/dL) | 89.4 ± 4.8 | 91.7 ± 5.7* |
| 2-Hour glucose (mg/dL) | 124.2 ± 17.9 | 125.4 ± 17.5 |
| Fasting insulin (μU/mL)a | 16.1 ± 11.7 | 18.5 ± 13.0 |
| 2-Hour insulin (μU/mL)a | 151.9 ± 126.3 | 186.6 ± 164.4 |
| Glucose AUC (mg/dL * min) | 258.1 ± 30.8 | 260.5 ± 32.3 |
| Insulin AUC (μU/mL * min)a | 328.2 ± 248.4 | 363.1 ± 230.3 |
| HOMA-IRa | 3.5 ± 2.5 | 4.1 ± 3.0 |
Pubertal status was defined as pre-puberty, puberty, and post puberty.
Sample size is 39. Significance from pair t-test and npaired signed rank test denoted as **p < 0.0001 and *p < 0.05.
3.2. Plasma PFASs were associated with clinical risk factors for type 2 diabetes
PFOS, PFOA, and PFHxS were detected in 97.5% of participants and geometric means were 12.22, 2.78, and 1.65 ng/ml, respectively (Table 2). PFASs concentrations showed moderate to high pairwise correlations, with the stronger correlation seen between PFOA and PFOS (Spearman r = 0.71) (Table 2). As shown in Table 3 and Fig. 1, for each ln-unit increase in PFOA and PFHxS concentrations, there was 30.6 mg/dL (95% CI: 8.8–52.4) and 10.2 mg/dL (95% CI: 2.7–17.7) increase in 2-hour glucose levels between the baseline and follow-up visit, respectively. Additionally, for each ln-unit increase in PFHxS concentrations, there was a 17.8 mg/dL increase in the glucose AUC (95% CI: 1.5–34.1). There was no evidence for effect modification by sex; however, there was evidence for effect modification by pubertal status when examining the relationships between PFHxS and glucose AUC (pinteraction = 0.03). For each ln-unit increase in PFHxS concentrations, there was a 46 mg/dL (95% CI: 16.3–75.5) increase in the glucose AUC among children who were in puberty or post-puberty (Tanner Stage 2–5) while this relationship was not observed among prepubertal children (Tanner Stage 1) (β = 63.8; 95% CI: −59.2–186.9). Lastly, there was no effect modification by BMI status (Pinteractions ≥ 0.2) and PFASs exposures were not significantly associated with other metabolic outcomes (i.e., fasting glucose, fasting insulin, and insulin AUC, HOMA-IR).
Table 3.
Adjusted associations between baseline polyfluoroalkyl substances (PFASs) plasma concentrations and changes in glucose homeostasis during follow up.
| Change in outcome | Estimated effects size between a 1-unit increase in ln PFASs (ng/mL) (95% CI) |
||
|---|---|---|---|
| ln PFHxS | ln PFOS | ln PFOA | |
| Model 1: clinical fasting glucose (mg/dL) | 1.1 (−2.1, 4.4) | 0.4 (−3.0, 3.9) | 4.6 (−4.8, 14) |
| Model 2: clinical fasting glucose (mg/dL) | 0.9 (−2.5, 4.2) | −0.2 (−3.7, 3.3) | 2.6 (−7.3, 12.6) |
| Model 1: fasting insulin (μU/mL)a | −1 (−7.1, 5) | −0.04 (−6.5, 6.4) | 8 (−8.9, 25) |
| Model 2: fasting insulin (μU/mL)a | −2.4 (−7.9, 3) | −1.2 (−7, 4.5) | 2.3 (−13.4, 18) |
| Model 1: 2-hour glucose (mg/dL) | 10.5 (2.8, 18.2) | 7.5 (−1.3, 16.3) | 33.6 (11.7, 55.4) |
| Model 2: 2-hour glucose (mg/dL) | 10.2 (2.7, 17.7) | 6.2 (−2.3, 14.8) | 30.6 (8.8, 52.4) |
| Model 1: HOMA-IRa | −0.1 (−1.5, 1.3) | −0.03 (−1.5, 1.5) | 2 (−1.9, 5.9) |
| Model 2: HOMA-IRa | −0.4 (−1.7, 0.8) | −0.3 (−1.6, 1) | 0.7 (−2.9, 4.3) |
| Model 1: 2-hour insulin (μU/mL)a | 33.1 (−26.9, 93.2) | 13.2 (−51.4, 77.8) | 103.7 (−64.4, 271.8) |
| Model 2: 2-hour insulin (μU/mL)a | 25.9 (−31.3, 83) | 5.8 (−54.5, 66.1) | 91.2 (−66.9, 249.4) |
| Model 1: glucose AUC (mg/dL * min) | 15.9 (0.3, 31.5) | 6.4 (−11.5, 24.2) | 26.8 (−20.6, 74.1) |
| Model 2: glucose AUC (mg/dL * min) | 17.8 (1.5, 34.1) | 4.8 (−13.6, 23.2) | 20.8 (−29.5, 71) |
| Model 1: insulin AUC (mg/dL * min)a | 43 (−39.1, 125) | −9.8 (−98.1, 78.5) | 67.2 (−166.5, 300.9) |
| Model 2: insulin AUC (mg/dL * min)a | 26.3 (−52.1, 104.8) | −30.5 (−111.4, 50.5) | −3.7 (−227.1, 219.8) |
Model 1: adjusted for sex, baseline social position (categorical), baseline outcome as well as baseline and change in age at follow-up. Model 2: Model 1 covariates plus further adjustment for pubertal status (categorical) as well as baseline and change in body fat percent at follow-up.
p values in bold denote statistical significance.
Sample size equals to 38.
Fig. 1.
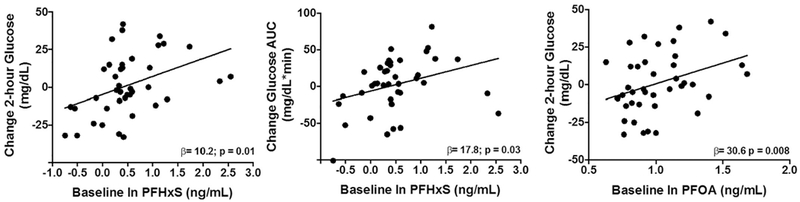
Associations between plasma polyfluoroalkyl substances (PFASs) concentrations and change in measures of glucose homeostasis.
Effect sizes (β) and p values shown were adjusted for sex, baseline social position (categorical), pubertal status (categorical), baseline outcome, as well as baseline and change in age and body fat percent at follow-up.
3.3. PFASs exposure was associated with plasma metabolites and metabolic pathways
To identify metabolic alterations associated with PFASs, we performed the metabolome-wide association study and identified 149, 298, and 17 metabolite features associated with plasma concentrations of PFOA, PFOS and PFHxS, respectively, at FDR < 20% (Fig. 2). We next performed the Mummichog pathway enrichment analysis with the input of all detected metabolite features correlated with PFOA, PFOS and PFHxS, and identified 24 metabolic pathways that were associated with PFASs exposure (Fig. 3). Exposure to PFASs was associated with dysregulation of multiple lipid metabolic pathways that included glycosphingolipid metabolism, fatty acid metabolism, de novo lipogenesis, and linoleic acid metabolism (Fig. 3). A series of amino acid metabolic pathways were also associated with PFASs exposure, including aspartate and asparagine, tyrosine, and arginine and proline metabolism (Fig. 3). Other significantly altered metabolic pathways included amino sugar metabolism, vitamins and cofactors (e.g., vitamin B3 and 9), as well as nitrogen metabolism.
Fig. 2.
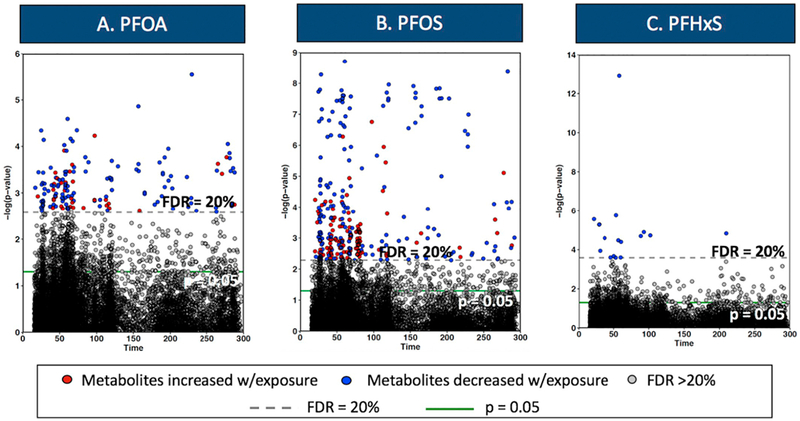
Metabolite features significantly altered in association with PFASs plasma concentrations.
Y axis represents the negative log10P for correlation of each metabolite feature with (A) PFOA, (B) PFOS and (C) PFHxS. X axis represents each metabolite in the function of retention time. Models adjusted for baseline age, sex, and social position (categorical). Metabolites above the dashed horizontal line and solid green line were significant at FDR < 20% and p < 0.05, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.
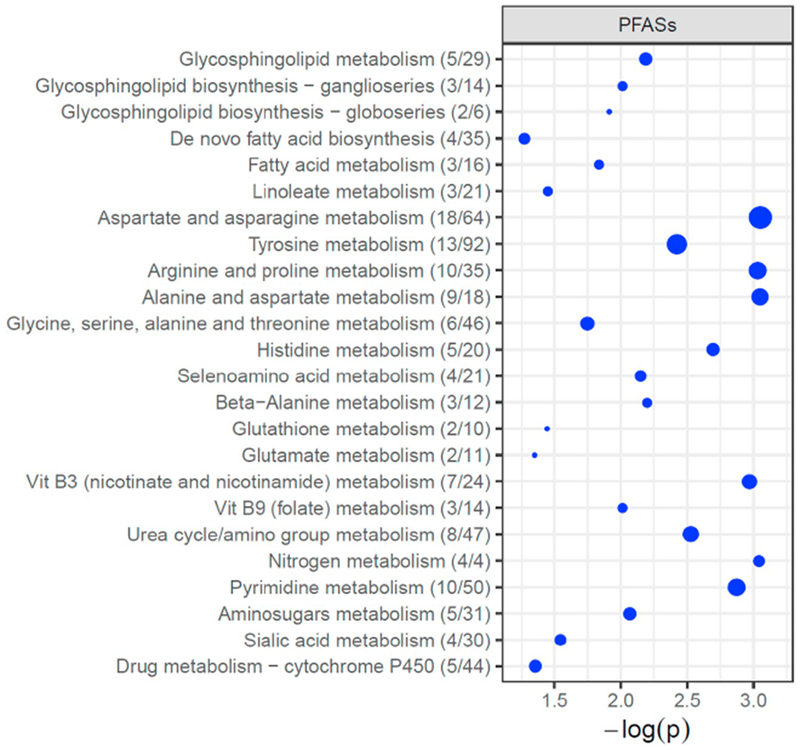
Metabolic pathways associated with plasma PFASs concentrations.
The vertical axis represents the pathways (blue circles) with circle radius representing the numbers of associated metabolite features, and horizontal axis represents the negative log10 (p-value) of each pathway. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.4. Identification of subgroups of children with alterations in glucose homeostasis using an integrated latent variable analysis
Two latent clusters were identified. “Cluster 2” was associated with increased 2 h hour glucose levels between the baseline and follow-up visit (Fig. 4, red line connecting “Cluster 2” and the outcome). The OR for increased 2 h glucose levels at follow up associated with this cluster was 8.63 (compared to “Cluster 1”). This “high-risk” cluster was also positively associated with the PFAS composite variable (blue line connecting PFAS to “Cluster 2”) and with an altered plasma metabolites pattern. This pattern was characterized by increased plasma levels (blue lines connecting “Cluster 2” to metabolites) of palmitic acid (de novo lipogenesis pathway), hydroperoxylinoleic acid (HPODE, linoleate metabolism), tyrosine and phenylalanine (tyrosine metabolism), and arginine (arginine and proline metabolism). It was also characterized by decreased plasma levels (grey lines connecting “Cluster 2” to metabolites) of sphingomyelin (glycosphingolipid pathway), linoleic acid (linoleate metabolism) and aspartate (aspartate and asparagine metabolism) (Fig. 4 and Supplementary Table 3). In order to characterize these clusters qualitatively, we assigned each child to one of two clusters based on an estimated probability >0.5 for membership within a cluster. Children assigned to the “high-risk” cluster (i.e. “Cluster 2”) had higher PFAS concentrations, associations reflective of the metabolites of previously mentioned to define “Cluster 2”, and a substantially increased risk for higher 2 h glucose levels than children in cluster 1 (Supplemental Table 3).
Fig. 4.
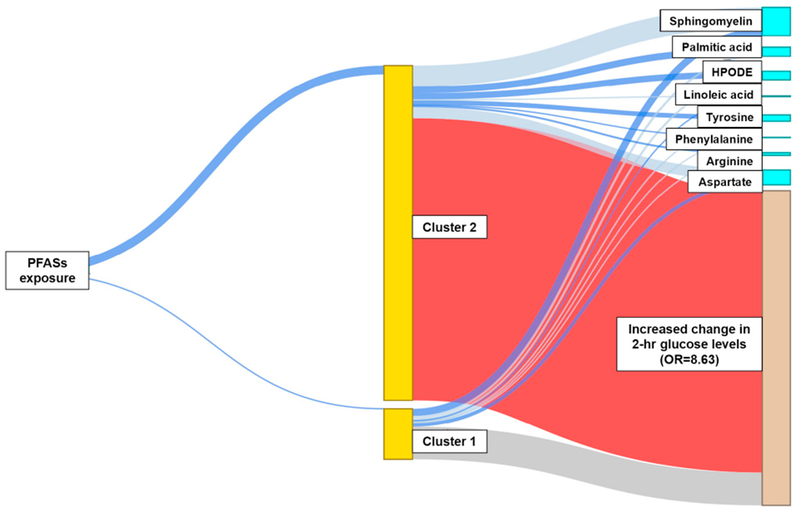
Structural integrated analysis of PFASs plasma concentrations and individual metabolites for the identification of a subgroup of children with increased risk for developing type 2 diabetes.
The thick blue line connecting “PFASs exposure” to “Cluster 2” indicates positive association, while thin blue line serves as the reference group. The blue lines connecting “Cluster” to metabolites suggest positive associations and grey lines suggest negative associations. The red line connecting “Cluster 2” and “Increased change in 2-hr glucose levels” represents that children in the latent “Cluster 2” were at higher risk for developing type 2 diabetes (OR = 8.63), compared to those in “Cluster 1” (reference). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
This is the first prospective study in Hispanic children to study environmental chemical exposures and alterations in glucose homeostasis using longitudinal clinical measures of insulin and glucose metabolism. We found that higher plasma PFASs concentrations were associated with dysregulated glucose levels following an oral glucose tolerance test. We also combined, for the first time, plasma PFASs chemical analysis and untargeted metabolomics analyses showing that PFASs exposure may be associated with changes in key metabolic pathways underlying type 2 diabetes pathophysiology such as metabolism of glycosphingolipids, fatty acid, linoleic acid, tyrosine, aspartate and asparagine, arginine and proline.
Several prospective studies in adults have shown that PFASs exposure may be associated with increased risk for type 2 diabetes (Halldorsson et al., 2012; Sun et al., 2018; Matilla-Santander et al., 2017). While longitudinal studies in children are scarce, a previous prospective study in Danish children (Domazet et al., 2016) found an association between PFOA exposure and decreased beta-cell function at age 15 years. Plasma concentrations of PFOS and PFOA in the Danish children were 3-times higher than the concentrations observed in the Hispanic youth in the current study. However, a recent pregnancy cohort study in the U.S. found that children with higher PFASs exposure had lower insulin resistance (Fleisch et al., 2017). The discrepancies in results among studies may be due to differences in study design, population demographics, or PFASs concentrations distributions. However, in the current study, PFASs concentrations were comparable to the average serum concentrations quantified for PFOS, PFOA and PFHxS in adolescents and/or Mexican Americans in NHANES between 1999 and 2010, and almost twice as high as the PFASs concentrations reported for the same NHANES subpopulations between 2011 and 2014, which is likely due to the decreasing environmental levels of these substances in U.S. and elsewhere (Fourth National Report on Human Exposure to Environmental Chemicals, 2017).
In the current study, PFASs exposure was only associated with the change in 2-hour glucose levels and the change in the glucose AUC but no other metabolic outcomes (e.g., change in fasting glucose and insulin levels, HOMA-IR). This may be due to the fact that the current study included otherwise healthy overweight and obese Hispanic youth. Further, it is possible that euglycemia in the fasting state was maintained despite important physiological changes in insulin resistance and β-cell function that may only be observed following a glucose challenge. Additionally, the consequences of insulin resistance may be more apparent after an oral glucose challenge when greater insulin levels are needed to maintain normoglycemia compared to the fasting state (Gerich, 2003). We also found that higher PFHxS concentrations were associated with an increase in the glucose AUC among children who were in puberty or post-puberty. Work by our group has shown reductions in insulin sensitivity and β-cell function during the pubertal transition (Goran and Gower, 2001). Therefore, any declines in insulin sensitivity and/or β-cell function that resulted from PFASs exposure may have been exacerbated by the normal declines that are seen during this transition. It is possible that discrepancies between findings in this study and prior work in adolescents (Lin et al., 2009; Domazet et al., 2016) may be due to failing to account for pubertal status.
The exact mechanisms underlying the associations between PFASs and diabetes-related traits remain uncertain. Rapid advancement in metabolomics technologies provides us a unique tool to examine the metabolic perturbations in response to environmental exposures (Salihovic et al., 2016; Andrianou et al., 2017). Consistent with another human study on PFAS (Salihovic et al., 2019), and other studies examining persistent organic pollutants (e.g. organochlorine pesticides) and alterations in plasma metabolome (Salihovic et al., 2016), we found that plasma PFASs concentrations were significantly associated with dysregulation of multiple lipid pathways (e.g., metabolism of glycosphingolipid, fatty acid, and linoleic acid). Importantly, alteration of such lipid metabolites was also reported to be associated with type 2 diabetes risk in metabolomics studies, as reviewed elsewhere (Guasch-Ferre et al., 2016; Padberg et al., 2014). Glycosphingolipids are suggested to modulate β-cell signaling pathways implicated in diabetic disease such as apoptosis, β-cells cytokine secretion, islet autoimmunity and insulin gene expression (Boslem et al., 2012; Janikiewicz et al., 2015; Aerts et al., 2011). In a targeted metabolomics study, sphingomyelin was found to be negatively associated with risk of type 2 diabetes (Floegel et al., 2013), which is consistent with our findings in the integrated analysis showing decreased plasma levels of sphingomyelin in the subgroups at high risk of type 2 diabetes. A representative metabolite in de novo lipogenesis, palmitic acid, can mediate insulin signaling pathway and cause insulin resistance through increase synthesis of deleterious complex lipids and impaired function of cellular organelles (Palomer et al., 2018; Ma et al., 2015). Linoleic acid and its oxidized product HPODE, play an important role in inflammation, while decreased linoleic acid is suggested to predict insulin resistance and diabetes risk (Guasch-Ferre et al., 2016; Padberg et al., 2014; Roberts et al., 2014).
In the current study, PFASs exposure was strongly associated with alterations in numerous amino acid metabolism, which is generally consistent with the concept highlighted in a most recent review paper suggesting altered circulating levels of amino acids may modulate the risk of complications related to diabetes (Kahl and Roden, 2018). Specifically, we reported that tyrosine metabolism was one of the most affected pathways associated with PFASs exposure. Increased aromatic amino acids, such as tyrosine and phenylalanine, has been consistently found to be closely associated with hyperglycemia, insulin resistance and risk of developing diabetes in adults (Guasch-Ferre et al., 2016; Wang et al., 2011; Würtz et al., 2013), although the mechanisms underlying these associations are not fully understood. Muscle cells cultured with a mixture of amino acids including aromatic (e.g. tyrosine) and branched chain amino acids (BCAAs) resulted in activation of the Mmammalian target of rapamycin (mTOR), impairment in insulin-stimulated phosphorylation of Akt/protein kinase B, and subsequently reduced glucose uptake (Tremblay and Marette, 2001). Although we did not find associations between PFASs exposure and alterations in the branched chain amino acids (BCAA) pathways, tyrosine can affect BCAA levels, since BCAA and amino acids compete for the same neutral amino acid transporter for cellular uptake (Adams, 2011; Fernstrom, 2005). We also found increased PFAS exposure was associated with the latent subgroup of participants at increased risk for developing type 2 diabetes, characterized by increased plasma arginine and decreased aspartate levels. These findings are in line with previous literature suggesting a positive association between arginine and risk of type 2 diabetes (Guasch-Ferre et al., 2016) and decreased aspartate and asparagine levels in adults developing type 2 diabetes (Palmer et al., 2015).
This study is strengthened by its longitudinal study design, the use of robust repeated clinical measures of insulin and glucose metabolism, and the use of novel statistical approaches to predict subgroups of Hispanic youth with increased susceptibility to type 2 diabetes based on their PFASs exposure and metabolomics profile. Detailed information regarding body fat percent and pubertal stage were also available and adjusted for in statistical models. Although we have adjusted for several factors that may affect glucose homeostasis, the latter is dependent on multiple exposures; thus, we cannot exclude the possibility of confounding from unmeasured variables such as diet. This proof-of-concept-study was designed in a relatively-small sample size, however, we showed markedly changes in glucose homeostasis and alterations in metabolic pathways associated with PFASs exposure. Although it would be informative to examine differences by weight status, the current study is limited for this type of analysis as it is focused primarily on obese children. We found that PFASs exposure was only associated with change in 2-hour glucose levels. These findings indicate that PFASs exposure may negatively affect β-cell function. However, effects on insulin resistance cannot be ruled out. Future studies should examine the effects of PFASs exposure on type 2 diabetes using robust methods to assess changes in insulin resistance and β-cell function (e.g., glucose clamp techniques and intravenous glucose challenge tests) (Bergman et al., 1979; DeFronzo et al., 1979). The study focused on Hispanic children and findings are of public health significance as Hispanics have a disproportionate burden of environmental exposures, high rates of type 2 diabetes in youth, and are underrepresented in clinical research (Lascar et al., 2018; Mayer-Davis et al., 2017).
5. Conclusions
In summary, this is the first prospective study to demonstrate that PFASs exposure was associated with longitudinal alterations in glucose homeostasis in overweight and obese Hispanic youth. Dysregulation of several lipid and amino acid pathways that have been linked with type 2 diabetes were also associated with PFASs exposure. Lastly, a novel integrated latent variable analysis demonstrated that the observed changes in glucose homeostasis were characterized by increased PFASs levels and altered plasma metabolite profiles. Findings from this proof-of-concept study suggest that PFASs may play an important role in the pathogenesis of type 2 diabetes; however, larger studies are needed to replicate findings and to fully elucidate the mechanisms explaining the diabetogenic effects of PFASs exposure.
Supplementary Material
Funding and acknowledgments
This work was supported by NIH P30ES007048 (Chatzi, Gilliland), NIEHS R21ES028903 (Chatzi), NIH R01DK59211 (Goran), NIEHS R00ES027853 (Alderete), NIEHS F32ES029828 (Jin), NIEHS K99ES027870 (Chen), R01-MH107205 (Jones and Walker), P30ES019776 (Jones and Walker), U2CES026560 (Jones and Walker), T32ES012870 (Walker), P01CA196569, R01CA140561, R01 ES016813 (Conti).
Abbreviations
- PFASs
perfluoroalkyl substances
- PFHxS
perfluorohexane sulfonic acid
- PFOA
perfluorooctanoic acid
- PFOS
perfluorooctane sulfonate
- EDCs
endocrine-disrupting chemicals
- PPARs
peroxisome proliferator activated receptors
- OGTT
oral glucose tolerance test
- HOMA-IR
homeostatic model assessment for insulin resistance
- AUC
area under the curve
- BMI
body mass index
- DEXA
dual-energy X-ray absorptiometry
- FDR
false discovery rate
- HRM
high-resolution metabolomics
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.envint.2019.02.047.
References
- Adams SH, 2011. Emerging perspectives on essential amino acid metabolism in obesity and the insulin-resistant state. Adv. Nutr 2 (6), 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aerts JM, Boot RG, van Eijk M, Groener J, Bijl N, Lombardo E, et al. , 2011. Glycosphingolipids and insulin resistance. Adv. Exp. Med. Biol 721, 99–119. [DOI] [PubMed] [Google Scholar]
- Alderete TL, Habre R, Toledo-Corral CM, Berhane K, Chen Z, Lurmann FW, et al. , 2017. Longitudinal associations between ambient air pollution with insulin sensitivity, beta-cell function, and adiposity in Los Angeles Latino children. Diabetes 66 (7), 1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Magdalena P, Quesada I, Nadal A, 2011. Endocrine disrupters in the etiology of type 2 diabetes mellitus. Nat. Rev. Endocrinol 7 (6), 346–353. [DOI] [PubMed] [Google Scholar]
- Andrianou XD, Charisiadis P, Makris KC, 2017. Coupling urinary trihalomethanes and metabolomic profiles of type II diabetes: a case-control study. J. Proteome Res 16 (8), 2743–2751. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol 57 (1), 289–300. [Google Scholar]
- Bergman RN, Ider YZ, Bowden CR, Cobelli C, 1979. Quantitative estimation of insulin sensitivity. Am. J. Phys 236 (6), E667–E677. [DOI] [PubMed] [Google Scholar]
- Boslem E, Meikle PJ, Biden TJ, 2012. Roles of ceramide and sphingolipids in pancreatic beta-cell function and dysfunction. Islets 4 (3), 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Tobin JD, Andres R, 1979. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am. J. Phys 237 (3), E214–E223. [DOI] [PubMed] [Google Scholar]
- Domazet SL, Grontved A, Timmermann AG, Nielsen F, Jensen TK, 2016. Longitudinal associations of exposure to perfluoroalkylated substances in childhood and adolescence and indicators of adiposity and glucose metabolism 6 and 12 years later: the European youth heart study. Diabetes Care 39 (10), 1745–1751. [DOI] [PubMed] [Google Scholar]
- Fernstrom JD, 2005. Branched-chain amino acids and brain function. J. Nutr 135 (6 Suppl) (1539s–46s). [DOI] [PubMed] [Google Scholar]
- Fleisch AF, Rifas-Shiman SL, Mora AM, Calafat AM, Ye X, Luttmann-Gibson H, et al. , 2017. Early-life exposure to perfluoroalkyl substances and childhood metabolic function. Environ. Health Perspect 125 (3), 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floegel A, Stefan N, Yu Z, Muhlenbruch K, Drogan D, Joost HG, et al. , 2013. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes 62 (2), 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourth National Report on Human Exposure to Environmental Chemicals. US Department of Health and Human Services, Centers for Disease Control and Prevention. [Google Scholar]
- Gerich JE, 2003. Clinical significance, pathogenesis, and management of postprandial hyperglycemia. Arch. Intern. Med 163 (11), 1306–1316. [DOI] [PubMed] [Google Scholar]
- Go YM, Walker DI, Liang Y, Uppal K, Soltow QA, Tran V, et al. , 2015. Reference standardization for mass spectrometry and high-resolution metabolomics applications to exposome research. Toxicol. Sci 148 (2), 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goran MI, Gower BA, 2001. Longitudinal study on pubertal insulin resistance. Diabetes 50 (11), 2444. [DOI] [PubMed] [Google Scholar]
- Goran MI, Bergman RN, Avila Q, Watkins M, Ball GD, Shaibi GQ, et al. , 2004. Impaired glucose tolerance and reduced beta-cell function in overweight Latino children with a positive family history for type 2 diabetes. J. Clin. Endocrinol. Metab 89 (1), 207–212. [DOI] [PubMed] [Google Scholar]
- Grandjean P, Clapp R, 2014. Changing interpretation of human health risks from perfluorinated compounds. Public Health Rep 129 (6), 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasch-Ferre M, Hruby A, Toledo E, Clish CB, Martinez-Gonzalez MA, Salas-Salvado J, et al. , 2016. Metabolomics in prediabetes and diabetes: a systematic reviewr and meta-analysis. Diabetes Care 39 (5), 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halldorsson TI, Rytter D, Haug LS, Bech BH, Danielsen I, Becher G, et al. , 2012. Prenatal exposure to perfluorooctanoate and risk of overw?eight at 20 years of age: a prospective cohort study. Environ. Health Perspect 120 (5), 668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Liu Y, Xu B, Gu L, Tang W, 2017. PFOA is associated with diabetes and metabolic alteration in US men: National Health and Nutrition Examination Survey 2003–2012. Sci. Total Environ 625, 566–574. [DOI] [PubMed] [Google Scholar]
- Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, et al. , 2017. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol 68, 3–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines EP, White SS, Stanko JP, Gibbs-Flournoy EA, Lau C, Fenton SE, 2009. Phenotypic dichotomy following developmental exposure to perfluorooctanoic acid (PFOA) in female CD-I mice: low doses induce elevated serum leptin and insulin, and overweight in mid-life. Mol. Cell. Endocrinol 304 (1-2), 97–105. [DOI] [PubMed] [Google Scholar]
- Janikiewicz J, Hanzelka K, Kozinski K, Kolczynska K, Dobrzyn A, 2015. Islet betacell failure in type 2 diabetes—within the network of toxic lipids. Biochem. Biophys. Res. Commun 460 (3), 491–496. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A, 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8 (1), 118–127. [DOI] [PubMed] [Google Scholar]
- Kahl S, Roden M, 2018. Amino acids - lifesaver or killer in patients with diabetes? Nat. Rev. Endocrinol 14 (8), 449–451. [DOI] [PubMed] [Google Scholar]
- Kuczmarski RJ, Ogden CL, Guo SS, Grummer-Strawn LM, Flegal KM, Mei Z, et al. , 2000. CDC growth charts for the United States: methods and development. Vital Health Stat 11 (246), 1–190 2002. [PubMed] [Google Scholar]
- Lascar N, Brown J, Pattison H, Barnett AH, Bailey CJ, Bellary S, 2018. Type 2 diabetes in adolescents and young adults. Lancet Diabetes Endocrinol 6 (1), 69–80. [DOI] [PubMed] [Google Scholar]
- Li S, Park Y, Duraisingham S, Strobel FH, Khan N, Soltow QA, et al. , 2013. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol 9 (7), e1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C-Y, Chen P-C, Lin Y-C, Lin L-Y, 2009. Association among serum per-fluoroalkyl chemicals, glucose homeostasis, and metabolic syndrome in adolescents and adults. Diabetes Care 32 (4), 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HS, Wen LL, Chu PL, Lin CY, 2018. Association among total serum isomers of perfluorinated chemicals, glucose homeostasis, lipid profiles, serum protein and metabolic syndrome in adults: NHANES, 2013–2014. Environ. Pollut 232, 73–79. [DOI] [PubMed] [Google Scholar]
- Lv Z, Li G, Li Y, Ying C, Chen J, Chen T, et al. , 2013. Glucose and lipid homeostasis in adult rat is impaired by early-life exposure to perfluorooctane sulfonate. Environ. Toxicol 28 (9), 532–542. [DOI] [PubMed] [Google Scholar]
- Ma W, Wu JH, Wang Q, Lemaitre RN, Mukamal KJ, Djousse L, et al. , 2015. Prospective association of fatty acids in the de novo lipogenesis pathway with risk of type 2 diabetes: the Cardiovascular Health Study. Am. J. Clin. Nutr 101 (1), 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WA, Tanner JM, 1969. Variations in pattern of pubertal changes in girls. Arch. Dis. Child. 44 (235), 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WA, Tanner JM, 1970. Variations in the pattern of pubertal changes in boys. Arch. Dis. Child. 45 (239), 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilla-Santander N, Valvi D, Lopez-Espinosa MJ, Manzano-Salgado CB, Ballester F, Ibarluzea J, et al. , 2017. Exposure to perfluoroalkyl substances and metabolic outcomes in pregnant women: evidence from the Spanish INMA birth cohorts. Environ. Health Perspect 125 (11), 117004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC, 1985. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28 (7), 412–419. [DOI] [PubMed] [Google Scholar]
- Mayer-Davis EJ, Dabelea D, Lawrence JM, 2017. Incidence trends of type 1 and type 2 diabetes among youths, 2002–2012. N. Engl. J. Med 377 (3), 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padberg I, Peter E, Gonzalez-Maldonado S, Witt H, Mueller M, Weis T, et al. , 2014. A new metabolomic signature in type-2 diabetes mellitus and its pathophysiology. PLoS One 9 (1), e85082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer ND, Stevens RD, Antinozzi PA, Anderson A, Bergman RN, Wagenknecht LE, et al. , 2015. Metabolomic profile associated with insulin resistance and conversion to diabetes in the Insulin Resistance Atherosclerosis Study. J. Clin. Endocrinol. Metab 100 (3), E463–E468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomer X, Pizarro-Delgado J, Barroso E, Vazquez-Carrera M, 2018. Palmitic and oleic acid: the Yin and Yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol. Metab 29 (3), 178–190. [DOI] [PubMed] [Google Scholar]
- Roberts LD, Koulman A, Griffin JL, 2014. Towards metabolic biomarkers of insulin resistance and type 2 diabetes: progress from the metabolome. Lancet Diabetes Endocrinol 2 (1), 65–75. [DOI] [PubMed] [Google Scholar]
- Salihovic S, Ganna A, Fall T, Broeckling CD, Prenni JE, van Bavel B, et al. , 2016. The metabolic fingerprint of p,p′-DDE and HCB exposure in humans. Environ. Int 88, 60–66. [DOI] [PubMed] [Google Scholar]
- Salihovic S, Fall T, Ganna A, Broeckling CD, Prenni JE, Hyotylainen T, et al. , 2019. Identification of metabolic profiles associated with human exposure to perfluoroalkyl substances. J. Expo. Sci. Environ. Epidemiol 29 (2), 196–205. [DOI] [PubMed] [Google Scholar]
- Simon-Manso Y, Lowenthal MS, Kilpatrick LE, Sampson ML, Telu KH, Rudnick PA, et al. , 2013. Metabolite profiling of a NIST Standard Reference Material for human plasma (SRM 1950): GC-MS, LC-MS, NMR, and clinical laboratory analyses, libraries, and web-based resources. Anal. Chem 85 (24), 11725–11731. [DOI] [PubMed] [Google Scholar]
- Soltow QA, Strobel FH, Mansfield KG, Wachtman L, Park Y, Jones DP, 2013. High-performance metabolic profiling with dual chromatography-Fourier-transform mass spectrometry (DC-FTMS) for study of the exposome. Metabolomics 9 (1 Suppl), S132–s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Zong G, Valvi D, Nielsen F, Coull B, Grandjean P, 2018. Plasma concentrations of perfluoroalkyl substances and risk of type 2 diabetes: a prospective investigation among U.S. women. Environ. Health Perspect 126 (3), 037001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Textor J, Hardt J, Knuppel S, 2011. DAGitty: a graphical tool for analyzing causal diagrams. Epidemiology 22 (5), 745. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Marette A, 2001. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J. Biol. Chem 276 (41), 38052–38060. [DOI] [PubMed] [Google Scholar]
- Uppal K, Soltow QA, Strobel FH, Pittard WS, Gernert KM, Yu T, et al. , 2013. xMSanalyzer: automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data. BMC Bioinf 14, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan HT, Zhao YG, Leung PY, Wong CK, 2014. Perinatal exposure to perfluorooctane sulfonate affects glucose metabolism in adult offspring. PLoS One 9 (1), e87137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. , 2011. Metabolite profiles and the risk of developing diabetes. Nat. Med 17 (4), 448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigensberg MJ, Cruz ML, Goran MI, 2003. Association between insulin sensitivity and post-glucose challenge plasma insulin values in overweight Latino youth. Diabetes Care 26 (7), 2094–2099. [DOI] [PubMed] [Google Scholar]
- Würtz P, Soininen P, Kangas AJ, Rönnemaa T, Lehtimäki T, Kähönen M, et al. , 2013. Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care 36 (3), 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Park Y, Johnson JM, Jones DP, 2009. apLCMS—adaptive processing of high-resolution LC/MS data. Bioinformatics 25 (15), 1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu N, Wei S, Li M, Yang J, Li K, Jin L, et al. , 2016. Effects of perfluorooctanoic acid on metabolic profiles in brain and liver of mouse revealed by a high-throughput targeted metabolomics approach. Sci. Rep 6, 23963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Lian Q, Chu Y, Hardy DO, Li XK, Ge RS, 2011. The inhibition of human and rat 11 beta-hydroxysteroid dehydrogenase 2 by perfluoroalkylated substances. J. Steroid Biochem. Mol. Biol 125 (1-2), 143–147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.