

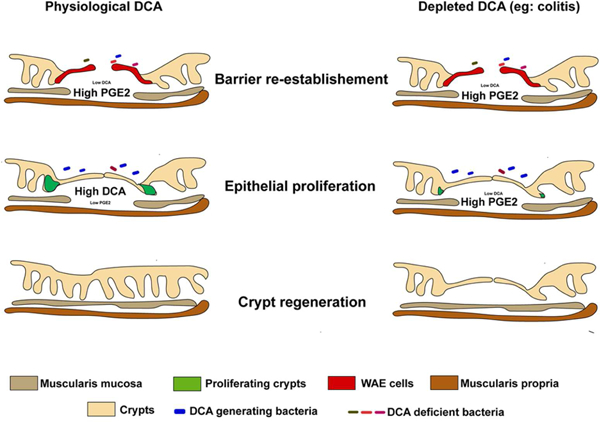
Summary
Colonic wound repair is an orchestrated process beginning with barrier re-establishment followed by wound channel formation and crypt regeneration. Elevated levels of Prostaglandin E2 (PGE2) promote barrier re-establishment; however, we found that persistently elevated PGE2 hinders subsequent repair phases. The bacterial metabolite deoxycholate (DCA) promotes transition through repair phases via PGE2 regulation. During barrier re-establishment, DCA levels are locally diminished in the wound, allowing enhanced PGE2 production and barrier reestablishment. However, during transition to the wound channel formation phase, DCA levels increase to inhibit PGE2 production and promote crypt regeneration. Altering DCA levels via antibiotic treatment enhances PGE2 levels but impairs wound repair, which is rescued with DCA treatment. DCA acts via its receptor, farnesoid X receptor, to inhibit the enzyme cPLA2 required for PGE2 synthesis. Thus, colonic wound repair requires temporally regulated signals from microbial metabolites to coordinate host associated signaling cascades.
Graphical Abstract
eTOC Blurb
Jain et.al discover that temporal regulation of a bacterial metabolite, deoxycholate, orchestrates phases of colonic repair. In the first phase, deoxycholate is depleted to allow for barrier reestablishment and re-emerges in the second phase to promote crypt regeneration. Mechanistically, deoxycholate promotes these transitions by modulating PGE2 levels.
Introduction
The intestinal epithelium acts as a physical barrier to separate the luminal contents from the underlying tissue compartment of the organ (Turner, 2009). Upon injury of this barrier, a cascade of coordinated events occurs in distinct phases involving multiple host cell types and molecules they produce that ensure efficient and complete repair (Leoni et al., 2015b). Important aspects of repair are the control of intestinal epithelial stem and progenitor proliferation in crypts of Lieberkühn, alteration of intestinal epithelial differentiation programs, and changes in cellular migration/tropism of epithelial cells and mobilizable populations of stromal and immune cells (Brown et al., 2007; Manieri et al., 2012; Miyoshi et al., 2012; Miyoshi et al., 2017). It is clear that the luminal microbiota can alter each of these host processes involved in repair, but the signaling pathways and coordination between host and microbes is not well understood (Leoni et al., 2015b).
To understand the processes and mechanisms of colonic wound repair, we and others have used endoscopy-guided biopsy forceps to remove focal areas (~1mm2) of the mucosa from the distal mouse colon (Leoni et al., 2015a; Seno et al., 2009). In contrast to chemical and infectious models of intestinal injury, this method allows for the investigation of mucosal repair with control over spatial and temporal variables. Studies of these biopsy-generated wounds have shown that colonic repair occurs in three distinct phases. In the initial barrier re-establishment phase, neutrophils migrate to the wound and into the colonic lumen and post-mitotic wound-associated epithelial cells (WAE) emerge from the adjacent crypts to cover the exposed wound bed (Manieri et al., 2012; Seno et al., 2009). The second phase is wound channel formation, which is characterized by extensive proliferation of epithelial stem cells within crypts that are adjacent to the wound bed. These channels emanate towards the center of the wound (Miyoshi et al., 2012). Finally, the third phase is crypt regeneration, which begins with Wnt5a generated by cells in the mesenchymal compartment that focally inhibit proliferation within the wound channel to initiate fission of wound channels into crypts (Miyoshi et al., 2012).
Multiple studies indicate that interactions between microbiota and host cells influence epithelial repair (Alam et al., 2016; Alam et al., 2014; Kaiko et al., 2016; Leoni et al., 2015b; 2004). Microbe-sensing receptors such as Toll-like receptors (TLRs) and formyl peptide receptors (FPRs) enhance wound closure by WAE cells in the colon (Alam et al., 2016; Leoni et al., 2013; Manieri et al., 2012). We have established that TLR-induced Prostaglandin-endoperoxide synthase 2 (PTSG2) expression is a key player in the early phase of wound repair (Manieri et al., 2012). In particular, PTGS2-expressing mesenchymal stem cells localize specifically in the colonic wound bed in close association with the crypts adjacent to the wound (Brown et al., 2007; Manieri et al., 2012). PGE2 secreted from these cells acts through its receptor PTGER4 on intestinal epithelial cells, to stimulate WAE differentiation (Miyoshi et al., 2017). Further support for the role of microbiota comes from a recent finding suggesting that an inflammatory environment during injury changes the microbiota composition near the site of the wound bed, including enrichment of Akkermansia muciniphilia, to enhance initial wound closure (Alam et al., 2016). Importantly, these studies have mostly focused on the mediators that aid the barrier re-establishment phase of wound repair (i.e. wound closure), whereas the mediators that mount an effective second phase of repair (crypt regeneration) remain unclear.
We previously found that enhanced PGE2 signaling in epithelial cells is critical for barrier re-establishment (Miyoshi et al., 2017). In the present study, we first demonstrated that the subsequent down modulation of PGE2 is necessary to permit the second phase of repair to proceed. We then sought to identify factors in the intestine that regulate the PGE2 switch. We considered that metabolites might be promising candidate factors for inhibition of PGE2 because these are small (<1.5 kD) molecules that can circumvent the mucus and epithelial barriers to engage the host. Metabolite libraries from the intestinal contents of mice and other hosts can now be determined by untargeted mass-spectroscopy approaches (Matsumoto et al., 2012). Such libraries are a powerful source of material for screens of host cell responses relevant to intestinal repair (Kaiko et al., 2016; Steed et al., 2017). Here, through a metabolite screen and in vivo validation using the biopsy injury model, we discovered that the temporal presence of a bacterial metabolite, deoxycholate (DCA), regulates the transition between phases of intestinal healing by controlling local PGE2 production.
Results
Reduction of PGE2 levels is critical for the transition from barrier re-establishment to wound channel/crypt regeneration phases of repair
Intestinal wound repair is a highly ordered process that occurs in sequential, defined phases. We use a model system of acute injury and repair whereby we make ~1 mm2 biopsy wounds in the distal colonic mucosa (Seno et al., 2009) that creates a functional breakdown of the epithelial barrier (S1A, B). The initial phase of repair is defined by barrier re-establishment that begins immediately after injury and typically extends through day 4 post-repair (Figure 1A). A defining feature of barrier re-establishment is the formation of a monolayer of post-mitotic WAE cells that are produced by stem/progenitor cells in crypts adjacent to wound (Figure S1A) (Manieri et al., 2012). WAE cell formation is dependent on PGE2 signaling to the epithelium through PTGER4 (Miyoshi et al., 2017). The next phase (days~4–8) is wound channel formation. This phase is defined by the epithelial stem cell expansion that emanates from wound adjacent crypts (Miyoshi et al., 2012) towards the center of the injury site (Figure S1A). A third phase, crypt regeneration, is defined by the subdivision of wound channels into new crypts that occurs at days 8–12 post-injury. Although PGE2 is clearly required for barrier re-establishment, the role of this molecule at later stages is not known. Comparison of PGE2 levels in colonic tissue from non-wounded and wounded areas at various times post-injury showed a striking pattern whereby PGE2 levels were elevated in wounds during barrier re-establishment (day 4 post-injury) and then returned to levels similar to non-wounded areas during the phases of wound channel formation and crypt regeneration (day 6–12 post-injury) (Figure 1B). We hypothesized that this decrease of PGE2 during the latter phases of repair was essential for the transition from barrier re-establishment to wound channel formation and crypt regeneration.
Figure 1. Diminished PGE2 levels are required for the transition to the crypt regeneration phase during intestinal wound repair.
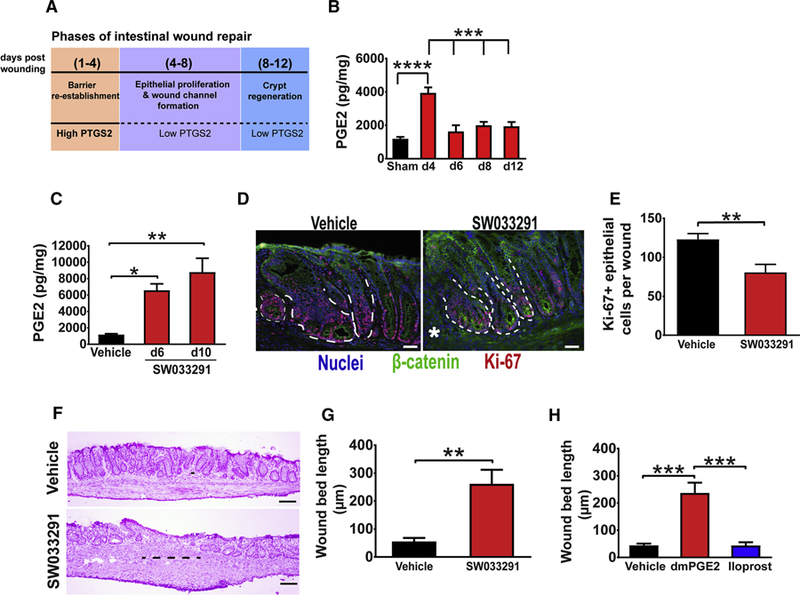
(A) Schematic representation of stages of intestinal wound repair. See also Figure S1. (B) PGE2 levels in WT wounds collected at the indicated days post-injury (n=17–36 wounds/group from 610 mice/group). Sham=colonic tissue from mice that underwent endoscopy but not biopsy injury. Significance was determined by one-way ANOVA and TUKEY’s post hoc test: ***p < 0.001, ****p < 0.0001. (C-G) Tissue from wounds of biopsy injured WT mice treated twice daily on days 4–10 post-injury with either vehicle or SW033291, an inhibitor of the prostaglandindegrading enzyme 15-PGDH. (C) PGE2 levels in wounds at day 6 and 10 post-injury (n=13–21 wounds/group from 5–6 mice/group). Significance was determined by one-way ANOVA and TUKEY’s post hoc test: *p<0.05, **p<0.01. (D) Representative images of sections of the three crypts adjacent to wound bed from indicated groups at day 6 post-injury and stained for Ki-67 (red, proliferating cells), β-catenin, (green, epithelial cells) and bis-benzimide (blue, nuclei). Dotted white lines represent the three crypts where Ki67 cells were counted and asterisks indicate the edge of the wound beds. Bars=40µm. (E) Average of Ki-67 positive cells per wound (n=17–18 wounds/group from 6–7 mice/group). Significance was determined by an unpaired Student’s t test: **p<0.01. (F) Representative images of H&E stained sections from wound beds of indicated groups at day 12 post-injury. Bars=100µm. The black line at the center of the wound represents wound bed length (largest distance between the crypts). (G) Wound bed length at day 12 post-injury from vehicle and SW033291 treated mice (n=14–18 wounds/group from 5–7 mice/group). Significance was determined by unpaired Student’s t test: **p<0.01. (H) Wound bed length at day 12 post-injury from mice treated twice daily on days 4–10 post-injury with either vehicle, dmPGE2, or the PGI2 analog Iloprost (n=14–17 wounds/group from 6–8 mice/group). Significance was determined by one-way ANOVA and TUKEY’s post hoc test: ***p<0.001. All values in B, C, E, G and H are displayed as mean ± SEM. See also Figure S2.
To test this hypothesis in vivo, we utilized two approaches to sustain elevated levels of PGE2 beyond the barrier re-establishment phase and during wound channel formation. For the first approach, C57BL/6J WT mice were treated beginning at day 4 post-injury with SW033291, an established inhibitor of the prostaglandin-degrading enzyme 15-PGDH (Zhang et al., 2015). Compared to vehicle-treated controls, wounds from SW033291-treated mice showed significantly elevated levels of PGE2 (Figure 1C). On day six post-injury, adjacent crypts showed diminished epithelial cell proliferation in SW033291-treated mice as compared to controls (Figure 1D–E). As a control, cleaved caspase 3 stained sections showed negligible amounts of apoptotic epithelial cells in adjacent crypts in both control and SW033291-treated mice (Figure S2A). The continued elevation of PGE2 had a dramatic effect on the crypt regeneration phase; at day 12 post-injury, the wound beds of SW033291-treated mice contained a large area that lacked crypts, while controls showed a near complete replacement of crypts at this stage of repair (Figure 1F–G). Importantly, mucosal sites distant from the original wound site in the SW033291-treated group did not show any overt signs of inflammation and/or injury (Figure S2B–D).
As a second, independent approach to sustain elevated PGE2 levels during the wound channel formation and crypt regeneration phases, mice were treated with dimethyl modified PGE2 (dmPGE2), which has an increased half-life in vivo relative to unmodified PGE2. We used a comparable dose and intraperitoneal injection schedule to that which rescued phenotypes in Ptgs2−/− mice (Brown et al., 2007; Manieri et al., 2012). Similar to SW033291-treated mice, dmPGE2-treated WT mice showed reduced epithelial proliferation in wound-adjacent crypts on day 6 post-injury (Figure S2E) as well as defective crypt regeneration on day 12 post-injury (Figures 1H and S2F) as compared to vehicle-treated controls. This effect was specific to PGE2, as the prostacyclin (PGI2) analog Iloprost did not elicit these phenotypes (Figure 1H). Taken together, these data support the hypothesis that the reduction of PGE2 levels during wound channel formation and crypt regeneration is permissive for the transition from the barrier reestablishment phase of repair to the phases that follow.
In vitro screen of bacterial metabolites identifies deoxycholate (DCA) as a potent suppressor of TLR2-induced PGE2 production
Rapid shifts in the bacterial microbiome occur at the site of intestinal wounds during intestinal mucosal wound repair, with specific taxa decreasing in relative abundance during the initial phase of repair and then rebounding to pre-injury abundances during later stages of repair (Alam et al., 2016). This timing is coincident with the observed shifts in wound PGE2 levels during repair and suggests that microbes could actively contribute to the transition between stages of repair. Microbial metabolites are small, secreted molecules that typically cross the host cellular barrier and can alter host signaling (Donia and Fischbach, 2015). Thus, we hypothesized that specific microbial metabolites would inhibit PGE2 production in the wound, thereby promoting transition through phases of repair.
Colonic mesenchymal stem cells (cMSCs) are a significant source of PGE2 during barrier re-establishment and MyD88 signaling in these cells is critical for elevated PGE2 production (Manieri et al., 2012). Thus we used cMSCs to develop an in vitro system to screen microbial metabolites for their effect on PGE2 production. We first defined the selective signaling upstream of MyD88 that stimulated PGE2 production by applying a panel of individual TLR ligands to cMSCs. The TLR2 ligands Pam3CSK4 and Pam2CSK4 significantly stimulated PGE2 production in wildtype (WT) cMSCs in a dose-dependent manner (Figure 2A and S3A–C). As a control, cMSCs from TLR2−/− mice showed no alteration in PGE2 levels (Figure S3D). The TLR2-PGE2 pathway was also relevant in vivo, as TLR2−/− mice did not elevate PGE2 in the wound site during the barrier re-establishment phase (Figure S3E); this was manifested by a lack of WAE cell formation at day 4 post-injury compared to WT controls (Figure S3F–G). Taken together, these findings indicate that TLR2 promotes the barrier re-establishment phase through PGE2 production.
Figure 2. In vitro screening of TLR2-stimulated cMSCs with microbial metabolites identified deoxycholate as a potent suppressor of PGE2 production.
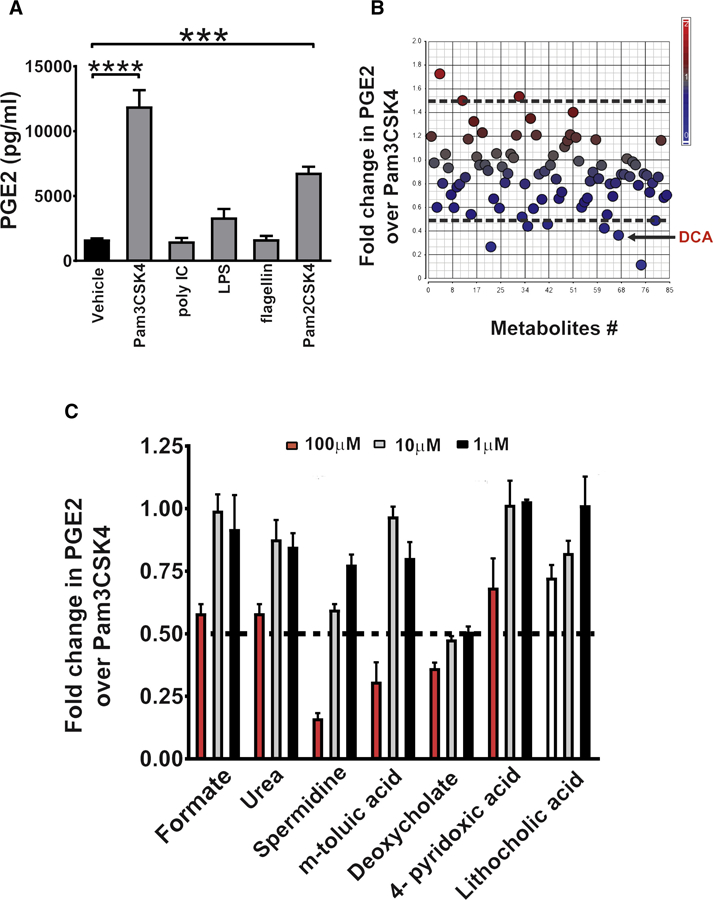
(A)cMSCs were cultured with vehicle or the indicated TLR ligands (100 ng/ml) for 24 hrs followed by quantification of PGE2 in the culture supernatants (n=3 experiments). Significance was determined by one-way ANOVA and TUKEY’s post hoc test: ***p<0.001, ****p<0.0001. See also Figure S3. (B-C) cMSCs treated with 100 ng/ml Pam3CSK4 in the presence or absence of individual microbial metabolites (Kaiko et al., 2016). (B) Scatter plot showing the fold-change in PGE2 production after treatment with 100 µm of each metabolite plus Pam3CSK4 versus vehicle plus Pam3CSK4 treated cells. The black arrow indicates DCA on the scatter plot. (C) Plot of dose-response for each metabolite (plus Pam3CSK4) that was identified in the primary screen and diminished PGE2 production (n=3 experiments). Relative PGE2 production expressed as fold-change compared to Pam3CSK4 plus vehicle treated cells. All values are displayed as mean ± SEM. See also Figure S3 and Table S1.
We utilized this model of TLR2-stimulated cMSCs to perform an in vitro screen of intestinal bacterial metabolites to search for repressors of PGE2 production. We screened a library of 84 metabolites that are abundant in the mouse colonic lumen and are either produced or induced by bacteria (Matsumoto et al., 2012) by applying them to cultured cMSCs and measuring PGE2 in the supernatant. The primary screen identified six metabolites that caused > 50% inhibition of TLR2-induced PGE2 production (Table S1 and Figure 2B). We chose 50% inhibition as a threshold value because 2-fold changes in PGE2 are functional in vivo (Zhang et al., 2015). Focusing on these six metabolites, we performed a secondary validation using three different concentrations of each metabolite and found that the secondary bile acid, deoxycholate (DCA), most potently suppressed PGE2 production in a dose dependent manner (Figure 2C). The 100µM dose of two other metabolites, spermidine and m-toluic acid, also demonstrated >50% decrease in PGE2 levels but these decreases were attributed to reduced cell viability (Figure S3H). Importantly, PGE2 inhibition occurred when DCA was present at concentrations similar to those detected in the colonic lumen of mice (Kaiko et al., 2016). We also measured PGE2 in the cMSC lysates following Pam3CSK4 induction and DCA treatment and observed a similar decrease in PGE2 (Figure S3I), implying that DCA affects PGE2 production. The secondary bile acids, DCA and lithocholic acid (LCA), are formed in the colon by the bacterial-dependent modifications of primary bile acids (Pavlidis et al., 2015). Although LCA was present in our metabolite library, it was not identified to inhibit >50% PGE2 production in the primary screen. We included it in the secondary screen and found that LCA was not as potent as DCA in inhibiting TLR2 dependent PGE2 production (Figure 2C).
Taken together, we devised a screen that utilized the robust response of cMSCs to TLR2 ligands to stimulate the production of PGE2. This screen identified a bacterial metabolite, DCA that effectively reversed the TLR2-mediated spike in PGE2 production from cMSCs. This finding sets up the hypothesis that DCA may act in vivo to mediate the transition through phases of repair, from barrier re-establishment to wound channel formation, by inhibition of PGE2 during the latter time point.
DCA inhibits PGE2 production in vivo to promote wound channel formation and crypt regeneration during colonic repair
We next investigated whether DCA could suppress PGE2 levels in vivo to promote crypt regeneration in the late repair phase. First, we determined the concentrations of DCA by mass spectrometry of colonic tissue taken from wounded and non-wounded areas at various times post-injury. DCA levels in wounds exhibited a reciprocal pattern to PGE2 levels in wounds (Figure 1B and 3A). DCA levels in wounds were lower on day 4 post-injury as compared to day 6 (Figure 3A). This reduction was restricted to the wound, as DCA levels on day 4 post-injury were not altered in mucosal sites distant to the wound bed (Figure S4A). Secondary bile acids, including DCA, are known to be produced primarily by the actions of members of the Clostridial cluster XIV that are vancomycin sensitive (Yoshimoto et al., 2013). Thus, we would expect that treatment of WT mice with vancomycin would greatly diminish DCA levels that would result in persistently increased PGE2 levels during repair. To test this idea, we biopsy-injured WT mice pre-treated with vancomycin for 2 weeks (Figure 3B). As expected, vancomycin-treated mice showed significantly lower amounts of fecal- and wound-associated DCA on day 6 post-injury as compared to controls (Figure 3C and S4B). In addition, wound PGE2 levels were significantly higher in vancomycin-treated mice versus controls at this time point (Figure 3D). Sustained PGE2 elevation in the wound bed following vancomycin treatment was associated with reduced epithelial proliferation in wound-adjacent crypts at day 6 as well as impaired crypt regeneration at day 12 post-injury (Figure S4C and 3E–F). The magnitude of these effects was similar to the above models where PGE2 levels were sustained with dmPGE2 or SW033291. Finally, the detrimental effects of vancomycin on repair could be rescued by additionally treating the mice with the selective inhibitor of PTGS2, NS-398, beginning at day 4 and continuing to day 10 postinjury (Figure 3G–H and S3D). These results demonstrate that loss of DCA contributes to impaired crypt regeneration due to sustained production of PGE2 in the wound.
Figure 3. Vancomycin treatment of WT mice depleted DCA in the wounds and impaired crypt regeneration in a PGE2-dependent manner.
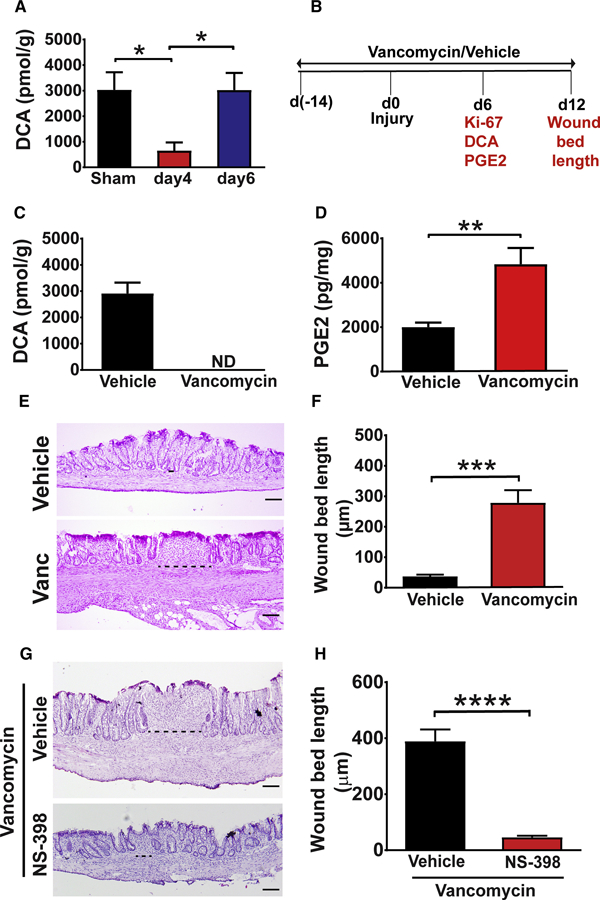
(A) Wound tissue from WT mice was collected on days 4 and 6 post-injury and analyzed for DCA levels by mass spectrometry (n=17–27 wounds/ group from 6–8 mice per group). Significance was determined by one-way ANOVA and TUKEY’s post hoc test: *p<0.05. Sham=colonic tissue from mice that underwent endoscopy but not biopsy injury. (B) Schematic representation of the procedures and analysis (in red text) measured on days 6 and 12 in C-F. See also Figure S4. (C) Wounds from mice treated with either vehicle or vancomycin were analyzed for DCA at day 6 post-injury, ND= not detected. (D) Wound PGE2 levels at day 6 post-injury from vehicle and vancomycin treated mice (n=19–21 wounds/group from 7–8 mice/group). Significance was determined by unpaired Student’s t test: **p<0.01. (E) Representative images of H&E stained sections (Bars=100µm) and (F) wound bed length at day 12 post-injury from indicated groups of mice (n=16–19 wounds/group from 6–9 mice/group). Significance was determined by unpaired Student’s t test: ***p<0.001. (G-H) Mice treated with vancomycin were biopsy injured, treated with either vehicle or NS-398 (twice daily from days 4–10 post-injury) and wounds were analyzed. (G) Representative images of H&E stained sections (Bars=100 µm) and (H) wound bed length at day 12 post-injury (n=14–19 wounds from 6–8 mice/group). Significance was determined by unpaired Student’s t test: ****p<0.0001. All values in A, C, D, F and H are displayed as mean ± SEM. See also Figure S4.
We next took two approaches to determine if restoration of DCA concentrations after the barrier re-establishment phase to physiological levels in vancomycin-treated mice could rescue defective crypt regeneration. First, DCA directly administered to the colonic lumen via daily rectal enemas (from days 4–10, Figure 4A), reversed the vancomycin-induced phenotype demonstrated by after barrier re-establishment to ensure complete crypt regeneration. To further test the role of timing, we performed DCA reconstitution from day 0-day 3 in vancomycin pre-treated mice and analyzed wounds at day 4 post-injury. We used a previously established method of analyzing wounds at day 4 from whole mount images (Miyoshi et al., 2017) and found that this treatment significantly impaired barrier re-establishment associated with reduced PGE2 levels (Figure S5C–E). Taken together, these findings confirm that the timing of DCA generation is crucial for transition from barrier re-establishment to crypt regeneration.
Figure 4. DCA reconstitution rescued vancomycin block in wound repair progression.
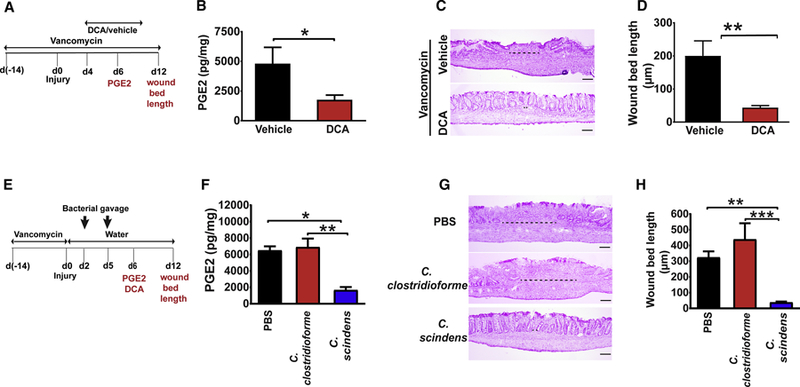
(A-D) Vancomycin-treated mice were biopsy injured and intra-rectally administered either DCA or vehicle (twice daily from day 4-day 10 post-injury). (A) Schematic for DCA reconstitution in vancomycin-treated mice with parameters measured on day 6 and 12 (in red text). (B) Wound PGE2 levels at day 6 post-injury (n=23–27 wounds/group from 7–8 mice/group). Significance was determined by unpaired Student’s t test: *p<0.05. (C) Representative images of H&E stained wound sections (Bars=100µm) and (D) wound bed length from indicated groups at day 12 postinjury (n=17–20 wounds/group from 8 mice/group). Significance was determined by unpaired Student’s t test: **p<0.01. See also Figure S5. (E-H) Vancomycin pre-treated mice were biopsy injured and gavaged twice with PBS, C. scindens or C. clostridioforme at day 2 and 5 postinjury. (E) Schematic for bacterial reconstitution in vancomycin mice with parameters (in red text) measured on day 6 and 12. (F) Wound PGE2 levels at day 6 post-injury from indicated groups of mice (n= 7–17 wounds from 3–5 mice/group). Significance was determined by one-way ANOVA and TUKEY’s post hoc test: *p<0.05, **p<0.01. (G) Representative images of H&E stained sections (Bars=100µm) and (H) wound bed length at day 12 post-injury (n=13–19 wounds/group from 7–8 mice/group). Significance was determined by one-way ANOVA and TUKEY’s post hoc test: ****p<0.0001. Values in B, D, F and H are displayed as mean ± SEM. See also Figure S5.
In the second approach, we restored DCA by gavaging vancomycin pre-treated mice with Clostridium scindens. C. scindens harbors bile acid inducible (bai) operon genes that contribute to 7-α dehydroxylation generating secondary bile acids (Ridlon et al., 2006). Mice were treated with vancomycin, biopsy-injured, provided a short washout period and then administered PBS, C. scindens or Clostridium clostridioforme-a member of cluster XIVa incapable of generating secondary bile acids (Dehoux et al., 2016; Studer et al., 2016) (Figure 4E). Similar to exogenous DCA, C. scindens colonization inhibited PGE2 levels in wounds and enhanced crypt regeneration in vancomycin treated mice (Figure 4F–H). Importantly, C. scindens colonization correlated with enhanced DCA generation (Figure S5F–G). C. clostridioforme, despite effective colonization (Figure S5F), neither induced DCA (Figure S5G) nor reversed the repair defect (Figure 4H). Taken together these findings demonstrate a functional relationship between microbes that produce the secondary bile acid DCA, PGE2 production and efficient colonic wound healing.
DCA acts via FXR to inhibit PGE2 production and promote crypt regeneration
Primary and secondary bile acids signal via a variety of receptors with DCA demonstrating strong affinities for farnesoid X receptor (FXR) and G protein-coupled bile acid receptor 1 (GPBAR1, also known as TGR5) (Schaap et al., 2014). To determine which receptor mediated the effects of DCA in intestinal repair, we utilized agonists for FXR and TGR5 in the in vitro TLR2-stimulated cMSC model and assessed PGE2 production. FXR agonist INT-747 significantly inhibited TLR2-dependent production of PGE2 from cMSCs (Figure 5A). However, the TGR5 agonist INT-777 did not significantly affect PGE2 production (Figure 5B). We next validated these results using cMSCs isolated from FXR−/− and TGR5−/− mice. We found that DCA did not inhibit TLR2-induced PGE2 in cMSCs from FXR−/− mice (Figure 5C). In contrast, DCA still inhibited PGE2 production in TLR2-stimulated TGR5−/− cMSCs, indicating that DCA preferentially utilized FXR to downregulate PGE2 in vitro (Figure 5D).
Figure 5. DCA inhibited PGE2 production from cMSCs via FXR in vitro.
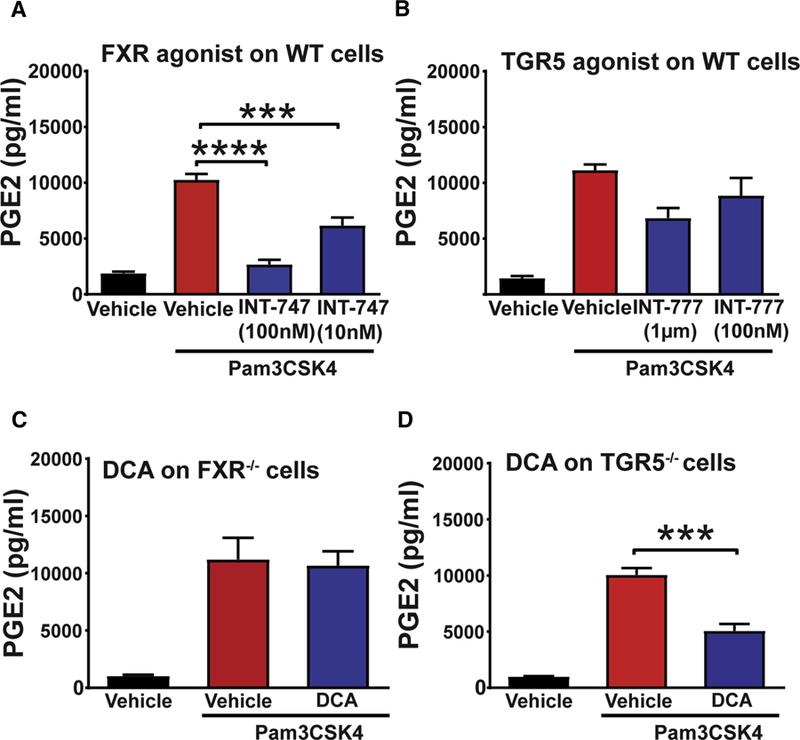
(A-B) WT cMSCs were cultured in DMEM with vehicle alone; with vehicle plus 100 ng/ml Pam3CSK4 and with Pam3CSK4 and indicated doses of agonists for (A) FXR (INT-747) or (B) TGR5 (INT-777). 24-hour culture supernatants were evaluated for PGE2 levels by ELISA (n=3 experiments). Significance was determined by one-way ANOVA and TUKEY’s post hoc test: ***p<0.001, ****p<0.0001. (C-D) FXR−/− and TGR5−/− cMSCS were cultured in DMEM with vehicle alone, vehicle plus 100ng/ml Pam3CSK4 and Pam3CSK4 plus DCA (10µM). PGE2 levels in the 24-hour culture supernatants of (C) FXR−/− and (D) TGR5−/− cMSCs as determined by ELISA (n=3 experiments). Significance was determined by one-way ANOVA and TUKEY’s post hoc test: ***p<0.001, All values are displayed as mean ± SEM.
Our hypothesis was that FXR agonism diminished PGE2 levels through effects on the expression of enzymes involved in PGE2 synthesis. We found that DCA treatment resulted in decreased protein levels of cytosolic phospholipase A2 (cPLA2), but did not alter the protein expression of PTGS-2, PTGS1, m-PEGS-2, or c-PEGS (Figure S6A–C). In the PGE2 synthesis pathway, cPLA2 liberates arachidonic acid (AA) from membrane phospholipids followed by conversion of AA to prostaglandin precursors by PTGS½ (Park et al., 2006). We then compared the actions of FXR and TGR5 agonists and found that an agonist for FXR, but not TGR5, inhibited the expression of cPLA2 similar to DCA (Figure S6D–E). In summary, these findings suggest that DCA downregulates cPLA2 through FXR to inhibit PGE2 production.
We further used both pharmacological and genetic approaches to test if DCA functions via FXR in vivo during repair. We utilized the vancomycin model and tested if low amounts of chenodeoxycholic acid (CDCA), a potent agonist of FXR (Lew et al., 2004; Wang et al., 1999), could rescue the phenotype of defective crypt regeneration. As expected, CDCA administration from day 4-day 10 resulted in efficient crypt regeneration compared to vancomycin controls (Figure S6F–G). In the second approach, FXR−/− and WT littermates were biopsy-injured and crypt regeneration was evaluated. At 6 days post-biopsy, FXR−/− wounds had elevated PGE2 levels compared to littermate controls (Figure 6A). Accordingly, FXR−/− mice showed inefficient crypt regeneration on day 12 that could be abolished by additional treatment with NS-398 (Figure 6B–C and S6H–I). The phenotype in FXR−/− mice was not due to dysregulated endogenous local DCA synthesis as both FXR−/− and littermate WT controls demonstrated equivalent levels of wound DCA (Figure 6D). Importantly, intrarectal administration of DCA did not rescue defective crypt regeneration in mice lacking FXR (Figure 6E–F). Collectively these findings suggest that DCA inhibits PGE2 production preferentially through FXR to promote transition into the second phase of repair for efficient crypt regeneration.
Figure 6. DCA promoted crypt regeneration via FXR in vivo.
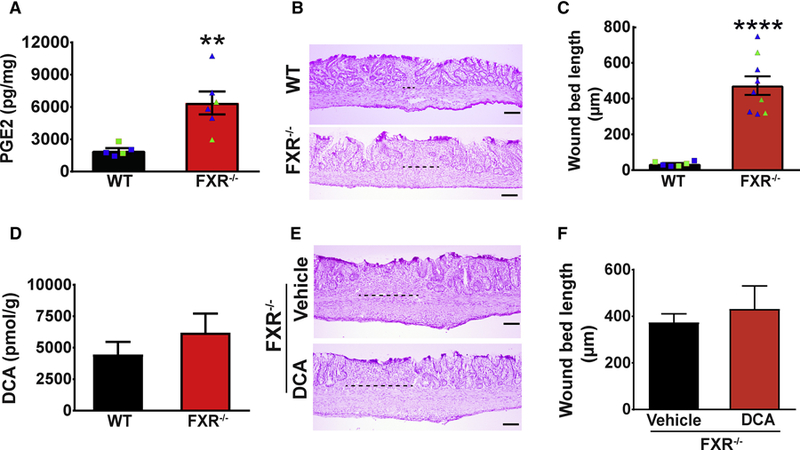
(A) Wound PGE2 levels at day 6 post-injury from FXR−/− and WT littermate controls (n=13–21 wounds/group from 5–6 mice/group). Significance was determined by unpaired Student’s t test: **p<0.01. Each dot represents data from a single mouse; blue indicates male and green represents female mice. (B) Representative images of H&E stained sections (Bars=100µm) and (C) wound bed length at day 12 post-injury from WT and FXR−/− mice (n=16–23 wounds/group from 6–9 mice/group). Significance was determined by unpaired Student’s t test: ****p<0.0001. Each dot represents data from a single mouse; blue indicates male and green represents female mice. (D) Wound DCA levels at day 6 post-injury from FXR−/− and WT mice (n=24–34 wounds/group from 9–10 mice/group). (E) Representative images of H&E stained sections (Bars=100µm) at day 12 postinjury from FXR−/− mice that were injured and intrarectally administered either DCA (100µM, twice daily enema from day 4-day 10) or equivalent volume of vehicle. (F) Wound bed length at day 12 post-injury from indicated groups (n=11–13 wounds from 5–6 mice/group). All values are displayed as mean ± SEM. See also Figure S6.
Discussion
Intestinal wound repair occurs in sequential phases: barrier re-establishment occurs first followed by wound channel formation and then crypt regeneration. A mouse model of colonic mucosal biopsy injury where repair can be tracked based on a specific location and wound initiation time has been important to establish these phases. A major unanswered question is the factor(s) that mediate phase transitions in repair. Here we investigated the transition from barrier re-establishment to wound channel formation and found that PGE2 levels in the wound bed must diminish to allow wound channel formation. Our overarching hypothesis is that intestinal microbes and/or their metabolic products mediate this transition by decreasing PGE2 levels in the wound. Thus, we performed a screen of intestinal microbial metabolites that are abundant in WT mice and found that deoxycholate (DCA); a secondary bile acid can mediate this transition by inhibiting PGE2 production in the wound. The results from additional experimental approaches supported this finding. First, antibiotics that deplete DCA inhibited this same phase transition. Following antibiotic treatment, the addition of either DCA or DCA-generating bacteria permitted this phase transition to occur. In a second approach, loss of function of the bile acid receptor FXR also inhibited this phase transition. Most importantly, addition of an exogenous PGE2 inhibitor rescued the transition defect observed in vancomycin and FXR−/− mice. Taken together, these findings suggest that bacterial metabolism that generates secondary bile acids is necessary to promote transition between stages of healing in the intestine in a PGE2 dependent manner.
We have previously shown that MyD88-dependent production of PGE2 from cMSCs during the barrier re-establishment phase is required for the formation WAE cells (Manieri et al., 2012). We found that of the TLR ligands, TLR½ and TLR2/6 agonists potently elicited PGE2 production from cMSCs in vitro. TLR2 is functional in vivo as TLR2−/− mice failed to increase PGE2 in the colonic wounds during the barrier re-establishment phase leading to a defect in repair during this phase. These findings support the necessity of TLR2 signaling for the elevation of PGE2 in colonic wounds during barrier reestablishment.
The production of PGE2 must be carefully regulated as elevated levels past the barrier reestablishment phase impaired stem cell proliferation leading to incomplete crypt regeneration. To understand the mechanism of PGE2 down regulation after the barrier re-establishment phase, it was important to identify factors that could inhibit TLR2-dependent production of PGE2 from cMSCs. Through a metabolite library screen, we found that DCA inhibits PGE2 production to ensure crypt regeneration. Levels of DCA in the tissue are diminished during the barrier reestablishment phase and re-emerge during wound channel formation phase. Importantly, this temporary alteration of the local microbiota composition is resolved with the formation of a WAE barrier. We propose the following temporal model of repair: 1) after injury, changes in microbiome composition at the wound site reduces DCA-generating bacteria, and TLR2 signaling in the stromal compartment induces the production of PGE2 (Manieri et al., 2012); 2) PGE2 stimulates WAE differentiation via EP4 that is expressed on intestinal epithelial cells (Miyoshi et al., 2017); 3) after WAE formation, DCA-generating bacteria reemerge at the wound site and inhibit TLR2-stimulated production of PGE2 through DCA thus 4) allowing for the transition to wound channel formation which has the hallmark of an epithelial stem cell expansion in adjacent crypts that is a precursor to new crypt formation (Miyoshi et al., 2012). Overall, our findings indicate that during healing after colonic mucosal injury the process of repair is controlled by concerted actions of specific microbes and their metabolites.
The role of PGE2 remains controversial with both beneficial and detrimental effects of PTGS2/PGE2 axis reported in experimental models of colitis (Paiotti et al., 2012). An important feature of the biopsy injury model is that it allows for the precise control of the timing and location of an injury site, which is permissive to define the role of PGE2 during certain phases of repair. Early in repair, a TLR2-dependent elevation of PGE2 is required to form the barrier, however PGE2 must be diminished later to allow for epithelial proliferation and crypt regeneration (Miyoshi et al., 2012; Miyoshi et al., 2017; Seno et al., 2009). This is because persistently elevated PGE2 results in reduced epithelial proliferation thereby limiting the formation of wound channels and subsequent crypt regeneration.
Secondary bile acids in excess of physiological concentrations can have cytotoxic effects in the colon (Pavlidis et al., 2015). However, physiological levels of secondary bile acids can be beneficial in the colon; both DCA and LCA derived from C. scindens directly inhibit the growth of Clostridium difficile and provide resistance against the pathogen (Buffie et al., 2015). This finding is medically relevant as fecal microbiota transplantation that restores DCA to levels of ~1mM (luminal) in humans is an effective therapy for C. difficile infection (Weingarden et al., 2016). Furthermore, physiological deoxycholate reduces Campylobacter jejuni-induced colitis in genetically susceptible mice possibly by modulating host inflammatory signaling but not by directly inhibiting the growth of C. jejuni (Sun et al., 2018). Bile acids can signal through both nuclear and membrane receptors to modulate inflammatory responses (Schaap et al., 2014). Our findings suggest that DCA acts primarily via FXR, a nuclear receptor, to promote transition from the barrier re-establishment phase to wound channel formation. This finding is consistent with previous studies showing benefits of FXR in chemical damage models of the mouse colon (Gadaleta et al., 2011; Vavassori et al., 2009). There are likely many cellular targets for FXR including the innate immune system. Our findings highlight a role of FXR that is permissive in intestinal epithelial expansion during repair. LCA, can also activate FXR, though we found a lesser effect on the inhibition of PGE2 production by cMSCs. This finding could be explained by the fact that different bile acids activate FXR in a gene-selective fashion demonstrating unique properties (Lew et al., 2004). Furthermore, in contrast to DCA, LCA is normally present in significantly lower quantities in the colonic lumen and also poorly absorbed by the colon, indicating that the cells might not be programmed to efficiently sense LCA(Sayin et al., 2013). Nevertheless, as DCA reconstitution restored the defect in the transition to the wound channel phase induced by vancomycin, we propose that DCA is a key effector of this transition.
cPLA2 is a rate limiting enzyme that converts membrane phospholipids to arachidonic acid-the substrate for PTGS2 (Park et al., 2006). In accordance, genetic deletion of cPLA2 results in reduced colonic PGE2 production during inflammation (Montrose et al., 2015). Mechanistically, we discovered that cPLA2 appears to be a proximate target of FXR. Indeed, FXR but not TGR5 activation, reduced cPLA2 expression that in turn reduced PGE2 production. This is consistent with other reports that have demonstrated that bile salts and FXR agonists can modulate phospholipase A2 activity leading to reduced production of prostaglandins (Komichi et al., 2005; Yu et al., 2007).Therefore we propose that reduced PGE2 production in the presence of DCA or FXR agonists is in part due to down-regulation of cPLA2.
Our findings have implications for diseases of the gastrointestinal tract that are associated with impaired wound healing. Generation of secondary bile acids is dependent on a small fraction of bacteria in the gut. It is estimated that only 0.0001% of the total colonic microbes are capable of metabolizing primary into secondary bile acids (Ridlon et al., 2006). Thus, proper representation of this consortium is paramount to intestinal health. Dysbiosis which a common feature of patients of inflammatory bowel disease (IBD) is associated with a loss of bacterial clusters that aid in generating secondary bile acids (Duboc et al., 2013; Frank et al., 2007; Morgan et al., 2012). In addition, multiple studies identified a lower abundance of secondary bile acid-generating microbes in IBD subjects versus controls (Duboc et al., 2013; Gnewuch et al., 2009; Kruis et al., 1986). Interestingly, mucosal inflammation in IBD patients is typically characterized by increased local PGE2 and decreased crypt density (Geboes and Dalle, 2002; Rampton et al., 1980; Wiercinska-Drapalo et al., 1999). Given our findings, we propose that DCA/FXR axis is an important determinant of the disease and its reconstitution might be an attractive therapeutic strategy for promoting repair in IBD.
MSCs are therapeutically attractive as they produce a range of factors necessary for repair such as PGE2, VEGF and IL-10 (Parekkadan and Milwid, 2010). MSCs derived from bone marrow or fat have shown efficacy as a treatment for perianal fistulas in patients of Crohn’s disease (Voswinkel et al., 2013); however, recent clinical trials with MSCs have been inconclusive (Martinez-Montiel Mdel et al., 2014), suggesting a requirement for in depth characterization of their mechanistic actions. Although other cell types including macrophages can express PTGS2 in the colon, we have found that cMSCs have the highest PTGS2 expression post-injury in the biopsy injury model (Brown et al., 2007; Manieri et al., 2012). Therefore, the ability of DCA to downregulate PGE2 production from cMSCs is highly relevant to intestinal healing. We cannot rule out that DCA also acts on other cell types that express FXR in the intestinal environment.
In conclusion, we demonstrate the importance of changes in local metabolite composition during healing and identify DCA as a key molecule that promotes crypt regeneration. Given that DCA and PGE2 are reciprocally regulated during efficient repair, our findings also assert the importance of proper timing of metabolite expression during the process of intestinal healing.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead Contact, Thaddeus Stappenbeck (stappenb@wustl.edu).
Experimental Models and Subject Details
Mice
WT, TLR2−/− and FXR−/− mouse strains were purchased from Jackson laboratories (Bar Harbor, ME). The TGR5−/− mouse strain was obtained from Dr. Bethany P. Cummings (Cornell University, Ithaca, NY) and is available at UCDAVIS KOMP Repository. All mouse strains were on a C57Bl/6J genetic background. Mice were housed under specific pathogen free conditions and had unrestricted access to autoclaved food and water. 12-hour light cycles were maintained throughout the experiments. All experiments were performed according to the protocols approved by animal studies committee at Washington University, St Louis. 7–10 week old and sex matched mice were used for all the experiments. All the biopsy experiments utilized littermate controls. In biopsy experiments involving WT and TLR2−/− mice, we utilized only male mice. In experiments comparing FXR−/− and WT (littermate controls), both male and female mice were used; colored symbols in the graph identify the sex of the animal.
cMSC culture
cMSCs were isolated from male or female WT and gene-deficient strains as described previously (Manieri et al., 2012; Walker et al., 2010). Colons were removed, flushed with PBS, minced with razor blade and incubated in 20 mL of low glucose DMEM (Life Technologies) containing 10mM HEPES (Corning), penicillin-streptomycin (Sigma), 5mM EDTA (Corning), 1mM DTT (Sigma) and 10% FBS (Sigma) for 20 minutes at 37°C. The tissue was then washed and suspended in low glucose DMEM containing collagenase I (Thermo Fisher Scientific) and DNase (Sigma) and incubated for 45 minutes at 37°C. Finally, the cells were washed, filtered and cultured onto tissue culture treated petri dishes at 37° C. After overnight culture, cells were washed with sterile PBS and fed fresh media. Media was subsequently changed every 2 days and cells were passaged when ~80% confluent. Cells isolated from 5 mice were pooled and used for each experiment.
Method Details
Colon biopsy and processing of wounds
Focal mucosal wounds (2–4 per mouse) were created in distal half of the mouse colon using biopsy forceps and a high resolution miniaturized colonoscopy system (Karl Storz) (Manieri et al., 2012; Miyoshi et al., 2017; Seno et al., 2009). Briefly, mice were anaesthetized and colons were inflated with PBS to remove fecal pellets. Three-French biopsy forceps were then inserted into the sheath adjacent to the camera and 3–5 full thickness areas were removed to create wounds. Mice were euthanized at day 1, 4, 6, 10 or 12 post-injury. Colons were opened longitudinally, pinned in OCT and visualized with an Olympus SZX12 stereo dissection microscope. Wounds along with 1–2mm of adjacent area were removed with a razor blade and embedded in OCT. Serial 5 µm-thick sections from the center of the wound were obtained using a cryostat and stored at −80°C until further analysis (Manieri et al., 2012; Seno et al., 2009). Biopsy injury was performed by investigators blinded to the treatments. Also, wounds were sectioned by an investigator who was not involved in the biopsy injury. Sections through the center of the wound bed were taken for all analysis (Manieri et al., 2012; Miyoshi et al., 2017; Seno et al., 2009). For whole mount analysis, dissected wounds were placed in OCT and visualized using an Olympus SZX12 stereo dissection microscope equipped with a digital camera and DP Controller software. Percent healing was calculated as described previously (Miyoshi et al., 2017); (1 – [wound area on day 4/original wound area] × 100).
For H&E staining, 5 µm-thick sections were fixed in 4% paraformaldehyde (Sigma) for 10 minutes, washed in tap water and stained with hematoxylin solution (EMD Millipore). After 1 minute, slides were washed and rinsed in distilled water, stained with eosin (Thermo Scientific) and dehydrated in Ethanol. Slides were passed through three changes of Xylene (Fisher Chemical) and mounted using Cytoseal (Thermo Scientific). Images were visualized and captured using an Olympus BX-51 microscope equipped with digital camera DP22. Wound bed length was calculated by measuring the distance between the two farthest crypts in section of the center of the wound bed using imaging software Olympus CellSens Standard.
For metabolite measurements, wounds were washed multiple times with PBS to remove fecal contamination, homogenized in RIPA buffer containing protease/phosphatase inhibitor (Thermo Scientific) followed by sonication (Qsonica). The samples were then centrifuged at 10,000xg for 15 mins, supernatants were collected and frozen at −80°C until further analysis.
Mouse treatments
The 15-hydroxyprostaglandin dehydrogenase (15-PGDH) inhibitor, SW003291 (Zhang et al., 2015), was purchased from Tocris, dissolved in DMSO (Sigma) and diluted in sterile saline. SW003291 was injected intraperitoneally (i.p.) in biopsy injured mice at 5mg/kg twice daily from day 4 until day 10 unless stated otherwise.
16,16-dimethyl prostaglandin E2 and Iloprost were purchased from Cayman Chemical. Both compounds were diluted in sterile 5% sodium bicarbonate immediately before use. Iloprost (200 µg/kg body weight) or 16,16-dimethyl prostaglandin E2 (50 µg/kg body weight) were injected i.p. twice daily into biopsy injured mice from day 4 until day 10 unless stated otherwise (Manieri et al., 2012).
Ptgs2 inhibitor, NS-398 (Cayman Chemical) was dissolved in DMSO to make a stock solution and diluted in saline immediately before use. NS-398 was administered intraperitoneally at a dose of 2.5mg/kg body weight, twice daily from day 4 until day 10 (Masferrer et al., 1994).
Chenodeoxycholic acid (CDCA, Sigma) was dissolved in DMSO and diluted in saline immediately before use. Sodium deoxycholate (Sigma) was dissolved in sterile water. For in vivo metabolite experiments, mice received intra-rectal administration of sterile water (vehicle), CDCA or sodium deoxycholate (100µM, Sigma) twice daily from day 4 until day 10 unless stated otherwise. The total volume administered by enema was 300µl.
For antibiotic experiments that did not involve bacterial reconstitution, mice were administered vancomycin in drinking water (Sigma, 0.5mg/ml) or Milli-Q water (vehicle) starting 2 weeks prior to the biopsy injury until the day of sacrifice. For studying the effects of DCA during barrier re-establishment phase, vancomycin treated mice were biopsy injured and administered DCA or vehicle from day 0-day 3 post-injury and sacrificed on day 4. In experiments studying the transition between phases of repair, the indicated groups of injured mice received either vehicle, DCA, CDCA or NS-398 from day 4 until day 10 and sacrificed on either day 6 or day 12. For bacterial reconstitution experiments, vancomycin was withdrawn after 2 weeks and mice were put on Milli-Q water. Mice were biopsy injured on the day of vancomycin withdrawal and bacteria or PBS were orally delivered on day 2 and day 5 post-vancomycin and indicated groups were sacrificed on either day 6 or 12. Clostridium scindens and Clostridium clostridioforme were obtained from ATCC. Individual strains of bacteria were grown in chopped media broth and orally administered at ~7×108 bacteria per mouse (Buffie et al., 2015; Steed et al., 2017).
Immunofluorescence
5 µm-thick sections were fixed in 4% paraformaldehyde for 10 minutes and blocked in 3% BSA/0.1% Triton-X for 30 minutes. The slides were then washed in PBS and incubated with primary antibody overnight at 4°C. This was followed by a 45-minute incubation with the secondary antibody and counterstaining with bis-benzimide (Hoechst 33258: Invitrogen). Slides were subsequently mounted in Fluoromount (Sigma).
The following antibodies were used for immunofluorescence: monoclonal mouse anti-βcatenin (1:2000, BD Transduction Laboratories), rabbit monoclonal anti-cleaved caspase-3 (1:300, Cell Signaling Technology), rabbit polyclonal Ki-67 (1:200, Abcam). Secondary antibodies conjugated to either Alexa Flour 488 or 594 were used at a dilution of 1:500. Images were visualized and captured using a Zeiss Axiovert inverted microscope equipped with an Axiocam MRM digital camera. For day 6 quantification, Ki67 positive epithelial cells (double positive for Ki67 and β-catenin) were counted in two high power fields (20X) in three crypts on both sides of the wound and averaged among all mice per group. For quantification on day 12, Ki67 positive epithelial cells were counted in areas distant to the wound in two high power fields and averaged among all mice per group. Ki-67 positive epithelial cells were quantified by two independent investigators blinded to the treatments.
Mass spectrometry
Stool and wounds were collected at indicated time points and frozen until further analysis. The stool and colon tissue samples were extracted at a concentration of 100 or 200 mg/mL in 80% methanol containing 1 µM of the internal standard, d4-deoxycholate in a tissue lyserII. Samples were clarified by centrifugation then filtered through 8.0 µm PES spin filters to remove particulates. Samples (one microliter) were loaded onto a 0.5 × 100 mM custom packed PLRPS column using 10 mM ammonium bicarbonate in water (A) and 10 mM ammonium bicarbonate in 95% acetonitrile (B). The gradient started at 2%B for three minutes followed by a ramp to 25%B over three minutes, then a ramp up to 100%B over two more minutes with a hold at 100%B for a minute then ramp back down to 2%B over one minute. The column was then reequilibrated for five minutes. Data were recorded in polarity switching mode with data dependent acquisition of the top 5 peaks for MS/MS per cycle using a Q-Exactive mass spectrometer set to a resolution setting of 70,000 (at m/z 200). The data were integrated using the QuanBroswer application of Xcalibu. The concentrations are calculated based on comparison to the internal standards and plotting against a calibration curve made from various concentrations of deoxycholate and static concentrations of internal standard d4 deoxycholate. All the mass spectrometry procedures including homogenization of the colon and stool samples was performed by independent investigators blinded to the treatment.
cMSC stimulation
In stimulation experiments, cMSCs were plated in 96-well plates at 20,000 cells/well in low glucose DMEM (containing 10mM HEPES, penicillin-streptomycin, 10% FBS) and allowed to adhere overnight. The following morning, cells were fed fresh media, stimulated with ligands and incubated at 37°C for 24 hours. For TLR screen, cMSCs were individually stimulated with Pam3CSK4, LPS, flagellin, Pam2CSK4, poly IC (Invitrogen) at indicated doses or vehicle (endotoxin free water, Invitrogen). For the metabolite screen, cMSCs were stimulated with 100ng/ml of Pam3CSK4 in the presence or absence of bacterial metabolites (100 µM). For secondary validation, metabolites at 100µM, 10µM and 1µM were used. For receptor agonists, cMSCs were stimulated with 100ng/ml Pam3CSK4 in the presence or absence of GW4064 (Tocris), INT747 (MedChem Express) or INT-777 (MedChem Express). 24-hour culture supernatants were collected from the treatments and PGE2 concentration was assayed. In some experiments, cells were washed with PBS and lysed in RIPA (Sigma) buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific). These lysates were then assayed for PGE2. All the experiments utilized cMSCs between passage number 5 and 8.
PCR to determine bacterial colonization
Stool was collected from the treated groups of mice at day 6 post-injury and DNA was isolated using the QIAamp DNA Stool Mini Kit (Qiagen). The following primers were used for PCR-based amplification (Nava et al., 2011; Steed et al., 2017; Wells et al., 2003; Yuli et al., 2005). baiCD forward (5′-GGWTTCAGCCCRCAGATGTTCTTTG-3′), baiCD reverse (5′-GAATTCCGGGTTCATGAACATTCTKCKAAG-3′ Clostridium clostridioforme forward (5’-GCCGCATGGCAGTGTGT-3’), Clostridium clostridioforme reverse (5′-ACCGGCCCGTCAGGGGGAT-3′), rpoB forward (5′-AACATCGGTTTGATCAAC-3′) and rpoB reverse (5′-CGTTGCATGTTGGTACCCAT-3′). The baiCD thermocyler conditions were as follows: 94°C for 2 minutes, followed by 35 cycles of 94°C for 20 s, 52°C for 30 s and 69°C for 90 s, ending with 68°C for 10 minutes. For C. clostridioforme, the following conditions were used: 35 cycles of 95°C for 20 seconds, 58°C for 60 s, and ending with 72°C for 5 minutes. For rpoB, the following conditions were used: 94°C for 3 minutes followed by 35 cycles of 94°C for 45 seconds, 50°C for 60 seconds, and 72°C for 90 seconds, ending with 60°C for 15 seconds. Bacterial DNA was isolated by an investigator blinded to the groups and PCR was run by another investigator who was not involved in DNA isolation.
MTT assay
After collecting the 24-hour culture supernatants from the secondary metabolite screen plate, cells were washed with sterile PBS and cell survival was assessed using the MTT cell proliferation assay kit from ATCC according to recommended instructions. The data were plotted as percentage viability compared to Pam3CSK4-treated wells.
PGE2 measurements
PGE2 concentration was measured using PGE2 ELISA kit (Cayman Chemical) according to the manufacturer’s instructions. Importantly, all treatments and corresponding controls were run on the same plate.
Immunoblot analysis
After 24 hr of treatment, cMSCs, were washed with PBS and lysed in RIPA (Sigma) buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific). Protein concentration was determined using the BCA protein assay kit (Thermo Fisher Scientific), subjected to SDSPAGE using any KD MINI-PROTEAN gels (Bio-Rad) and electro transferred to nitrocellulose membranes (Bio-Rad). Membranes were incubated in Blocking One solution (Nacalai tesque) for 1 hour at room temperature followed by overnight incubation at 4°C with primary antibodies. Membranes were further incubated with HRP conjugated secondary antibodies (1:10,000) for 1 hour at room temperature. SuperSignal Femto kit (Thermo Fisher Scientific) was used for detection with autoradiography film (Midsci). The following primary antibodies were used for immunoblotting: rabbit anti-COX2 (1:1000, Thermo Fisher Scientific), rabbit anti-Cox1 (1:1000, Cell Signaling Technologies), rabbit anti-cPLA2 (1:1000, Cell Signaling Technologies), rabbit anti-Prostaglandin E Synthase-2, microsomal (1:200), rabbit anti-Prostaglandin E Synthase (cytosolic) (1:200, Cayman), mouse anti-β-Actin (1:10,000, Sigma). Band intensity was calculated using ImageJ (NIH) software, normalized to loading control and expressed as fold change over vehicle control.
In vivo permeability assay
Mice were fasted (no food and water) for 4 hours and then administered 4KDa FITC-dextran (Sigma, 44 mg/100 g body weight dose) by oral gavage. After three hours, mice were anesthetized, blood was collected via retro orbital bleeding and serum was separated by centrifugation. Serum samples were diluted two-fold in PBS and fluorescence intensity was measured at an excitation wavelength of 485nm. FITC was serially diluted in PBS to prepare a standard curve and serum concentration was calculated.
Quantification and Statistical analysis
Statistical analysis
Data are presented as mean ± SEM. Parametric or nonparametric statistical tests were applied as appropriate after testing for normal distribution of data. Comparisons between two groups that did not meet the assumptions of a parametric t test were analyzed by Mann-Whitney test. Comparisons between multiple groups (>2) with normal data distributions were analyzed by oneway ANOVA with Tukey’s post hoc test. A significant difference was defined as p<0.05. All statistical analysis was performed using GraphPad Prism version 7.02. The exact value of ‘n’ within figures is indicated in figure legends. For biopsy experiments, ‘n’ represents the total number of wounds per group. Each cell culture stimulation and perturbation experiment was performed with pooled cells from five different mice and ‘n’ represents number of experiments.
Supplementary Material
Highlights.
PGE2 promotes barrier re-establishment but inhibits crypt regeneration in the colon
DCA and PGE2 levels are temporally and reciprocally regulated during colonic repair
DCA acts via its receptor FXR to inhibit PGE2 production and promote crypt regeneration
DCA/FXR inhibits cPLA2 involved in PGE2 synthesis to suppress PGE2 production
Acknowledgements
We thank Drs. Ta-Chiang Liu and William Stenson for critical comments, Dr. Bradley Evans (Danforth Plant Science Center) for mass spectroscopy analysis and Dr. Gerard E. Kaiko for metabolite screen analysis. The SHINE program (Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT) provided support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare that no conflict of interest exists.
References
- Alam A, Leoni G, Quiros M, Wu H, Desai C, Nishio H, Jones RM, Nusrat A, and Neish AS (2016). The microenvironment of injured murine gut elicits a local pro-restitutive microbiota. Nat Microbiol 1, 15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A, Leoni G, Wentworth CC, Kwal JM, Wu H, Ardita CS, Swanson PA, Lambeth JD, Jones RM, Nusrat A, et al. (2014). Redox signaling regulates commensal-mediated mucosal homeostasis and restitution and requires formyl peptide receptor 1. Mucosal Immunol 7, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SL, Riehl TE, Walker MR, Geske MJ, Doherty JM, Stenson WF, and Stappenbeck TS (2007). Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury. J Clin Invest 117, 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehoux P, Marvaud JC, Abouelleil A, Earl AM, Lambert T, and Dauga C (2016). Comparative genomics of Clostridium bolteae and Clostridium clostridioforme reveals species-specific genomic properties and numerous putative antibiotic resistance determinants. BMC Genomics 17, 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia MS, and Fischbach MA (2015). HUMAN MICROBIOTA. Small molecules from the human microbiota. Science 349, 1254766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboc H, Rajca S, Rainteau D, Benarous D, Maubert MA, Quervain E, Thomas G, Barbu V, Humbert L, Despras G, et al. (2013). Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62, 531–539. [DOI] [PubMed] [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, and Pace NR (2007). Molecularphylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 104, 13780–13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadaleta RM, van Erpecum KJ, Oldenburg B, Willemsen EC, Renooij W, Murzilli S, Klomp LW, Siersema PD, Schipper ME, Danese S, et al. (2011). Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 60, 463–472. [DOI] [PubMed] [Google Scholar]
- Geboes K, and Dalle I (2002). Influence of treatment on morphological features of mucosal inflammation. Gut 50 Suppl 3, III37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnewuch C, Liebisch G, Langmann T, Dieplinger B, Mueller T, Haltmayer M, Dieplinger H, Zahn A, Stremmel W, Rogler G, et al. (2009). Serum bile acid profiling reflects enterohepatic detoxification state and intestinal barrier function in inflammatory bowel disease. World J Gastroenterol 15, 3134–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, Pearce EL, Oltz EM, and Stappenbeck TS (2016). The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 165, 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komichi D, Tazuma S, Nishioka T, Hyogo H, and Chayama K (2005). A nuclear receptor ligand down-regulates cytosolic phospholipase A2 expression to reduce bile acid-induced cyclooxygenase 2 activity in cholangiocytes: implications of anticarcinogenic action of farnesoid X receptor agonists. Dig Dis Sci 50, 514–524. [DOI] [PubMed] [Google Scholar]
- Kruis W, Kalek HD, Stellaard F, and Paumgartner G (1986). Altered fecal bile acid pattern in patients with inflammatory bowel disease. Digestion 35, 189–198. [DOI] [PubMed] [Google Scholar]
- Leoni G, Alam A, Neumann PA, Lambeth JD, Cheng G, McCoy J, Hilgarth RS, Kundu K, Murthy N, Kusters D, et al. (2013). Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. J Clin Invest 123, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni G, Neumann PA, Kamaly N, Quiros M, Nishio H, Jones HR, Sumagin R, Hilgarth RS, Alam A, Fredman G, et al. (2015a). Annexin A1-containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. J Clin Invest 125, 1215–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni G, Neumann PA, Sumagin R, Denning TL, and Nusrat A (2015b). Wound repair: role of immune-epithelial interactions. Mucosal Immunol 8, 959–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew JL, Zhao A, Yu J, Huang L, De Pedro N, Pelaez F, Wright SD, and Cui J (2004). The farnesoid X receptor controls gene expression in a ligand- and promoter-selective fashion. J Biol Chem 279, 8856–8861. [DOI] [PubMed] [Google Scholar]
- Manieri NA, Drylewicz MR, Miyoshi H, and Stappenbeck TS (2012). Igf2bp1 is required for full induction of Ptgs2 mRNA in colonic mesenchymal stem cells in mice. Gastroenterology 143, 110–121 e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Montiel Mdel P, Gomez-Gomez GJ, and Flores AI (2014). Therapy with stem cells in inflammatory bowel disease. World J Gastroenterol 20, 1211–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, and Seibert K (1994). Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci U S A 91, 3228–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Kibe R, Ooga T, Aiba Y, Kurihara S, Sawaki E, Koga Y, and Benno Y (2012). Impact of intestinal microbiota on intestinal luminal metabolome. Sci Rep 2, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, and Stappenbeck TS (2012). Wnt5a potentiates TGFbeta signaling to promote colonic crypt regeneration after tissue injury. Science 338, 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, VanDussen KL, Malvin NP, Ryu SH, Wang Y, Sonnek NM, Lai CW, and Stappenbeck TS (2017). Prostaglandin E2 promotes intestinal repair through an adaptive cellular response of the epithelium. EMBO J 36, 5–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montrose DC, Nakanishi M, Murphy RC, Zarini S, McAleer JP, Vella AT, and Rosenberg DW (2015). The role of PGE2 in intestinal inflammation and tumorigenesis. Prostaglandins Other Lipid Mediat 116-117, 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13, R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava GM, Friedrichsen HJ, and Stappenbeck TS (2011). Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J 5, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiotti AP, Marchi P, Miszputen SJ, Oshima CT, Franco M, and Ribeiro DA (2012). The role of nonsteroidal antiinflammatory drugs and cyclooxygenase-2 inhibitors on experimental colitis. In Vivo 26, 381–393. [PubMed] [Google Scholar]
- Parekkadan B, and Milwid JM (2010). Mesenchymal stem cells as therapeutics. Annu Rev Biomed Eng 12, 87–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Pillinger MH, and Abramson SB (2006). Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol 119, 229–240. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Powell N, Vincent RP, Ehrlich D, Bjarnason I, and Hayee B (2015). Systematic review: bile acids and intestinal inflammation-luminal aggressors or regulators of mucosal defence? Aliment Pharmacol Ther 42, 802–817. [DOI] [PubMed] [Google Scholar]
- Rampton DS, Sladen GE, and Youlten LJ (1980). Rectal mucosal prostaglandin E2 release and its relation to disease activity, electrical potential difference, and treatment in ulcerative colitis. Gut 21, 591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, and Hylemon PB (2006). Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47, 241–259. [DOI] [PubMed] [Google Scholar]
- Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, Angelin B, Hyotylainen T, Oresic M, and Backhed F (2013). Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17, 225–235. [DOI] [PubMed] [Google Scholar]
- Schaap FG, Trauner M, and Jansen PL (2014). Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 11, 55–67. [DOI] [PubMed] [Google Scholar]
- Seno H, Miyoshi H, Brown SL, Geske MJ, Colonna M, and Stappenbeck TS (2009). Efficient colonic mucosal wound repair requires Trem2 signaling. Proc Natl Acad Sci U S A 106, 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steed AL, Christophi GP, Kaiko GE, Sun L, Goodwin VM, Jain U, Esaulova E, Artyomov MN, Morales DJ, Holtzman MJ, et al. (2017). The microbial metabolite desaminotyrosine protects from influenza through type I interferon. Science 357, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer N, Desharnais L, Beutler M, Brugiroux S, Terrazos MA, Menin L, Schurch CM, McCoy KD, Kuehne SA, Minton NP, et al. (2016). Functional Intestinal Bile Acid 7alpha-Dehydroxylation by Clostridium scindens Associated with Protection from Clostridium difficile Infection in a Gnotobiotic Mouse Model. Front Cell Infect Microbiol 6, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Winglee K, Gharaibeh RZ, Gauthier J, He Z, Tripathi P, Avram D, Bruner S, Fodor A, and Jobin C (2018). Microbiota-derived Metabolic Factors Reduce Campylobacteriosis in Mice. Gastroenterology. Turner, J.R. (2009). Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 9, 799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavassori P, Mencarelli A, Renga B, Distrutti E, and Fiorucci S (2009). The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 183, 6251–6261. [DOI] [PubMed] [Google Scholar]
- Voswinkel J, Francois S, Gorin NC, and Chapel A (2013). Gastro-intestinal autoimmunity: preclinical experiences and successful therapy of fistulizing bowel diseases and gut Graft versus host disease by mesenchymal stromal cells. Immunol Res 56, 241–248. [DOI] [PubMed] [Google Scholar]
- Walker MR, Brown SL, Riehl TE, Stenson WF, and Stappenbeck TS (2010). Growth factor regulation of prostaglandin-endoperoxide synthase 2 (Ptgs2) expression in colonic mesenchymal stem cells. J Biol Chem 285, 5026–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Chen J, Hollister K, Sowers LC, and Forman BM (1999). Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 3, 543–553. [DOI] [PubMed] [Google Scholar]
- Weingarden AR, Dosa PI, DeWinter E, Steer CJ, Shaughnessy MK, Johnson JR, Khoruts A, and Sadowsky MJ (2016). Changes in Colonic Bile Acid Composition following Fecal Microbiota Transplantation Are Sufficient to Control Clostridium difficile Germination and Growth. PLoS One 11, e0147210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JE, Williams KB, Whitehead TR, Heuman DM, and Hylemon PB (2003). Development and application of a polymerase chain reaction assay for the detection and enumeration of bile acid 7alphadehydroxylating bacteria in human feces. Clin Chim Acta 331, 127–134. [DOI] [PubMed] [Google Scholar]
- Wiercinska-Drapalo A, Flisiak R, and Prokopowicz D (1999). Mucosal and plasma prostaglandin E2 in ulcerative colitis. Hepatogastroenterology 46, 2338–2342. [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. (2013). Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. [DOI] [PubMed] [Google Scholar]
- Yu BZ, Apitz-Castro RJ, Jain MK, and Berg OG (2007). Role of 57–72 loop in the allosteric action of bile salts on pancreatic IB phospholipase A(2): regulation of fat and cholesterol homeostasis. Biochim Biophys Acta 1768, 2478–2490. [DOI] [PubMed] [Google Scholar]
- Yuli S, Chengxu L, and Sydney MF (2005). Multiplex PCR for rapid differentiation of three species in the “Clostridium clostridioforme group”. FEMS Microbiol Lett 244, 391–395. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Desai A, Yang SY, Bae KB, Antczak MI, Fink SP, Tiwari S, Willis JE, Williams NS, Dawson DM, et al. (2015). TISSUE REGENERATION. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science 348, aaa2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.