

Abstract
The inhibition of the mitogen-activated protein kinases signalling pathway through combined use of BRAF and MEK inhibitors (BRAFi+MEKi) represents an established therapeutic option in patients with BRAF-mutated, advanced melanoma. These efficient therapies are well tolerated with mostly moderate and reversible side effects and a discontinuation rate due to adverse events of 11.5%–15.7%. Median duration of therapy ranges between 8.8 and 11.7 months. Based on data from confirmatory trials, safety profiles of three BRAFi+MEKi combinations were reviewed, that is, dabrafenib plus trametinib, vemurafenib plus cobimetinib and encorafenib plus binimetinib. Many adverse events are class effects, such as cutaneous, gastrointestinal, ocular, cardiac and musculoskeletal events; some adverse events are substance associated. Fever (dabrafenib) and photosensitivity (vemurafenib) are the most common and clinically prominent examples. Other adverse events are less frequent and the association to one substance is less strong such as anaemia, facial paresis (encorafenib), neutropenia (dabrafenib), skin rash, QTc prolongation and increased liver function tests (vemurafenib). This narrative review provides recommendations for monitoring, adverse event evaluation and management focusing on the clinically relevant side effects of the three regimens.
Keywords: melanoma, BRAF, MEK, adverse events, safety
Introduction
Combination therapy with BRAF plus MEK inhibitors (BRAFi+MEKi) offers an effective therapy in BRAF-mutated metastatic1–6 and, for adjuvant therapy, in BRAF-mutated stage III melanoma.7
Investigator-assessed response rates in BRAF-mutated, metastatic melanoma were 64%–67% for dabrafenib+trametinib (D+T),2 3 68% for vemurafenib+cobimetinib (V+C)5 and 75% for encorafenib+binimetinib (E+B)6; landmark survival analyses showed 2-year overall survival rates of 53% for D+T,8 48% for V+C9 and 58% for E+B.10 However, direct comparison of these findings cannot be made since the trials show differences in patient populations; for example, for V+C, a significantly higher number of patients with elevated lactate dehydrogenase levels was included.11
Side effects occurring in all three BRAFi+MEKi combinations were assessed during the confirmatory clinical trials.1–6 Some events can be attributed to BRAFi adverse reactions12–15 and some to MEKi adverse reactions.16–19 Class effects of MEKi include the induction of a papulopustular exanthema in almost all treated patients, neuroretinal detachment, muscular problems, hypertension and ventricular ejection fraction decrease. Certain side effects are drug associated and may also occur with the respective combination; examples comprise phototoxicity,14 15 20 21 fever12 22 or transient facial paresis.23
With extensive adverse event (AE) data sets for E+B becoming publicly available now,6 the safety profiles of D+T, V+C and E+B are reviewed here with recommendations for monitoring, evaluation and management of the most common or most critical AE.
Methods and materials
The tolerability of the three combination regimens D+T, V+C and E+B is described here by following safety parameters: (1) AE frequencies (all grades and grade 3–4); (2) selected AEs of special interest; (3) serious AE rates; (4) deaths that occurred within 90 days after end of treatment and that are considered ‘related’ or ‘possibly related’ to study treatment and (5) dose reduction, dose interruption and dose discontinuation rates in relation to AEs of any causality.
Event rates for the three combinations were extracted from reports of the pivotal, confirmative phase III trials COMBI-V, COMBI-D, coBRIM and COLUMBUS (Part I); rates were determined by the common terminology criteria for AEs of the US National Cancer Institute, V.4.0. A recently published indirect treatment comparison of D+T versus V+C in previously untreated patients with melanoma provided methodical guidance and updated safety data for the COMBI-V trial24; the comparison focused exclusively on trials with vemurafenib (dose regimen 960 mg twice daily) used as a control arm for null-hypothesis testing.
All incidence variables for tolerability were collected from publicly available data sets. Recent data sets were used in this narrative review to compile AE frequency, considering the cut-off dates for safety analyses, length of AE observation and proportion of patients still under therapy (see online supplementary file 1 for additional information). Follow-up times for AE data sets analysed in our paper, calculated as period from day of last-patient-included until safety-data cut-off-date, were 19.8 months (D+T),24 25 18.5 months (V+C)9 26 and 16.5 months (E+B).6
esmoopen-2019-000491supp001.tif (212.1KB, tif)
This retrospective, non-experimental analysis of anonymised, pooled patient data did not require institutional review board approval. Standard recommendations to enhance the quality of evidence-based judgements were followed as previously described.27 28
Pharmacological drug profiles
The various BRAFi and MEKi differ in kinase-binding properties, structural and pharmacodynamic characteristics. These parameters influence the inhibitory potency towards the V600-mutated BRAF kinase and determine off-target effects.29
For the predominant BRAFV600E-mutant kinase, the half maximal inhibitory concentration (IC50)—a measure of the potency of a drug to inhibit a specific biological or biochemical function—is 0.65 nM for dabrafenib, 10 nM for vemurafenib and 0.35 nM for encorafenib. All three BRAFi were also active against other BRAF mutations as well as against wild-type BRAF. Encorafenib inhibited most cell lines at an IC50 of <40 nmol/L; slightly higher concentrations of dabrafenib (<100 nmol/L), but significantly higher concentrations of vemurafenib (<1000 nmol/L) were required in the same assay to inhibit proliferation of most cell lines with BRAF mutations.23
The dissociation half-life (t½diss), a measure describing target inhibition and its durability, is relevant to determine drug-dosing intervals. In vitro investigations on drug–receptor interactions indicated that t½diss for encorafenib is with >30 hour considerably longer than for dabrafenib (2 hour) or vemurafenib (0.5 hour), resulting in longer lasting pharmacodynamic target inhibition of encorafenib.23
For the MEKi trametinib, IC50 values for in vitro inhibition of MEK1 and MEK2 range between 0.7 and 0.9 nM.30 The IC50 of cobimetinib for MEK1 is with 0.95 nM much lower than for MEK2 (199 nM).30 In target inhibitory assays, binimetinib was a potent inhibitor of MEK1 and MEK2 with an enzyme IC50 of 12–46 nM.30
Bioavailability, a measure how much of the administered drug reaches systemic circulation, was 95% and 85% for BRAFi dabrafenib and encorafenib, respectively.23 31–33 For vemurafenib with its low solubility and permeability, it is unknown.20 For MEKi trametinib, cobimetinib and binimetinib, bioavailability was 72%,34 35 46%18 and 50%,36 respectively.
Further pharmacokinetic properties of the BRAFi and MEKi, as determined in their early clinical trials,18 20 23 34–36 are outlined in figure 1. Due to their differing bioavailability and pharmacokinetics, drug doses and administration schedules vary: 150 mg dabrafenib are administered twice daily 1 hour before or 2 hour after the morning and evening meal (ie, 75 mg capsules 2-0-2), vemurafenib twice daily (960 mg per dose; 240 mg tablets 4-0-4), and encorafenib once daily in the morning or evening independent of food intake (450 mg; 75 mg capsules 6-0-0 or 0-0-6).
Figure 1.
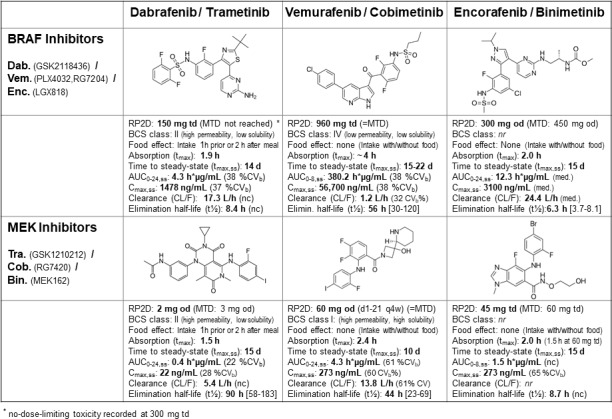
Structural and population pharmacokinetic properties (single drug) of BRAF indicator dabrafenib, vemurafenib and encorafenib and of MEK indicator trametinib, cobimetinib and binimetinib. AR, accumulation ratio; AUC 0–24, area under the curve for 0–24 h (AUC 0–8 : 0-8 h); BCS, biopharmaceuticsclassification system; C max, maximum concentration; C 24, concentration after 24 h; d, days; h, hours; MTD, maximum tolerated dose; od, once daily; RP2D, recommended phase 2 dose; t 1/2 eff, effective half-life, calculated as –0.693*tau/(ln[1–{1/AR}]); td, twice daily; t max, time taken to reach maximum concentration, reported as median. In brackets: (%CV b), between-subject coefficient of variation; (nc), not calculated; (nr), not reported; […], range. All reported values are means, if not indicated otherwise (i.e. median).
Administration of trametinib (2 mg; 2 mg tablets 1-0-0) as well as of cobimetinib (60 mg; 20 mg tablets 3-0-0) is once daily, whereas binimetinib is taken twice daily (45 mg per dose; 15 mg tablets 3-0-3). In combination, D+T and E+B are administered continuously. V+C is administered in combination for 21 days plus 7 days rest for cobimetinib.4
Toxicity profiles of BRAFi and MEKi monotherapies
For dabrafenib, treatment-related fever has been identified in 20% of patients in its first-in-human phase I trial22; no dose–response correlation was noted for fever of grade 2 or higher in the monotherapy trials, but pharmacokinetic analyses conducted in the context of the BRAFi+MEKi combination studies showed a possible association between fever and exposure to the hydroxy-dabrafenib metabolite and, to a lesser extent, to dabrafenib.3 Common toxicities included cutaneous side effects, arthralgia, fatigue and headache.13
Most treatment-related side effects of vemurafenib appeared to be proportional to dose and exposure of the drug20; the photosensitivity induced by vemurafenib is considered to be a property of the chemical structure of the drug, not related to its BRAF-inhibiting activity.22 Common side effects in early monotherapy trials were arthralgia, nausea, fatigue, rash, cutaneous squamous-cell carcinoma, pruritus and palmar–plantar dysesthesia.20
Most frequent drug-related AEs occurring with encorafenib monotherapy included myalgia, nausea, palmoplantar erythrodysesthesia, arthralgia, alopecia and hyperkeratosis. Transient Bell’s palsy, the most common disorder affecting a single nerve and associated with facial nerve weakness/paralysis, was reported in 8% of patients treated with encorafenib, whereas it has rarely been reported in association with other BRAFi.33
Trametinib showed an early onset of dose-limiting toxic effects, as observed in trametinib’s first-in-human trial including 206 patients34 with two cases of rash, one case of diarrhoea and three cases of central serous retinopathy. As two of the three ocular toxicity events arose either 1 day after a loading dose or within days of administration of the highest once-daily dose, this treatment-related effect was thought to be potentially related to the Cmax of trametinib.34 Similarly, a reported papulopustular exanthema adverse reaction emerging after cessation of combined D+T treatment was proposed to be associated with trametinib’s long half-life37 (figure 1).
Cobimetinib’s most frequent AEs attributed in its phase I monotherapy trial were diarrhoea, rash, fatigue, oedema, nausea and vomiting.18
Dose-limiting toxicities of binimetinib in early monotherapy trials were single events of therapy-resistant papulopustular rash and of central serous-like retinopathy, respectively. Other frequent treatment-related AEs included gastrointestinal (GI) (diarrhoea, nausea) and skin disorder as well as peripheral oedema, increased creatine phosphokinase (CPK) and retinal disorders.38
Toxicity profile of BRAFi+MEKi combinations
Overall AE and serious AE frequencies for the three combinations D+T, V+C and E+B and the respective median drug exposure times are displayed in table 1. Although comparison across trials has its limitations, that is, with respect to different methodological AE evaluation across trials, AE rates in the respective vemurafenib comparator arms are nearly identical.6 9 24 AEs of any grade occurred in almost all patients treated with the combination therapy. In patients treated with a BRAFi+MEKi, grade 3–4 AEs occurred in 46%–56% for D+T (COMBI-D, COMBI-V), 69% for V+C (coBRIM) and 58% for E+B (COLUMBUS, Part I).
Table 1.
Overall frequencies of adverse events in the safety populations of the pivotal clinical trials and median drug exposure times and rates
| Combination regimen of interest | Dabrafenib+Trametinib (D+T) |
Vemurafenib+Cobimetinib (V+C) | Encorafenib+Binimetinib (E+B) |
||||||
| Date of (safety) analysis | 13 Mar 201524 25 | 12 Jan 20152 | 19 Sep 201424 | 19 May 20166 | |||||
| Study | COMBI-V | COMBI-D | coBRIM | COLUMBUS Part 1 | |||||
| Arms | D+T | V | D+T | D+placebo | V+C | V+placebo | E+B | V | E |
| N° pts (ITT) rand (N° safety population) | 352 (350) | 352 (349) | 211 (209) | 212 (211) | 247 (247) | 248 (246) | 192 (192) | 191 (186) | 194 (192) |
| Daily dose (mg) | 300/2 | 1920 | 300/2 | 300 | 1920/60 | 1920 | 450/90 | 1920 | 300 |
| Events (frequencies) | |||||||||
| AE (all grades, any causality) (%) | 345 (98.6) | 345 (98.9) | 203 (97.1) | 205 (97.2) | 244 (98.8) | 240 (97.6) | 188 (97.9) | 185 (99.5) | 191 (99.5) |
| AE (°3–4) (%) | 195 (55.7) | 228 (65.3) | 95 (45.5) | 106 (50.2) | 171 (69.2) | 143 (58.1) | 111 (57.8) | 118 (63.4) | 127 (66.1) |
| AE leading to death (‘fatal AE’; (°5)) (%) | 4 (1.1) | 4 (1.2) | 5 (2.4) | 1 (0.5) | 5 (2.0) | 3 (1.2) | 3 (1.7) | 2 (1.1) | 2 (1.0) |
| AE leading to discontinuation (%) | 55 (15.7) | 48 (13.8) | 24 (11.5) | 14 (6.6) | 37 (15.0) | 20 (8.1) | 24 (12.5) | 31 (16.7) | 27 (14.1) |
| AE leading to dose reduction (%) | 192 (54.9) | 197 (56.4) | 59 (28.2) | 29 (13.7) | 110 (44.5) | 87 (35.4) | 22 (11.5) | 42 (22.6) | 52 (27.1) |
| AE leading to dose interruption (%) | 118 (56.5) | 78 (37.0) | 88 (45.8) | 98 (52.7) | 122 (63.5) | ||||
| SAE (all grades, any causality) (%) | 151 (43.1) | 136 (39.0) | 88 (42.1) | 78 (37.0) | 85 (34.4) | 64 (26.0) | 66 (34.4) | 69 (37.1) | 65 (33.9) |
| Dose exposure rates/times (months) | |||||||||
| Median duration of therapy (range) | 10.0 (0–21) | 6.0 (0–18) | 11.0 (0–30) | 8.0 (0–32) | 8.8 (0–18) | 5.7 (0–17) | 11.7 (0–27) | 6.2 (0–28) | 7.2 (0–26) |
| Median dose intensity (in %) | 97.3%+100% | 96.1% | 98.2%+100% | 99.9%+100% | 94.4%+96.6% | 96.7%+99.0% | 100%+99.6% | 94.5% | 86.2% |
AE, adverse events; B, binimetinib;C, cobimetinib;D, dabrafenib;E, encorafenib;ITT, intention-to-treat;SAE, serious adverse event;T, trametinib;V, vemurafenib; pts rand., patients randomized.
Median duration of therapy ranged between 10.0 and 11.0 months for D+T, 8.8 months for V+C and 11.7 months for E+B, with median dose intensities ranging between 94% and 100% for all substances.
The detailed AE frequencies of D+T (COMBI-V), V+C (coBRIM) and E+B (COLUMBUS Part I) for relevant organ systems are listed in table 2. To facilitate the analysis and interpretation of the data, AE frequencies for the combination regimen versus frequencies of the respective vemurafenib monotherapy arms of each trial6 9 24 were compared: the respective shifts, expressed as an increase or decrease in BRAFi+MEKi AE rates compared with vemurafenib, are displayed in figure 2.
Table 2.
Frequencies of AE of combination therapy arms, as observed in the safety population of pivotal clinical trials comparing BRAFi +MEKi combinations versus vemurafenib6 9 24
| Combination regimen of interest | Dabrafenib+Trametinib (D+T) |
Vemurafenib+Cobimetinib (V+C) |
Encorafenib+Binimetinib (E+B) |
| Data cut-off dates (safety analysis) | 13 Mar 2015 | 30 Sep 2015 | 19 May 2016 |
| Median follow-up time | 19.8 months | 18.5 months | 16.6 months |
| Study | COMBI-V | coBRIM | COLUMBUS Part 1 |
| N° pts rand. (intention to treat) (safety population) | 352 (350) | 247 (247) | 192 (192) |
| Daily dose (mg) | 300/2 | 1920/60 | 450/90 |
| CTC AE grade | Any | 3–4 | Any | 3–4 | Any | 3–4 |
| Dermatological events, including new skin neoplasms | ||||||
| Rash* | 84 (24.0) | 3 (0.9) | 101 (40.9) | 13 (5.3) | 27 (14.1) | 2 (1.0) |
| Rash maculopapular | 13 (3.7) | 2 (0.6) | 38 (15.4) | 18 (7.3) | 3 (1.6) | 0 |
| Dry skin | 33 (9.4) | 0 | 38 (15.4) | 2 (0.8) | 27 (14.1) | 0 |
| Pruritus | 36 (10.3) | 0 | 49 (19.8) | 3 (1.2) | 21 (10.9) | 1 (0.5) |
| Erythema | 35 (10.0) | 0 | 26 (10.5) | 0 | 13 (6.8) | 0 |
| Dermatitis acneiform* | 23 (6.6) | 0 | 34 (13.8) | 6 (2.4) | 6 (3.1) | 0 |
| Alopecia | 23 (6.6) | 0 | 41 (16.6) | 1 (0.4) | 26 (13.5) | 0 |
| Hyperkeratosis | 18 (5.1) | 0 | 25 (10.1) | 1 (0.4) | 27 (14.1) | 1 (0.5) |
| Palmoplantar keratoderma | – | – | 5 (2.0) | 0 | 17 (8.9) | 0 |
| Palmo–plantar erythrodysesthesia*† | 14 (4.0) | 0 | 17 (6.9) | 0 | 13 (6.8) | (0) |
| Actinic keratosis | 5 (1.4) | 0 | 13 (5.3) | 8 (3.2) | – | – |
| Keratosis pilaris* | 4 (1.1) | 0 | 9 (3.6) | 0 | 9 (4.7) | 0 |
| Photosensitivity reaction* | 15 (4.3) | 0 | 84 (34.0) | 1 (0.4) | 8 (4.2) | 1 (0.5) |
| Sunburn | 3 (0.9) | 0 | 37 (15.0) | 2 (0.8) | 0 | 0 |
| (Cutaneous) squamous cell carcinoma* | 5 (1.4) | 5 (1.4) | 10 (4.0) | 9 (3.6) | 5 (2.6) | 0 |
| Keratocanthoma* | 2 (0.6) | 2 (0.6) | 4 (1.6) | 3 (1.2) | 4 (2.1) | 0 |
| Skin papilloma* | 8 (2.3) | 0 | 17 (6.9) | 0 | 12 (6.3) | 0 |
| Basal cell carcinoma* | 3 (0.9) | 2 (0.6) | 15 (6.1) | 14 (5.7) | 3 (1.6) | 0 |
| Gastrointestinal events | ||||||
| Diarrhoea* | 120 (34.3) | 4 (1.1) | 150 (60.7) | 16 (6.5) | 70 (36.4) | 5 (2.6) |
| Nausea | 126 (36.0) | 1 (0.3) | 105 (42.5) | 3 (1.2) | 79 (41.1) | 3 (1.6) |
| Vomiting | 107 (30.6) | 4 (1.1) | 63 (25.5) | 4 (1.6) | 57 (29.7) | 3 (1.6) |
| Abdominal pain | 39 (11.1) | 1 (0.3) | 27 (10.9) | 1 (0.4) | 32 (16.7) | 5 (2.6) |
| Abdominal pain upper | 33 (9.4) | – | 12 (4.9) | 0 | 23 (12.0) | 2 (1.0) |
| Constipation | 54 (15.4) | 0 | 27 (10.9) | 0 | 42 (21.9) | 0 |
| General disorders (and symptoms/disorders of central nervous system) | ||||||
| Fatigue | 110 (31.4) | 4 (1.1) | 91 (36.8) | 11 (4.5) | 55 (28.6) | 4 (2.1) |
| Asthenia | 61 (17.4) | 5 (1.4) | 47 (19.0) | 5 (2.0) | 35 (18.2) | 3 (1.6) |
| Fever* | 193 (55.1) | 16 (4.6) | 71 (28.7) | 3 (1.2) | 35 (18.2) | 7 (3.6) |
| Peripheral oedema/swelling*‡ | 48 (13.7) | 1 (0.3) | 34 (13.8) | 0 | 3 (1.6%) | 0 |
| Headache* | 112 (32.0) | 4 (1.1) | 44 (17.8) | 1 (0.4) | 42 (21.8) | 3 (1.6) |
| Dizziness* | 34 (9.7) | 1 (0.3) | 15 (6.1) | 0 | 24 (12.5) | 3 (1.6) |
| Investigations/laboratory examinations | ||||||
| ALT level increased | 49 (14.0) | 9 (2.6) | 65 (26.3) | 28 (11.3) | 21 (10.9) | 10 (5.2) |
| AST level increased | 42 (12.0) | 5 (1.4) | 60 (24.3) | 22 (8.9) | 16 (8.3) | 4 (2.1) |
| γ-GT level increased | 38 (10.9) | 19 (5.4) | 54 (21.9) | 36 (14.6) | 29 (15.1) | 18 (9.4) |
| Blood AP increased | 26 (7.4) | 7 (2.0) | 42 (17.0) | 12 (4.9) | 16 (8.3) | 1 (0.5) |
| Blood CPK level increased | 10 (2.9) | 6 (1.7) | 87 (35.2) | 30 (12.1) | 44 (22.9) | 13 (6.8) |
| Blood creatinine level increased | 15 (4.3) | 0 | 37 (15.0) | 3 (1.2) | 12 (6.3) | 2 (1.0) |
| Lipase level increased | – | – | 9 (3.6) | 8 (3.2) | 4 (2.1) | 3 (1.6) |
| Hyperglycaemia* | 17 (4.9) | 8 (2.3) | 8 (3.2) | 1 (0.4) | 9 (4.7) | 4 (2.1) |
| Hyponatremia* | 16 (4.6) | 15 (4.3) | 13 (5.3) | 7 (2.8) | 2 (1.0) | 1 (0.5) |
| Anaemia | 26 (7.4) | 7 (2.0) | 39 (15.8) | 4 (1.6) | 29 (15.1) | 8 (4.2) |
| Neutropenia* | 32 (9.1) | 17 (4.9) | 3 (1.2) | 0 | 5 (2.6) | 2 (1.0) |
| Musculoskeletal events | ||||||
| Arthralgia | 93 (26.6) | 3 (0.9) | 94 (38.1) | 6 (2.4) | 49 (25.5) | 1 (0.5) |
| Pain in extremity | 45 (12.9) | 4 (1.1) | 29 (11.7) | 3 (1.2) | 21 (10.9) | 2 (1.0) |
| Myalgia | 66 (18.8) | 0 | 37 (15.0) | 1 (0.4) | 26 (13.5) | 0 |
| Cardiovascular events | ||||||
| QT interval prolongation (ECG)* | 5 (1.4) | 2 (0.6) | 11 (4.5) | 3 (1.2) | 0 | 0 |
| Ejection fraction decreased* | 29 (8.3) | 13 (3.7) | 29 (11.7) | 5 (2.0) | 11 (5.7) | 2 (1.0) |
| Hypertension | 103 (29.4) | 54 (15.4) | 39 (15.8) | 15 (6.1) | 21 (10.9) | 11 (5.7) |
| Ocular events | ||||||
| Vision blurred | 17 (4.9) | 0 | 28 (11.3) | 0 | 30 (15.6) | 0 |
| Chorioretinopathy* | 2 (0.6) | 0 | 32 (13.0) | 2 (0.8) | 5 (2.6) | 2 (1.0) |
| Retinal detachment* | – | – | 22 (8.9) | 5 (2.0) | 15 (7.8) | 1 (0.5) |
| Pulmonary events | ||||||
| Cough | 77 (22.0) | 0 | 23 (9.3) | 0 | 16 (8.3) | 1 (0.5) |
| Pneumonia* | 2 (0.6) | 0 | 6 (2.4) | 3 (1.2) | 3 (1.6) | 3 (1.6) |
| Pulmonary embolism* | 7 (2.0) | 7 (2.0) | 2 (0.8) | 2 (0.8) | 6 (3.1) | 2 (1.0) |
| Renal events | ||||||
| Acute kidney injury* | 4 (1.1) | 4 (1.1) | 7 (2.8) | 3 (1.2) | 3 (1.6) | 2 (1.0) |
| Dehydration* | 15 (4.3) | 6 (1.7) | 11 (4.5) | 5 (2.0) | 11 (4.5) | 5 (2.0) |
Listed are AE with event frequencies (of any grade) ≥10%, and/or with event frequencies (≥2% for grade ≥3 AE) and frequency independent, clinically relevant ‘AE of specific interest’.
Values in bold indicates CTC grade 3 and grade 4 toxicity reported; values in italics indicates different data cut-off dates for safety analysis (D+T only).
*D+T: values and frequencies reported at initial safety analysis (14 April 2014) only,2 extended listings are published in the European Public Assessment Report (EMA/589140/2015, dated 2 Septmber 2015).
†D+T: term ‘hand–foot syndrome’ was reported, including the terms palmo–plantar erythrodysesthesia, planto–palmar hyperkeratosis and palmoplantar keratoderma.
‡E+B: frequencies for peripheral swelling were reported.
–, not reported; AE, adverse event; ALT, alanine aminotransferase;AP, alkaline phosphatase;AST, aspartate aminotransferase;CPK, creatine phosphokinase;CTC, common toxicity criteria;GT, γ-gamma-glutamyl transferase; pts rand., patients randomised.
Figure 2.
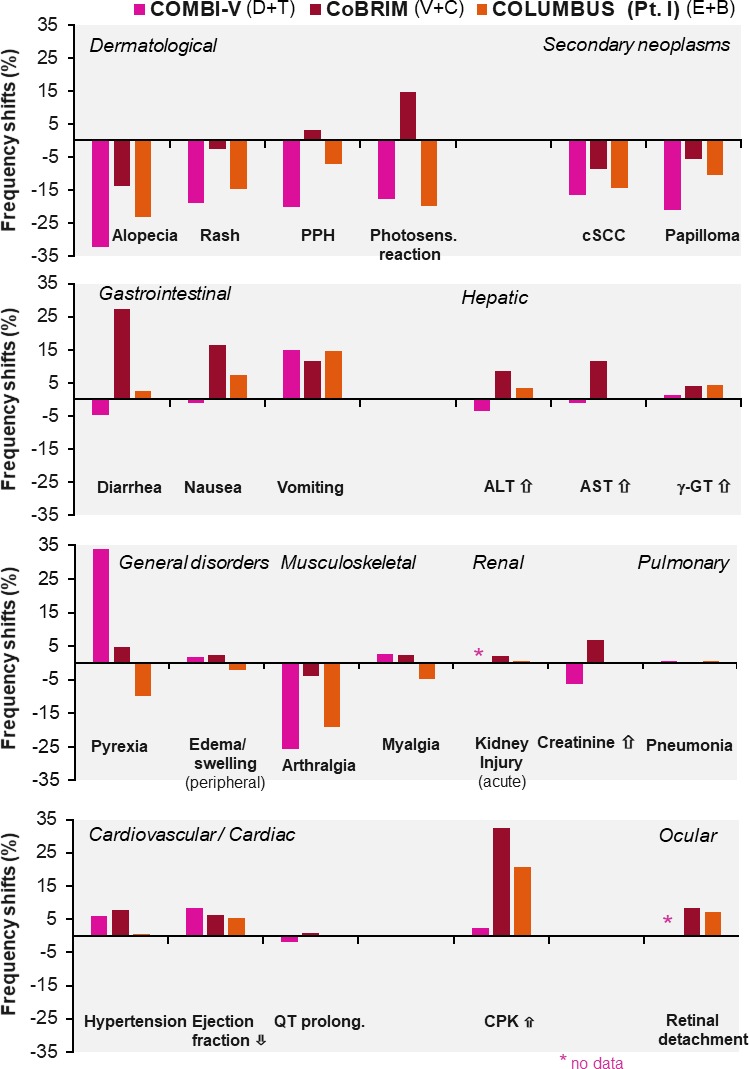
Differences in adverse event frequencies: combination therapies versus vemurafenib monotherapy. CPK, creatine phosphokinase; cSCC, cutaneous squamous cell carcinoma; PPH, palmoplantar hyperkeratosis.
Remarkably, most cutaneous side effects, particularly secondary neoplasms (cutaneous squamous cell carcinoma and its specific variant keratoacanthoma and skin papilloma), decreased considerably compared with BRAF monotherapy (figure 2); this effect is explained in the literature by the suppression of the paradoxical activation of the mitogen-activated protein kinases (MAPK) pathway in BRAF wild-type cells in various tissues through MEKi coadministration.39 On the other side, an increase of GI side effects, particularly vomiting, is noted due to the addition of the MEKi. Increases in cardiac and ocular side effects are related to MEKi coadministration as well.
Therapy related, organ class-specific AEs
Dermatological events, secondary skin neoplasms
The most frequent cutaneous side effects are rash, itching, dry skin, hair loss, photosensitivity reaction, keratinocytic proliferation and panniculitis.
Different conditions such as maculopapular exanthema, papulopustular exanthema or even eczema are often summed up by ‘rash’; however, a differentiation is desirable to apply the most adequate treatment approach. Rash was most often reported for V+C with 41% and was less common for D+T (24%) and for E+B (14%) (table 2). Although ‘rash’ is often a low-grade AE, severe and even life-threatening side effects of the skin have been reported, including erythema exsudativum multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis, drug rash with eosinophilia and systemic symptoms, drug-induced hypersensitivity syndrome and acute generalised exanthematous pustulosis.40
Dry skin (xerosis cutis) and itching (pruritus) are side effects that occur regularly among all three combinations with a frequency of 10%–20% (table 2). Pruritus is often the result of xerosis cutis.
In 34% of patients treated with V+C, ultraviolet A (UVA)-mediated photosensitivity reactions with erythema, blistering and painful burning were reported, whereas this occurred only in 4% of patients treated with D+T or E+B. Radiosensitivity was observed in patients treated concomitantly with radiotherapy and BRAFi/MEKi, mainly with vemurafenib.41 Effectiveness does not seem to be reduced by interruption of BRAF therapy during radiotherapy, as shown for vemurafenib.42
BRAFi+MEKi therapy associated ‘alopecia’ generally means diffuse hair loss. Interestingly, curly hairs often found under BRAFi monotherapy are not observed under BRAFi+MEKi therapy.
Keratinocytic proliferations including keratosis pilaris, actinic keratoses, cutaneous squamous cell carcinoma (cSCC), keratoacanthoma and skin papilloma were observed in up to 7% of patients treated with BRAFi+MEKi (table 2). Keratosis pilaris with disseminated small hyperkeratotic follicular papules on the face or proximal extremities was reported in 4%–7% of patients (table 2). With regard to hand–foot syndrome, there is a spectrum of clinical variants: palmoplantar erythrodysesthesia (PPE) with inflammatory and painful lesions not restricted to pressure points and palmoplantar hyperkeratosis (PPH, also called palmoplantar keratoderma) with hyperkeratotic and painful lesions at pressure points. They are also called type I and type II hand–foot syndrome.43 Within the clinical studies, this was not clearly differentiated (table 2). While encorafenib seems to induce both PPE and PPH more often than vemurafenib or dabrafenib, the BRAFi+MEKi combination therapy is well tolerated: mainly PPH occurs.44 Frequency of PPH is lowered with D+T and E+B, compared with vemurafenib (figure 2).
D+T, V+C or E+B induce benign acanthotic skin papillomas, keratoacanthomas and well-differentiated cSCC in 2%–7%, 1%–2% and 1%–4% of patients, respectively (table 2); these rates are lower than the AE rates induced by BRAFi monotherapy (figure 2). New primary melanomas were observed in less than 1% of all patients treated with BRAFi+MEKi. Moreover, panniculitis with painful erythematous subcutaneous nodules predominantly located on the extremities and buttocks—which can occur with or without fever, arthralgia or joint swelling—have been described under combined BRAFi+MEKi with unknown frequency.45
Cutaneous side effects are usually well treatable and should not immediately lead to dose reduction or discontinuation of therapy. Exanthema, xerosis cutis and pruritus can be successfully treated by regular application of moisturisers containing urea or glycerine or topical application of class II–III glucocorticoids. Severe cases of exanthema require systemic steroids, dose interruptions or permanent discontinuation.
For ultraviolet (315–380 nm: UVA) mediated photosensitivity, strict avoidance of UVA and sun protection with UVA filter-containing sun screen and protective clothing (including hat, sunglasses) is crucial even behind windows since UVA can penetrate the window glass. Sunburn can be treated with topical steroids and possibly by non-steroidal anti-inflammatory drugs (NSAIDs).
Keratosis pilaris and PPH can be treated with creams containing urea or salicylic acid and in cases of inflammation with topical steroids. Patients with PPH should avoid pressure and friction. Panniculitis can be treated symptomatically with non-steroidal anti-inflammatory drugs (eg, etoricoxib), topical steroids and compression; some severe cases require systemic steroids and temporary dose interruption. Keratoacanthomas, cSCC and new primary melanomas should be surgically removed.
GI events
GI toxicities are commonly seen during therapy with BRAFi+MEKi and include diarrhoea and nausea and vomiting and can be accompanied by abdominal pain and GI bleeding. Frequencies of GI AE are, with shifts, for example, for diarrhoea up to 27%, higher for the combination of BRAFi+MEKi compared with either monotherapy (figure 2). The underlying pathophysiological mechanisms are not completely understood. The MAPK pathway is activated via EGFR in GI normal mucosa and there is evidence that this pathway is a negative regulator of chloride secretion. Inhibitors of the EGFR pathway could therefore increase chloride secretion and thereby induce secretory diarrhoea.46
Alternative aetiologies for GI toxicities such as progressive disease or infection (eg, Clostridium difficile infection or other bacterial/viral pathogens) need to be ruled out; the time of onset is of diagnostic relevance to assess whether the AE is treatment related. A cytomegalovirus DNA PCR performed in blood can diagnose cytomegalovirus infection or reactivation. For persistent grade 2 diarrhoea, colonoscopy with colonic biopsies can be considered. All grade diarrhoea occurred much more frequently in patients treated with V+C (61%), compared with D+T or E+B (34% and 36%); nausea (36%–41%) and vomiting (26%–31%), however, occurred at similar frequencies (table 2).
Management includes rehydration since vomiting and diarrhoea can lead to dehydration, hypotension and in severe cases to kidney failure. Grades 1 and 2 diarrhoeas may be managed with antidiarrhoeal medications including loperamide and octreotide, oral hydration and electrolyte supplements. In grade >3 cases, BRAFi/MEKi therapy should be withheld in addition to symptomatic treatment. Common GI adverse reactions usually resolve within a few days after cessation of treatment. Systemic treatment and prophylaxis of nausea and vomiting should follow established guidelines.
Hepatic AE manifest as asymptomatic increase of liver function tests, mainly aspartate aminotransferase (AST), alanine aminotransferase (ALT) and gamma-glutamyl transferase, rarely bilirubin. They are very frequent (table 2). The very common frequency of AST and ALT events may be related to the addition of the MEKi (figure 2). Regular laboratory controls are required with treatment interruption in case of elevations of grade 3 or higher. In some cases with grade 2 elevation, specifically with simultaneous elevated bilirubin levels, treatment may be paused earlier. Other causes of acute liver injury such as infection should be ruled out.
General disorders and haematological events
Fever, fatigue and peripheral oedema are very frequent with BRAFi+MEKi therapy. Fever is one of the main symptoms under D+T treatment, occurring in more than half of the patients (table 2) and is more frequent than under BRAFi monotherapy with vemurafenib (21%) or dabrafenib (28%).3 24 However, the grade >3 fever rate remains below 5%. E+B and V+C can also induce fever, but less frequent (ie, in 18%–29% of patients). Fatigue can accompany this AE or may occur independently, with similar frequencies throughout the three combinations (28%–37%).
Fever occurs early after treatment start—mainly within the first 4 weeks.26 47 A dose interruption of both drugs is recommended if the fever exceeds 38.5°C. The aetiology of BRAFi+MEKi induced fever is still unclear, and an infectious cause should be excluded. No baseline clinical characteristics predict fever, and it is not associated with treatment outcome.47 The fever can be treated with antipyretics such as ibuprofen and paracetamol and, if not effective, with low-dose corticosteroids. To avoid hypovolaemia with hypotension and possible acute renal failure, sufficient fluid substitution is important. As soon as the fever ceased for at least 24 hours, BRAFi+MEKi therapy can be restarted.48 Patients experience a median of two events of fever, with 21% of patients having >4 events.47 A dose reduction is recommended if fever recurs. However, clinical experience shows that short ‘drug holidays’ (about 2–7 days) are much more effective and the full dose can be maintained.48 In recurrent cases, intermittent treatment might be an option to avoid additional fever events.
Peripheral oedema is a typical side effect of the MEKi (table 2). Mostly extremities are affected, but facial oedema especially of the eye lids is also common. In mild cases, symptomatic treatment with compression therapy and head elevation while sleeping is adequate after exclusion of other causes, for example, hypalbuminaemia or reduced kidney function.
Haematological events occur in the form of disorders in the different cell subsets of the peripheral blood, most frequently anaemia with around 15% for V+C and E+B and 7% for D+T. Severe anaemia occurs in 2.0, 1.6 and 4.2% for D+T, V+C and E+B, respectively. In contrast, D+T induces neutropenia more frequently, followed by E+B and rarely occurring in V+C (table 2). In general, neutropaenia occurs with a late onset (up to 2 months after treatment initiation) and resolves without dose modifications. All other reported differential blood count changes such as thrombocytopenia, lymphopenia and eosinophilia occur with AE frequencies below 5% and are rarely severe.
Musculoskeletal/rheumatic events
Musculoskeletal/rheumatic side effects primarily include arthralgia, myalgia and vasculitis.
Arthralgia—the main musculoskeletal event—is a very common AE associated with BRAF monotherapy. In combination, incidences of arthralgia were lowered for D+T, V+C and E+B (figure 2): arthralgia of grade 1–2 remains, however, a very common AE (table 2). Grade ≥3 arthralgia events occur at frequencies of 0.5%–2%; drug withdrawal or adjustment may be considered. Arthritis also appears to occur more frequently in BRAF monotherapy compared with combination therapy9 and can be managed with dose reduction and corticosteroid treatment.49
Myalgia is another very common AE, occurring with BRAFi+MEKi in 14%–19% (table 2); frequencies are similar for the different BRAFi (figure 2).
Vasculitis is mainly described in single patients treated with BRAFi as cutaneous side effects in the context of panniculitis,45 50–53 and as leukocytoclastic vasculitis,45 54 but also involving the kidney as glomerulonephritis55 56 and the eye as retinal vasculitis.57
For treatment of mild symptoms of arthralgia or myalgia, NSAIDs or low-dose corticosteroids can be applied. For more accentuated musculoskeletal/rheumatic side effects that require intra-articular or high-dose steroids, rheumatologists should be consulted.
Severe AEs, for example, myositis or vasculitic organ-threatening manifestations require moderate to high-dose (1 mg/kg) corticosteroids; BRAFi/MEKi therapy discontinuation can be considered. If symptoms persist, corticosteroid-sparing therapies like leflunomide or methotrexate can be applied. Based on findings from in vitro and in vivo models, in which leflunomide prevents melanoma growth in combination with BRAFi+MEKi,58 leflunomide might be preferred.
Cardiovascular events
Cardiovascular side effects have been described for BRAFi and MEKi including QT-prolongation, cardiomyopathy with reduced pump function and hypertension. While QT prolongation is mainly an issue in treatment with BRAFi, decreased ejection fraction has been described for MEKi, however, with different frequencies. The MAPK pathway in cardiomyocytes is a protective signalling pathway and its inhibition interferes with intramyocytic repair mechanisms by inhibition of extracellular signal-regulated kinases 1/2.59 Immunotherapy-mediated subclinical cardiotoxicity or damage induced by radiotherapy may therefore increase the risk for significant left ventricular systolic dysfunction or even heart failure induced by concomitant or subsequent use of BRAFi and MEKi.60 However, despite fatal events considered to be due to arrhythmias or sudden cardiac death (10/139 in an analysis of the FDA Adverse Event Reporting System database60 61), most of the cardiac side effects can be adequately managed and are reversible.
QT prolongation was observed in up to 3%–7% of patients treated with vemurafenib and 2% treated with V+C5 62 and has been shown to be dose dependent.63 Grade ≥3 QT interval prolongation occurred in 1% of patients treated with vemurafenib monotherapy or with V+C.9 Such negative effects on QT prolongation were not seen with dabrafenib or encorafenib, considered to be due to an additional fluorinated phenyl ring.6 64 65 The MEKi trametinib caused no QT prolongation.66 Importantly, other factors like electrolyte disturbances, long QT syndrome and concomitant medications can potentiate the AE.
Thus, electrolyte dysbalances, for example, due to diarrhoea, should be corrected (including magnesium) and other QT-prolonging drugs (eg, pantoprazole, ciprofloxacin) omitted.67 ECG should be assessed before therapy and monthly during the first 3 months, then every 12 weeks; doses should be withheld in case of QTc >500 ms, or more than 60 ms increase from baseline.
It is important to detect left ventricular dysfunction in treatment with BRAFi+MEKi, since it often results in discontinuation of therapy. The degree of left ventricular dysfunction can range from asymptomatic changes, best diagnosed by echocardiographic strain analysis, to severe cardiac failure. Although BRAFi+MEKi were not included in the European Society for Medical Oncology guidelines on cardiotoxicity, the guideline outlines the diagnostic procedures including cardiac MRI and multigated acquisition scans.68
Grade ≥3 decrease in ejection fraction (ie, <40% or decrease of >20% from baseline) was reported in 4%, 2% and 1% in D+T, V+C, and E+B, respectively (table 2). Myocardial dysfunction is modified by genetic factors and impaired myocardial function before initiating cancer treatment, arterial hypertension, >65 years of age, body mass index >30 kg/m2 and radiotherapy increase the risk. The onset of left ventricular dysfunction after application of MEKi or BRAFi+MEKi therapy ranges from 2 weeks to 5 months and 1–13 months, respectively, and resolved in the majority of cases.61
Thus, risks and benefits of BRAFi+MEKi therapy should be carefully evaluated in patients with significant heart disease, and hypertension should be controlled before initiation of therapy. Ejection fraction, troponin and the N-terminal prohormone of brain natriuretic peptide (NT-proBNP) should be checked at therapy start. While troponin indicates myocardiac damage and thus represents an early marker, NT-proBNP is associated with myocardial insufficiency and a marker of chronic heart failure. Further controls can be adapted based on risk factors, symptoms and CPK findings. Management includes withholding treatment in case of an ejection fraction reduction of >10%, with rechallenge at a lower dose and discontinuation in case of ejection fraction reduction of >20%. Symptomatic treatment, for example, with beta-blockers, can be given as advised by the cardiologist.
Arterial hypertension can be caused by BRAFi; for BRAFi+MEKi combinations, the MEKi also contributes to this side effect. The incidence for combination therapies with D+T, V+C and E+B were 29%, 16%, and 11%, respectively (table 2). Antihypertensive treatment should be pursued according to the existing guidelines.
Ocular events
Fluid accumulation (oedema) in the retina resulting in a serous neuroretinal detachment (SND) is a regular AE during treatment with BRAFi+MEKi (table 2). Although this AE is sometimes called serous retinopathy, chorioretinopathy, macular oedema or retinal pigment epithelial detachment, the term SND is preferable since the other terms refer to established diagnoses, with in part different clinical manifestations.69 SND associated with BRAFi+MEKi is often asymptomatic, but may rarely cause transient visual disturbances, that is, blurred vision, reduced visual acuity, dyschromatopsia and photophobia.70 The diagnosis is made with optical coherence tomography (OCT); the oedema is often bilateral and multifocal.69 71
After initiation of BRAF+MEK inhibition, SND occurs early within hours, days or weeks and is mostly caused by the MEKi.69 71 Frequency of diagnosis depends on time and method of the ophthalmology examinations with an incidence of 13% or 8% of patients in the V+C and E+B trials, where regular OCT examinations were included (table 2). The pathogenetic mechanism is currently still unknown. SND is usually transient, but long-term experience is limited and retinal atrophy without functional relevance has been described.72 Regular controls without any treatment are usually sufficient; however, in severe cases, dose interruption or reduction of the MEKi might be necessary.
Ocular inflammation (uveitis, conjunctivitis) is rarely induced by BRAFi. Here, topical steroids are usually sufficient for treatment; in severe cases, dose interruption or reduction of the BRAFi might be necessary.73 In single cases, retinal vein occlusions have been described both during BRAFi and BRAFi+MEKi therapy.73 In these cases, study protocols required discontinuation of BRAFi+MEKi and ophthalmological treatment. However, the frequency of retinal vein occlusions is similar in patients not treated with BRAFi+MEKi74; thus, the relation and the therapeutic consequences are unclear.
Taken together, ophthalmologic AEs (in particular SND) are frequent but often asymptomatic and transient; during treatment, ophthalmologic checks including OCT should be performed depending on visual disturbances.
Pulmonary events
Lung toxicity under BRAFi+MEKi therapy is infrequent. Cough, pneumonitis, sometimes accompanied by fever and pulmonary embolism may occur commonly.75–77 Grade 3/4 events of interstitial lung disease or pneumonitis, subsumed rather under the term pneumonia in clinical studies, occurred in up to 2% of the cases (table 2). Pneumonitis is thought to be mainly caused by the MEKi.
In case of cough, shortness of breath or abnormal auscultation, chest CT should be performed. Lung infiltrates sometimes show a discrepancy to patient-reported symptoms. When an infectious origin has been ruled out via bronchoalveolar lavage, high dose of corticosteroids should be applied and tapered over time. While in severe cases, the MEKi has to be permanently discontinued, reinduction of the BRAFi after recovery is possible.
Renal events
Combined BRAFi+MEKi therapy can cause renal impairment, mostly as increase of serum creatinine. Acute renal failure or electrolyte disorders have been reported for all combination therapy78 79 (table 2). A recent systematic review of BRAFi-mediated nephrotoxicity including pathology reports of kidney biopsies indicated tubule-interstitial damage with an acute and chronic component.80–83
In cases of higher grade acute kidney injury, BRAFi +MEKi therapy should be interrupted and reintroduced after improvement of renal function at a lower dose. As therapy with D+T frequently causes fever that may lead to dehydration of the patient—in cases of insufficient oral fluid intake—pre-renal insufficiency may occur that ameliorates after rehydration of the patient.
Discussion
BRAFi+MEKi combinations are highly effective in the therapy of metastatic melanoma84 85 and D+T has additionally been shown to prolong overall survival in the adjuvant setting.7 However, AEs occur in almost all (ie, ≥97%) of patients treated with D+T, V+C or E+B (table 1), with grade 3–4 AE rates of 46%–56%, 69% and 58%, respectively. Knowledge of side effect profiles, monitoring and management strategies are important to tailor the therapy to patients, diagnose side effects early, keep discontinuation rates low and guide patients through occurrences of such effects. This review is based on the available safety data from phase III clinical studies: our group’s recommendations for the monitoring of side effects are provided in table 3, our recommendations for the management of clinically relevant and/or very frequent AEs of BRAFi +MEKi therapy are summarised in figure 3.
Table 3.
Recommendations for routine* monitoring of BRAF/MEK combination therapy
| Test | Therapy start | Monthly control | Quarterly control |
| Blood count | |||
| Differential blood count | x | x | |
| Clinical chemistry | |||
| Electrolytes (Na, K, Ca, Mg) | x | x | |
| Creatinine | x | x | |
| CPK | x | x | |
| Troponin | x | (x)† | x |
| NT-proBNP | x | (x)† | x |
| Liver transaminases (AST, ALT, γ-GT) | x | x | |
| Bilirubin | x | x | |
| Examinations (non-laboratory tests) | |||
| Skin inspection | x | x‡ | x |
| Visual acuity control § | x | (x)¶ | |
| Ocular OCT | (x)¶ | ||
| Blood pressure | x | x | |
| ECG | x | x‡ | x |
| Echocardiography | x | (x)† | |
*In patients/situations without clinical particularities.
†In case of clinically abnormal signs (eg, heart, chest) or increasing CPK.
‡In months 1 and 2.
§Visual acuity (at therapy start) to be checked/noted (ie, in patient history).
¶in case of patient-reported visual disturbances.
γ-GT, gamma-glutamyl transferase;ALT, alanine transaminase;AST, aspartate transaminase;CPK, creatine phosphokinase;LDH, lactate dehydrogenase;NT-proBNP, N-terminal prohormone of brain natriuretic peptide;OCT, optical coherence tomography; x, test recommended; (x), test optional.
Figure 3.
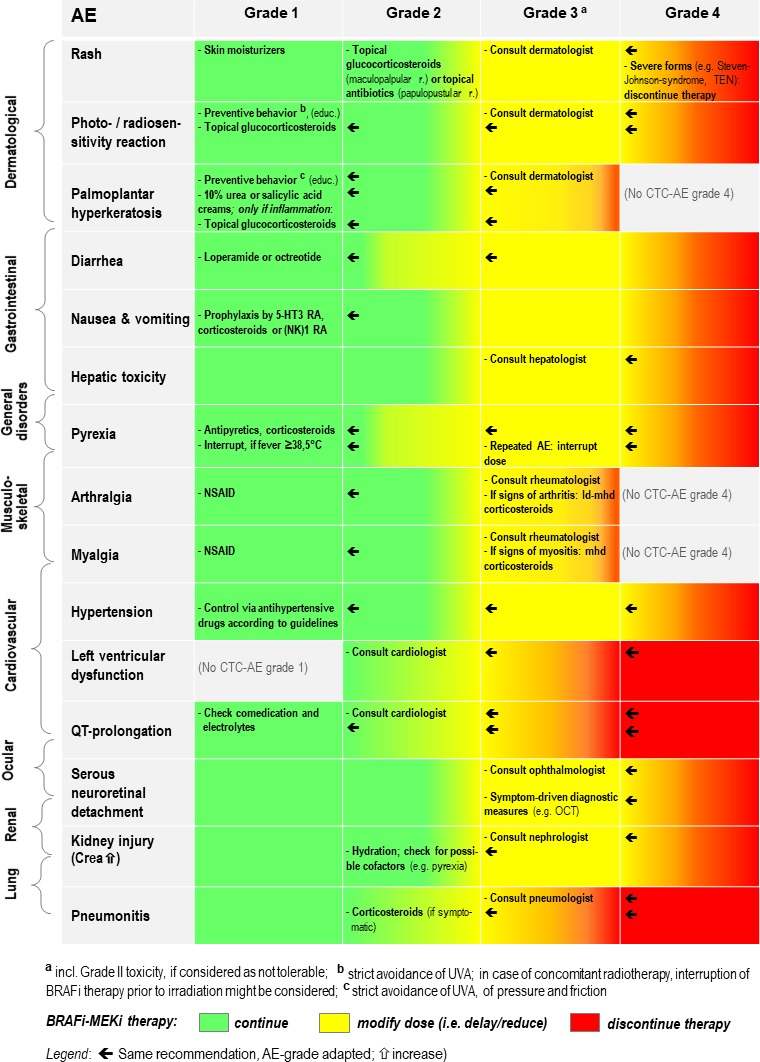
Recommendations for the management of clinically relevant and/or very frequent AE of BRAFi+MEKi therapy. Disclaimer: Official recommendations for the management of the three combinations might differ (please consider first the detailed specifications and recommendations as outlined in the summary of product characteristics (European Union) or in the respective, official prescribing information documents (USA and other countries outside European Union)). CTC-AE, common terminology criteria for adverse events; Crea, creatinine; ld, low-dose; mhd, middle-to-high-dose; NSAID, nonsteroidal anti-inflammatory drugs; OCT, optical coherence tomography; RA, receptor antagonist; TEN, toxic epidermal necrolysis; UVA, ultraviolet A (radiation).
While some of the side effects are class effects, others are substance-specific. Differences in drug tolerability might partly be explained by their individual pharmacokinetic (figure 1) and pharmacodynamics characteristics.
Class-effects of BRAFi include gastrointestinal side effects, increases in transaminases and arthralgia as well as cutaneous toxicities with formation of secondary neoplasms.20 22 23 For MEKi, class-effects encompass gastrointestinal side effects, increases in transaminases, oedema, ocular, cardiovascular AEs and cutaneous toxicity with the occurrence of papulopustular rash.86–89 The frequency of papulopustular rash, described in MEKi monotherapy trials,18 34 36 is decreased in combination regimens (figure 2). This is similar for the cutaneous side effects of BRAFi which are reduced by the suppression of the ‘paradoxical activation’ of the MAPK pathway, when adding a MEKi to the BRAFi. A systematic review indicates higher risk ratios for all grade diarrhoea, decreased ejection fraction and fever as well as a higher risk ratio for high grade diarrhoea when comparing BRAFi+MEKi versus BRAFi monotherapy.90
Substance specific, very common side effects include fever (for D+T) and photosensitivity (for V+C), attributed to the pharmacological properties of the BRAF inhibitors dabrafenib and vemurafenib. They were already identified during initial clinical development in BRAFi monotherapy trials.12–15 20 22 Differences in side effect profiles can be used to adjust prescription to the individual patient, but also to switch between BRAFi+MEKi combinations when side effects occur.
Because frequencies of documented side effects also depend on the design of a given clinical study, its definition of monitoring intervals and its methods to assess safety parameters, comparisons across studies have limited validity. Since there are no head-to-head trials comparing D+T, V+C and E+B, the indirect comparison against vemurafenib is currently the best approximation (figure 2). The only published indirect treatment comparison, comparing BRAFi+MEKi combinations so far, concluded that D+T shows a more favourable toxicity profile than V+C.24
Like the published indirect treatment comparison between D+T and V+C, which itself provides a less strong evidence than a randomised head-to-head trial could do, our review has certain limitations. Since the confirmatory trials COMBI-V (D+T), CoBRIM (V+C) and COLUMBUS Part I (E+B) ended at different time points and had follow-up intervals of variable length (see online supplementary file 1), there is some analysis bias in favour of E+B, authorised for use in the USA and the European Union since July and September 2018, respectively, only. In addition, some publication bias must be considered, as case reports and case series, describing, for example, rare and very rare side effects as discussed in our review, have been issued so far mainly for D+T and V+C: late or additional cases reporting on a new BRAFi or a newly approved combination might maybe be less easily publishable too. By choosing—as the main source for the appraisal of AE and serious AE frequencies (tables 1 and 2)—three confirmatory trials comparing all against a common comparator, we attempted, however, to diminish analysis and publication bias.
In conclusion, BRAF plus MEK inhibitor therapy is an effective and safe therapy, if monitored adequately. Therefore, the treating physicians need to be familiar with side effect management to reduce morbidity and mortality as well as premature treatment discontinuation.
Acknowledgments
The authors vouch for the content of this publication and confirm that recommendations made by consensus are matching their viewpoints. The corresponding author prepared the initial draft of the manuscript with editorial and writing assistance, all authors and particularly the first author contributed to the subsequent drafts. All authors contributed to manuscript writing, reviewed the drafted manuscript and approved the final version as well as the submission. Editorial and medical writing assistance was provided by Dr Markus Hartmann from Ecc-Oncology, funded by Pierre Fabre Pharma GmbH. Pierre Fabre Pharma GmbH did not influence the content of the manuscript, nor did the authors receive financial compensation for authoring this manuscript.
Footnotes
Contributors: All authors made essential contributions to the conception of the article and drafted one or several paragraphs and/or chapters, were revising the content of the manuscript critically, agreed on final approval of the version and agreed to be accountable for all aspects of the work. The first author consolidated the draft manuscript in conjunction with the corresponding author who also co-ordinated the manuscript drafting and finalisation process.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: LH: Personal fees and advisory role: Amgen, Bristol-Myers Squibb, MSD, Novartis, Roche, Pierre Fabre, Sanofi; Travel grants: Bristol-Myers Squibb, MSD; Grants: Novartis. TE: Consultancy fees from Amgen, Bristol-Myers Squibb, Celgene, Merck KGaA, MSD, Pierre Fabre, Novartis, Roche, Sanofi. MF: Consultant/advisory role: Bristol-Myers Squibb, Merck KGaA, MSD, Novartis, Roche, Sanofi. JH: Personal fees: Bristol-Myers Squibb, MSD, Novartis, Pfizer, Roche; Scientific Support/Grants: Bristol-Myers Squibb. DHS: None. JL: Speaker fees: AbbVie, Bristol-Myers Squibb, Celgene, Janssen-Cilag, MSD, Pfizer, Roche, Sanofi, USB; Consultant fees: AbbVie, AstraZeneca, Bristol-Myers Squibb, Celgene, Hospira, Janssen-Cilag, Leo Pharma, Lilly, Novartis, Roche, Sanofi. MP: None. AV: Consultancy and speaker fees: Amgen, Array, Merck KGaA, Novartis, Roche; Research grants: none. LZ: Consultant/advisory role: Bristol-Myers Squibb, MSD, Novartis, Roche, Sanofi; Honoraria: Bristol-Myers Squibb, Merck KGaA, MSD, Pierre Fabre, Novartis, Roche; Support for scientific meetings: Amgen, Bristol-Myers Squibb, MSD, Novartis, Pierre Fabre. RG: Honoraria: Almirall Hermal, Amgen, AstraZeneca, Bristol-Myers Squibb, Incyte, Leo Pharma, Merck Serono, MSD, Novartis, Pierre Fabre, Pfizer, Roche, Sanofi, SUN; Research funding: Johnson & Johnson, Novartis, Pfizer.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med Overseas Ed 2012;367:1694–703. 10.1056/NEJMoa1210093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372:30–9. 10.1056/NEJMoa1412690 [DOI] [PubMed] [Google Scholar]
- 3.Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88. 10.1056/NEJMoa1406037 [DOI] [PubMed] [Google Scholar]
- 4.Ribas A, Gonzalez R, Pavlick A, et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAF(V600)-mutated melanoma: a phase 1b study. Lancet Oncol 2014;15:954–65. 10.1016/S1470-2045(14)70301-8 [DOI] [PubMed] [Google Scholar]
- 5.Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76. 10.1056/NEJMoa1408868 [DOI] [PubMed] [Google Scholar]
- 6.Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2018;19:603–15. 10.1016/S1470-2045(18)30142-6 [DOI] [PubMed] [Google Scholar]
- 7.Long GV, Hauschild A, Santinami M, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med 2017;377:1813–23. 10.1056/NEJMoa1708539 [DOI] [PubMed] [Google Scholar]
- 8.Schadendorf D, Long GV, Stroiakovski D, et al. Three-year pooled analysis of factors associated with clinical outcomes across dabrafenib and trametinib combination therapy phase 3 randomised trials. Eur J Cancer 2017;82:45–55. 10.1016/j.ejca.2017.05.033 [DOI] [PubMed] [Google Scholar]
- 9.Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol 2016;17:1248–60. 10.1016/S1470-2045(16)30122-X [DOI] [PubMed] [Google Scholar]
- 10.Dummer R, Ascierto PA, Gogas H, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2018;19:1315–27. 10.1016/S1470-2045(18)30497-2 [DOI] [PubMed] [Google Scholar]
- 11.Petrelli F, Ardito R, Merelli B, et al. Prognostic and predictive role of elevated lactate dehydrogenase in patients with melanoma treated with immunotherapy and BRAF inhibitors: a systematic review and meta-analysis. Melanoma Res 2019;29:1–12. 10.1097/CMR.0000000000000520 [DOI] [PubMed] [Google Scholar]
- 12.Ascierto PA, Minor D, Ribas A, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol 2013;31:3205–11. 10.1200/JCO.2013.49.8691 [DOI] [PubMed] [Google Scholar]
- 13.Hauschild A, Grob J-J, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65. 10.1016/S0140-6736(12)60868-X [DOI] [PubMed] [Google Scholar]
- 14.Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 2012;366:707–14. 10.1056/NEJMoa1112302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16. 10.1056/NEJMoa1103782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim KB, Kefford R, Pavlick AC, et al. Phase II Study of the MEK1/MEK2 Inhibitor Trametinib in Patients With Metastatic BRAF -Mutant Cutaneous Melanoma Previously Treated With or Without a BRAF Inhibitor. JCO 2013;31:482–9. 10.1200/JCO.2012.43.5966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14. 10.1056/NEJMoa1203421 [DOI] [PubMed] [Google Scholar]
- 18.Rosen LS, LoRusso P, Ma WW, et al. A first-in-human phase I study to evaluate the MEK1/2 inhibitor, cobimetinib, administered daily in patients with advanced solid tumors. Invest New Drugs 2016;34:604–13. 10.1007/s10637-016-0374-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol 2013;14:249–56. 10.1016/S1470-2045(13)70024-X [DOI] [PubMed] [Google Scholar]
- 20.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363:809–19. 10.1056/NEJMoa1002011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemurafenib therapy. N Engl J Med 2012;366:480–1. 10.1056/NEJMc1113752 [DOI] [PubMed] [Google Scholar]
- 22.Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. The Lancet 2012;379:1893–901. 10.1016/S0140-6736(12)60398-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delord J-P, Robert C, Nyakas M, et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma. Clin Cancer Res 2017;23:5339–48. 10.1158/1078-0432.CCR-16-2923 [DOI] [PubMed] [Google Scholar]
- 24.Daud A, Gill J, Kamra S, et al. Indirect treatment comparison of Dabrafenib plus trametinib versus vemurafenib plus cobimetinib in previously untreated metastatic melanoma patients. J Hematol Oncol 2017;10 10.1186/s13045-016-0369-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robert C, Karaszewska B, Schachter J, et al. 3301 two year estimate of overall survival in COMBI-v, a randomized, open-label, phase III study comparing the combination of Dabrafenib (D) and trametinib (T) with vemurafenib (Vem) as first-line therapy in patients (PTS) with unresectable or metastatic BRAF V600E/K mutation-positive cutaneous melanoma. Eur J Cancer 2015;51 10.1016/S0959-8049(16)31820-2 [DOI] [Google Scholar]
- 26.Dréno B, Ribas A, Larkin J, et al. Incidence, course, and management of toxicities associated with cobimetinib in combination with vemurafenib in the coBRIM study. Ann Oncol 2017;28:1137–44. 10.1093/annonc/mdx040 [DOI] [PubMed] [Google Scholar]
- 27.Eigentler TK, Hassel JC, Berking C, et al. Diagnosis, monitoring and management of immune-related adverse drug reactions of anti-PD-1 antibody therapy. Cancer Treat Rev 2016;45:7–18. 10.1016/j.ctrv.2016.02.003 [DOI] [PubMed] [Google Scholar]
- 28.Hassel JC, Heinzerling L, Aberle J, et al. Combined immune checkpoint blockade (anti-PD-1/anti-CTLA-4): evaluation and management of adverse drug reactions. Cancer Treat Rev 2017;57:36–49. 10.1016/j.ctrv.2017.05.003 [DOI] [PubMed] [Google Scholar]
- 29.Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting Raf kinase in human cancer. Nat Rev Cancer 2017;17:676–91. 10.1038/nrc.2017.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng Y, Tian H. Current development status of MEK inhibitors. Molecules 2017;22 10.3390/molecules22101551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falchook GS, Long GV, Kurzrock R, et al. Dose selection, pharmacokinetics, and pharmacodynamics of BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res 2014;20:4449–58. 10.1158/1078-0432.CCR-14-0887 [DOI] [PubMed] [Google Scholar]
- 32.Ouellet D, Gibiansky E, Leonowens C, et al. Population pharmacokinetics of Dabrafenib, a BRAF inhibitor: effect of dose, time, covariates, and relationship with its metabolites. J Clin Pharmacol 2014;54:696–706. 10.1002/jcph.263 [DOI] [PubMed] [Google Scholar]
- 33.Koelblinger P, Thuerigen O, Dummer R. Development of encorafenib for BRAF-mutated advanced melanoma. Curr Opin Oncol 2018;30:1–33. 10.1097/CCO.0000000000000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Infante JR, Fecher LA, Falchook GS, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol 2012;13:773–81. 10.1016/S1470-2045(12)70270-X [DOI] [PubMed] [Google Scholar]
- 35.Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol 2012;13:782–9. 10.1016/S1470-2045(12)70269-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bendell JC, Javle M, Bekaii-Saab TS, et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br J Cancer 2017;116:575–83. 10.1038/bjc.2017.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol 2015;151:1103–9. 10.1001/jamadermatol.2015.1745 [DOI] [PubMed] [Google Scholar]
- 38.Koelblinger P, Dornbierer J, Dummer R. A review of binimetinib for the treatment of mutant cutaneous melanoma. Future Oncol 2017;13:1755–66. 10.2217/fon-2017-0170 [DOI] [PubMed] [Google Scholar]
- 39.Holderfield M, Nagel TE, Stuart DD. Mechanism and consequences of Raf kinase activation by small-molecule inhibitors. Br J Cancer 2014;111:640–5. 10.1038/bjc.2014.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen C-B, Wu M-Y, Ng CY, et al. Severe cutaneous adverse reactions induced by targeted anticancer therapies and immunotherapies. Cancer Manag Res 2018;10:1259–73. 10.2147/CMAR.S163391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hecht M, Zimmer L, Loquai C, et al. Radiosensitization by BRAF inhibitor therapy-mechanism and frequency of toxicity in melanoma patients. Ann Oncol 2015;26:1238–44. 10.1093/annonc/mdv139 [DOI] [PubMed] [Google Scholar]
- 42.Hecht M, Meier F, Zimmer L, et al. Clinical outcome of concomitant vs interrupted BRAF inhibitor therapy during radiotherapy in melanoma patients. Br J Cancer 2018;118:785–92. 10.1038/bjc.2017.489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Degen A, Alter M, Schenck F, et al. The hand-foot-syndrome associated with medical tumor therapy - classification and management. J Dtsch Dermatol Ges 2010;8:652–61. 10.1111/j.1610-0387.2010.07449.x [DOI] [PubMed] [Google Scholar]
- 44.Graf NP, Koelblinger P, Galliker N, et al. The spectrum of cutaneous adverse events during encorafenib and binimetinib treatment in B-rapidly accelerated fibrosarcoma-mutated advanced melanoma. J Eur Acad Dermatol Venereol 2019;33 10.1111/jdv.15363 [DOI] [PubMed] [Google Scholar]
- 45.Mössner R, Zimmer L, Berking C, et al. Erythema nodosum-like lesions during BRAF inhibitor therapy: report on 16 new cases and review of the literature. J Eur Acad Dermatol Venereol 2015;29:1797–806. 10.1111/jdv.13039 [DOI] [PubMed] [Google Scholar]
- 46.Keely SJ, Barrett KE. P38 mitogen-activated protein kinase inhibits calcium-dependent chloride secretion in T84 colonic epithelial cells. Am J Physiol Cell Physiol 2003;284:C339–C348. 10.1152/ajpcell.00144.2002 [DOI] [PubMed] [Google Scholar]
- 47.Menzies AM, Ashworth MT, Swann S, et al. Characteristics of pyrexia in BRAFV600E/K metastatic melanoma patients treated with combined dabrafenib and trametinib in a phase I/II clinical trial. Ann Oncol 2015;26:415–21. 10.1093/annonc/mdu529 [DOI] [PubMed] [Google Scholar]
- 48.Atkinson V, Long GV, Menzies AM, et al. Optimizing combination dabrafenib and trametinib therapy in BRAF mutation-positive advanced melanoma patients: guidelines from Australian melanoma medical oncologists. Asia Pac J Clin Oncol 2016;12 Suppl 7:5–12. 10.1111/ajco.12656 [DOI] [PubMed] [Google Scholar]
- 49.Babacan T, Türkbeyler IH, Balakan O, et al. A case of vemurafenib-induced polyarhritis in a patient with melanoma: how to manage it? Int J Rheum Dis 2017;20:398–401. 10.1111/1756-185X.12396 [DOI] [PubMed] [Google Scholar]
- 50.Maldonado-Seral C, Berros-Fombella JP, Vivanco-Allende B, et al. Vemurafenib-associated neutrophilic panniculitis: an emergent adverse effect of variable severity. Dermatol Online J 2013;19. [PubMed] [Google Scholar]
- 51.Zimmer L, Livingstone E, Hillen U, et al. Panniculitis with arthralgia in patients with melanoma treated with selective BRAF inhibitors and its management. Arch Dermatol 2012;148:357–61. 10.1001/archdermatol.2011.2842 [DOI] [PubMed] [Google Scholar]
- 52.Ueno M, Namiki T, Inui K, et al. Neutrophilic panniculitis with vasculitis in a melanoma patient treated with vemurafenib: a case report and its management. Int J Dermatol 2017;56:e163–5. 10.1111/ijd.13566 [DOI] [PubMed] [Google Scholar]
- 53.Chaminade A, Conte H, Jouary T, et al. BRAF inhibitors-induced panniculitis: a cutaneous side effect mimicking subcutaneous melanoma metastasis. J Eur Acad Dermatol Venereol 2015;29:392–3. 10.1111/jdv.12397 [DOI] [PubMed] [Google Scholar]
- 54.Novoa RA, Honda K, Koon HB, et al. Vasculitis and panniculitis associated with vemurafenib. J Am Acad Dermatol 2012;67:e271–2. 10.1016/j.jaad.2012.05.019 [DOI] [PubMed] [Google Scholar]
- 55.Maanaoui M, Saint-Jacques C, Gnemmi V, et al. Glomerulonephritis and granulomatous vasculitis in kidney as a complication of the use of BRAF and MEK inhibitors in the treatment of metastatic melanoma: a case report. Medicine 2017;96:e7196 10.1097/MD.0000000000007196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mirouse A, Savey L, Domont F, et al. Systemic vasculitis associated with vemurafenib treatment: case report and literature review. Medicine 2016;95:e4988 10.1097/MD.0000000000004988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niro A, Recchimurzo N, Sborgia A, et al. Bilateral ischemic retinal vasculitis in metastatic cutaneous melanoma patient treated with dabrafenib and trametinib: a case report. Ocul Immunol Inflamm 2018;26:783–5. 10.1080/09273948.2016.1261166 [DOI] [PubMed] [Google Scholar]
- 58.Hanson K, Robinson SR, Al-Yousuf K, et al. The anti-rheumatic drug, leflunomide, synergizes with MEK inhibition to suppress melanoma growth. Oncotarget 2018;9:3815–29. 10.18632/oncotarget.23378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lips DJ, Bueno OF, Wilkins BJ, et al. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation 2004;109:1938–41. 10.1161/01.CIR.0000127126.73759.23 [DOI] [PubMed] [Google Scholar]
- 60.Banks M, Crowell K, Proctor A, et al. Cardiovascular effects of the MEK inhibitor, trametinib: a case report, literature review, and consideration of mechanism. Cardiovasc Toxicol 2017;17:487–93. 10.1007/s12012-017-9425-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shah RR, Morganroth J. Update on cardiovascular safety of tyrosine kinase inhibitors: with a special focus on QT interval, left ventricular dysfunction and overall risk/benefit. Drug Saf 2015;38:693–710. 10.1007/s40264-015-0300-1 [DOI] [PubMed] [Google Scholar]
- 62.Flaherty L, Hamid O, Linette G, et al. A single-arm, open-label, expanded access study of vemurafenib in patients with metastatic melanoma in the United States. Cancer J 2014;20:18–24. 10.1097/PPO.0000000000000024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim G, McKee AE, Ning Y-M, et al. FDA approval summary: vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin Cancer Res 2014;20:4994–5000. 10.1158/1078-0432.CCR-14-0776 [DOI] [PubMed] [Google Scholar]
- 64.Nebot N, Arkenau H-T, Infante JR, et al. Evaluation of the effect of dabrafenib and metabolites on QTc interval in patients with BRAF V600-mutant tumours. Br J Clin Pharmacol 2018;84:764–75. 10.1111/bcp.13488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bronte E, Bronte G, Novo G, et al. What links BRAF to the heart function? New insights from the cardiotoxicity of BRAF inhibitors in cancer treatment. Oncotarget 2015;6:35589–601. 10.18632/oncotarget.5853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patnaik A, Tolcher A, Papadopoulos KP, et al. Phase 1 study to evaluate the effect of the MEK inhibitor trametinib on cardiac repolarization in patients with solid tumours. Cancer Chemother Pharmacol 2016;78:491–500. 10.1007/s00280-016-3090-y [DOI] [PubMed] [Google Scholar]
- 67.Haverkamp W, Mönnig G, Schulze-Bahr E, et al. Physician-induced torsade de pointes--therapeutic implications. Cardiovasc Drugs Ther 2002;16:101–9. 10.1023/A:1015797214679 [DOI] [PubMed] [Google Scholar]
- 68.Curigliano G, Cardinale D, Suter T, et al. Cardiovascular toxicity induced by chemotherapy, targeted agents and radiotherapy: ESMO clinical practice guidelines. Ann Oncol 2012;23 Suppl 7:vii155–66. 10.1093/annonc/mds293 [DOI] [PubMed] [Google Scholar]
- 69.Francis JH, Habib LA, Abramson DH, et al. Clinical and morphologic characteristics of MEK inhibitor-associated retinopathy: differences from central serous chorioretinopathy. Ophthalmology 2017;124:1788–98. 10.1016/j.ophtha.2017.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de la Cruz-Merino L, Di Guardo L, Grob J-J, et al. Clinical features of serous retinopathy observed with cobimetinib in patients with BRAF-mutated melanoma treated in the randomized coBRIM study. J Transl Med 2017;15 10.1186/s12967-017-1246-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Urner-Bloch U, Urner M, Stieger P, et al. Transient MEK inhibitor-associated retinopathy in metastatic melanoma. Ann Oncol 2014;25:1437–41. 10.1093/annonc/mdu169 [DOI] [PubMed] [Google Scholar]
- 72.Urner-Bloch U, Urner M, Jaberg-Bentele N, et al. MEK inhibitor-associated retinopathy (MEKAR) in metastatic melanoma: Long-term ophthalmic effects. Eur J Cancer 2016;65:130–8. 10.1016/j.ejca.2016.06.018 [DOI] [PubMed] [Google Scholar]
- 73.Choe CH, McArthur GA, Caro I, et al. Ocular toxicity in BRAF mutant cutaneous melanoma patients treated with vemurafenib. Am J Ophthalmol 2014;158:831–7. 10.1016/j.ajo.2014.07.003 [DOI] [PubMed] [Google Scholar]
- 74.Rogers S, McIntosh RL, Cheung N, et al. The prevalence of retinal vein occlusion: pooled data from population studies from the United States, Europe, Asia, and Australia. Ophthalmology 2010;117:313–9. 10.1016/j.ophtha.2009.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Welsh SJ, Corrie PG. Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Ther Adv Med Oncol 2015;7:122–36. 10.1177/1758834014566428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Daud A, Tsai K. Management of Treatment‐Related adverse events with agents targeting the MAPK pathway in patients with metastatic melanoma. The Oncologist 2017;22:823–33. 10.1634/theoncologist.2016-0456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schmitt L, Schumann T, Löser C, et al. Vemurafenib-induced pulmonary injury. Onkologie 2013;36:10–16. 10.1159/000355643 [DOI] [PubMed] [Google Scholar]
- 78.Teuma C, Pelletier S, Amini-Adl M, et al. Adjunction of a MEK inhibitor to Vemurafenib in the treatment of metastatic melanoma results in a 60% reduction of acute kidney injury. Cancer Chemother Pharmacol 2017;79:1043–9. 10.1007/s00280-017-3300-2 [DOI] [PubMed] [Google Scholar]
- 79.Launay-Vacher V, Zimner-Rapuch S, Poulalhon N, et al. Acute renal failure associated with the new BRAF inhibitor vemurafenib: a case series of 8 patients. Cancer 2014;120:2158–63. 10.1002/cncr.28709 [DOI] [PubMed] [Google Scholar]
- 80.Teuma C, Perier-Muzet M, Pelletier S, et al. New insights into renal toxicity of the B-Raf inhibitor, vemurafenib, in patients with metastatic melanoma. Cancer Chemother Pharmacol 2016;78:419–26. 10.1007/s00280-016-3086-7 [DOI] [PubMed] [Google Scholar]
- 81.Wanchoo R, Devoe C, Jhaveri KD. BRAF inhibitors - do we need to worry about kidney injury? Expert Opin Drug Saf 2016;15:1–3. 10.1517/14740338.2016.1164139 [DOI] [PubMed] [Google Scholar]
- 82.Jhaveri KD, Sakhiya V, Fishbane S. Nephrotoxicity of the BRAF inhibitors vemurafenib and dabrafenib. JAMA Oncol 2015;1:1133–4. 10.1001/jamaoncol.2015.1713 [DOI] [PubMed] [Google Scholar]
- 83.Wanchoo R, Jhaveri KD, Deray G, et al. Renal effects of BRAF inhibitors: a systematic review by the cancer and the kidney international network. Clin Kidney J 2016;9:245–51. 10.1093/ckj/sfv149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mai R, Zhou S, Zhong W, et al. Therapeutic efficacy of combined BRAF and MEK inhibition in metastatic melanoma: a comprehensive network meta-analysis of randomized controlled trials. Oncotarget 2015;6:28502–12. 10.18632/oncotarget.4375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pasquali S, Chiarion-Sileni V, Rossi CR, et al. Immune checkpoint inhibitors and targeted therapies for metastatic melanoma: a network meta-analysis. Cancer Treat Rev 2017;54:34–42. 10.1016/j.ctrv.2017.01.006 [DOI] [PubMed] [Google Scholar]
- 86.Abdel-Rahman O, ElHalawani H, Ahmed H, et al. Risk of selected gastrointestinal toxicities in cancer patients treated with MEK inhibitors: a comparative systematic review and meta-analysis. Expert Rev Gastroenterol Hepatol 2015;9:1433–45. 10.1586/17474124.2015.1087847 [DOI] [PubMed] [Google Scholar]
- 87.Yang Y, Liu Y-H, Sun X, et al. Risk of peripheral edema in cancer patients treated with MEK inhibitors: a systematic review and meta-analysis of clinical trials. Curr Med Res Opin 2017;33:1663–75. 10.1080/03007995.2017.1349657 [DOI] [PubMed] [Google Scholar]
- 88.Alves C, Ribeiro I, Penedones A, et al. Risk of ophthalmic adverse effects in patients treated with MEK inhibitors: a systematic review and meta-analysis. Ophthalmic Res 2017;57:60–9. 10.1159/000446845 [DOI] [PubMed] [Google Scholar]
- 89.Abdel-Rahman O, ElHalawani H, Ahmed H. Risk of selected cardiovascular toxicities in patients with cancer treated with MEK inhibitors: a comparative systematic review and meta-analysis. J Glob Oncol 2015;1:73–82. 10.1200/JGO.2015.000802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abdel-Rahman O, ElHalawani H, Ahmed H. Doublet BRAF/MEK inhibition versus single-agent BRAF inhibition in the management of BRAF-mutant advanced melanoma, biological rationale and meta-analysis of published data. Clin Transl Oncol 2016;18:848–58. 10.1007/s12094-015-1438-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
esmoopen-2019-000491supp001.tif (212.1KB, tif)