

Abstract
Precision oncology aims to distinguish which patients are eligible for a specific treatment in order to achieve the best possible outcome. In the last few years, genetic screens have shown their potential to find the new targets and drug combinations as well as predictive biomarkers for response and/or resistance to cancer treatment. In this review, we outline how precision oncology is changing over time and describe the different applications of genetic screens. Finally, we present some practical examples that describe the utility and the limitations of genetic screens in precision oncology.
Keywords: precision oncology, synthetic lethality, mechanisms of resistance, targeted therapy, genetic screens
Advantages and limitations of precision oncology
Precision medicine is defined as administering the right medicine at the right dose at the right time to the right patient. Despite being used in different medical fields, it is most commonly applied to oncology. Prasad and Gale analysed the use of precision oncology in the biomedical literature by classifying 50 articles over three time intervals.1 Between 2005 and 2010, the term precision oncology was mainly used to describe the use of targeted therapies such as epidermal growth factor receptor (EGFR) inhibitors or BCR/ABL1 inhibitors like gefitinib/erlotinib and imatinib. In 2013, precision oncology was used to describe the use of therapies based on specific biomarkers, like the administration of crizotinib for patients with lung cancer whose tumour had an EML-ALK rearrangement. By 2016, the definition of precision oncology referred to the use of next generation sequencing to guide the treatment choice. Regardless of this evolution in terminology, precision oncology has always been referred to the use of a certain drug based on molecular aberrations carried by the tumour. Historically, treatment decisions were made based on the histology of the tumour while nowadays are also based on mutation analysis.2 Genomic projects, like The Cancer Genome Atlas,3 have identified the main drivers of most solid and haematological malignancies, thus improving the diagnostic and the classification process, as well as the therapeutic approaches (table 1).
Table 1.
List of current approved molecular-driven treatments
| Disease | Gene | Drug |
| CML | ABL | Imatinib |
| Resistant CML | mutant ABL | Dasatinib |
| HES | PDGFRa | Imatinib |
| CMML | PDGFRb | Imatinib |
| Myelofibrosis | JAK2 | Ruxolitinib |
| AML | FLT3 | Quizartinib |
| Gastrointestinal stromal tumour | KIT | Imatinib |
| Lung cancer | EGFR | Erlotinib, Gefitinib |
| Kidney cancer | VEGFR | Sunitinib, Sorafenib |
| Breast cancer | HER2 | Trastuzumab/Pertuzumab |
| Lung cancer | ALK | Crizotinib |
| Melanoma | BRAF | Vemurafenib/Trametinib |
| Ovarian cancer | BRCA | Olaparib |
| Gastric cancer | HER2 | Trastuzumab |
AML, acute myeloid leukaemia; CML, chronic myeloid leukaemia; CMML, chronic myelomnocytic leukaemia; HES, hypereosinophilic syndrome.
The observation that many genomic aberrations are recurrent across multiple cancer types has led to the design of both basket and umbrella trials. The inhibition of HER2 in breast, gastric and colon cancer is, in this context, a successful example4–6 Molecular profiling of tumours has clearly shown to be beneficial for treatment decision-making. Indeed, several trials have shown that an individualised approach based on the molecular profiling of the tumour, can result in a better progression-free survival (PFS) when compared with the PFS of the previous regimen received by the patients.7–9 This benefit has been observed in adult as well as in paediatric cohorts.10 11 Nevertheless, in spite of these encouraging results, none of these trials were randomised. The molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA) trial, which is the only completed randomised phase II basket trial for precision oncology, showed that the use of molecularly targeted agents outside their indications did not improve the clinical outcome of heavily pretreated patients as compared with the treatment according to clinician’s choice.12 However, the effect of targeted anticancer drugs outside their approved indications is still under evaluation in big international precision oncology initiatives like the Target Agent and Profiling Utilization Registry, the Molecular Analysis of Therapy Choice and the Secured Access to Innovative Therapies Programme.13–15
There are several other limitations that interfere with a broader success of precision oncology. For example, from the DNA-sequencing data, we have learnt that fewer than 10% of patients with advanced cancer have a simple actionable mutation.16 17 Moreover, although some targets might appear to be interesting, the activity of drugs that inhibit them can be limited. Davis et al18 clearly showed that most drugs that entered the market in the period between 2009 and 2013 did not show a benefit in overall survival (OS) or in quality of life after 3 years follow-up.
Furthermore, we know from basket trials that the histological context can be an important determinant of response to targeted agents.19–21 This means that we cannot completely ignore the histology of the tumour nor the molecular context in which the mutation has been detected. Second, even if the tumour depends on that aberration, meaning that we can block the tumour growth specifically, mechanisms of acquired resistance might still emerge,22 23 which again may be tissue specific. Finally, the use of gene expression profiling is not yet a clinical standard, drugs targeting a specific aberration might not be effective, combinatorial approaches might be toxic to the patient and tumours are heterogeneous in space and time.
One way to overcome these limitations is to further dissect the biology of cancer aberrations by using synthetic lethal interaction approaches. In this review, we will describe the different approaches using functional genetic screens and their applications in precision medicine. We will summarise the current evidence showing that synthetic lethality can help to understand some of the limitations and lead to improve the success rate of precision oncology. Most importantly, we will also highlight the limitations of such approaches and the difficulties to translate preclinical findings into clinical practice.
Synthetic lethality and genetic screens
The understanding of cancer biology as well as advances in precision oncology heavily relies on preclinical research. Approaches that exploit synthetic lethality can help understand cancer vulnerabilities, mechanisms of primary and secondary resistance to treatment, the role of specific aberrations (mutations, amplifications, gene silencing) and their dependence on the tissue context.
Synthetic lethality is described as a phenomenon where a deficiency of two genes leads to cell death, but the deficiency of either one does not impair cell viability24 25 (figure 1). The deficiency can be due to a loss-of-function mutation, epigenetic silencing or pharmacological inhibition of the protein.
Figure 1.
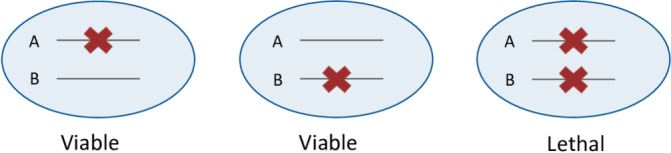
Synthetic lethality is defined as a phenomenon where a loss of either gene A or gene B is tolerated by the cell, but the loss of A and B is lethal.
The first clinically relevant example of a synthetic lethal interaction in cancer was the one between mutations in the genes encoding BRCA 1 and 2 and inhibition of the enzymes of the Poly (ADP-ribose) polymerase (PARP) family of enzymes. Its discovery came from the observation that PARP null mice are viable, but rely heavily on BRCA-mediated homologous recombination to repair the DNA damage. This led to the hypothesis that the inverse was true as well, meaning that BRCA deficient cells would depend more on PARP. This hypothesis-driven approach of predicting a synthetic lethal interaction turned out to be true. Actually, tumours that harbour BRCA1 or 2 loss-of-function mutations are especially sensitive to PARP inhibitors.26 27 These results were followed by the investigation and approval of PARP inhibitors for the treatment of patients with a germline BRCA1/2 mutated ovarian and breast cancer.
This interaction is an example of genotype-specific synthetic lethality, where a mutation in a tumour cell causes dependency on another pathway in order to maintain viability. When the compensatory pathway is inhibited, either genetically or pharmacologically, viability is impaired. Those types of interactions are of great importance for cancer treatment, since they offer selective targeting of mutated cancer cells over normal cells. In addition, drug-specific synthetic lethality can be exploited to identify rational combinational treatment. In this case, a combination of two drugs can be more effective than each of the drugs alone.28
A valuable tool to discover novel synthetic lethal interactions are functional genetic screens. Screens can offer an unbiased insight into complex biological processes, identify cancer vulnerabilities and biomarkers of resistance and sensitivity to the specific treatment. A genetic screen can be performed only with the help of techniques that allow large-scale gene perturbations, for example, RNA interference (RNAi), clustered regularly interspaced palindromic repeats (CRISPR) or transposons.
The first breakthrough technology that allowed for a systematic screening of multiple thousands of genes was RNAi using small interfering RNA to degrade selected transcripts.29 30 Although revolutionary at the time, the biggest drawback of this technology was the transient and unstable silencing. A significant improvement was achieved by the introduction of the short hairpin RNA (shRNA) technologies, that was characterised by a more stable and durable knockdown, and by the possibility to pool the shRNAs which simplified the screening procedure.31–33 Despite the success of shRNA-based genetic screens, also this technology had its drawbacks, mainly off-target effects.34
Lastly, CRISPR technology got adapted for precise genome editing in mammalian cells. The CRISPR-Cas9 system, which was originally discovered in bacteria as a form of primitive immune system to protect against viral infections,35 consists of two parts: an endonuclease Cas9 and a single-guide RNA (sgRNA) molecule. When they are both present in the cell, they form a complex, which is guided to the target genomic DNA location by the sgRNA. Next, Cas9 cuts the DNA resulting in a double strand break. As the cells try to repair the double strand break, small insertions and deletions (indels) can arise at the break site. These indels can lead to loss-of-function mutation in the targeted gene. Therefore, the knockout of the gene is a direct consequence of error-prone DNA-repair mechanisms and not due to the double strand break. By using a variety of sgRNAs that target any gene in the genome, we can create knockout mutations in every gene. In addition, the Cas9 protein has been modified, allowing to also perform transcriptional silencing known as CRISPR interference (CRISPRi) or activation (CRISPRa) screens.36 37 The main limitations of CRISPR screens include big difference in efficiency between sgRNAs leading to variable editing and mismatch tolerance, also producing some off-target effects.38 In comparison to shRNA, CRISPR shows better on-target activity and is nowadays widely used for screening.39
In addition to RNAi and CRISPR, transposons can also be used to disrupt genes. Transposons have been modified to allow the performance of insertional mutagenesis screens. In this type of screens, the enzyme transposase randomly cuts and pastes the transposon sequences across the genome, thus disrupting genes. The most widely used transposon systems are PiggyBac and Sleeping Beauty systems.40 Even if this system is less effective for studying recessive phenotypes, the use of a haploid cell line HAP1, that contains only one copy of each gene,41 can circumvent this limitation. Overview of their molecular mechanisms is depicted in figure 2.
Figure 2.

Molecular mechanisms of genetic perturbations (A) siRNA molecule is transiently transfected into the cell, where it binds and thus silences the target mRNA molecule. (B) shRNA is introduced in the cell trough viral infection. Upon stable integration into the genomic DNA, it is processed into an siRNA that silences the target mRNA. (C) CRISPR system is generally introduced in the cell trough viral infection. Upon stable integration into the genomic DNA, both Cas9 and the sgRNA are expressed. The endonuclease Cas9 and a sgRNA form, therefore, a complex causing a double-strand DNA break at a target location. Mistakes during DNA repair can cause mutations at the break site. (D) Upon viral infection, transposon and transposase enzyme integrate into the genomic DNA and lead to random insertions in the genome, thus disrupting genes. CRISPR, clustered regularly interspaced palindromic repeats;mRNA, messenger RNA; sgRNA, single guide RNA; siRNA, small interfering RNA; shRNA, short hairpin RNA.
Independent of the technology used, genetic screens can answer a variety of biological questions by changing the setup and read-out of the screen. To identify novel synthetic lethal interactions with a certain gene alteration we can perform synthetic lethal or ‘drop-out’ screens.
We can make use of isogenic cell line pairs or large panels of cell lines where one group carries the mutation while the other one does not.42 In addition to loss-of-function mutations, those aberrations also include gain-of-function mutations, gene amplifications, overexpression, gene signatures and epigenetic changes.43–46 Moreover, we can use drop-out screens to find genes whose loss can confer sensitivity to a certain drug treatment, thus uncovering mechanisms of primary resistance. The potential clinical utility of those screens lies in discovering new combinational treatment strategies that overcome primary resistance47 or identification of predictive biomarkers of response that can be used to select the group of patients that is most likely to benefit from that treatment.48 In contrast to drop-out screens, positive selection screens or ‘enrichment screens’ can be used to identify mechanisms of secondary resistance to a certain drug and identify which genes upon loss confer resistance to the specific treatment (figure 3).49 Besides genetic screens, other approaches can help in uncovering synthetic lethal interactions and finding new combinations of treatment, for example, drug screens and computational approaches.50 51
Figure 3.

Functional genetic screens (A) Drop-out screen, an isogenic cell line pair infected with the CRISPR library, selected and cultured alongside. Next, gRNA or shRNA barcodes are recovered, and the abundance of barcodes between the cell lines and the reference samples is compared. (B) Resistance screen where a cell line that is sensitive to the test treatment is used. After infection and selection, the cell population is split to treated and control arm. Next, barcodes are recovered from remaining cells and compared between reference sample, treated and untreated samples. CRISPR, clustered regularly interspaced palindromic repeats; gRNA, guide RNA; shRNA, short hairpin RNA; WT, wild type.
One of the major advantages of functional genetic screens is that they can be applied to any biological process. However, they do require an extensive in vitro and in vivo validation, as well as clinical trials before a novel finding can be translated into clinical practice.
Applying genetic screens to precision oncology
Targeted therapies like Braf and MEK inhibitors revolutionised the treatment of BRAF (V600E) metastatic melanoma and have been shown to be active in other malignancies as well.52 Paradoxically, although the same point mutation occurs in about 8%–10% of colorectal cancer (CRCs), these tumours do not respond to the BRAF (V600E) inhibitor (vemurafenib) when used as single agent.20 The mechanism underlying this unresponsiveness has been elegantly uncovered using a synthetic lethal screen. In particular, Prahallad et al47 performed a drop-out screen in BRAF (V600E) CRC cell lines looking for kinases that could sensitise cells to vemurafenib. With this approach, they discovered a feedback reactivation of EGFR on BRAF inhibition in BRAF (V600E) CRC cells as the driver of unresponsiveness to such treatment. These results led to the hypothesis that BRAF inhibitors need to be administered in combination with EGFR inhibitors to effectively kill these tumours. This hypothesis has been extensively validated both in vitro and in vivo. Most importantly, the results of this preclinical work have led to the design of several clinical trials, where BRAF (V600E) metastatic CRC (mCRC) patients have been treated either with a dual combination of Braf and EGFR inhibitors or a triple combination of a BRAF, EGFR and MEK or PI3K inhibitors.53–55 The results of these studies have clearly shown that the dual and triple blockade improved response rates and outcome as compared with BRAF inhibition alone. Simultaneously, a phase 1b study and a phase II study evaluated the combination of targeted therapies with chemotherapy in a three-drug regimen of vemurafenib, cetuximab and irinotecan.56 57 The addition of a BRAF inhibitor showed an increase of response rate and PFS when compared with the standard combination of anti-EGFR treatment and chemotherapy. Finally, the BEACON CRC58 is the first phase III trial that compares the triple combination (BRAF, MEK and EGFR inhibitors) versus dual combination (BRAF and EGFR inhibitor) versus a control arm (EGFR inhibitor and chemotherapy) as second or third-line treatment for BRAF (V600E) mCRC patients. Recently, an update of the safety lead of the study confirmed the triple combination to be safe. Clinical activity was characterised by 48% of overall response rate and efficacy by an improved PFS and OS as compared with standard of care.59 Based on these data, the US Food and Drug Administration (FDA) has granted breakthrough therapy designation of the triple combination as second or third-line treatment to patients affected by BRAF (V600E) mCRC.60 Nevertheless, the responses and the outcome benefit were not observed in the entire cohort of patients enrolled and secondary resistance occurred. Recently, CRCs have been classified into four distinct consensus molecular subtypes (CMS): CMS1 characterised by microsatellite instability and immune infiltration (14%), CMS2, known as canonical with Wingless/int1 (WNT) and myelocytomatosis oncogene (MYC) signalling activation (37%), CMS3 harbouring metabolic dysregulation (13%) and CMS4 with mesenchymal characteristics (23%).61 Notably, BRAF (V600E) mutations are present across all four different CRC molecular subtypes.
In addition, Oddo et al62 described that mutations leading to reactivation of the MAPK pathway represent the major mechanisms of secondary resistance. Therefore, tumour heterogeneity and clonal evolution could partially explain the heterogeneous response to both, the dual and triple blockade, observed in those clinical trials.
Prior to the CMS classification61 two independent groups found BRAF (V600E) colon cancers (CCs) to be characterised by a distinct gene expression profile when compared with KRAS-mutant and KRAS-BRAF double wild type (WT2) CCs. These tumours were defined as BRAF-mutant like by a transcriptional signature.63 64 To note, this gene signature identified BRAF (V600E) CCs and subsets of KRAS-mutant (30%) and WT2 (13%) CCs. The relevance of this transcriptional signature relies on the fact that the BRAF-mutant like tumours harbour similar poor prognosis regardless of the presence of BRAF (V600E) mutation.63 64 The signature has been further validated in a larger cohort of BRAF (V600E) CC patients65 and its biological implication has been investigated by using a synthetic lethal screen. By performing a drop-out screen, Vecchione et al46 identified RANBP2 to be synthetic lethal with the BRAF-like signature in CC cell lines. Further investigation of the function of this protein in CC cell lines led to the hypothesis that BRAF-like CC cells lines could be more vulnerable to antimitotic agents. This concept was extensively validated in vitro and in vivo models and is currently under investigation in the Motricolor consortium.66 Immediately after this finding, a prospective multicentre phase II clinical study started, where chemorefractory BRAF (V600E) mCRC patients were treated with vinorelbine67 A total of 20 patients were enrolled. Unfortunately, no responses were observed, with only one stable disease reported. In contrast, Masuishi et al68 reported tumour shrinkage in four BRAF (V600E) mCRC patients treated with eribulin as third and fifth line of treatment. Based on the results of these four cases, the BRAVERY study is now investigating the activity of eribulin as second line treatment in BRAF (V600E) mCRC.69The hypothesis generated from Vecchione et al46 is still far from being applicable, highlighting how complex is to translate preclinical findings into clinical practice.
An example of enrichment screen performed by Berns et al70 identified loss of phosphatase and tensin homolog (PTEN) as well as activating mutations in PIK3CA to induce resistance to trastuzumab in HER2 amplified breast cancer cell lines. These findings were further validated in a small cohort of HER2 amplified patients with breast cancer where both PIK3CA mutations and low PTEN expression correlated with poor prognosis after trastuzumab treatment. Moreover, similar preclinical results were obtained by other independent groups and with different HER2 inhibitors supporting the relevance of these discovery.71 72 Based on these results, the role of PIK3CA mutations and loss of PTEN in HER2 amplified patients with breast cancer treated with anti-HER2 antibody has been investigated, both in neoadjuvant and metastatic setting. The combined analysis of the GeparQuattro, GeparQuinto and GeparSixto trials showed PIK3CA mutant HER2 amplified breast tumours to have reduced pathological complete response (pCR) when compared with PIK3CA WT tumours73 Similarly, Majewski et al74 found lower pCR rate on trastuzumab and lapatinib monotherapy or in combination in PIK3CA mutant HER2 amplified patients with early breast cancer versus PIK3CA WT tumours. Additionally, PIK3CA mutated HER2 positive patients with metastatic breast cancer treated with capecitabine and lapatinib showed lower PFS compared with PIK3CA WT HER2 positive patients.75 To increase statistical power, Loibl et al76 performed a pooled analysis including approximately 1000 HER2 amplified patients with breast cancer whose PIK3CA status was known and were treated with anti-HER2 antibody. They confirmed PIK3CA mutant tumours to have lower chances to achieve a pCR when treated with HER2 blockade. Interestingly, this is especially significant in hormone receptor (HR) positive group as compared with the HR negative group. Importantly, none of the studies observed differences in outcome between PIK3CA mutant and PIK3CA WT tumours. Finally, the biomarker analysis of the NeoSphere study77 found only PIK3CA mutations in exon 9 to be associated with resistance to HER2 blockade. Overall, even if the results of the preclinical genetic screen are clear and robust, PIK3CA mutations are not used as predictive biomarker yet. Furthermore, other clinical variables might be considered to better understand its role in predicting resistance to HER2 blockade in breast cancer.
More recently, a group of researchers discovered loss of E-Cadherin, a frequently mutated gene in breast (>13%) and gastric cancer (>14%), to be synthetic lethal with ROS1 inhibitors, such as crizotinib. Authors used lobular breast cancer models for a perturbation screen with a focused library and a compound screen with drugs that are either approved in the clinic or that are being tested. As a result, E-cadherin loss became a potential biomarker for treatment with ROS1 inhibitors in a significant subset of patients with poor prognosis.78 Currently, a phase II clinical trial is testing crizotinib as a monotherapy in diffuse gastric cancer as well as crizotinib in combination with fulvestrant in lobular breast cancer.79
Another example of frequent genetic alteration that cannot be selectively targeted yet is KRAS mutations. The development of MEK inhibitors became a promising option for the treatment of these aggressive tumours. Lamentably, KRAS mutant tumours harbour different mechanisms of primary resistance to those inhibitors. In an attempt to identify genes whose loss could synergise with MEK inhibitors in KRAS mutant cancer cells, Corcoran et al performed a loss-of-function genetic screen. They identified Bcl-XL, a member of BH-3 antiapoptotic family, to be synthetic lethal with MEK inhibitors in KRAS mutant cell lines. These data were further validated in preclinical models by using a Bcl-XL inhibitor (navitoclax).80 At present, a clinical trial is recruiting patients with advanced or metastatic solid tumours to test MEK inhibitor (trametinib) in combination with navitoclax.81
In the last few years, checkpoint inhibitors have shown encouraging results that have changed the therapeutic approach of certain tumours, like non-small cell lung cancer, melanoma and microsatellite instable mCRC.82–84 In spite of this success, the efficacy and responsiveness to anti PD1, PD-L1 and CTLA-4 varies among different tumour types and across individual patients. Therefore, establishment of predictive biomarkers for checkpoint blockades as well as identification of novel targets for cancer immunotherapy are key to maximise therapeutic benefits. In this context, the use of genetic screens could be of great support. For example, by using a pooled loss-of-function in vivo genetic CRISPR-Cas9 screen to unravel genes responsible for sensitivity and resistance, Manguso et al demonstrated that loss of PTPN2 in cancer cells enhances interferon-γ-mediated effects on antigen presentation and growth suppression, thus increasing the efficacy of immunotherapy in a mouse transplantable tumour model.85 Similarly, another group performed an enrichment genome-scale CRISPR/Cas9 screen in coculture with activated cytotoxic CD8 +T-lymphocytes seeking for genes whose loss evoke resistance to adaptive immune response. The authors identified the expression of five negative regulators of the MAPK pathway as responsible for resistance to immunotherapy.86 On the opposite, loss of genes belonging to the SWI/SNF complex, the nuclear factor κB (NF-κB) pathway and metabolic pathway were shown to confer sensitivity to immunotherapy in a mouse melanoma model. Patel et al87 confirmed that loss of genes with a role in antigen presentation pathway as well as in interferon-γ signalling are responsible for immunotherapy resistance. Among the validated genes, they identified that loss of APLNR reduces the efficacy of adoptive cell transfer and checkpoint blockade by interacting with JAK1, thus, modulating interferon-γ responses. Finally, Mezzadra et al88 used an haploid genetic screen to seek for regulators of PD-L1 protein. They identified CMTM4 and CMTM6 as new potential target to block the PD-1 pathway. Altogether, these data highlight the importance of genetic screens to unveil mechanisms of responsiveness to immunotherapy as well as new potential targets to exploit therapeutically. Nevertheless, none of those results have been validated in the clinic yet. A schematic overview of the preclinical findings and the clinical studies reported above is depicted in table 2.
Table 2.
From bench to the bedside
| Preclinical findings | Clinical trials | Clinical practice changing |
| EGFR loss is synthetic lethal with BRAF (V600E) in CRC in vivo and in vitro models47 | 53–59 | FDA breakthrough therapy designation60 |
| BRAF-like CCs are vulnerable to antimitotic agents46 | 66–68 | Controversial data. Waiting for further studies |
| PTEN loss and PIK3CA mutations confer resistance to trastuzumab in HER2 amplified breast cancer cell lines70 | 73–77 | Not yet |
| E-cadherin loss is synthetic lethal with ROS1 inhibitors in lobular breast cancer preclinical models78 | 79 | Trial not yet recruiting |
| Loss of BCL-XL is synthetic lethal with MEK inhibition in KRAS mutant preclinical models80 | 81 | Trial ongoing |
| Loss of PTPN2 synergises with immunotherapy in mouse transplantable tumour models85 | No trials ongoing nor retrospective analysis of already closed trials | Not yet |
| Identification of biomarkers of response and resistance to immunotherapy in a mouse melanoma model86 87 | No trials ongoing nor retrospective analysis of already closed trials | Not yet |
| Identification of novel targets for immunotherapy88 | No trials ongoing nor retrospective analysis of already closed trials | Not yet |
Depicts preclinical findings followed by clinical trials and clinical practice implementation.
CCs, colon cancers; CRC, colorectal cancer; EGFR, epidermal growth factor receptor; FDA, Food and Drug Administration.
Future directions
Precision oncology is based on molecular profile of cancer cells. Defining genetic alterations helps to establish a precise molecular diagnosis of the tumour and to predict the course of the disease. Moreover, it allows the administration of a tailored therapy in accordance to the genomic aberrations carried by that individual tumour. The development of targeted therapies requires several years of intense multidisciplinary effort, from understanding the cancer biology to testing a new drug in a phase III study. Nevertheless, large phase III clinical trials are often not feasible for rare tumour subtypes. In this context, a possible solution are basket trials, which can accelerate the translation into clinical practice. Moreover, several limitations need to be considered during this complex process, like unpredicted toxicity of combinatorial treatments, tumour evolution, cancer heterogeneity, context dependency and the tumour microenvironment. In addition, due to the ever increasing number of FDA-approved cancer drugs, the number of possible drug combinations increases exponentially. This poses a conundrum that can only be solved by upfront selection of the most potent combinations. We have argued here that genetic screens can be a useful tool to identify such powerful drug combinations. A second potential clinical use regards the notion that not all patients treated with a specific drug will benefit from it. As we have discussed as well, genetic screens can help to identify biomarkers of response or resistance.
After almost two decades from the introduction of the RNAi technology in human cancer cells, we are starting to witness the benefits of the use of genetic screens. As a result, we see new therapies being implemented for some malignancies that were untreatable before. Indeed, some clinical trials are finding strong correlation in what has been described in vitro.47 In addition, organoids and in vivo screens are now being exploited as techniques to study the complex interplay between the tumour and its stroma. Although in vivo screens are technically a huge challenge, they have become a valuable tool, especially when looking for targets that are related with the immune system.
The technology that allows genome-wide screens has become easily available, cheaper and relatively simple to implement. As a consequence, there has been an exponential increase in the number of screens performed. Even though there are indications that functional genetic screens can play a role in clinically relevant discoveries, there are a lot of hurdles to deal with, when translating the preclinical observation from a genetic screen into clinical practice.
First, genetic screens are often long and complicated. It should also be remembered that complete removal of a protein from a cell is not necessarily the same as pharmacological inhibition of the protein, as proteins can also have scaffolding functions. Thus, nor CRISPR or shRNA technologies can simulate drug inhibition. On the one hand, shRNAs are prone to off-target effects. On the other hand, CRISPR screens have less off-target effects, however, drugs seldom inhibit a protein for the full 100%, which is the result of a CRISPR knockout. Moreover, even if a genetic screen unveils a new target, the development of small molecule inhibitors needed to clinically validate can often take years. Therefore, a great number of genetic screens with potential clinical utility still remain to be proven relevant for the patients.
Second, to overcome limitations like context dependency, heterogeneity and tumour evolution, the use of a comprehensive and integrated analysis can be of great help. Combining genetic approaches with cell line analysis and patient data, when available, could help to overcome these problems in order to focus on clinical relevant targets.89
As a result, we need to design smarter and better screens, to maximise the outcomes while minimising the costs. Since screens can be adapted to answer a wide variety of questions, we can use them to investigate complex biological processes. We can use different reporter systems for phenotype selection,90 or knock-in of a selection marker to a target locus.91 Now, we can design screens that are not focused only on cell death or proliferation. For example, a flow cytometry-based read-out allows separation of the population of cells based on any protein for which an antibody is available. Currently, CRISPR technology offers a diverse toolkit to modify gene expression. In addition, CRISPRa and CRISPRi, introduction of diverse point mutations or epigenetic reprogramming is possible. Subsequently, it is expected that screens adopting these technologies will offer novel insights into the complex biology of the cancer cell in the near future.
Another important aspect that applies to precision oncology is that the phenotype and behaviour of a certain tumour might be the consequence of the activity of multiple genes. For example, it may not be the aberration in gene X that plays a role in that specific tumour context, but rather the combination with other gene aberrations. To better model this, we need to develop systems that allow perturbations of more than one gene at the time. In that respect, there have been significant improvements in the last years to develop screens that allow screening for interactions, both with shRNAs and CRISPR.92–94 Additionally, a dual system that combines activation of transcription with knockout has recently been developed, which can further expand our understanding of genetic interactions.95 Furthermore, we can couple pooled genetic screens with single cell RNA sequencing, for example, Perturb-seq, which allows immediate transcriptional profiling of genetically diverse populations.96–98
In conclusion, genetic screens have already shown to be a relevant tool to find new therapeutic options and to predict treatment response. Nevertheless, it is an early technology that we are still improving. Therefore, optimising and integrating this technology with other analysis would potentially bring us to the new era of precision oncology.
Acknowledgments
We would like to thank Professor René Bernards and Professor Ulrich Keilholz for fruitful discussions and critical reading of the manuscript.
Footnotes
AM-S and ZP contributed equally.
Contributors: All authors of this review have directly participated in its planning and execution, have read and approved the final version submitted.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: LV is a member of the GI connect group, spouse is employee and shareholder of Bayer AG.
Patient consent for publication: Not required.
Provenance and peer review: Commissioned; externally peer reviewed.
References
- 1.Prasad V, Gale RP. What precisely is precision Oncology—and will it work? ASCO post, 2017. [Google Scholar]
- 2.Gerlinger M, Rowan AJ, Horswell S, et al. . Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med Overseas Ed 2012;366:883–92. 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.National Cancer Institute The Cancer genome atlas. Available: https://cancergenome.nih.gov/ [Accessed 15 Feb 2019].
- 4.Baselga J, Perez EA, Pienkowski T, et al. . Adjuvant trastuzumab: A milestone in the treatment of HER-2-positive early breast cancer. Oncologist 2006;11(suppl_1):4–12. 10.1634/theoncologist.11-90001-4 [DOI] [PubMed] [Google Scholar]
- 5.Sartore-Bianchi A, Trusolino L, Martino C, et al. . Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:738–46. 10.1016/S1470-2045(16)00150-9 [DOI] [PubMed] [Google Scholar]
- 6.Bang Y-J, Van Cutsem E, Feyereislova A, et al. . Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687–97. 10.1016/S0140-6736(10)61121-X [DOI] [PubMed] [Google Scholar]
- 7.Von Hoff DD, Stephenson JJ, Rosen P, et al. . Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. JCO 2010;28:4877–83. 10.1200/JCO.2009.26.5983 [DOI] [PubMed] [Google Scholar]
- 8.Prager GW, Unseld M, Waneck F, et al. . Results of the extended analysis for cancer treatment (exact) trial: a prospective translational study evaluating individualized treatment regimens in oncology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoefflin R, Geißler A-L, Fritsch R, et al. . Personalized clinical decision making through implementation of a molecular tumor board: a German single-center experience. JCO Precis Oncol 2018;2:1–16. 10.1200/PO.18.00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim ES, Herbst RS, Wistuba II, et al. . The battle trial: personalizing therapy for lung cancer. Cancer Discov 2011;1:44–53. 10.1158/2159-8274.CD-10-0010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harttrampf AC, Lacroix L, Deloger M, et al. . Molecular screening for cancer treatment optimization (MOSCATO-01) in pediatric patients: a single-institutional prospective molecular stratification trial. Clin Cancer Res 2017;23:6101–12. 10.1158/1078-0432.CCR-17-0381 [DOI] [PubMed] [Google Scholar]
- 12.Le Tourneau C, Delord J-P, Gonçalves A, et al. . Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol 2015;16:1324–34. 10.1016/S1470-2045(15)00188-6 [DOI] [PubMed] [Google Scholar]
- 13.ASCO The targeted agent and profiling utilization registry. Available: https://www.tapur.org/ [Accessed 11 Apr 2019].
- 14.National Cancer Institute NCI-MATCH trial (molecular analysis for therapy choice). Available: https://www.cancer.gov/about-cancer/treatment/clinical-trials/nci-supported/nci-match [Accessed 04 Nov 2019].
- 15.Buzyn A, Blay J-Y, Hoog-Labouret N, et al. . Equal access to innovative therapies and precision cancer care. Nat Rev Clin Oncol 2016;13:385–93. 10.1038/nrclinonc.2016.31 [DOI] [PubMed] [Google Scholar]
- 16.Group E-ACR, Others Executive summary: interim analysis of the NCI-MATCH trial. ECOG-ACRIN Cancer Research Group. [Google Scholar]
- 17.Meric-Bernstam F, Brusco L, Shaw K, et al. . Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. JCO 2015;33:2753–62. 10.1200/JCO.2014.60.4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis C, Naci H, Gurpinar E, et al. . Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: retrospective cohort study of drug approvals 2009-13. BMJ 2017;359 10.1136/bmj.j4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chapman PB, Hauschild A, Robert C, et al. . Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16. 10.1056/NEJMoa1103782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopetz S, Desai J, Chan E, et al. . Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF -Mutated Colorectal Cancer. JCO 2015;33:4032–8. 10.1200/JCO.2015.63.2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyman DM, Puzanov I, Subbiah V, et al. . Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med 2015;373:726–36. 10.1056/NEJMoa1502309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagle N, Emery C, Berger MF, et al. . Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. JCO 2011;29:3085–96. 10.1200/JCO.2010.33.2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgillo F, Della Corte CM, Fasano M, et al. . Mechanisms of resistance to EGFR-targeted drugs: lung cancer. ESMO Open 2016;1:e000060 10.1136/esmoopen-2016-000060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobwansky TH. Genetics of natural populations: recombination and variability in populations of Drosophila pseudoobscura. Genetics 1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nijman SMB. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett 2011;585:1–6. 10.1016/j.febslet.2010.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farmer H, McCabe N, Lord CJ, et al. . Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–21. 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- 27.Bryant HE, Schultz N, Thomas HD, et al. . Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434:913–7. 10.1038/nature03443 [DOI] [PubMed] [Google Scholar]
- 28.Brunen D, Bernards R. Drug therapy: exploiting synthetic lethality to improve cancer therapy. Nat Rev Clin Oncol 2017;14:331–2. 10.1038/nrclinonc.2017.46 [DOI] [PubMed] [Google Scholar]
- 29.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002;296:550–3. 10.1126/science.1068999 [DOI] [PubMed] [Google Scholar]
- 30.Berns K, Hijmans EM, Mullenders J, et al. . A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004;428:431–7. 10.1038/nature02371 [DOI] [PubMed] [Google Scholar]
- 31.Burgess DJ, Doles J, Zender L, et al. . Topoisomerase levels determine chemotherapy response in vitro and in vivo. PNAS 2008;105:9053–8. 10.1073/pnas.0803513105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silva JM, Marran K, Parker JS, et al. . Profiling essential genes in human mammary cells by multiplex RNAi screening. Science 2008;319:617–20. 10.1126/science.1149185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlabach MR, Luo J, Solimini NL, et al. . Cancer proliferation gene discovery through functional genomics. Science 2008;319:620–4. 10.1126/science.1149200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson AL, Bartz SR, Schelter J, et al. . Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol 2003;21:635–7. 10.1038/nbt831 [DOI] [PubMed] [Google Scholar]
- 35.Makarova KS, Grishin NV, Shabalina SA, et al. . A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 2006;1 10.1186/1745-6150-1-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilbert LA, Horlbeck MA, Adamson B, et al. . Genome-scale CRISPR-mediated control of gene repression and activation. Cell 2014;159:647–61. 10.1016/j.cell.2014.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konermann S, Brigham MD, Trevino AE, et al. . Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015;517:583–8. 10.1038/nature14136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fortin J-P, Tan J, Gascoigne KE, et al. . Multiple-gene targeting and mismatch tolerance can confound analysis of genome-wide pooled CRISPR screens. Genome Biol 2019;20 10.1186/s13059-019-1621-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evers B, Jastrzebski K, Heijmans JPM, et al. . CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat Biotechnol 2016;34:631–3. 10.1038/nbt.3536 [DOI] [PubMed] [Google Scholar]
- 40.O'Donnell KA. Advances in functional genetic screening with transposons and CRISPR/Cas9 to illuminate cancer biology. Curr Opin Genet Dev 2018;49:85–94. 10.1016/j.gde.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carette JE, Guimaraes CP, Varadarajan M, et al. . Haploid genetic screens in human cells identify host factors used by pathogens. Science 2009;326:1231–5. 10.1126/science.1178955 [DOI] [PubMed] [Google Scholar]
- 42.Berns K, Caumanns JJ, Hijmans EM, et al. . ARID1A mutation sensitizes most ovarian clear cell carcinomas to Bet inhibitors. Oncogene 2018;37:4611–25. 10.1038/s41388-018-0300-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mair B, Kubicek S, Nijman SMB. Exploiting epigenetic vulnerabilities for cancer therapeutics. Trends Pharmacol Sci 2014;35:136–45. 10.1016/j.tips.2014.01.001 [DOI] [PubMed] [Google Scholar]
- 44.Chan N, Pires IM, Bencokova Z, et al. . Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res 2010;70:8045–54. 10.1158/0008-5472.CAN-10-2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steckel M, Molina-Arcas M, Weigelt B, et al. . Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res 2012;22:1227–45. 10.1038/cr.2012.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vecchione L, Gambino V, Raaijmakers J, et al. . A vulnerability of a subset of colon cancers with potential clinical utility. Cell 2016;165:317–30. 10.1016/j.cell.2016.02.059 [DOI] [PubMed] [Google Scholar]
- 47.Prahallad A, Sun C, Huang S, et al. . Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012;483:100–3. 10.1038/nature10868 [DOI] [PubMed] [Google Scholar]
- 48.Ding Y, Gong C, Huang D, et al. . Synthetic lethality between HER2 and transaldolase in intrinsically resistant HER2-positive breast cancers. Nat Commun 2018;9 10.1038/s41467-018-06651-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang S, Hölzel M, Knijnenburg T, et al. . MED12 controls the response to multiple cancer drugs through regulation of TGF-β receptor signaling. Cell 2012;151:937–50. 10.1016/j.cell.2012.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jerby-Arnon L, Pfetzer N, Waldman YY, et al. . Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell 2014;158:1199–209. 10.1016/j.cell.2014.07.027 [DOI] [PubMed] [Google Scholar]
- 51.Sinha S, Thomas D, Chan S, et al. . Systematic discovery of mutation-specific synthetic lethals by mining pan-cancer human primary tumor data. Nat Comms 2017;8 10.1038/ncomms15580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dummer R, Hauschild A, Lindenblatt N, et al. . Cutaneous melanoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2015;26(Suppl 5):v126–32. 10.1093/annonc/mdv297 [DOI] [PubMed] [Google Scholar]
- 53.Corcoran RB, Atreya CE, Falchook GS, et al. . Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600–Mutant Colorectal Cancer. JCO 2015;33:4023–31. 10.1200/JCO.2015.63.2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Geel RMJM, Tabernero J, Elez E, et al. . A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF -Mutant Colorectal Cancer. Cancer Discov 2017;7:610–9. 10.1158/2159-8290.CD-16-0795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corcoran RB, André T, Atreya CE, et al. . Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAFV600E-Mutant Colorectal Cancer. Cancer Discov 2018;8:428–43. 10.1158/2159-8290.CD-17-1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hong DS, Morris VK, El Osta B, et al. . Phase Ib study of vemurafenib in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with BRAFV600E mutation. Cancer Discovery 2016;6:1352–65. 10.1158/2159-8290.CD-16-0050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kopetz S, McDonough SL, Lenz H-J, et al. . Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). JCO 2017;35(15_suppl):3505 10.1200/JCO.2017.35.15_suppl.3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clinical trials Beacon (CRC). Available: https://clinicaltrials.gov/ct2/show/NCT02928224 [Accessed 15 Feb 2019].
- 59.Kopetz S, Grothey A, Yaeger R, et al. . Updated results of the beacon CRC safety lead-in: Encorafenib (ENCO) + binimetinib (BINI) + cetuximab (CETUX) for BRAFV600E-mutant metastatic colorectal cancer (mCRC). JCO 2019;37(4_suppl):688 10.1200/JCO.2019.37.4_suppl.688 [DOI] [Google Scholar]
- 60.The ASCO Post FDA grants breakthrough therapy designation for Encorafenib plus binimetinib and cetuximab in BRAF V600E–Mutant metastatic colorectal cancer. Available: http://www.ascopost.com/News/59160 [Accessed 15 Feb 2019].
- 61.Guinney J, Dienstmann R, Wang X, et al. . The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350–6. 10.1038/nm.3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oddo D, Sennott EM, Barault L, et al. . Molecular landscape of acquired resistance to targeted therapy combinations in BRAF-mutant colorectal cancer. Cancer Res 2016;76:4504–15. 10.1158/0008-5472.CAN-16-0396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Popovici V, Budinska E, Tejpar S, et al. . Identification of a poor-prognosis BRAF-Mutant–Like population of patients with colon cancer. J Clin Orthod 2012;30:1288–95. [DOI] [PubMed] [Google Scholar]
- 64.Tian S, Simon I, Moreno V, et al. . A combined oncogenic pathway signature of BRAF, KRAS and PI3KCA mutation improves colorectal cancer classification and cetuximab treatment prediction. Gut 2013;62:540–9. 10.1136/gutjnl-2012-302423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Veld S, Duong KN, Snel M. A computational workflow translates a 58-Gene signature to a formalin-fixed, paraffin-embedded sample-based companion diagnostic for personalized treatment of the BRAF-Mutation-Like subtype of colorectal cancers. High Throughput 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Motricolor Molecularly guided trials with specific treatment strategies in patients with advanced newly molecular defined subtypes of colorectal cancer. Available: http://www.motricolor.eu/ [Accessed 15 Feb 2019].
- 67.Cremolini C, Pietrantonio F, Tomasello G, et al. . Vinorelbine in BRAF V600E mutated metastatic colorectal cancer: a prospective multicentre phase II clinical study. ESMO Open 2017;2:e000241 10.1136/esmoopen-2017-000241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Masuishi T, Taniguchi H, Sugiyama K, et al. . Eribulin in BRAF V600E-mutant metastatic colorectal cancer: case series and potential rationale. Ann Oncol 2018;29:1330–1. 10.1093/annonc/mdy107 [DOI] [PubMed] [Google Scholar]
- 69.Masuishi T, Taniguchi H, Kotani D, et al. . BRAVERY study: a multicenter phase II study of eribulin in patients with BRAF V600E mutant metastatic colorectal cancer (EPOC1701). Annals of oncology. Available: https://academic.oup.com/annonc/article/29/suppl_8/mdy281.153/5140513 [Accessed 11 Apr 2019]. [DOI] [PMC free article] [PubMed]
- 70.Berns K, Horlings HM, Hennessy BT, et al. . A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007;12:395–402. 10.1016/j.ccr.2007.08.030 [DOI] [PubMed] [Google Scholar]
- 71.Nagata Y, Lan K-H, Zhou X, et al. . PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004;6:117–27. 10.1016/j.ccr.2004.06.022 [DOI] [PubMed] [Google Scholar]
- 72.Eichhorn PJA, Gili M, Scaltriti M, et al. . Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res 2008;68:9221–30. 10.1158/0008-5472.CAN-08-1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Loibl S, von Minckwitz G, Schneeweiss A, et al. . PIK3CA Mutations Are Associated With Lower Rates of Pathologic Complete Response to Anti–Human Epidermal Growth Factor Receptor 2 (HER2) Therapy in Primary HER2-Overexpressing Breast Cancer. JCO 2014;32:3212–20. 10.1200/JCO.2014.55.7876 [DOI] [PubMed] [Google Scholar]
- 74.Majewski IJ, Nuciforo P, Mittempergher L, et al. . PIK3CA Mutations Are Associated With Decreased Benefit to Neoadjuvant Human Epidermal Growth Factor Receptor 2–Targeted Therapies in Breast Cancer. JCO 2015;33:1334–9. 10.1200/JCO.2014.55.2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baselga J, Lewis Phillips GD, Verma S, et al. . Relationship between tumor biomarkers and efficacy in EMILIA, a phase III study of trastuzumab emtansine in HER2-positive metastatic breast cancer. Clin Cancer Res 2016;22:3755–63. 10.1158/1078-0432.CCR-15-2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loibl S, Majewski I, Guarneri V, et al. . PIK3CA mutations are associated with reduced pathological complete response rates in primary HER2-positive breast cancer: pooled analysis of 967 patients from five prospective trials investigating lapatinib and trastuzumab in early stage epithelial ovarian carcinoma. Gynecol Oncol 2004;93:301–6.15099937 [Google Scholar]
- 77.Bianchini G, Kiermaier A, Bianchi GV, et al. . Biomarker analysis of the NeoSphere study: pertuzumab, trastuzumab, and docetaxel versus trastuzumab plus docetaxel, pertuzumab plus trastuzumab, or pertuzumab plus docetaxel for the neoadjuvant treatment of HER2-positive breast cancer. Breast Cancer Res 2017;19 10.1186/s13058-017-0806-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bajrami I, Marlow R, van de Ven M, et al. . E-Cadherin/ROS1 inhibitor synthetic lethality in breast cancer. Cancer Discov 2018;8:498–515. 10.1158/2159-8290.CD-17-0603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clinical Trials ROS1 targeting with crizotinib in advanced E-cadherin negative, ER positive lobular breast cancer or diffuse gastric Cancer study (ROLo). Available: https://clinicaltrials.gov/ct2/show/NCT03620643 [Accessed 15 Feb 2019].
- 80.Corcoran RB, Cheng KA, Hata AN, et al. . Synthetic lethal interaction of combined Bcl-xL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell 2013;23:121–8. 10.1016/j.ccr.2012.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Clinical trials Trametinib and Navitoclax in treating patients with advanced or metastatic solid tumors. Available: https://clinicaltrials.gov/ct2/show/NCT02079740 [Accessed 15 Feb 2019].
- 82.Lugowska I, Teterycz P, Rutkowski P. Immunotherapy of melanoma. Wo 2018;2018:61–7. 10.5114/wo.2018.73889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Planchard D, Popat S, Kerr K, et al. . Metastatic non-small cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2018;29(Suppl 4):iv192–237. 10.1093/annonc/mdy275 [DOI] [PubMed] [Google Scholar]
- 84.Overman MJ, Lonardi S, Wong KYM, et al. . Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch Repair–Deficient/Microsatellite Instability–High metastatic colorectal cancer. JCO 2018;36:773–9. 10.1200/JCO.2017.76.9901 [DOI] [PubMed] [Google Scholar]
- 85.Manguso RT, Pope HW, Zimmer MD, et al. . In vivo CRISPR screening identifies PTPN2 as a cancer immunotherapy target. Nature 2017;547:413–8. 10.1038/nature23270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pan D, Kobayashi A, Jiang P, et al. . A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science 2018;359:770–5. 10.1126/science.aao1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Patel SJ, Sanjana NE, Kishton RJ, et al. . Identification of essential genes for cancer immunotherapy. Nature 2017;548:537–42. 10.1038/nature23477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mezzadra R, Sun C, Jae LT, et al. . Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 2017;549:106–10. 10.1038/nature23669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bester AC, Lee JD, Chavez A, et al. . An integrated genome-wide CRISPRa approach to Functionalize lncRNAs in drug resistance. Cell 2018;173:649–64. 10.1016/j.cell.2018.03.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang L, Leite de Oliveira R, Wang C, et al. . High-throughput functional genetic and compound screens identify targets for senescence induction in cancer. Cell Reports 2017;21:773–83. 10.1016/j.celrep.2017.09.085 [DOI] [PubMed] [Google Scholar]
- 91.Breslow DK, Hoogendoorn S, Kopp AR, et al. . A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat Genet 2018;50:460–71. 10.1038/s41588-018-0054-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Horlbeck MA, Xu A, Wang M, et al. . Mapping the genetic landscape of human cells. Cell 2018;174:953–67. 10.1016/j.cell.2018.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shen JP, Zhao D, Sasik R, et al. . Combinatorial CRISPR–Cas9 screens for de novo mapping of genetic interactions. Nat Methods 2017;14:573–6. 10.1038/nmeth.4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kampmann M, Bassik MC, Weissman JS. Functional genomics platform for pooled screening and generation of mammalian genetic interaction maps. Nat Protoc 2014;9:1825–47. 10.1038/nprot.2014.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boettcher M, Tian R, Blau JA, et al. . Dual gene activation and knockout screen reveals directional dependencies in genetic networks. Nat Biotechnol 2018;36:170–8. 10.1038/nbt.4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dixit A, Parnas O, Li B, et al. . Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 2016;167:1853–66. 10.1016/j.cell.2016.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Adamson B, Norman TM, Jost M, et al. . A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 2016;167:1867–82. 10.1016/j.cell.2016.11.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Datlinger P, Rendeiro AF, Schmidl C, et al. . Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods 2017;14:297–301. 10.1038/nmeth.4177 [DOI] [PMC free article] [PubMed] [Google Scholar]