

SUMMARY
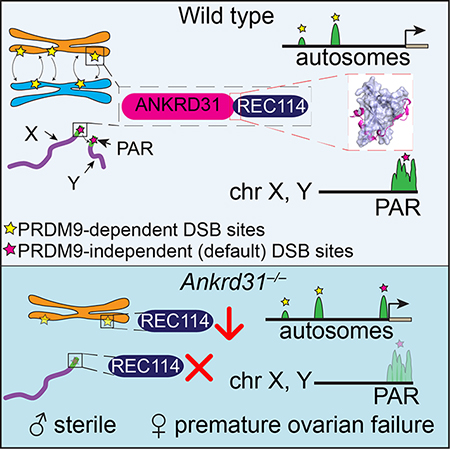
Double-strand breaks (DSBs) initiate the homologous recombination that is crucial for meiotic chromosome pairing and segregation. Here we unveil mouse ANKRD31 as a lynchpin governing multiple aspects of DSB formation. Spermatocytes lacking ANKRD31 have altered DSB locations and fail to target DSBs to sex chromosomes’ pseudoautosomal regions (PAR). They also have delayed/fewer recombination sites but, paradoxically, more DSBs, suggesting DSB dysregulation. Unrepaired DSBs and pairing failures—stochastic on autosomes, nearly absolute on X and Y—cause meiotic arrest and sterility in males. Ankrd31-deficient females have reduced oocyte reserves. A crystal structure defines a pleckstrin homology (PH) domain in REC114 and its direct intermolecular contacts with ANKRD31. In vivo, ANKRD31 stabilizes REC114 association with the PAR and elsewhere. Our findings inform a model that ANKRD31 is a scaffold anchoring REC114 and other factors to specific genomic locations, thereby regulating DSB formation.
eTOC blurb
Boekhout et al. discover ANKRD31 as a REC114 interactor and key player in meiotic recombination. ANKRD31 acts as molecular scaffold to regulate double-strand break formation and promote X–Y recombination. An atomic resolution structure illuminates conserved features of REC114–ANKRD31 interaction including an unexpected pleckstrin homology domain in REC114.
Graphical Abstract
INTRODUCTION
Meiotic recombination initiates with programmed DSBs made by SPO11 and accessory proteins, most of which are conserved (Lam and Keeney, 2014; Robert et al., 2016). Obligate mouse SPO11 partners include meiosis-specific REC114, MEI4, and IHO1 (orthologs of yeast Rec114, Mei4, and Mer2) (Kumar et al., 2010; Stanzione et al., 2016; Tesse et al., 2017; Kumar et al., 2018). We have a broad-strokes picture of the network of interactions connecting these proteins to one another and to chromosomes (Lam and Keeney, 2014), but mechanistic detail is lacking. Moreover, it is unclear if the catalog of relevant vertebrate proteins is complete.
DSB number and timing are controlled by intersecting negative feedback circuits. In one circuit, the DSB-activated kinase ATM inhibits further DSB formation locally on the same chromatid and on the sister chromatid (Lange et al., 2011; Garcia et al., 2015). This and other circuits collaborate to ensure that sufficient DSBs are made to support homologous pairing and recombination without a deleterious excess of DNA damage, but the molecular players supporting this regulation are not fully known (Cooper et al., 2014; Keeney et al., 2014).
DSB locations are also controlled, with PRDM9 a critical contributor in most mammals. PRDM9 binds DNA sequence-specifically and methylates nearby nucleosomes on histone H3 lysines 4 and 36, thereby targeting SPO11 (Grey et al., 2018). In the absence of PRDM9, SPO11 instead targets “default” sites, often where PRDM9-independent H3K4 trimethylation occurs such as promoters (Brick et al., 2012). Some sites are also targeted independently of PRDM9 in normal meiosis, perhaps most prominently the pseudoautosomal region (PAR), the small segment where the X and Y chromosomes share homology and must recombine to ensure sex chromosome segregation in male meiosis (Rouyer et al., 1986; Brick et al., 2012). The downstream factors that connect PRDM9-dependent hotspot designation to SPO11 activity remain to be identified (Grey et al., 2018). PRDM9-independent SPO11 targeting is likewise poorly understood.
Here we implicate ANKRD31 (ankyrin repeat domain-containing 31) as a critical component of the DSB-targeting and control machinery. ANKRD31 is essential for male meiosis because it regulates the timing, number, and location of DSBs globally and is crucial for DSB formation in the PAR. ANKRD31 stabilizes association of pro-DSB factors with chromosomes by a direct interaction with REC114 PH domain elucidated by crystallography.
RESULTS
ANKRD31 is a meiosis-specific REC114-interactor
We used mouse REC114 and MEI4 as baits in a yeast two-hybrid screen with a cDNA library from juvenile mouse testes. Among other hits, the screen yielded partial clones of Ankrd31 when REC114 was the bait (Figures 1A and 1B) (STAR Methods). We focused on ANKRD31 because of its rapid sequence divergence, which is often seen for meiotic proteins (Keeney, 2008) (Figures S1A and S1B).
Figure 1. ANKRD31 is a meiosis-specific REC114-interacting protein.

(A) Yeast two-hybrid interactions of REC114 with ANKRD31 and MEI4. Cells express the indicated Gal4 activating domain (AD) and binding domain (BD) fusions. EV, empty vector. “Selection” indicates aureobasidin to detect reporter activation.
(B) Domain structure of mouse ANKRD31 (above) and conservation plot for vertebrate homologs (below, (Wheeler et al., 2014)). Positions of frameshift mutation (asterisk), yeast two-hybrid (Y2H) clones, antigen for antibody production, and conserved domains (CD1–CD5) are indicated.
(C) IP and immunoblotting (IB) of ANKRD31 from whole-testis extracts.
(D) Ankrd31 exon map and ENCODE long RNA-sequencing reads from adult testis. Sequence context for the CRISPR-Cas9 guide RNA (gRNA in blue) and frameshift mutations (em1 and em2) is shown.
(E) Expression time course of ANKRD31 protein (top, IP/IB) and RNA (below, reverse-transcription quantitative PCR, normalized to B2M) in juvenile mouse testes.
(F) ANKRD31 localizes to chromatin. Spread spermatocytes were stained for SYCP3, ANKRD31 (guinea pig antiserum), and REC114. Arrowheads indicate examples of ANKRD31 blobs. Zoomed images show ANKRD31 colocalization with REC114 in small foci and blobs. Scale bars are 10 μm.
(G) ANKRD31 focus counts at different meiotic stages (n = 2 mice).
(H) Colocalization of ANKRD31 with REC114.
Red lines in panels E, G, and H are means.
See also Figure S1.
ANKRD31 has two ankyrin-repeat domains (ARDs) with three ankyrin repeats apiece (Figure 1B). Ankyrin repeats are a ~33-amino-acid alpha-helical motif often involved in protein-protein interactions (Li et al., 2006). We identified likely orthologs only in vertebrates (STAR Method). Alignments identified a conserved domain (CD) overlapping each ARD plus three CDs in the C-terminal third (Figure 1B). CD5 interacts directly with REC114 (detailed below); functions of the others are unknown.
Adult testis extracts probed by immunoprecipitation (IP) and immunoblotting with polyclonal antisera revealed a protein near the predicted size (206 kDa) that was not detected in Ankrd31 mutants described below (Figure 1C). Ankrd31 expression appears to be largely meiosis-specific as judged by protein and mRNA presence in adult testis but not somatic tissues (Figures 1D, S1C and S1D), and by their accumulation during the first meiotic wave in juvenile mice (Figure 1E).
ANKRD31 is chromatin-associated, based on immunostaining of spread spermatocyte chromosomes (Figure 1F). Early in prophase I, aligned sister chromatids begin to form an SYCP3-containing protein axis from which chromatin loops emanate (leptonema). Axes elongate and homologous chromosomes begin to align and form stretches of synaptonemal complex (SC), a tripartite structure comprising the homologous axial elements and the central region connecting them (zygonema). The SC elongates until it juxtaposes each homolog pair along their lengths (pachynema), then disassembles (diplonema).
At leptonema, ANKRD31 formed numerous foci mostly on chromosome axes (average of 210 per cell; Figure 1F). Focus numbers decreased progressively through zygonema, particularly in synapsed regions (Figures 1F, 1G and S1E). Little background staining was seen in the Ankrd31 mutant, indicating antibody specificity (Figures S1F and S1G). Several heavily-stained ANKRD31 blobs were also detected (arrowheads in Figure 1F). These correspond to the PARs of the X and Y chromosomes (readily identified by chromosome morphology in late zygonema; Figure 1F) and other regions that have PAR-like sequences containing arrays of the mo-2 minisatellite (Acquaviva et al., 2019). ANKRD31 colocalized with REC114, both in small foci and in mo-2-associated blobs (Figures 1F, 1H, and S1E), supporting that these proteins interact physically in vivo. ANKRD31 also colocalizes with MEI4, MEI1, and IHO1 (Acquaviva et al., 2019).
Ankrd31 mutation causes male sterility from spermatogenic arrest
We used CRISPR-Cas9 to generate endonuclease-mediated (em) mutations in exon 3. Two alleles had single-nucleotide insertions at the same position (Figure 1D). Indistinguishable phenotypes were observed where tested, so for simplicity the Ankrd31em1 allele (hereafter Ankrd31−) is shown unless indicated otherwise. Lack of IP/immunoblot signal from Ankrd31−/− testes (Figure 1C) suggests these are null or severe loss-of-function alleles. Distinct mutations by Tóth and colleagues gave similar phenotypes (Papanikos et al., 2018).
Heterozygotes had normal fertility and Mendelian transmission of the mutations (Figure S2A). Histopathology turned up no somatic defects in homozygous mutants (STAR Methods), suggesting ANKRD31 is dispensable in non-meiotic cells. However, Ankrd31−/− males were sterile: none of the 4 animals tested sired offspring when bred with wild-type females for 16 weeks, and testes were 37% of the size in littermate controls (Figures 2A and S2B).
Figure 2. Ankrd31 in male and female fertility.

(A) Reduced testis size. Sections from adult testes are at left (4 mos); quantification is at right (red lines, mean ± s.d.).
(B) Defective spermatogenesis. Bouin’s-fixed and periodic acid Schiff (PAS)-stained seminiferous tubule sections from adult testes are shown (5 mos). Se, Sertoli cells; Sg, spermatogonia; Sc, spermatocytes; rSt, round spermatids; eSt, elongated spermatids.
(C) Increased apoptosis. Images show adult (4 mos) testis sections stained with TUNEL and hematoxylin. Each point on the scatter plot is the measurement from one animal; red lines are means. Bar graph details Ankrd31−/− apoptotic tubules by spermatocyte stage (mean and s.d. for three experiments). MI, metaphase I; P, pachytene; ?, ambiguous.
(D) Reduced oocyte reserve and premature ovarian failure. Ovary sections at 4 and 32 dpp were immunostained for MVH to mark oocytes (brown stain); insets for 32 dpp ovaries highlight primordial follicles (arrows). Samples at 8 mos were Bouin’s fixed and PAS-stained. The graph shows oocyte counts summed across every third serial section. Red lines are means.
In panels A and C, the results of two-tailed t tests are indicated (ns, not significant (p > 0.05).
See also Figure S2.
Meiotic recombination defects cause sterility and hypogonadism because of spermatocyte apoptosis (de Rooij and de Boer, 2003). Testis sections from control animals had the full array of spermatogenic cells including spermatocytes and round and elongated spermatids, as expected (Figure 2B). In contrast, Ankrd31−/− tubules contained spermatogonia and primary spermatocytes but were largely if not completely devoid of postmeiotic cells (Figure 2B). TUNEL staining detected more frequent apoptosis (Figures 2A and 2C). In some apoptotic tubules (~23%), dying cells were pachytene spermatocytes (Figure 2C) as in mutants lacking or unable to repair DSBs (e.g., Spo11−/− or Dmc1−/−) (Barchi et al., 2005). However, pachytene arrest was only partially penetrant, as a higher percentage of apoptotic tubules instead contained metaphase I cells (~65%, Figure 2C).
Reduced oocyte reserve and premature ovarian failure in Ankrd31−/− females
Young adult Ankrd31−/− females were fertile (Figure S2A), but the partial pachytene arrest in males led us to consider that similar defects might occur in females. Oocytes initiate meiosis during fetal development and finish pachynema around birth, then arrest and form follicles. Meiotic prophase defects cause elevated oocyte loss (Hunter, 2017), so we examined ovaries to test if ANKRD31 deficiency causes more oocyte culling (Figures 2D and S2C).
As expected, wild type had abundant oocytes or follicles at 1 and 4 days post partum (dpp) and a mix of resting (primordial) and growing follicles at 32 dpp. In contrast, Ankrd31−/− females had greatly reduced oocyte numbers at all ages examined. Growing follicles were observed at 32 dpp, so ANKRD31 is dispensable for follicle development per se, but there were fewer primordial follicles (Figure 2D insets). Exhaustion of this smaller reserve caused reduced ovary size by ~8 mos (Figure 2D).
ANKRD31 deficiency causes stochastic synapsis failures
To understand ANKRD31 function, we focused on the male phenotype. The mixed arrest—partial in pachynema and more complete in metaphase I—suggested that the protein might contribute to multiple processes so we addressed each arrest in detail. Pachytene apoptosis can be triggered by persistent DSBs or by failure to silence the sex chromosomes, both of which can also be associated with defects in homologous synapsis (Royo et al., 2010; Pacheco et al., 2015).
We tracked synapsis by immunostaining for SYCP3 and the SC central region protein SYCP1 (de Vries et al., 2005). In Ankrd31−/−, leptonema and zygonema appeared normal and most pachytene cells had complete autosome synapsis, so ANKRD31 is dispensable for formation of axial elements and SC (Figures 3A, 3B and S3A). However, 14% of pachytenelike cells displayed a mix of fully synapsed autosomes plus asynaptic chromosomes and/or chromosome tangles, i.e., a mix of synapsed and unsynapsed axes with partner switches indicative of nonhomologous synapsis (Figures 3A, 3B and 3C). Such synapsis defects can cause apoptosis (Kauppi et al., 2013). Other abnormalities were also observed, including chromosome end associations (Figure S3B).
Figure 3. Dysregulated recombination in Ankrd31−/− spermatocytes.

(A) Synapsis defects in pachynema. Above, representative spreads; below, quantification of autosomal synapsis defects (“aberrant” cells with asynapsis and/or chromosome tangles) from two animals of each genotype (mean and range, n is number of cells counted).
(B) Spermatocyte stages based on SYCP3 and γH2AX staining (pre-leptonema, leptonema, early zygonema, late zygonema, pachynema, diplonema, diakinesis; aberrant pachytene-like cells with autosome synapsis defects were tallied separately). Bars are mean and range of two animals.
(C) Expansion microscopy showing SC partner switches in a pachytene-like Ankrd31−/− spermatocyte.
(D) ANKRD31 is required for normal REC114 localization. Micrographs are matched exposures. Arrowheads indicate REC114 blobs, absent in Ankrd31−/−. Each point in the left graph is the focus count for one cell (red lines, mean ± s.d.). The box plot at right summarizes data from Figure S3C. Boxes indicate median, 25th and 75th percentiles; whiskers indicate 10th and 90th percentiles; outliers are not shown. Blobs in wild type are summarized separately (tan boxes). Intensities of smaller foci were lower in Ankrd31−/− (open boxes) than wild type (black boxes).
(E–H) Altered numbers and timing of recombination foci. Each point in F–H is the count from one cell (totals are below graphs) from 3 animals of each genotype, except RPA in Ankrd31+/− (2 mice). Red lines are means ± s.d.
(I) γH2AX time course. Arrowheads are sex bodies.
(J) Reduced γH2AX intensity in Ankrd31−/−. Each point is the immunofluorescent signal from a single spread nucleus. For each of two independent experiments with a mutant and wild-type pair, values were normalized to the mean from leptonema in wild type. Red lines are medians, cell totals are below.
In panels A, B, and F–H, “aberrant” refers to pachytene-like cells with unsynapsed autosomal regions. In panels A, D, F, G, H, and J the results of two-tailed Mann Whitney tests are shown: ns, not significant (p > 0.05), *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001.
Scale bars, 10 μm except panel C.
See also Figures S3 and S4.
ANKRD31 deficiency causes dysregulated recombination
Because we isolated ANKRD31 as a REC114 interactor and because Ankrd31−/− synaptic defects resemble those in mice with fewer DSBs (Kauppi et al., 2013), we hypothesized that ANKRD31 contributes to DSB formation. We therefore examined behaviors of pre- and post-DSB recombination proteins.
Ankrd31−/− spermatocytes had fewer and less intense axial foci of REC114 genome-wide throughout prophase I, and REC114 no longer formed blobs on the PAR or other regions (Figures 3D, S3C and S3D). This functional dependency strengthens the conclusion that ANKRD31 and REC114 interact in vivo. MEI1 and MEI4 localization were similarly affected, but not IHO1 (Acquaviva et al., 2019).
To assess recombination, we immunostained chromosomes for RAD51 and DMC1 (Figures 3E–G, S4A and S4B). In normal meiosis, foci of these proteins appear in leptonema, accumulate to maximal levels in early zygonema, then decline as DSB repair proceeds, persisting longer on the non-homologous parts of X and Y.
In Ankrd31−/−, fewer foci were present at leptonema (one fourth or fewer of the number in wild type). Foci were maximal in early zygonema with a lower than normal average number but a wide range, such that many cells were similar to wild type (more than 100 foci per cell) while others continued to have substantially fewer foci. As in wild type, focus numbers declined as chromosomes synapsed, but foci accumulated to high levels on unsynapsed axes (Figure 3E and “aberrant” class in Figures 3F and 3G). Importantly, however, pachytene cells with apparently normal synapsis also had elevated RAD51 and DMC1 foci (Figures 3F and 3G), so the increase is not simply because of synaptic errors. Earlier stage cells also had fewer foci of the ssDNA-binding protein RPA (Figures 3H and S4C). Seeing fewer RPA foci in early cells suggests that the reduction in RAD51 and DMC1 foci is caused by a delay and/or reduced efficiency of forming cytologically observable sites containing resected DSBs, not simply because of defective loading of the strand exchange proteins. Elevated RPA foci were also seen in pachytene cells with normal autosome synapsis, and they accumulated in cells with unsynapsed autosomes (Figure 3H). Ankrd31+/− heterozygotes had normal DMC1 and RPA focus numbers (Figures 3F and 3H), but a weak intermediate phenotype for RAD51 both at early stages (modest reduction at leptonema and zygonema) and later (modest elevation at pachynema) (Figure 3G), possibly indicating a mild haploinsufficient effect.
Reducing DSB numbers to half the normal level causes pairing and synaptic failure (Kauppi et al., 2013), so the reduced numbers of RAD51- and DMC1-associated recombination sites during the critical pairing periods of leptonema and zygonema may explain the synapsis problems. Accumulation of numerous foci on unsynapsed axes is a hallmark of DSBs to continuing to form past the time they normally would have ceased after homolog engagement (Kauppi et al., 2013).
We also examined phosphorylation of histone H2AX (γH2AX), which is formed rapidly by ATM in response to DSBs then disappears progressively as recombination proceeds until it is largely gone from autosomes in pachynema (Figure 3I) (Mahadevaiah et al., 2001; Barchi et al., 2005). γH2AX also forms on the silenced X and Y chromosomes in the sex body (Mahadevaiah et al., 2001). Ankrd31−/− spermatocytes had pan-nuclear γH2AX staining in leptonema and zygonema, but less than wild type (Figures 3I and 3J). Most γH2AX signal dissipated as autosomes synapsed, leaving brightly stained sex bodies (Figure 3I). Asynaptic regions retained γH2AX (Figure 4A), as expected (Mahadevaiah et al., 2001). We infer that ANKRD31 is dispensable for ATM activation by DSBs, but there appears to be less total ATM activity at early stages in keeping with the reduced DMC1, RAD51, and RPA foci.
Figure 4. Persistent γH2AX, more DSBs, and metaphase arrest from achiasmate sex chromosomes.

(A) Persistent γH2AX. Ankrd31−/− cells with γH2AX flares and/or larger patches are shown at left. Graph quantifies γH2AX patterns from 3 mice of each genotype (cell totals indicated above). Bars are means and s.d.
(B) SPO11-oligo complexes. Upper panel, representative autoradiograph of SPO11-oligo complexes IP’d from adult testis extracts, radiolabeled with terminal transferase and [α32P] dCTP, and separated by SDS-PAGE. Asterisk, nonspecific labeling. Radioactive signals were background-corrected and normalized to the littermate control. Graph summarizes multiple experiments from adults (normalized to heterozygous controls) and juveniles (normalized to wild type). Red lines are medians. P values are from two-sided one-sample t tests. See also legend to Figure S4D.
(C) Ankrd31−/− seminiferous tubule showing metaphase I spermatocytes with unaligned chromosomes (arrowheads).
(D) Defective sex chromosome pairing and synapsis. Left, representative pachytene cells labeled by immuno-FISH for SYCP3 and a PAR-specific probe. Right, quantification of X–Y pairing at pachynema, based on immunofluorescence for SYCP3 and γH2AX, for 3 animals of each genotype (cell totals indicated above). Bars are means and s.d.
(E) MLH1 foci. Left, pachytene cells. Graphs show MLH1 focus counts and breakdown of MLH1 foci per chromosome from 3 animals of each genotype (p value from Mann-Whitney test).
Scale bars in B and C, 10 μm.
See also Figure S4.
Ankrd31−/− pachytene cells with apparently normal autosome synapsis also frequently displayed discrete flares of persistent γH2AX (Figure 4A). This result and elevated DMC1, RAD51, and RPA foci (Figures 3F–H), suggests that many cells contain incompletely repaired DSBs. Persistent DSBs at pachynema can trigger apoptosis (Pacheco et al., 2015), so this may account for some of the pachytene death in Ankrd31−/−.
Finally, we evaluated DSB formation by labeling SPO11-oligonucleotide complexes (Lange et al., 2011). Surprisingly, we observed an increase in Ankrd31−/− adults (median 1.4-fold, Figure 4B). Juveniles at 12 dpp had a variable pattern, with some mice showing elevation as in adults and others showing levels similar to control littermates (Figures 4B and S4D). At this age, testes are enriched for leptonema and zygonema, with few or no pachytene cells expected (Bellve et al., 1977). Importantly, none of the animals showed the reduction in SPO11-oligo complexes that would be predicted if DSB frequencies were reduced in line with the diminished recombination protein foci in early stage spermatocytes (Figure S4D legend).
To summarize, recombination foci, γH2AX, and SPO11-oligo complexes demonstrate that DSBs are made, so Ankrd31−/− does not simply phenocopy Rec114−/− (Kumar et al., 2018). DSB-associated recombination foci and γH2AX are delayed and reduced, but SPO11-oligo complexes are not reduced and appear instead to be elevated, suggesting that more total DNA cleavage events occur. Ways to reconcile these apparently contradictory findings are addressed in Discussion. The results strongly argue that DSB formation and recombination are substantially dysregulated in the absence of ANKRD31. Furthermore, partially penetrant pachytene apoptosis is likely accounted for by a combination of persistent DSBs and synaptic defects.
Sex chromosome recombination relies on ANKRD31
Metaphase I arrest is typical of mutants that can complete DSB repair but that fail to generate crossovers on some or all chromosomes (Odorisio et al., 1998). Indeed, Ankrd31−/− metaphase I cells frequently had unaligned chromosomes (Figure 4C). Moreover, the sex chromosomes were unpaired in 92% of otherwise normal-looking pachytene cells (Figures 4D and S3A). By comparison, autosomes showed only a modest defect: pachytene cells with complete autosome synapsis had a small increase in the number of MLH1 foci (means of 23.7 in wild type and 24.8 in Ankrd31−/−; Figure 4E). A slight increase in the number of chromosomes lacking an MLH1 focus was seen, but this was not statistically significant (p = 0.105, Fisher’s exact test).
These results indicate that those cells that progress past pachynema not only successfully pair and synapse autosomes but also efficiently generate autosomal crossovers and chiasmata, thus ensuring proper biorientation at metaphase I (with perhaps infrequent exceptions). In contrast, the X and Y almost always fail to pair, synapse, and recombine; we infer that this leads to achiasmate sex chromosomes at metaphase I, which are known to trigger apoptosis (Odorisio et al., 1998). Thus, while ANKRD31 contributes to recombination and pairing on autosomes, it is essential on sex chromosomes in males.
ANKRD31 shapes the DSB landscape
The data thus far indicated that Ankrd31−/− male sterility was at least partially attributable to dysregulated DSB formation. Is this strictly an effect on DSB number and timing, or are DSB locations also affected? To answer this question, we mapped DSBs genome wide by sequencing DMC1-bound ssDNA (ssDNA sequencing, or SSDS (Brick et al., 2018)) from testes of adults and 12-dpp juveniles. At the younger age, DSB signal is principally from leptonema and zygonema (Bellve et al., 1977) and thus unlikely to be affected by loss of pachytene cells or hyperaccumulation of DSBs in unsynapsed regions.
Ankrd31−/− mutants displayed little or none of the high-level SSDS signal normally found in the PAR or in PAR-proximal hotspots (Figure 5A). This profound defect (confirmed by cytology of PAR-associated RPA foci (Acquaviva et al., 2019)) is probably sufficient to account for the highly penetrant failures in sex chromosome pairing, synapsis, and recombination.
Figure 5. Altered DSB landscape in Ankrd31−/− males.

(A) Ankrd31−/− mutants are unable to target the PAR and PAR-adjacent hotspots for high-level DSB formation. SSDS maps from adults or juveniles were smoothed with a 1 kb sliding window in 0.1 kb steps. Only part of the PAR (green) is present in the mm10 assembly.
(B) An autosomal segment illustrating increased use of default hotspots. Shading highlights a few of the PRDM9-directed (gray) and default (yellow) hotspots; weaker hotspots are not highlighted. The bottom two tracks show H3K4me3 coverage for the same region.
(C) Metaplots of SSDS averages around PRDM9 hotspots (black, defined in wild-type B6 mice) and default hotspots (yellow, defined in Prdm9−/− mice).
(D) Overlap of hotspot calls in juveniles of the indicated genotype with either PRDM9 (gray) or default (yellow) hotspots.
(E) Metaplots of H3K4me3 around PRDM9 hotspot centers.
B6 wild-type and Prdm9−/− SSDS data are from Brick et al. (2012).
See also Figure S5.
PAR DSBs are largely independent of PRDM9 (Brick et al., 2012) (Figure 5A) and have other requirements distinct from autosomes (Kauppi et al., 2011; Smagulova et al., 2013), so a PAR defect does not predict that SPO11 targeting to PRDM9 sites would be affected.
Surprisingly, however, Ankrd31-deficient animals also showed a highly unusual pattern globally: they used many of the PRDM9-targeted hotspots found in B6 mice but also used default (PRDM9-independent) hotspots (Figures 5B–D). Heterozygotes appeared largely normal but had slightly increased SSDS signal when averaged over default hotspots, suggesting subtle haploinsufficiency (Figure 5C). Although many default hotspots used in Ankrd31−/− were of modest strength, there were also many strong ones (Figures 5B and S5A), such that 28% of SSDS fragments were in default hotspots. Furthermore, even the shared hotspots often differed in strength in Ankrd31−/− (Figure S5B).
Mixed use of PRDM9-directed and default hotspots indicates that ANKRD31 promotes but is not essential for PRDM9 targeting of SPO11 activity. We considered that ANKRD31 might function upstream of PRDM9, i.e., that mutants partially lose PRDM9 function. However, Prdm9 mRNA levels were normal (Figure S5C) and H3K4me3 appeared unaffected at PRDM9 and default sites (Figures 5B and 5E). These results imply that PRDM9 expression, binding to chromatin, and histone methylation are effectively normal in the absence of ANKRD31. By process of elimination, we suggest that ANKRD31 functions downstream of PRDM9 (see Discussion).
These findings reveal that ANKRD31 substantially shapes the landscape of recombination initiation. In doing so, it has two distinct roles. First, it is essential for preferential DSB formation in PAR, and is thus crucial for male sex chromosome recombination. Second, it promotes but is not essential for SPO11 cleavage at PRDM9-targeted locations. Thus, paradoxically, absence of ANKRD31 eliminates use of PRDM9-independent sites in the PAR but also increases the use of PRDM9-independent (default) sites globally. Scenarios to reconcile this paradox are described below (Discussion).
Structure of an ANKRD31–REC114 complex
The N-terminal two-thirds of REC114 (aa 1–147) and the C-terminal tail of ANKRD31 (aa 1810–1857) were necessary and sufficient for yeast two-hybrid interaction (Figures 6A and 6B). We obtained crystals for recombinant REC114 aa 1–158 (REC114N) complexed with ANKRD31 aa 1808–1857 (ANKRD31C) and solved the 2.8-Å structure by single-wavelength anomalous diffraction (Figures 6C and 6D and Table 1).
Figure 6. Structure of REC114N in complex with ANKRD31C.

(A, B) Identification of minimal interacting domains in ANKRD31 and REC114 by yeast two-hybrid assay.
(C) Domain organization (to scale) of REC114 and C-terminal portion of ANKRD31. Red arrow connects interacting segments.
(D) Front and back views of the overall structure of a REC114N–ANKRD31C heterodimer.
(E and F) Structure-based sequence alignment of REC114N (E) and ANKRD31C (F) orthologs. Secondary structure elements are according to mouse proteins. Dashed lines indicate invisible residues in the structure. Residues involved in intramolecular (red triangles) and intermolecular (blue triangles) interactions are indicated. Boxed residues highlight conservation. The six N-terminal SSMs are mapped above the REC114N sequence. Species: Mus musculus, Homo sapiens, Danio rerio, Arabidopsis thaliana, Saccharomyces cerevisiae, Egretta garzetta, Chelonia mydas, Rhincodon typus, Pogona vitticeps.
(G) Surface conservation of REC114 PH domain. Conservation for 138 aligned sequences from UNIREF-90 was mapped on the surface of mouse REC114 PH domain using ConSurf (Ashkenazy et al., 2016).
See also Figures S6.
Table 1.
Data collection and structure refinement statistics.
| Data collection | |
| Beamline | APS-24ID-C |
| Wavelength (Å) | 0.9791 |
| Space group | P43212 |
| Cell parameters | |
| a, b, c (Å) | 71.81, 71.81, 154.98 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 50.0–2.80 (2.90–2.80)* |
| Rmerge (%) | 8.2 (77.5) |
| Average I/σ(I) | 23.3 (1.9) |
| Completeness (%) | 99.9 (99.6) |
| CC (1/2) | 0.981 |
| Average redundancy | 7.5 (6.3) |
| No. of reflections | 10,273 |
| Refinement | |
| Rwork/Rfree (%) | 22.2/27.4 |
| No. of non-H atoms | 2612 |
| Average B factor (Å2) | |
| REC114N | 84.8 |
| ANKRD31C | 90.5 |
| R.m.s. deviations | |
| Bond length (Å) | 0.011 |
| Bond angles (°) | 1.157 |
| Ramachandran plot (%) | |
| Favored | 95.1 |
| Allowed | 4.9 |
| Outliers | 0 |
Values in parentheses are for the highest resolution shell.
The crystal asymmetric unit contained a pair of REC114N–ANKRD31C heterodimers (Figure S6A) that superpose well (0.47 Å r.m.s.d.; Figure S6B). Contacts across heterodimers (Figure S6A) are also present in higher order crystal lattice symmetry units (Figure S6C), so they are likely due to crystal packing. This conclusion is reinforced by molecular weights of monomeric REC114N and the REC114N–ANKRD31C heterodimer in solution (Figure S6D).
REC114N adopts a β-sandwich fold, consisting of two nearly orthogonal antiparallel β-sheets, that is closed at one end by a C-terminal amphipathic α-helix (α1) (Figure 6D). The N-terminal β-sheet is formed by β1 and β4–β6 and the C-terminal sheet comprises β2, β3, and β7–β9. These secondary structure elements form a hydrophobic core that stabilizes the β-sandwich. The loop connecting β2 and β3 is invisible in the density map, suggesting flexibility.
A structural homology search revealed that REC114N is similar to the N-terminal domain of coactivator-associated methyltransferase 1 (CARM1) (Troffer-Charlier et al., 2007), which belongs to the PH domain superfamily. The PH superfamily shares a similar fold without significant sequence similarity. Structure-based alignment suggested that REC114 orthologs contain similar PH domains (Figure 6E). Indeed, the secondary structure elements of REC114N correspond closely to previously identified short signature sequence motifs (SSMs) shared among REC114 orthologs (Maleki et al., 2007; Kumar et al., 2010; Tesse et al., 2017) (Figures 6E and S6E). Additional conserved regions emerged from the structure-based alignment (Figure 6E). Hydrophobic residues in β4 and β5 along with other residues forming the hydrophobic core are highly conserved and S56 at the end of β3 forms hydrogen bonds with L22, K23, S57 and L63, which contributes to the stability at the protein surface (Figure S6F). Other conserved hydrophilic residues include R27, E83 and R114, which are involved in both intramolecular hydrogen-bond interactions and intermolecular contacts with ANKRD31C described below. By contrast, the variable loops connecting the β-strands are poorly conserved (Figure 6E).
ANKRD31C wraps extensively around REC114N, burying ~1,800 Å2 of surface area (Figure 6G). Almost all ANKRD31C residues, including the ultimate C terminus, are involved in the interaction with REC114N (Figures 6F and 7A) and can be well traced in the structure. To outline the intermolecular contacts, and for simplicity, we subdivided ANKRD31C intermolecular contacts into three segments (S1, S2 and S3; Figures 6F and 7B–7I). Segment S1 (aa 1812–1824) forms a turn that strategically positions side chains for mainly hydrophobic contacts with the N-terminal β-sheet of REC114N (Figure 7B), with this interaction disrupted by the Y1818A/L1819A double mutation (Figure 7H). The importance of the participating hydrophobic residues on the REC114N N-terminal β-sheet was also confirmed by the loss of binding for the dual mutation F74A/L81A (Figures 7G and 7I). The turn of S1 is stabilized by two intramolecular main-chain hydrogen bonds formed by P1816-L1819 and H1817-Q1820 segments (Figure 7C) and the β-strand at the C terminus of S1 is hydrogen bonded with β6 of REC114N (Figure 7D).
Figure 7. Intermolecular contacts between REC114N and ANKRD31C,

(A) Front (above) and side (below) views of the REC114N-ANKRD31C heterodimer.
(B-F) Detailed views of interfaces between REC114N and ANKRD31C S1 (B, C, D), S2 (E), S3 (F) segments in the heterodimeric complex. Hydrogen bonds and salt bridges are shown as black dashed lines. See legend to Figure S6 for additional discussion.
(G) Yeast two-hybrid assay confirming interaction defects of the indicated mutant REC114 proteins.
(H and I) Mutational analysis of ANKRD31C (H) and REC114N (I) residues involved in binding, assayed by pull-down assay with GST-tagged ANKRD31C. SDS-PAGE gels were stained with Coomassie Blue.
See also Figure S6.
Segment S2 (aa 1825–1832) interacts with the open end of the β-sandwich (Figure 7E). E1831 is the key residue involved in bridging contacts with REC114N, with the E1831 side chain anchored by two salt bridges and one hydrogen bond. The intramolecular hydrogen bond was formed with the main-chain amino group of S1828, and the salt bridges are formed with the side chain of highly conserved R27 of REC114N (Figures 6E and 7E). The R27 side chain is further stabilized by a hydrogen-bond and salt bridge network formed by the nearly invariant Y25, L82, E83 and R114 (Figures 6E and 7E), which also contributes to the hydrophobic core of the N-terminal and C-terminal β-sheets. Furthermore, the Q1830 main-chain carboxyl group of ANKRD31C hydrogen bonds with L30 from REC114N, stabilizing the interaction of E1831 with R27. Mutating ANKRD31C E1831 (E1831A) or REC114N R27 (R27A) dramatically reduced binding (Figures 7G–7I).
Segment S3 (aa 1833–1857) primarily comprises two amphipathic α-helixes that pack against a relatively hydrophobic groove on the C-terminal β-sheet of REC114N (Figure 7F). The importance of these hydrophobic interactions was confirmed by the disruptive effect of the W1842A mutant of ANKRD31C and the F28A/L104A dual mutant of REC114N (Figures 7G–7I). L1833 forms main-chain hydrogen bonds with S113 of REC114N at the site where ANKRD31C passes through the C-terminal β-sheet between β2 and β9. The side chain of Y1845 hydrogen bonds with strictly conserved W51 of REC114N, which stabilizes the conformation of the first αhelix in the S3 segment. The C-terminal W1857 residue inserts into a hydrophobic pocket of REC114N formed by the aliphatic side chains of R24, R117, Q119 and V53 (Figure 7F), which are strictly conserved in vertebrate REC114 orthologs (Figure 6E). This pocket is hydrophilic at the surface but hydrophobic in the interior. The aromatic ring of W1857 also forms a cation-π interaction by packing against the ammonium group of R117 and the aromatic side chain of invariant W51 from REC114N. The interaction is further stabilized by the hydrogen bond between S1856 and R117 (Figure 7F).
Interaction-critical residues of ANKRD31C were highly conserved (Figure 6F), but interestingly, we also observed conservation of REC114N residues that interact with ANKRD31C (Figures 6E and 6G). Invariant W51 contributes to complex formation by hydrogen bonding with Y1845 and cation-π packing with W1857 of ANKRD31C (Figure 7F). Other hydrophobic residues on the surface of REC114N involved in the contacts with ANKRD31C, including F28, F74, L81, and L104, are also conserved, even in plants and yeast (Figure 6E).
DISCUSSION
Our search for new meiotic recombination components uncovered mouse ANKRD31 as a direct interaction partner of REC114, with the principles underlying molecular recognition elucidated from a crystal structure of the REC114N–ANKRD31C complex. ANKRD31 is required for normal assembly of REC114 complexes on chromosomes and for normal location, timing and number of DSBs genome wide and especially in the PAR. Absence of ANKRD31 causes catastrophic failure of spermatogenesis and greatly diminished oogenesis. Our results agree well with independent findings of Tóth, de Massy and colleagues (Papanikos et al., 2018).
Paradoxical mutant phenotypes shed light on ANKRD31 functions
Ankrd31−/− mutants present two paradoxes: 1) They had fewer/delayed sites marked by recombination proteins but also more DSBs as measured by SPO11-oligo complexes. 2) They had increased use of some PRDM9-independent sites (default sites genome-wide) but also failed to use others in the PAR. Reconciling these seemingly contradictory findings provides insights into the mechanism and regulation of DSB formation.
A simple way to reconcile the first paradox is if individual recombination foci often contain clusters of DSBs in Ankrd31−/−. Such clusters occur occasionally in wild-type yeast and frequently in mutants lacking the ATM ortholog Tel1 (Garcia et al., 2015). Because of the reduced number and intensity of REC114 foci, we favor that Ankrd31−/− mutants make fewer, less efficient DSB-forming machines and thus have delayed and/or fewer recombination sites, but we further propose that these machines are less tightly controlled and often generate multiple DSBs. In this model, ANKRD31 is a lynchpin of DSB regulation with both DSB-promoting and -suppressing activities. A nonexclusive alternative for reduced recombination foci is defective DSB resection, but this would not explain increased SPO11-oligo complexes, diminished REC114 assemblies, or reduced γH2AX levels.
Ankrd31−/− mutants partially phenocopy Atm−/− for increased SPO11-oligo complexes (Lange et al., 2011; Pacheco et al., 2015), so ANKRD31 may contribute to ATM-dependent inhibition of DSBs. ANKRD31 may be a direct ATM target because it has 24 SQ/TQ sites, the ATM phosphorylation motif. Or ANKRD31 may act upstream of ATM by promoting kinase activation by DSBs. The reduced γH2AX could reflect such a role in ATM activation, or could simply reflect reduced numbers of sites where ATM is being activated.
For the second paradox, we propose that ANKRD31 has distinct roles in targeting SPO11 activity to different parts of the genome, but that these roles are related through recruitment of REC114. ANKRD31 colocalizes with REC114 in large assemblies in the PAR and in smaller foci genome wide. The large REC114 assemblies are eliminated in the absence of ANKRD31, whereas the smaller foci are still present but become fainter and less numerous. A separate study shows that the PAR undergoes dramatic structural reorganization during meiosis—the chromosome axes elongate and sister axes split apart—and that these behaviors require ANKRD31-dependent accumulation of REC114 and other pro-DSB factors (Acquaviva et al., 2019). It is thus possible that ANKRD31 recognizes a cis-acting feature of the PAR itself and recruits REC114 and other proteins by direct interaction. We hypothesize that the ability to recruit REC114 has allowed ANKRD31 to acquire a highly specialized function in the PAR. PRDM9 is mostly irrelevant for this targeting, at least in mice (Brick et al., 2012; Hinch et al., 2014).
Elsewhere in the genome, where PRDM9 is the major arbiter of hotspot positions, ANKRD31 is important but not essential for SPO11 targeting. One possibility is that ANKRD31 directly links SPO11 to PRDM9-established marks, a process that remains poorly understood (Grey et al., 2018). Regardless of the underlying mechanism, however, our results definitively establish that the two modes of PRDM9-independent targeting (default hotspots vs. the PAR) are mechanistically distinct because they respond in opposite ways to ANKRD31 deficiency.
ANKRD31 in male and female fertility
Metaphase I arrest in Ankrd31−/− males is likely attributable to failure of sex chromosome recombination tied to PAR dysfunction, but achiasmate autosomes may also contribute. The pachytene arrest appears to reflect a mixture of problems. Specifically, we propose that reduced DSB formation during a critical window early in prophase I (leptonema through early zygonema) leads to stochastic defects in pairing and synapsis. Altered DSB locations and formation of clustered DSBs may also contribute. Cells with synaptic defects die in pachytene from a recombination checkpoint, sex body failure, or both (Royo et al., 2010; Pacheco et al., 2015). We further surmise that some pachytene cells die from persistent DSBs (evidenced by γH2AX flares and recombination protein foci) even though they achieve full synapsis. A repair defect could be indirectly caused by dysregulated DSB formation, e.g., cutting of both sister chromatids, or ANKRD31 could have a direct repair-promoting function.
Oocytes do not have the same arrest behavior as spermatocytes in the face of synaptic defects (Hunter, 2017), and the two X chromosomes do not rely on the PAR for pairing and chiasma formation. However, oocytes with persistent DSBs are eliminated (Hunter, 2017). A plausible hypothesis, therefore, is that similar molecular defects contribute to oocyte loss in Ankrd31−/− females as cause pachytene cell death in males. If so, then the sexually dimorphic Ankrd31−/− phenotype tracks with known sex differences both for quality control surveillance and for the unique constraints on male sex chromosome recombination.
ANKRD31 as a modular scaffold to regulate DSB formation
The unexpected PH domain (also shown by Kumar et al., 2018) explains previously recognized sequence conservation in REC114 orthologs (Maleki et al., 2007; Kumar et al., 2010; Tesse et al., 2017) by demonstrating that most SSMs are key secondary structure elements. Many well conserved residues contribute to PH domain folding, but others are surface exposed (Figure 6G). Some of the latter mediate interactions with ANKRD31C but others likely mediate interactions with other proteins and/or the REC114 C-terminal region.
The ANKRD31C peptide wraps around REC114N and makes extensive contacts, yielding a stable heterodimer. The ANKRD31C sequence and its REC114-interacting residues are conserved, implying conservation of the interaction. More intriguing, many residues in REC114N that interact with ANKRD31C are also highly conserved, even in species such as A. thaliana and S. cerevisiae that do not have an obvious ANKRD31 ortholog. We speculate that ANKRD31 is more widely conserved than currently apparent, or that ANKRD31 employs a REC114 surface that is evolutionarily constrained because it binds a different partner(s) in other species (perhaps also in competition with ANKRD31 in vertebrates).
Additional conserved domains in ANKRD31 include the two ARDs and the two conserved domains (CD3 and CD4) between ARD-2 and ANKRD31C. ARDs are often involved in protein-protein interactions, including direct interactions with nucleosomes (Saredi et al., 2016), so it is likely that the large ANKRD31 protein acts as a modular scaffold that anchors REC114 to other proteins and to meiotic chromosomes.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by, the Lead Contact Scott Keeney at Memorial Sloan Kettering Cancer Center (skeeney@ski.mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were maintained and euthanized under U.S.A. federal regulatory standards and experiments were approved by the Memorial Sloan Kettering Cancer Center (MSKCC) Institutional Animal Care and Use Committee. Animals were fed regular rodent chow with ad libitum access to food and water.
Endonuclease mediated alleles (em1, em2) were generated by the MSKCC Mouse Genetics core facility by targeting exon 3. A guide RNA cassette with sequence (5′-GTTGTTGCTGGCTCTTAGTG) was cloned into pU6T7 vector (Romanienko et al., 2016). In vitro-transcribed guide RNA (100 ng/μl) and Cas9 (50 ng/μl) were microinjected using conventional techniques (Romanienko et al., 2016) into pronuclei of CBA/J × C57BL/6J F2 hybrid zygotes generated by crossing CBAB6F1/J hybrid females with C57BL/6J males. Genomic DNA from founder animals was amplified with primers ANKRD31-A and ANKRD31-B and digested with T7 endonuclease I to identify potential indel-carrying animals. All primer sequences are listed in Supplemental Table S1.
To determine the spectrum of mutant alleles in T7-positive Ankrd31em founder mice, the targeted region was amplified by PCR of tail-tip DNA (ANKRD31-A and ANKRD31-B primers) and sequenced on the Illumina MiSeq platform (Illumina Inc, San Diego, California) at the MSKCC Integrated Genomics Operation. Reads were aligned to mouse genome assembly GRCm37/mm9 and variants were identified using Genome Analysis Toolkit version 2.8–1-g932cd3a (McKenna et al., 2010; DePristo et al., 2011; Van der Auwera et al., 2013). Variants with a minimum variant frequency of 0.01 were annotated using VarScan v2.3.7 software (Koboldt et al., 2012).
Founder mice were crossed to C57BL/J6 mice purchased from Jackson laboratories to obtain germline transmission, then heterozygous animals were backcrossed to C57BL/J6 a further ≥3 generations. Experimental animals were generated by crossing Ankrd31+/− heterozygous males with either Ankrd31+/− heterozygous or Ankrd31−/− homozygous females. Genotyping was performed by PCR on genomic DNA with primers 16G008 and 16G011, followed with digestion by AflII (NEB, R0520L) which recognizes em1 (+A) allele or with HrpCh4IV (NEB, R0619L), which recognizes em2 (+C) allele. The PCR product is 339 bp and restriction digestion products are 264 and 75 bp. The systematic names of the alleles generated in this study are Ankrd31em1Sky and Ankrd31em2Sky.
METHOD DETAILS
Yeast two-hybrid screen
Mouse Mei4 and Rec114 cDNA were amplified by PCR and cloned into pGBKT7 vector (Clontech). Briefly, PCR products were amplified from testis cDNA library and digested by EcoRI (NEB) and NdeI (NEB) and then cloned into linearized pGBKT7. pGBKT7-Mei4 or pGBKT7-Rec114 plasmids were transformed into Y2HGold yeast two-hybrid strain following manufacturer’s instructions (Clontech, 630439). Single colonies were picked to verify protein expression by immunoblotting and to ensure that neither pGBKT7-Mei4 nor pGBKT7-Rec114 auto-activated Gal4-driven reporters promoters or were toxic to yeast.
The yeast two-hybrid library was prepared from cDNA generated from whole-testis extracts of 12-dpp males using the Make Your Own “Mate & Plate” Library System according to the manufacturer’s instructions (Clontech, 630490). We estimated that the library contained ~106 independent clones.
The yeast-two hybrid screen was performed using Matchmaker Gold Yeast Two-Hybrid System according to the manufacturer’s instructions (Clontech, 630489). Briefly, individual clones of Y2H bait strains containing pGBKT7-Mei4 or pGBKT7-Rec114 were inoculated in liquid synthetic dextrose medium lacking tryptophan (SD-Trp) and grown to OD600 of ~0.8, then cells were pelleted and resuspended in SD-Trp at 108 cells/ml. Bait strains were mixed with aliquots of the library strain and incubated in 2× YPDA medium with 50 μg/ml kanamycin for 20–24 hours at 30°C, shaking at 40 rpm. Cultures were checked for the presence of zygotes by light microscopy, and then cells were plated on 50 plates (150 mm × 15 mm) of SD lacking tryptophan and leucine and containing X-α-galactose and aureobasidin A (SD-Trp/Leu/X-α-gal/AbA), and incubated for 5 days at 30°C. Approxi mately 106 zygotes were screened for each bait. Those that formed a blue colony were re-streaked on fresh SD-Trp/Leu/X-α-gal/AbA plates. If also positive in this second growth test, they were subjected to PCR amplification as instructed in Matchmaker Insert Check PCR Mix 2 (Clontech, 630497). The plasmids were then recovered from yeast and transformed into E. coli and sequenced, using Easy Yeast Plasmid Isolation Kit (Clontech, 630467).
The screen with pGBKT7-Mei4 as bait yielded clones containing sequences from Rec114 and Med10 (one clone each). Recovery of REC114 when MEI4 was the bait is consistent with known interactions in mouse (Kumar et al., 2010) and yeasts (Arora et al., 2004; Miyoshi et al., 2012). The screen with pGBKT7-Rec114 yielded Ankrd31, Sohlh1, Coro1b, Phka2, Rps20, Sin3b, Ctnna1, and Hsp90ab (one clone each except Ankrd31 (two independent clones)).
Presumptive full-length Ankrd31 cDNA (5.9 kb) cloned from testis matched NCBI reference RNA XM_006517797.1, comprising 26 exons and encoding a predicted protein of 206 kDa (Figure 1B).
Targeted yeast two-hybrid assays
Genes were amplified from cDNA generated from testes of 12-dpp C57Bl/6J mice as described above. Genes of interest were amplified and cloned into either pGADT7 or pGBKT7. Mating of bait- and prey-containing strains (Y187 and Y2HGold yeast strains) and selection on SD-Trp/Leu/X-α-gal/AbA plates were performed following manufacturer’s instructions as described above (Clontech). Primers used for cloning are listed in Supplemental Table S1.
Analysis of sequence conservation
Amino acid sequence divergence (fraction of residues changed) and Ka and Ks values were downloaded from HomoloGene Release 68 (April 2014), https://www.ncbi.nlm.nih.gov/homologene. Estimated times to last common ancestor were obtained from http://www.timetree.org/ (Hedges et al., 2015) (accessed July 26, 2015).
For each HomoloGene entry with mammalian orthologs (n = 19,498) we generated a least-squares linear regression line fitting amino acid sequence divergence as a function of time since last common ancestor, forcing an intercept of zero. The slope of this line (multiplied by 100) is the divergence rate (percent amino acid changes per Myr) plotted in Figure S1B. We also calculated the Ka/Ks ratio for each pairwise comparison between mammalian species within a HomoloGene entry, and took the median value among these as the representative value for that HomoloGene entry. This is the median Ka/Ks ratio plotted in Figure S1B.
We identified likely ANKRD31 orthologs across the vertebrate subphylum including cartilaginous fish (e.g., ghost shark Callorhinchus milii), but not in more distant taxa. Likely orthologs were identified by BLAST searches using full-length human ANKRD31 as the query, or using just the C-terminal 70 amino acids. From a collection of over 150 homologs including representatives from mammals, reptiles, and birds, we generated a multiple sequence alignment of the C-terminal 70 amino acids using MUSCLE (Edgar, 2004) in MegAlign Pro (Lasergene v 14.1.0), then used this alignment as input for a HHMER3 search (http://hmmer.org/). This search again identified ANKRD31 homologs in vertebrates, including one in Gasterosteus aculeatus (three-spined stickleback; UniProt G3N8J6), but failed to find any significant hits in fungi. When the stickleback protein was used as a BLAST query, we identified additional homologs from fish that had not turned up in the earlier searches. Interestingly, the fish homologs all have only one ARD, which by multiple sequence alignment appeared to be a better match for ARD-2 in the mammalian proteins.
Antibody generation
All antibodies are listed in Supplemental Table S2. REC114 antibodies used here were described previously (Stanzione et al., 2016) and their generation is detailed further elsewhere (Acquaviva et al., 2019). To raise polyclonal antibodies against ANKRD31, a fragment of Ankrd31 coding sequence (corresponding to a.a. 1–324) was cloned into pETDuet™−1 (Millipore) using the In-Fusion cloning kit (Clontech, Takara). This expression vector was used to produce 6×His-ANKRD311–134. Briefly, E. coli strain BL21 was transformed with the vector and a single colony was picked to inoculate LB overnight at 37°C. The overnight culture was diluted to ~0.04 (OD600) and once OD600 had reached ~0.6, the protein expression was induced by addition of 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) for 2 hrs at 37°C. Cells were pelleted by centrifugation at 6000 g, 15 min. Pellets were resuspended in lysis buffer (50 mM HEPES-NaOH pH 8.0, 300 mM NaCl, 0.1 mM dithiothreitol (DTT), 10% glycerol and 20 mM imidazole) and stored at −80°C. To purify the ANKRD 31 fragment, the pellet was thawed at room temperature and placed on ice and all further steps were executed at 4°C. The sample was sonicated 8 times 15 seconds on-off, 20% duty cycle. The solution was cleared by centrifugation at 19,000 rpm, 1 hr. Equilibrated Ni-NTA Agarose (QIAGEN, 30230) was added to the cleared protein lysate and incubated for 1 hr, then the lysate-resin slurry was transferred into Poly-prep chromatography columns (BioRad, 731–1550) and washed with lysis buffer several times. The protein was eluted with elution buffer (lysis buffer + 500 mM imidazole) and fractions containing the ANKRD31 fragment were combined and subjected to size-exclusion chromatography. Sample was loaded on a Superdex 200 column equilibrated with 50 mM HEPES-NaOH pH 8.0, 300 mM NaCl, 1 mM DTT, 10% glycerol and 5 mM EDTA and the fractions under the main peak were collected and analyzed on SDS-PAGE. Purified protein fractions were combined and concentrated (Millipore), then frozen in liquid nitrogen and stored at –80°C. Purified protein fragment was used to imm unize two rabbits and two guinea pigs by Pocono Rabbit Farm & Laboratory. Polyclonal anti-ANKRD31 antibodies were enriched from serum using NAb™ Protein A Plus Spin Columns (Thermoscientific, 89952).
Polyclonal antibody specificity was tested by immunofluorescent staining of meiotic chromosome spreads and immunoprecipitation followed by immunoblotting of adult testis extracts from wild type and Ankrd31-deficient animals (Figures 1C, S1D, and S1F). Furthermore, our immunostaining results agree well with those reported for independent antibody preparations (Papanikos et al., 2018).
Total mRNA extraction, cDNA library generation and RT-qPCR
Testis tissue from Ankrd31+/+ and Ankrd31−/− animals was dissected and frozen on dry ice. Total mRNA was extracted using RNeasy Plus Mini Kit (QIAGEN, 74134) following the manufacturer’s instructions. Superscript™ III First-Strand Synthesis SuperMix (Invitrogen, 18080400) was used with oligo dT primers to generate testis cDNA, which was diluted 1:10 to be used in RT-qPCR carried out using LightCycler 480 SYBR Green I Master (Roche, 4707516001). Amplification products were detected on the LightCycler 480 II Real-Time PCR instrument (Roche). LightCycler 480 Software was used to quantify products by absolute quantification analysis using the second derivative maximum method. All reactions were done in triplicate and the mean of crossing point (Cp) value were used for the analysis. Cp values were normalized to the value obtained for B2M reactions (ΔCp). Then, the differences between knockout and wild-type samples were calculated for each primer set (ΔΔCp) and the fold change (knockout vs wild type) was calculated as 2−ΔΔCp. Primers used for RT-qPCR are listed in Supplemental Table S1.
Immunoprecipitation and immunoblot analysis of ANKRD31
Dissected testes were placed in an Eppendorf tube and snap frozen on liquid nitrogen and stored at −80°C. The frozen tissue was resuspen ded in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP40) supplemented with protease inhibitors (Roche Mini tablets). The tissue was disrupted with a plastic pestle. The homogenized extract was supplemented with 10 mM MgCl2 and Benzonase nuclease (EMD Millipore (70664–3), 28 unit/μl) and incubated with end-over-end rotation for 1 hr at 4°C. The samples were centrifuged at 15,000 rpm for 20 min. The clear lysate was transferred to a new tube and used for the immunoprecipitation. Whole-cell extract was pre-cleared with protein A/G Dynabeads (Thermofisher, 1004D, 1001D) by end-over-end rotation for 1 hr at 4°C. Antibodies (home-made, rabbit anti-ANKRD31 or guinea pig anti-ANKRD31, 1–2 μg) were added to precleared lysates and incubated overnight with end-over-end rotation at 4°C. Protein A/G Dynabeads were added to the tubes and incubated for 1 hr with end-over-end rotation, at 4°C. Beads were washed three times with RIPA buffer, resuspended in 1× NuPAGE LDS sample buffer (Invitrogen) with 50 mM DTT, and incubated 10 min at 70°C to elute immunoprecipitated proteins.
For immunoblotting, samples were separated on 3–8% Tris-Acetate NuPAGE precast gels (Life Technologies) at 150 V for 70 min. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes by wet transfer method in Tris-Glycine-20% methanol, at 120 V for 40 min at 4°C. Membranes were blocked with 5% n on-fat milk in 1× phosphate buffered saline (PBS)-0.1% Tween (PBS-T) for 30 min at room temperature on an orbital shaker. Blocked membranes were incubated with primary antibodies (guinea pig anti-ANKRD31, 1:4000) 1 hr at room temperature or overnight at 4°C. Membranes were washed with PBS-T for 30 min at room temperature, then incubated with HRP-conjugated secondary antibodies (rabbit anti-guinea pig IgG (Abcam), 1:10,000) for 1 hr at room temperature. Membranes were washed with PBS-T for 15 min and the signal was developed by ECL Prime (GE Healthcare).
Spermatocyte chromosome spreads
Testes were dissected and deposited after removal of the tunica albuginea in 50 ml Falcon tubes containing 2 ml TIM buffer (104 mM NaCl, 45 mM KCl, 1.2 mM MgSO4, 0.6 mM KH2PO4, 6.0 mM sodium lactate, 1.0 mM sodium pyruvate, 0.1% glucose). 200 μl collagenase (20 mg/ml in TIM buffer) was added, and left shaking at 550 rpm for 55 min at 32°C. Every 15 min, tissue was checked and carefully pulled further apart to enhance digestion. After incubation, TIM buffer was added to a final volume of 15 ml, followed by centrifugation for 1 min at 600 rpm at room temperature. Supernatant was decanted and this wash and centrifuge procedure was repeated 3 times. Separated tubules were resuspended in 2 ml TIM, with 200 μl trypsin (7 mg/ml in TIM) and 20 μl DNase I (400 μg/ml in TIM buffer) and incubated for 15 min. at 32°C at 550 rpm in a thermomixer. 500 μl trypsin inhibitor (20 mg/ml in TIM) and 50 μl DNase I solution were added and mixed. A wide mouthed plastic Pasteur pipette was used to disperse the tissue further by pipetting up and down for 2 min. The Pasteur pipette was first used to pipet a 2% BSA solution in PBS to coat the plastic and minimize cell loss. Cells were passed through a 70-μm cell strainer into a new 50 ml Falcon tube. TIM was added to a final volume of 15 ml and mixed. Cells were centrifuged for 5 min at 1200 rpm. Supernatant was decanted, 15 μl DNase I solution was added and gently mixed, followed by 15 ml TIM. Washing with TIM and resuspension in presence of DNase I was repeated 3 times. Single-cell suspension was pelleted and resuspended in TIM according to original weight (~200 mg in 400 μl). 10 μl of cell suspension was added to 90 μl of 75 mM sucrose solution, flicked three times and incubated for 8 min at room temperature. Superfrost glass slides were divided in two squares by use of Immedge pen, each square received 100 μl 1% paraformaldehyde (PFA) (freshly dissolved in presence of NaOH at 65°C, 0.15% Triton, pH 9.3, cleared through 0.22 μm filter) and 45 μl of cell suspension was added per square, swirled three times, and dried in a closed slide box for 3 hr, followed by drying with half-open lid 1.5 hr at room temperature. Slides were washed in a Coplin jar 2 × 3 min in milli-Q water on a shaker, 1 × 5 min with 0.4% PhotoFlow, air dried and stored in aluminum foil in −80°C.
Prophase I substages of nuclei on meiotic spreads were defined based on behavior of chromosome axes stained with SYCP3 antibodies. Leptonema was defined as short SYCP3 stretches without evidence of synapsis as assessed by thickening of the SYCP3 signal. Early zygonema was defined as longer stretches of SYCP3 and some synapsis, while late zygonema was defined as having substantial (>70%) synapsis. Pachynema was defined by synapsis of all autosomes. For mutant cells, X and Y chromosomes were mostly unsynapsed at pachynema, while in wild type this was rare. Pachytene-like cells in the mutant consisted of two subclasses of cells: i) Those in which most autosomes were completely synapsed from end to end, but a few chromosomes (usually between one and four) showed complete asynapsis. ii) Those with mixes of homologous and non-homologous synapsis apparent as ‘tangles’ involving more than two chromosomal axes. In cells with asynapsis but no tangles, X and Y could often be identified by axial length, but in cells with tangles the X and Y were often not readily identifiable.
Immunostaining
Slides of spread spermatocytes were blocked for 30 min at room temperature in 100 ml solution containing 1× PBS with 0.05% Tween-20 and 3 mg/ml bovine serum albumin (BSA). Slides were incubated with primary antibody overnight in a humid chamber at 4°C. Slides were washed 3 × 10 min in PBS–0.05% Tween, then incubated with secondary antibody 45 min at 37°C in a humid chamber. Slides were washed 3 × 5 min in the dark on a shaker with PBS–0.05% Tween, before air drying and mounting with Vectashield containing DAPI. All primary and secondary antibodies used here are listed in Supplemental Table S2.
PAR FISH
All following steps were performed in the dark to prevent loss of fluorescent signal. After regular staining for immunofluorescence, slides were re-fixed in 2% PFA in PBS for 10 min at room temperature. Slides were rinsed once in PBS, and washed for 4 min in PBS at room temperature. Slides were washed with 70% ethanol for 4 min, followed by 4 min wash with 90% ethanol, and final wash with 100% ethanol for 5 min. Slides were air dried vertically for 5 min. Slides were incubated with 15 μl of PAR probe BAC RP24–500I4 and coverslips were sealed with rubber cement (Weldwood contact cement). Slides were denatured for 7 min at 80°C, followed by overnight incubation (>14 hr) in a humid chamber at 37°C. Coverslips were removed, and slides were rinsed in 0.1× SSC buffer, washed in 0.4× SSC, 0.3% NP-40 for 5 min, followed by a wash in PBS–0.05% Tween-20. Slides were washed once in H2O, dried and mounted in Hardset Vectashield (without DAPI).
Expansion microscopy
Expansion microscopy was performed by the MSKCC Molecular Cytology Core Facility based on published methods (Tillberg et al., 2016). Slides of spread spermatocyte chromosomes were prepared and stained as described above. Slides were treated with Acryloyl X-SE (AcX) (0.05 mg/ml diluted in PBS) overnight at room temperature. Samples were washed twice for 15 min with PBS, followed by treatment with monomer solution (8.6% (w/v) sodium acrylate, 2.5% (w/v) acrylamide, 0.1% (w/v) N,N′-methylenebisacrylamide, 11.7% (w/v) NaCl in PBS) for 30 min at 4°C. Gelation solution was prepared from chilled monomer solution on ice by mixing in the following order 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (4-hydroxy-TEMPO) inhibitor solution (final concentration 0.01% (w/v)), 10% tetramethylethylenediamine TEMED (final concentration 0.2% (w/v)). 10% APS solution (final concentration 0.2% (w/v)) was added to initiate the gelling process. The solution was briefly vortexed, and the sample was placed in imaging spacer, used as a well on a glass slide coated with SigmaCoat. The well was sealed with a cover glass and incubated for 2 hr at 37°C in a humidified chamber. Gelled samples were transferred to a 6-well glass bottom plate and treated with digestion buffer (50 mM Tris-HCl pH 8.0, 23 mM EDTA, 0.5% Triton X-100, 0.8 M guanidine HCl) containing freshly added proteinase K (final concentration 200 μg/ml). The sample was digested for 3 hr at 60°C, or until the gel detached from the slide. The container was sealed to prevent drying out during digestion. Digestion buffer was removed and replaced with PBS, sample was stained with DAPI for 30 min at room temperature. DAPI solution was removed and sample was washed in excess of dH2O, three times, 30 min each to expand the sample. Water was removed, and the sample was embedded in 2% agarose in dH2O. Samples were imaged after agarose was fully solidified.
Histology
Fixation, tissue processing, and staining were performed as described (Jain et al., 2018). Testes from adult mice were fixed overnight in 4% PFA at 4°C, or in Bouin’s fixative for 4 to 5 hr at room temperature. Bouin’s fixed testes were washed in water for 1 hr at room temperature, followed by five 1-hr washes in 70% ethanol at 4°C. Wild-type and mutant ovaries were fixed in 4% PFA overnight at 4°C. PFA-fixed tissues were was hed 4 × 5 minutes in water at room temperature. Fixed tissues were stored in 70% ethanol for up to 5 days prior to embedding in paraffin and sectioning (5 μm for testes, 8 μm for ovaries). The tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems) and antigen retrieval was performed with CC1 buffer (Ventana Medical Systems). Sections were blocked for 30 min with Background Buster solution (Innovex), followed by avidin-biotin blocking for 8 min (Ventana Medical Systems). Periodic acid Schiff (PAS) staining and immunohistochemical TUNEL assay were performed by the MSKCC Molecular Cytology Core Facility using the Autostainer XL (Leica Microsystems, Wetzlar, Germany) automated stainer for PAS with hematoxylin counterstain, and using the Discovery XT processor (Ventana Medical Systems, Oro Valley, Arizona) for TUNEL. Ovaries were fixed with 4% PFA overnight and serially sectioned at 8 μm. Sections were incubated with anti-VASA for 5 hr, followed by 60 min incubation with biotinylated goat anti- rabbit (Vector Labs, cat# PK6101) at 1:200 dilution. The detection was performed with DAB detection kit (Ventana Medical Systems) according to manufacturer’s instruction. Slides were counterstained with hematoxylin and coverslips were mounted with Permount (Fisher Scientific).
Histological examination of somatic tissues
Gross histopathological analysis of major organs and tissues was performed by the MSKCC Laboratory of Comparative Pathology for the following male mice: two Ankrd31 mutants and two heterozygous littermates aged 28 weeks. The following females were analyzed: one Ankrd31 mutant and one wild-type littermate aged 24 weeks; one Ankrd31 mutant and one heterozygous littermate aged 15 weeks. Histological examination of the following tissues was performed: diaphragm, skeletal muscle, sciatic nerve, heart/aorta, thymus, lung, kidneys, salivary gland, mesenteric lymph nodes, stomach, duodenum, pancreas, jejunum, ileum, cecum, colon, adrenals, liver, gallbladder, spleen, uterus, ovaries, cervix, urinary bladder, skin of dorsum and subjacent brown fat, skin of ventrum and adjacent mammary gland, thyroid, parathyroid, esophagus, trachea, stifle, sternum, coronal sections of head/brain, vertebrae and spinal cord. Tissues were fixed in 10% neutral buffered formalin and bones were decalcified in formic acid solution using the Surgipath Decalcifier I (Leica Biosystems, Wetzlar, Germany) for 48 hr. Samples were routinely processed in alcohol and xylene, embedded in paraffin, sectioned (5 μm), and stained with hematoxylin and eosin. Mutant males examined had marked degeneration of seminiferous tubules with aspermia. In heterozygous littermates testes appeared normal, but some minimal germ cell exfoliation in seminiferous tubules was observed, and minimal intraluminal cell debris in epididymides, which were considered likely to be naturally occurring for this age and genetic background. All follicle types were absent in the ovaries of the 24 week old homozygous knockout; the mutant female of 15 weeks appeared to have a reduced number of oocytes and follicles (not quantified). Some obesity associated conditions were observed, such as moderate hepatic lipidosis, but these were not attributable to Ankrd31 genotype. All other findings in mutants were considered incidental and/or age-related.
Image acquisition
Images of spread spermatocytes were acquired on a Zeiss Axio Observer Z1 Marianas Workstation, equipped with an ORCA-Flash 4.0 camera, illuminated by an X-Cite 120 PC-Q lightsource, with either 63× 1.4 NA oil immersion objective or 100× 1.4 NA oil immersion objective. Marianas Slidebook (Intelligent Imaging Innovations, Denver Colorado) software was used for acquisition.
Imaging of the expanded sample was performed on a Zeiss LSM 880 confocal microscope with a 63× 1.4 NA oil immersion objective. Z-stacks were acquired with 488 and 561 laser lines used for excitation. Zeiss Airyscan detector has been used for imaging to increase the resolution of the 4× expanded sample further. All imaging data was acquired at optimal pixel sizes and optimal axial intervals. Data were processed using the Zen software (Carl Zeiss, Jena, Germany).
Whole slides (histology), either PAS or TUNEL stained, were scanned and digitized with the Panoramic Flash Slide Scanner (3DHistech, Budapest, Hungary) with a 20× 0.8 NA objective (Carl Zeiss, Jena, Germany). High resolution images of PAS and IHC images were acquired with a Zeiss Axio Imager microscope using a 63× 1.4 NA oil immersion objective (Carl Zeiss, Jena, Germany).
Quantification of SPO11-oligo complexes
We purified SPO11 oligos from testes of adults (>8 weeks of age) or 12-dpp juvenile mice as previously described (Lange et al., 2011; Pan et al., 2011). Testis were decapsulated, flash frozen in liquid nitrogen and stored at −80°C. Testes were homogenized with prechilled plastic pestles in lysis buffer (1% Triton X-100, 25 mM HEPES-NaOH pH 7.4, 5 mM EDTA) containing EDTA-free protease inhibitors, cleared by centrifugation for 25 min at 4°C, 100,000 rpm, followed by transfer of supernatant to low binding Eppendorf tubes. Supernatants were incubated with mouse anti-SPO11–180 antibody (3 μg per immunoprecipitation (IP) sample) before protein A-agarose beads (1 hr 4°C end-over-end rotation) were added for an additional 3 hr at 4°C. SPO11 IP samples were eluted in Laemmli sample buffer for 4 min at 95°C. Beads from the SPO11 IP were washed 3 times with IP buffer (1% Triton X-100, 150 mM NaCl, 15 mM Tris-HCl pH 8.0), and washing steps were consolidated into eluates. Combined IP eluate and washes were diluted with 5 volumes of IP buffer before performing a second round of IP (SPO11 IP2). Anti-Mouse SPO11–180 antibody was added (3 μg per sample) for 1 hr at 4°C with end-over-end rotation, before addition of protein A-agarose beads and further mixing overnight. SPO11 IP2 samples were washed 3 times with IP wash buffer and 2 times with 1× NEB4 before radiolabeling with terminal deoxynucleotidyl transferase and [α−32P] CTP in NEB buffer 4 for 1 hr at 37°C in a thermomixer at 300 rpm. Beads were washed 5 times with IP buffer before elution with 2× Laemmli sample buffer. Proteins were separated by SDS-PAGE on 8% Invitrogen BOLT gels, 125 V for 90 min. Proteins were transferred to PVDF membrane via semidry transfer in 210 mM Tris, 103 mM glycine, 0.04% SDS, pH 9.5) for 17 min at 17 V. Radiolabelled species were detected and quantified after 48 hr exposure with Fuji phosphor screens and quantified in ImageJ.
SSDS and H3K4me3 ChIP
DMC1-SSDS was performed as described (Brick et al., 2018) except that lysis was performed in RIPA buffer (10 mM Tris-HCl pH 8, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1 % SDS, 140 mM NaCl) instead of lysis buffer containing 1% SDS. Therefore, the dialysis step was omitted. 24 μg of DMC1 antibody (C-20, sc-8973, discontinued) was used to pull down the nucleoprotein filaments.
H3K4me3 ChIP was performed as described (Brick et al., 2012) except that MNase treatment was substituted for sonication. Testes were dissected and placed in cold PBS, then were moved to 10 cm tissue culture dish (Gibco), tunica albuginea was removed and testes were placed on a rocker in a 15 ml Falcon tube containing 10 ml 1% freshly made paraformaldehyde (PFA) in MilliQ H2O for 10 min at room temperature. One ml of 1.25 M glycine was added for 5 min after which samples were put on ice. Fixed tissue was homogenized in a 15 ml Dounce homogenizer by 10 strokes with the tight pestle. Samples were filtered through a 70-μm cell strainer into a 15 ml Falcon tube, centrifuged for 5 min at 4°C at 900 g. The supernatant was removed, the pellet resuspended in 10 ml PBS and centrifuged at 900 g for 5 min at 4°C. Pellets were resuspended in 10 ml hypotonic lysis buffer (HLB) (10 mM Tris-HCl pH 8.0, 1 mM KCl, 1.5 mM MgCl2 and 250 μl 40 Protease inhibitor (Roche 11836170001)). Samples were left on rocker for 30 min at 4°C, followed by homogenization with 15 ml Dounce homogenizer. Samples were centrifuged for 10 min at 10,000 g at 4°C. The pellet containing nuclei was resuspended in 1 ml MNase buffer (50 mM Tris-HCl pH 8.0, 1 mM CaCl2, 4 mM MgCl2, 4% NP-40) and 27 μl 40x protease inhibitor. Samples were transferred to low protein binding Eppendorf tubes (#022431081). MNase (150 units per sample) was added (USB affymetrix, J70196-ZCR) and incubated for 5 min at 37°C. 21 μl 0.5 M EDTA was added (final concentration 10 mM) and incubated for 5 min at 4°C. Samples were spun down at 15,000 rpm in table top centrifuge at 4°C for 10 min. Supernatant was transferred to new Eppendorf tubes and centrifuged again. Supernatant was split into two identical volumes in new tubes and diluted 1:3 with RIPA buffer (10 mM Tris-HCl pH 8, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1 % SDS, 140 mM NaCl). 10 μg of H3K4me3 antibody (Abcam, ab8580) was added per sample (5 μg per tube) and incubated with end-over-end rotation overnight at 4°C. 100 μl of Protein G Dynabeads per tube were aliquoted and washed 3× with RIPA buffer using magnetic holder. Beads were pelleted, resuspended in 50 μl of digested chromatin sample and transferred to tube with the rest of the chromatin sample, followed by 2 hrs incubation at 4°C with end-over-end rotation. Beads were collected using a magnetic holder, the supernatant removed, and beads were subject to washes with 1.2 ml of each of the following buffers in order: low salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 150 mM NaCl), high salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 500 mM NaCl), LiCl immune complex wash buffer (0.25 M LiCl, 1% IGEPAL-CA630, 0.5% sodium deoxycholate, 1mM EDTA, 10 mM Tris-HCl pH 8.0), and 2 washes of TE buffer (10 mM Tris-HCl, 1 mM EDTA pH 7.4). Beads were pelleted and supernatant removed. 150 μl of elution buffer (1% SDS, 100 mM NaHCO3 in MilliQ water) was added and beads were flicked, before incubation at 65°C for 30 min, shaking in a thermomixer at 500 rpm. Beads were pelleted, supernatant was transferred to new tube, in which previously split samples were now combined. 12 μl of 5 M NaCl was added to each sample and incubated overnight (18–20 hr) at 65°C. Each sample received 6 μl of 0.5 M EDTA, 12 μl of 1 M Tris-HCl pH 6.5 and 5 μl of proteinase K (20 mg/ml) followed by 2 hr incubation at 45°C. Samples were cleaned up using the Qiagen MinElute column following manufacturer instructions, with the alteration that samples were diluted with 7 volumes of PB buffer and passed over columns at a volume of 700 μl, each time with 1 min binding and 1 min centrifugation until all sample had been applied. Columns were washed with 750 μl PE buffer, and samples were finally eluted using 12 μl EB buffer. Immunoprecipitated DNA was diluted to 50 μl using Ultrapure H2O and was quantified by PicoGreen and the size was evaluated on a High Sensitivity BioAnalyzer chip. When possible, fragments between 100 and 600 bp were size selected using a MPure XP beads (Beckman Coulter catalog # A63882) and Illumina libraries were prepared using the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) according to the manufacturer’s instructions with 8–10 cycles of PCR. Barcoded libraries were sequenced on a Hiseq 2500 in rapid mode in a 50 bp x 50 bp paired-end run, using the HiSeq Rapid SBS Kit v2 (Illumina). An average of 59 million paired reads was generated per sample.
Protein expression and purification for crystallography
The mouse ANKRD31 (residues 1808–1857, ANKRD31C) and REC114 (residues 1–158, REC114N) were cloned into a modified RSFduet-1 vector (Novagen) with an N-terminal His6SUMO tag on ANKRD31C and no tag on REC114N. ANKRD31C and REC114N were coexpressed in E. coli strain BL21(DE3) RIL (Stratagene). The cells were grown at 37°C until OD600 reached 0.8, then the media was cooled to 16°C and IPTG was added to a final concentration of 0.35 mM to induce protein expression overnight at 16°C. The cells were harvested by centrifuge at 4°C and disrupted by son ication in Binding buffer (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 20 mM imidazole) supplemented with 1 mM PMSF (phenylmethylsulfonyl fluoride) and 3 mM β-mercaptoethanol. After centrifugation, the supernatant containing complexes of ANKRD31C and REC114N was loaded onto 5 ml HisTrap Fastflow column (GE Healthcare). After extensive washing with Binding buffer, the complex was eluted with Binding buffer supplemented with 500 mM imidazole. The His6-SUMO tag was removed by Ulp1 protease digestion during dialysis against Binding buffer and separated by reloading onto HisTrap column. The flow-through fraction was further purified by HiTrap Q FF column and Superdex 75 16/60 column (GE Healthcare). The pooled fractions were concentrated to 5 mg/ml in crystallization buffer (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 1 mM DTT). For the selenomethionine (SeMet) derivative protein, the cells were grown in M9 medium supplemented with lysine, threonine, phenylalanine, leucine, isoleucine, valine, and Se-methionine. GST-tagged ANKRD31C proteins also have C-terminal His6 tag and were first purified by HisTrap Fastflow column. Mutants were generated by the QuikChange Mutagenesis Kit (Stratagene). The SeMet derivative protein, GST-tagged protein and all the mutants were purified as described above.
Crystallization, data collection and structure determination
Crystals of SeMet derivative REC114N-ANKRD31C complex were grown from a solution containing 0.1 M Bis-Tris pH 5.5, 25% PEG 3350 using the hanging drop vapor diffusion method at 20°C. For data collection, the crystals were fla sh frozen (100 K) and collected on NE-CAT beam line 24ID-C at the Advanced Photo Source (APS), Argonne National Laboratory. The diffraction data were processed with HKL2000 (Otwinowski et al., 2003). The structure of REC114N-ANKRD31C complex was solved by the single-wavelength anomalous diffraction method using PHENIX (Adams et al., 2002). The automatic model building was carried out using the program PHENIX AutoBuild (Adams et al., 2002). The resulting model was completed manually using COOT (Emsley et al., 2010) and PHENIX refinement (Adams et al., 2002). The statistics of the diffraction and refinement data are summarized in Table 1. All molecular graphics were generated with the PyMOL program (https://pymol.org/2/). The sequence alignments were generated with Multalin (Corpet, 1988) and ESPript (Robert and Gouet, 2014).
GST pull-down assay
50 ng GST-tagged ANKRD31C and mutants were incubated with 50 μL Glutathione Sepharose 4B beads (GE heathcare) at 4°C for 4 h. T he beads were washed two times with 1 ml crystallization buffer and then 100 ng of pre-purified REC114N and mutants were added and incubated for 2 h at 4°C. An additional three washe s were applied and each sample was analyzed with SDS-PAGE.
SEC-MALS
SEC-MALS experiments were performed by using an Äkta-MALS system. Proteins (500 μl) at a concentration of 2.5 mg/ml were loaded on Superdex 75 10/300 GL column (GE Healthcare) and eluted with crystallization buffer at a flow rate of 0.3 ml/min. The light scattering was monitored by a miniDAWN TREOS system (Wyatt Technologies) and concentration was measured by an Optilab T-rEX differential refractometer (Wyatt Technologies). Molecular masses of proteins were analyzed using the Astra program (Wyatt Technologies).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Statistical analyses were performed using R version 3.4.4 and RStudio version 1.1.442 (R Core Team, 2018), and Graphpad Prism 7. Statistical parameters and tests are reported in the figures and corresponding figure legends. Sample size, statistical test used, means and error bars are defined on figures and/or in figure legends. No statistical tests was used to predetermine sample size. One of the experiments quantifying SPO11-oligo complexes in 12 dpp animals was performed blinded as to genotype of the animals. No data from experiments were excluded from analysis. In cases where outliers were removed for plotting purposes, none of the data points were removed from the statistical analysis.
Image analysis
Single cells were manually cropped and analyzed with semi-automated scripts in Fiji (Schindelin et al., 2012). Scripts are available on Github: https://github.com/Boekhout/ImageJScripts. Briefly, for detection of number of foci, images were auto-thresholded on SYCP3 staining, which was used as a mask to use ‘Find Maxima’ to determine number of RPA, DMC1 or RAD51 foci. Images were manually inspected to determine that there were no obvious defects in determining SYCP3 axis, axis from neighboring cells were counted, artifacts were present or clear foci were missed by the script. For detection of ANKRD31 and REC114 foci, overlap and intensity with SYCP3 was detected with a thresholded algorithm regardless of meiotic stage, and a difference of Gaussian (DoG) blurs was used to isolate REC114 or ANKRD31 foci. All images were manually inspected upon analysis.
For γH2AX intensity analysis, leptotene and early zygotene cells were identified by SYCP3 staining, regions of interest were manually drawn and used to measure both the integrated density of a cell of interest, and for a region containing no cells for background subtraction. Integrated density was normalized to mean integrated intensity of wild type leptotene cells per experiment to combine different experiments.
Supplementary Material
Highlights.
REC114 directly interacts with ANKRD31, a novel factor required for normal fertility
ANKRD31 influences the distribution of double-strand breaks genome wide
ANKRD31 is essential for the recombination between X and Y chromosomes
A crystal structure reveals a PH domain in REC114 and its contacts with ANKRD31
ACKNOWLEDGMENTS
We thank Attila Tóth and Bernard de Massy for sharing unpublished information; Keeney lab members C. Claeys Bouuaert, N. Mohibullah, M. van Overbeek, and S. Kim for experimental advice; and MSKCC core facilities, supported by NIH cancer center core grant P30 CA008748: Mouse Genetics (P. Romanienko and W. Mark), Molecular Cytology (K. Manova), Integrated Genome Operation (A. Viale and N. Mohibullah), Bioinformatics (N. Socci), and the Laboratory of Comparative Pathology (S. Monette). This work utilized computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). X-ray diffraction studies were conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, supported by NIGMS grant P41 GM103403 and U.S. Department of Energy grant DE-AC0206CH11357. The Pilatus 6M detector on 24-ID-C beamline is funded by a NIHORIP HEI grant (S10 RR029205). MB was supported in part by a Rubicon fellowship from the Netherlands Organization for Scientific Research. LA was supported in part by a Lalor Foundation fellowship. This work was supported in part by NIGMS grant R35 GM118092 (SK), the Howard Hughes Medical Institute (SK), and the Maloris Foundation (DJP).
Footnotes
DECLARATION OF INTEREST
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
The atomic coordinates have been deposited in the Protein Data Bank (PDB) (accession number 6NXF). SSDS and H3K4me3 ChIP-seq sequencing data have been deposited in the Gene Expression Omnibus (GEO) repository under accession number GSE118913. Raw image data for figure panels have been deposited on Mendeley (http://dx.doi.org/10.17632/r8znwwkvzw.1). Image analysis scripts are available on Github: https://github.com/Boekhout/ImageJScripts
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Acquaviva L, Boekhout M, Karasu ME, Brick K, Pratto F, van Overbeek M, Kauppi L, Camerini-Otero RD, Jasin M, and Keeney S (2019). Ensuring meiotic DNA break formation in the mouse pseudoautosomal region. bioRxiv DOI: 10.1101/536136. [DOI] [PMC free article] [PubMed]
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, and Terwilliger TC (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Arora C, Kee K, Maleki S, and Keeney S (2004). Antiviral protein Ski8 is a direct partner of Spo11 in meiotic DNA break formation, independent of its cytoplasmic role in RNA metabolism. Mol Cell 13, 549–559. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, and Ben-Tal N (2016). ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44, W344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballow D, Meistrich ML, Matzuk M, and Rajkovic A (2006). Sohlh1 is essential for spermatogonial differentiation. Dev Biol 294, 161–167. [DOI] [PubMed] [Google Scholar]
- Barchi M, Mahadevaiah S, Di Giacomo M, Baudat F, de Rooij DG, Burgoyne PS, Jasin M, and Keeney S (2005). Surveillance of different recombination defects in mouse spermatocytes yields distinct responses despite elimination at an identical developmental stage. Mol Cell Biol 25, 7203–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellve AR, Cavicchia JC, Millette CF, O’Brien DA, Bhatnagar YM, and Dym M (1977). Spermatogenic cells of the prepuberal mouse. Isolation and morphological characterization. J Cell Biol 74, 68–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, and Petukhova GV (2012). Genetic recombination is directed away from functional genomic elements in mice. Nature 485, 642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick K, Pratto F, Sun CY, Camerini-Otero RD, and Petukhova G (2018). Analysis of meiotic double-strand break initiation in mammals. Methods Enzymol 601, 391–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TJ, Wardell K, Garcia V, and Neale MJ (2014). Homeostatic regulation of meiotic DSB formation by ATM/ATR. Exp Cell Res 329, 124–131. [DOI] [PubMed] [Google Scholar]
- Corpet F (1988). Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16, 10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij DG, and de Boer P (2003). Specific arrests of spermatogenesis in genetically modified and mutant mice. Cytogenet Genome Res 103, 267–276. [DOI] [PubMed] [Google Scholar]
- de Vries FA, de Boer E, van den Bosch M, Baarends WM, Ooms M, Yuan L, Liu JG, van Zeeland AA, Heyting C, and Pastink A (2005). Mouse Sycp1 functions in synaptonemal complex assembly, meiotic recombination, and XY body formation. Genes Dev 19, 1376–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V, Gray S, Allison RM, Cooper TJ, and Neale MJ (2015). Tel1(ATM)-mediated interference suppresses clustered meiotic double-strand-break formation. Nature 520, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey C, Baudat F, and de Massy B (2018). PRDM9, a driver of the genetic map. PLoS Genet 14, e1007479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SB, Marin J, Suleski M, Paymer M, and Kumar S (2015). Tree of life reveals clock-like speciation and diversification. Mol Biol Evol 32, 835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinch AG, Altemose N, Noor N, Donnelly P, and Myers SR (2014). Recombination in the human pseudoautosomal region PAR1. PLoS Genet 10, e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter N (2017). Oocyte quality control: Causes, mechanisms, and consequences. Cold Spring Harb Symp Quant Biol 82, 235–247. [DOI] [PubMed] [Google Scholar]
- Jain D, Puno MR, Meydan C, Lailler N, Mason CE, Lima CD, Anderson KV, and Keeney S (2018). ketu mutant mice uncover an essential meiotic function for the ancient RNA helicase YTHDC2. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi L, Barchi M, Baudat F, Romanienko PJ, Keeney S, and Jasin M (2011). Distinct properties of the XY pseudoautosomal region crucial for male meiosis. Science 331, 916–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi L, Barchi M, Lange J, Baudat F, Jasin M, and Keeney S (2013). Numerical constraints and feedback control of double-strand breaks in mouse meiosis. Genes Dev 27, 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S (2008). Spo11 and the formation of DNA double-strand breaks in meiosis In Recombination and Meiosis, Lankenau DH, ed. (Heidelberg, Springer-Verlag; ), pp. 81–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Lange J, and Mohibullah N (2014). Self-organization of meiotic recombination initiation: general principles and molecular pathways. Annu Rev Genet 48, 187–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Bourbon HM, and de Massy B (2010). Functional conservation of Mei4 for meiotic DNA double-strand break formation from yeasts to mice. Genes Dev 24, 1266–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Oliver C, Brun C, Juarez-Martinez AB, Tarabay Y, Kadlec J, and de Massy B (2018). Mouse REC114 is essential for meiotic DNA double-strand break formation and forms a complex with MEI4. Life Sci Alliance 1, e201800259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam I, and Keeney S (2014). Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb Perspect Biol 7, a016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange J, Pan J, Cole F, Thelen MP, Jasin M, and Keeney S (2011). ATM controls meiotic double-strand-break formation. Nature 479, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Mahajan A, and Tsai MD (2006). Ankyrin repeat: A unique motif mediating proteinprotein interactions. Biochemistry 45, 15168–15178. [DOI] [PubMed] [Google Scholar]
- Mahadevaiah SK, Turner JM, Baudat F, Rogakou EP, de Boer P, Blanco-Rodriguez J, Jasin M, Keeney S, Bonner WM, and Burgoyne PS (2001). Recombinational DNA doublestrand breaks in mice precede synapsis. Nat Genet 27, 271–276. [DOI] [PubMed] [Google Scholar]
- Maleki S, Neale MJ, Arora C, Henderson KA, and Keeney S (2007). Interactions between Mei4, Rec114, and other proteins required for meiotic DNA double-strand break formation in Saccharomyces cerevisiae. Chromosoma 116, 471–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi T, Ito M, Kugou K, Yamada S, Furuichi M, Oda A, Yamada T, Hirota K, Masai H, and Ohta K (2012). A central coupler for recombination initiation linking chromosome architecture to S phase checkpoint. Mol Cell 47, 722–733. [DOI] [PubMed] [Google Scholar]
- Odorisio T, Rodriguez TA, Evans EP, Clarke AR, and Burgoyne PS (1998). The meiotic checkpoint monitoring synapsis eliminates spermatocytes via p53-independent apoptosis. Nat Genet 18, 257–261. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Borek D, Majewski W, and Minor W (2003). Multiparametric scaling of diffraction intensities. Acta Crystallogr A 59, 228–234. [DOI] [PubMed] [Google Scholar]
- Pacheco S, Marcet-Ortega M, Lange J, Jasin M, Keeney S, and Roig I (2015). The ATM signaling cascade promotes recombination-dependent pachytene arrest in mouse spermatocytes. PLoS Genet 11, e1005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Sasaki M, Kniewel R, Murakami H, Blitzblau HG, Tischfield SE, Zhu X, Neale MJ, Jasin M, Socci ND, et al. (2011). A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell 144, 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanikos F, Clement JAJ, Testa E, Ravindranathan R, Grey C, Dereli I, Bondarieva A, Valerio-Cabrera S, Stanzione M, Schleiffer A, et al. (2018). ANKRD31 regulates spatiotemporal patterning of meiotic recombination initiation and ensures recombination between heterologous sex chromosomes in mice. bioRxiv DOI: 10.1101/423293. [DOI] [PubMed]
- R Core Team (2018). R: A language and environment for statistical computing. (Vienna, Austria, R Foundation for Statistical Computing; ). [Google Scholar]
- Robert T, Vrielynck N, Mezard C, de Massy B, and Grelon M (2016). A new light on the meiotic DSB catalytic complex. Semin Cell Dev Biol 54, 165–176. [DOI] [PubMed] [Google Scholar]
- Robert X, and Gouet P (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanienko PJ, Giacalone J, Ingenito J, Wang Y, Isaka M, Johnson T, You Y, and Mark WH (2016). A vector with a single promoter for in vitro transcription and mammalian cell expression of CRISPR gRNAs. PLoS One 11, e0148362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouyer F, Simmler MC, Johnsson C, Vergnaud G, Cooke HJ, and Weissenbach J (1986). A gradient of sex linkage in the pseudoautosomal region of the human sex chromosomes. Nature 319, 291–295. [DOI] [PubMed] [Google Scholar]
- Royo H, Polikiewicz G, Mahadevaiah SK, Prosser H, Mitchell M, Bradley A, de Rooij DG, Burgoyne PS, and Turner JM (2010). Evidence that meiotic sex chromosome inactivation is essential for male fertility. Curr Biol 20, 2117–2123. [DOI] [PubMed] [Google Scholar]
- Saredi G, Huang H, Hammond CM, Alabert C, Bekker-Jensen S, Forne I, ReveronGomez N, Foster BM, Mlejnkova L, Bartke T, et al. (2016). H4K20me0 marks postreplicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature 534, 714718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin YH, Ren Y, Suzuki H, Golnoski KJ, Ahn HW, Mico V, and Rajkovic A (2017). Transcription factors SOHLH1 and SOHLH2 coordinate oocyte differentiation without affecting meiosis I. J Clin Invest 127, 2106–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagulova F, Brick K, Pu Y, Sengupta U, Camerini-Otero RD, and Petukhova GV (2013). Suppression of genetic recombination in the pseudoautosomal region and at subtelomeres in mice with a hypomorphic Spo11 allele. BMC Genomics 14, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanzione M, Baumann M, Papanikos F, Dereli I, Lange J, Ramlal A, Trankner D, Shibuya H, de Massy B, Watanabe Y, et al. (2016). Meiotic DNA break formation requires the unsynapsed chromosome axis-binding protein IHO1 (CCDC36) in mice. Nat Cell Biol 18, 1208–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson WJ, and Vacquier VD (2002). The rapid evolution of reproductive proteins. Nat Rev Genet 3, 137–144. [DOI] [PubMed] [Google Scholar]
- Tesse S, Bourbon HM, Debuchy R, Budin K, Dubois E, Liangran Z, Antoine R, Piolot T, Kleckner N, Zickler D, et al. (2017). Asy2/Mer2: an evolutionarily conserved mediator of meiotic recombination, pairing, and global chromosome compaction. Genes Dev 31, 1880–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillberg PW, Chen F, Piatkevich KD, Zhao Y, Yu CC, English BP, Gao L, Martorell A, Suk HJ, Yoshida F, et al. (2016). Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat Biotechnol 34, 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troffer-Charlier N, Cura V, Hassenboehler P, Moras D, and Cavarelli J (2007). Functional insights from structures of coactivator-associated arginine methyltransferase 1 domains. EMBO J 26, 4391–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43, 11.10.11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TJ, Clements J, and Finn RD (2014). Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinformatics 15, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.