

Abstract
Here, we describe a diene-containing noncanonical amino acid (ncAA) capable of undergoing fast and selective normal electron-demand Diels–Alder (DA) reactions following its incorporation into antibodies. A cyclopentadiene derivative of lysine (CpHK) served as the reactive handle for DA transformations and the substrate for genetic incorporation. CpHK incorporated into antibodies with high efficiency and was available for maleimide conjugation or self-reaction depending on position in the amino acid sequence. CpHK at position K274 reacted with the maleimide drug-linker AZ1508 at a rate of ~79 M−1s−1 to produce functional antibody–drug conjugates (ADCs) in a one-step process. Incorporation of CpHK at position S239 resulted in dimerization, which covalently linked antibody heavy chains together. The diene ncAA described here is capable of producing therapeutic protein conjugates with clinically validated and widely available maleimide compounds, while also enabling proximity-based stapling through a DA dimerization reaction.
Keywords: antibody–drug conjugate, Diels–Alder reaction, diene, non-canonical amino acid, proximity-driven reaction
Graphical Abstract
Genetic code expansion technology enables manipulation of proteins beyond the limitations of naturally occurring amino acids, yielding new functions and properties that can be used to study metabolic processes and to produce therapeutic entities.[1] Noncanonical amino acids (ncAAs) bearing novel functional groups can be incorporated into proteins using prokaryotic or eukaryotic expression systems.[2] To date, ncAAs have been synthesized and applied to achieve functions such as: 1) covalent coupling of small-molecule reporters or drugs to proteins; 2) manipulate protein properties such as hydrophobicity or fluorescence; and 3) linking protein units together in a proximity-based manner.[3]
A key advancement in this area has been the development of ncAAs bearing functional groups that produce reactive proteins that can be altered, making them attractive therapeutic targets. For example, production of reactive proteins for covalent modification has led to development of ncAA-bearing compounds such as azide, alkyne, norbornene, acetylphenyl alanine, cyclopropene, and others.[4] These ncAAs have enabled site-specific covalent attachment of entities to proteins, development of dye-labeled reporter systems in vitro and in vivo, and production of therapeutic agents such as antibody-drug conjugates (ADCs).[5] On the other hand, several strategies to enable unnatural proximity-dependent reactivity in proteins have also been described, capitalizing on a complementary pair of canonical and noncanonical amino acids to covalently link protein units together. These pairs include noncanonical amino acids that bear thiol-reactive bromo, fluoro, or fluorobenzene functional groups[6]; lysine-reactive aryl carbamate groups[7]; and lysine-, histidine-, and tyrosine-reactive fluorosulfate groups.[8] The general coupling strategy employed in these systems is to introduce a single noncanonical amino acid into the protein sequence along with the appropriate canonical amino acid reaction partner in close proximity in the folded or assembled protein structure. Although the strategies outlined above represent powerful and elegant tools for bioconjugation and protein engineering, they all have certain drawbacks. Most notably, reactive ncAA compounds developed to date are designed for a specific, single-use application; that is, small-molecule coupling or proximity-dependent stapling. Given the extensive effort associated with the development of ncAAs, we envisioned the development of a new ncAA consisting of a single compound capable of multifunctionality: conjugation and/or self-dimerization. Although other ncAAs chemistries could theoretically be used for both bioconjugation or self-dimerization, to our knowledge this approach has never been explored.
In the current work, we demonstrate that a cyclic diene ncAA platform enables two distinct Diels–Alder (DA) coupling reactions on proteins: 1) attachment of maleimide-based drugs to antibodies in a single-step process; and 2) proximity-driven self-DA reactions that link protein subunits together. Importantly, the selectivity can be controlled by the position in the amino acid sequence. The DA reactive ncAA was prepared by functionalization of a lysine core unit with cyclopentadiene to yield CpHK. CpHK efficiently incorporated into antibodies at an amber stop codon (TAG) in a Chinese hamster ovary (CHO) cell transient expression system by co-transfecting plasmids bearing the antibody gene and the Methanosarcina mazei pyrrolysine (pyl) tRNA synthetase/tRNA [PylRS/tRNA(Pyl)] pair. We show that the electron-rich diene on CpHK was stable throughout the 11-day antibody expression process and that the isolated antibody product retained reactivity with maleimide. Furthermore, we demonstrate that a DA dimerization reaction occurs when CpHKs can interact due to their proximity in the folded and assembled protein structure. Cyclopentadiene was selected as a ncAA functional group based on our previous studies aimed to design new conjugation strategies that take advantage of clinically used and widely available maleimide compounds.[9] That work evaluated the reactivity of several cyclic dienes with a maleimide dienophile in aqueous conditions. Excellent reactivity of cyclopentadiene with maleimide, its compact 5-membered ring structure (analogous to the side chain end group of pyrrolysine), and known dimerization properties led us to evaluate cyclopentadiene as a novel ncAA functional group. A detailed discussion of the DA reaction and considerations for use in bioconjugation can be found in our previous publication.[9] To date, only dienes with slow DA kinetics have been incorporated into ncAAs, but these are not able to be used for efficient conjugation with maleimide or dimerization.[10] As such, our approach demonstrates the utility of the classic DA reaction for production of bioconjugates and protein engineering applications.
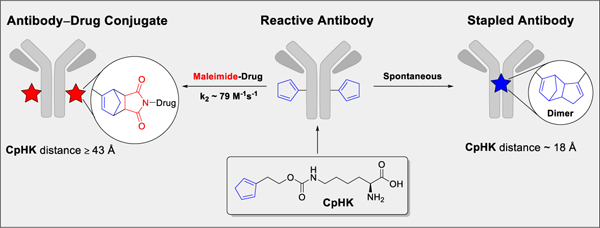
One critical concern in producing antibodies with a highly reactive diene is stability and biocompatibility during the expression process. To experimentally investigate diene chemistry for ncAA applications, CpHK was synthesized and incorporated into antibodies using genetic expansion technology based on amber stop-codon suppression and the M. mazei PylRS/tRNA(Pyl)] pair (Figure 1, see supporting information).[11] Antibody heavy-chain positions S239 and K274 (EU numbering) were selected for incorporation of CpHK following analysis of the known crystal structure of the human IgG1 antibody Fc fragment to estimate distances between amino acid α-carbons and orientation of side chains using PyMOL (Schrodinger LLC) and the crystal structure from PDB entry 1FC1.[12] In the fully folded and assembled antibody molecule, heavy-chain amino acid α-carbons of amino acids at position S239CpHK (1) are within ~18 Å from each other and side chains are likely able to interact and enable DA dimerization, whereas amino acids α-carbons at heavy chain position K274CpHK (2) are ~43 Å and side chains are likely facing away, thus preventing dimerization and enabling reaction with maleimide for bioconjugation (Figure 1).
Figure 1.

Production of antibodies incorporating cyclopentadiene (CpHK) and structure of positions K274 and S239 in the human IgG1 antibody Fc region. a) Process for antibody expression. b) Distance between amino acid α-carbons and orientation of serine and lysine side chains at positions S239 and K274 in an assembled antibody structure. Amino acid α-carbons are shown as colored spheres and approximate orientation of native lysine and serine side chains are shown as yellow arrows. The antibody Fab region is not shown. Only the 2-substituted isomer of cyclopentadiene is shown.
Antibodies were transiently expressed in CHO cells following transfection with three plasmids encoding 1) anti-EphA2 mAb (termed 1C1) containing an amber TAG stop codon replacing the natural codon at heavy-chain position S239 or K274; 2) nine copies of tRNA (Pyl) under control of the U6 snRNA promoter;[13] and 3) the M. mazei PylRS comprising a Y306A/Y384F double mutation[14] under control of the cytomegalovirus promoter. Transfected cells were cultured in media supplemented with CpHK (2 mM) for 11 days under standard antibody expression conditions. To our gratification, antibody recovery following purification with protein A for 1 and 2 were 138 and 139 mg/L respectively (Table S1), which is higher than titers reported for expression of azide or cyclopropene ncAAs (approximately 30–80 mg/L) using a similar transient expression system.[4d, 15] High ncAA titers also correlated with high CHO cell viability (>80% measured at the time of harvest). Antibody products contained high monomer content, with 94% and 91% monomer for antibodies 1 and 2, respectively (Figure S7). Moreover, CpHK itself was well tolerated, stable, and biocompatible, as evidenced by high cell viability throughout expression, lack of formation of DA adducts with natural metabolites, or degradation of the diene unit as determined by mass spectrometry (MS) (vide infra). These results demonstrate that the CpHK functional group is robust and suitable for applications that demand exposure to complex biological milieu and metabolic processes for extended periods of time.
Characterization of intact mAb products by MS showed a single species was obtained for both 1C1 S239CpHK (1) and 1C1 K274CpHK (2) (Figure 2 and Figure S8), corresponding to the expected molecular weight after mutation of serine or lysine to CpHK. To our delight, analysis of the reduced antibody products by MS and SDS-PAGE showed that mAbs 1 and 2 were fundamentally different (Figure 2), suggesting that site-selective multifunctional chemistry was occurring. Specifically, the wild-type (WT) antibody and antibody 2 denatured into heavy and light chains after reduction with TCEP (MS) or dithiothreitol (SDS-PAGE), whereas antibody 1 denatured into light chains and a higher molecular weight species of ~100 kDa. MS analysis of reduced antibody confirmed the higher molecular weight species to be 100,708.78 Da, corresponding to heavy-chain dimer containing two CpHK amino acids (Figure 2).
Figure 2.

Characterization of reduced monoclonal antibody (mAb) products bearing CpHK at position S239 (1) or K274 (2) by SDS-PAGE (top) and deglycosylated mass spectrometry (bottom). WT -wild-type mAb. Only one possible isomer of dicyclopentadiene dimer is shown.
Antibody product 1 was further evaluated by MS to confirm the mechanism of dimerization as formation of a DA adduct. First, antibody was digested with IdeS to remove the Fd region of the antibody by proteolytic cleavage at position 236. Next, the Fc fragment containing the 239 position was further digested with trypsin to generate a 12 amino acid fragment containing S239CpHK (Figure 3a). HPLC analysis showed that two species contained the expected peptide fragments at 97.3% (dimer) and 2.7% (monomer) relative abundance. Dimerized peptide was discerned from monomer peptide by the total mass and charge state. For example, the common peak at ~497 amu represents a +3 ion, with 1/3 m/z isotope spacing for the monomer peptide fragment whereas for the dimer peptide fragment this is a +6 ion, with 1/6 m/z isotopic spacing. Evaluation of the MS/MS profile of both monomer and dimer species revealed a species at 185.133 amu unique to the dimer peptide. The mass of this species unique to the dimer peptide corresponds to the DA adduct liberated by fragmentation of CpHK at the carbamate bond (Figures 3g and S9), thus providing direct evidence of dimer formation.
Figure 3.

Evaluation of the CpHK Diels-Alder adduct in antibody 1. A) Enzymatic digestion of 1 to generate a peptide fragment containing S239CpHK. B) RPHPLC analysis of the digestion product showing the extracted ion chromatogram at the m/z of the peptide fragment. C-D) MS analysis of monomer and dimer peptide fragments with the ionization states indicated. E-F) Zoomed mass spectra of the 497 amu peak common to both monomer and dimer peptide fragments. G) MS/MS analysis of the peptide dimer fragment. Note that the carbamate of CpHK was cleaved back to lysine under these conditions. Peptide containing CpHK degraded to lysine and the proposed Diels-Alder adduct are indicated with *.
We have reasoned that the ability of CpHK to form covalent DA adducts through dimerization appears to be proximity driven for three reasons: 1) no free, unincorporated CpHK was detected coupled to antibodies 1 or 2; 2) CpHK-containing antibodies did not show high levels of aggregation due to antibody-antibody intermolecular DA reactions; and 3) dimerization occurred only at position S239CpHK and not at position K274CpHK. Observation of the CpHK self-reaction at antibody position S239 complements recent efforts to develop compounds for proximity-dependent reactions in proteins. However, this approach is unique in the sense that CpHK is self-reactive and also has the ability to efficiently react with maleimide for bioconjugation (vide infra). The CpHK proximity-based self-reaction described here represents the first homogeneous ncAA stapling strategy and offers the advantages of being bioorthogonal, spontaneous, and unaffected by canonical amino acids.
To demonstrate the utility of the DA reaction of CpHK with maleimide for bioconjugation, antibodies 1 and 2 were reacted with the drug-linker AZ1508, containing a tubulysin warhead and maleimide-based linker, to produce ADCs (Scheme 1). Antibody 1 did not react, which is consistent with the stapled heavy-chain dimer structure whereas 2 reacted in nearly quantitative conversion (96%, 1 hour, 2 eq maleimide per diene) to produce ADC 3, resulting in a drug:antibody ratio (DAR) of 1.91 determined by mass spectrometry (Figures S10–S12). Further analysis of reaction kinetics of 2+AZ1508 yielded a second-order rate constant of 78.5 M−1s−1 (Figures 4a, S14), which is consistent with our previous report of k2 = 77 M−1s−1 for linker-based dienemaleimide reactions on antibodies.[9] The CpHK-maleimide reaction rate is faster than other ncAA reactions used to produce ADCs including strain-promoted azide-alkyne cycloaddition (SPAAC, k2 ~1.4 M−1s−1),[16] p-phenylenediamine-catalyzed oxime ligation (k2 = 4.7 M−1s−1),[17] and inverse electron-demand DA (IEDDA, k2 = 7 M−1s−1)[18] but slower than thiol-maleimide (k2 ~500–700 M−1s−1).[19] Drug conjugation via a DA adduct did not cause aggregation as the measured monomer content was 95% in the recovered ADC (Figure S13). Importantly, ADC 3 was stable in serum, with no drug deconjugation detected after incubation for 7 days at 37 °C (Figure 4b), which is also consistent with our findings with linker-based DA ADCs.[9] Analysis of ADC activity against receptor-positive PC3 cancer cells and receptor-negative PC3F2 cells in vitro confirmed that ADC 3 was potent and selective (Figure 4c), the observed 10 ng/mL EC50 activity of 3 in PC3 cells is within the expected range for a tubulin-inhibitor DAR2 ADC against this target (Table S2).[20] In contrast, the antibody product recovered following reaction of antibody 1+AZ1508 showed no cytotoxic activity, again confirming that this construct contained no conjugated drug due to the diene self-reaction preventing drug attachment. These conjugation studies thus demonstrated that CpHK was capable of efficiently reacting with maleimide when incorporated at a distant (non-dimerizing) position, whereas CpHK was not capable of conjugating with maleimide when incorporated at a dimerizing position.
Scheme 1.

One-step synthesis of ADCs via maleimide-diene coupling.
Figure 4.
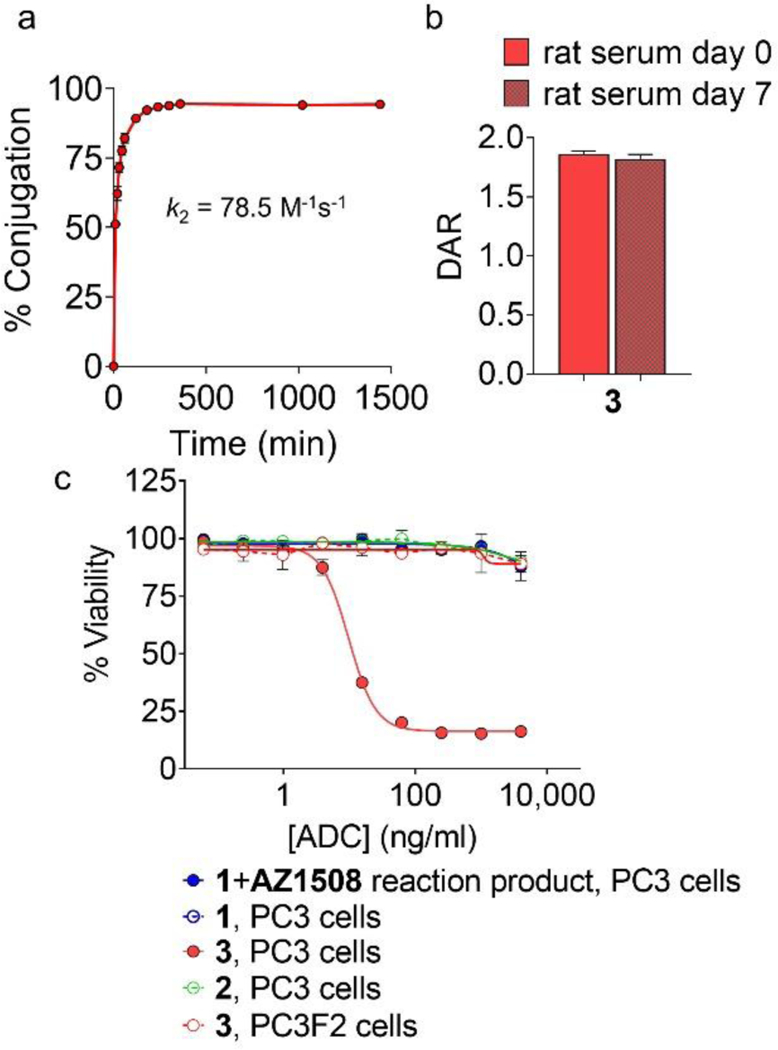
Evaluation of ADCs. a) Conversion of antibody 2 to ADC 3 following addition of 1 molar equivalent of AZ1508 (phosphate-buffered saline, 10% v/v dimethyl sulfoxide, pH 5.5, room temperature, n = 3 ± SD). b) Stability of ADC 3 following incubation in rat serum for 7 days at 37°C, n = 3 ± SD. c) In vitro activity of ADC products against EphA2 receptor-positive PC3 cancer cells and receptor-negative PC3F2 cancer cells.
In summary, the current work demonstrates that the classic DA reaction can be applied in the context of ncAAs to produce therapeutically relevant bioconjugates and enable new protein engineering strategies. Our evaluation of CpHK within antibodies and ADCs allowed insight into the biocompatibility, stability, reactivity with maleimide, and dimerization properties of this diene. CpHK enabled us to evaluate unique properties that have not yet been developed by ncAA platforms: the ability to rapidly react with a dienophile for bioconjugation and the ability to dimerize in a proximity-dependent manner. In that regard, CpHK is analogous to cysteine in its reaction properties (i.e., reaction with maleimide and dimerization) with the added benefit that both DA reaction products are irreversible under physiological conditions. Development of the CpHK ncAA bioconjugation platform to complement maleimide chemistry allows rapid formation of stable bioconjugates with widely available maleimide compounds in a simple, one-step reaction. Dimerization of CpHK through proximity-driven DA reactions enables a unique bioorthogonal stapling strategy controlled by the diene positions in the folded protein. Altogether, the dual functionality of CpHK provides a new tool for producing stable bioconjugates and engineered proteins through simple and straightforward processes.
Supplementary Material
Acknowledgements
This work was financially supported by AstraZeneca. We would also like to thank Linda Xu for laboratory support and Raghothama Chaerkady for assistance with mass spectrometry analysis. A.H.S. thanks the Natural Sciences and Engineering Research Council of Canada (NSERC) for a postgraduate scholarship (PGS-D). Nuclear magnetic resonance instrumentation was supported by a Shared Instrumentation Grant (1S10OD012077–01A1) from the National Institutes of Health. Assistance with manuscript preparation from Deborah Shuman is gratefully acknowledged.
Footnotes
Experimental Section
All experimental details can be found in Supporting Information.
Supporting information for this article is given via a link at the end of the document.
Conflict of Interest Statement
F.H., J.L., K.R., V. O., S. M., J. H., M.M., H.W., C.G., and R.J.C. are all employees of AstraZeneca and work to develop ADCs for clinical use. A patent application has been filed related to this technology.
References
- [1].a) Sun SB, Schultz PG, Kim CH, ChemBioChem 2014, 15, 1721–1729; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Johnson JA, Lu YY, Van Deventer JA, Tirrell DA, Curr. Opin. Chem. Biol 2010, 14, 774–780; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Coin I, Curr. Opin. Chem. Biol 2018, 46, 156–163. [DOI] [PubMed] [Google Scholar]
- [2].a) Hohsaka T, Sisido M, Curr. Opin. Chem. Biol 2002, 6, 809–815; [DOI] [PubMed] [Google Scholar]; b) Wang L, Xie J, Schultz PG, Annu. Rev. Biophys. Biomol. Struct, Vol. 35, 2006, pp. 225–249; [DOI] [PubMed] [Google Scholar]; c) Dumas A, Lercher L, Spicer CD, Davis BG, Chem. Sci 2015, 6, 50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Lang K, Chin JW, Chem. Rev 2014, 114, 4764–4806; [DOI] [PubMed] [Google Scholar]; b) Nguyen T-A, Cigler M, Lang K, Angew. Chem., Int. Ed 2018, 57, 14350–14361. [DOI] [PubMed] [Google Scholar]
- [4].a) Nguyen DP, Lusic H, Neumann H, Kapadnis PB, Deiters A, Chin JW, J. Am. Chem. Soc 2009, 131, 8720–8721; [DOI] [PubMed] [Google Scholar]; b) Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, Chin JW, Nat. Chem 2012, 4, 298–304; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wangt L, Zhang Z, Brock A, Schultz PG, Proc. Natl. Acad. Sci. U. S. A 2003, 100, 56–61; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Oller-Salvia B, Kym G, Chin JW, Angew Chem Int Ed Engl 2018, 57, 2831–2834; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kim CH, Axup JY, Schultz PG, Curr. Opin. Chem. Biol 2013, 17, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lim SI, Kwon I, Crit. Rev. Biotechnol 2016, 36, 803–815. [DOI] [PubMed] [Google Scholar]
- [6].a) Cigler M, Müller TG, Horn-Ghetko D, von Wrisberg MK, Fottner M, Goody RS, Itzen A, Müller MP, Lang K, Angew. Chem., Int. Ed 2017, 56, 15737–15741; [DOI] [PubMed] [Google Scholar]; b) Xiang Z, Ren H, Hu YS, Coin I, Wei J, Cang H, Wang L, Nat. Methods 2013, 10, 885–888; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Embaby AM, Schoffelen S, Kofoed C, Meldal M, Diness F, Angew. Chem., Int. Ed. Engl 2018, 57, 8022–8026. [DOI] [PubMed] [Google Scholar]
- [7].Xuan W, Shao S, Schultz PG, Angew. Chem., Int. Ed 2017, 56, 5096–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang N, Yang B, Fu C, Zhu H, Zheng F, Kobayashi T, Liu J, Li S, Ma C, Wang PG, Wang Q, Wang L, J. Am. Chem. Soc 2018, 140, 4995–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].St. Amant AH, Lemen D, Florinas S, Mao S, Fazenbaker C, Zhong H, Wu H, Gao C, Christie RJ, Read de Alaniz J, Bioconjugate Chem 2018, 29, 2406–2414. [DOI] [PubMed] [Google Scholar]
- [10].a) Schmidt MJ, Summerer D, Angew. Chem., Int. Ed 2013, 52, 4690–4693; [DOI] [PubMed] [Google Scholar]; b) Torres J, PhD Thesis, North Carolina State University (USA), 2014. [Google Scholar]
- [11].a) Wan W, Tharp JM, Liu WR, Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844, 1059–1070; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Brabham R, Fascione MA, ChemBioChem 2017, 18, 1973–1983. [DOI] [PubMed] [Google Scholar]
- [12].Deisenhofer J, Biochemistry 1981, 20, 2361–2370. [PubMed] [Google Scholar]
- [13].Mukai T, Kobayashi T, Hino N, Yanagisawa T, Sakamoto K, Yokoyama S, Biochem. Biophys. Res. Commun 2008, 371, 818–822. [DOI] [PubMed] [Google Scholar]
- [14].Yanagisawa T, Ishii R, Fukunaga R, Kobayashi T, Sakamoto K, Yokoyama S, Chem. Biol. (Oxford, U. K.), 2008, 15, 1187–1197. [DOI] [PubMed] [Google Scholar]
- [15].Vanbrunt MP, Shanebeck K, Caldwell Z, Johnson J, Thompson P, Martin T, Dong H, Li G, Xu H, D’Hooge F, Masterson L, Bariola P, Tiberghien A, Ezeadi E, Williams DG, Hartley JA, Howard PW, Grabstein KH, Bowen MA, Marelli M, Bioconjugate Chem 2015, 26, 2249–2260. [DOI] [PubMed] [Google Scholar]
- [16].Zimmerman ES, Heibeck TH, Gill A, Li X, Murray CJ, Madlansacay MR, Tran C, Uter NT, Yin G, Rivers PJ, Yam AY, Wang WD, Steiner AR, Bajad SU, Penta K, Yang W, Hallam TJ, Thanos CD, Sato AK, Bioconjugate Chem 2014, 25, 351–361. [DOI] [PubMed] [Google Scholar]
- [17].Wendeler M, Grinberg L, Wang X, Dawson PE, Baca M, Bioconjugate Chem 2014, 25, 93–101. [DOI] [PubMed] [Google Scholar]
- [18].Yang J, Seckute J, Cole CM, Devaraj NK, Angew. Chem., Int. Ed 2012, 51, 7476–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Saito F, Noda H, Bode JW, ACS Chem. Biol 2015, 10, 1026–1033; [DOI] [PubMed] [Google Scholar]; Christie RJ, Fleming R, Bezabeh B, Woods R, Mao S, Harper J, Joseph A, Wang Q, Xu ZQ, Wu H, Gao C, Dimasi N, J. Controlled Release 2015, Part B. 220, 660–670. [DOI] [PubMed] [Google Scholar]
- [20].Jackson D, Gooya J, Mao S, Kinneer K, Xu L, Camara M, Fazenbaker C, Fleming R, Swamynathan S, Meyer D, Senter PD, Gao C, Wu H, Kinch M, Coats S, Kiener PA, Tice DA, Cancer Res 2008, 68, 9367–9374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.