

Abstract
Background:
The prefrontal cortex (PFC) integrates information from multiple inputs to exert “top down” control allowing for appropriate responses in a given context. In psychiatric disorders such as posttraumatic stress disorder (PTSD), PFC hyperactivity is associated with inappropriate fear in safe situations. We previously reported a form of muscarinic acetylcholine receptor (mAChR)-dependent long-term depression (LTD) in the PFC that we hypothesize is involved in appropriate fear responding and could serve to reduce cortical hyperactivity following stress. However, it is unknown if this LTD occurs at fear-related inputs.
Methods:
Using optogenetics with extracellular and whole-cell electrophysiology, we assessed the effect of mAChR activation on the synaptic strength of specific PFC inputs. We used selective pharmacological tools to assess the involvement of M1 mAChRs in conditioned fear extinction in control mice and in the stress-enhanced fear learning (SEFL) model.
Results:
M1 mAChR activation induced LTD at inputs from the ventral hippocampus and basolateral amygdala but not the mediodorsal nucleus of the thalamus. We found that systemic M1 mAChR antagonism impaired contextual fear extinction. Treatment with an M1 PAM enhanced contextual fear extinction consolidation in SEFL-conditioned mice.
Conclusions:
M1 mAChRs dynamically modulate synaptic transmission at two PFC inputs whose activity is necessary for fear extinction and M1 mAChR function is required for proper contextual fear extinction. Furthermore, an M1 PAM enhanced the consolidation of fear extinction in the SEFL model suggesting M1 PAMs may provide a novel treatment strategy to facilitate exposure therapy in the clinic for the treatment of PTSD.
Keywords: M1 muscarinic receptor, synaptic plasticity, prefrontal cortex, posttraumatic stress disorder, fear extinction, positive allosteric modulator
Introduction
The prefrontal cortex (PFC) integrates information from a diverse set of cortical and subcortical sources (1, 2) and is a central structure involved in higher-order cognitive functions (3, 4). Normal function of the PFC is critical for “top down” processing of internal and external signals to inhibit inappropriate thoughts, emotions, and actions, and allows for relevant behavioral responses in appropriate contexts (5–7). To properly integrate synaptic information and facilitate executive functions, input to the PFC undergoes dynamic regulation via mechanisms of synaptic plasticity including long-term potentiation and long-term depression (LTD) of synaptic strength. These forms of synaptic plasticity are commonly considered the molecular correlates of learning and memory (8–10) and are critical in directing PFC activity to guide emotional and behavioral responses (5, 11).
The PFC plays a critical role in extinction of fear conditioning by integrating information from the ventral hippocampus (vHipp) and the basolateral amygdala (BLA), key regions for encoding conditioned fear and regulating emotional responses to fearful stimuli (11, 12). Interestingly, multiple studies suggest that exposure to acute or repeated stress can induce disruptions in PFC function (6, 7) and can dramatically inhibit normal fear extinction (13). Stress-induced loss of fear extinction can impair recovery from trauma and is thought to play a critical role in sustaining pathological fear in post-traumatic stress disorder (PTSD) patients (13).
Preclinical and clinical studies suggest that cholinergic projections to the PFC from the basal forebrain play important roles in the extinction of fear learning (14). Acetylcholine (ACh) acts in large part through the five subtypes of muscarinic acetylcholine receptors (mAChRs), M1-M5, of which the primarily Gαq-coupled M1 and Gαicoupled M4 subtypes are the most abundant in the PFC (15). mAChRs are involved in working memory (16), attention (17), as well as appropriate fear (18) and emotional responses (19). These roles of mAChRs in the PFC have been studied primarily using non-selective pan-mAChR antagonists such as scopolamine (16, 18, 19), but the relative contribution of each subtype to cognitive and affective functions has remained elusive, in part due to a dearth of subtype-selective compounds. We and others have recently developed selective ligands for mAChR subtypes, including highly selective agonists (20), antagonists (21), and positive allosteric modulators (PAMs) (9, 22–26) for the M1 mAChR. Using these new tools, along with genetic manipulations (27, 28), we recently reported that M1 mAChR activation induces a form of LTD in the rodent prelimbic (PL) PFC (9). This is especially interesting in light of studies suggesting that PFC neurons display robust firing during states of high fear, and that depression of excitatory inputs to the PFC may be important for fear extinction learning (29–31). This raises the possibility that M1 LTD could play a role in mAChR regulation of fear extinction learning. If so, this could provide important new insights that are relevant for the treatment of PTSD and other disorders in which fear extinction learning is disrupted. However, the PFC receives input from multiple subcortical areas (1, 32) and it is not known whether M1 LTD is expressed at synapses in the vHipp-PFC-BLA circuit that have been implicated in fear conditioning and extinction learning.
We now report a series of studies in which we found that M1 mAChR activation induces LTD at the vHipp-PFC and BLA-PFC synapses but not at synapses from the mediodorsal nucleus of the thalamus (MDT). Further studies utilizing viral-mediated deletion of M1 from pyramidal cells revealed that vHipp-PFC mAChR LTD requires postsynaptic M1 in PFC pyramidal neurons. Interestingly, selective blockade of M1 impaired contextual fear extinction. Finally, we found that an M1 PAM was able to reverse deficits in contextual fear extinction in a rodent model of PTSD, implying that M1 PAMs may have clinical efficacy as an adjunct to exposure therapy. These results are especially exciting in light of the development of M1 PAMs as potential therapeutics for psychiatric and neurodegenerative disorders.
Materials and Methods
Animal Use
C57BL/6J mice were acquired from Jackson Laboratories (ME). Chrm1loxP/loxP mice (28) were bred in-house. Experiments were performed in group-housed 8-12 week old mice (2–5/cage) on a 12hr light cycle (lights on at 6:00a.m.) and given access to food/water ad libitum. All experimental protocols were approved by the Vanderbilt Institutional Animal Care and Use Committee. Viral injections were performed as previously described (10, 33) with AAV5-CaMKIIa-ChR2-eYFP, AAV5-CaMKIIa-Cre-mCherry, and AAV5-CaMKIIa-mCherry from UNC Viral Core, NC.
Electrophysiology
Extracellular field and whole-cell recordings were performed as previously described (9, 10). Briefly, PFC slices were prepared using NMDG-based cutting/recovery solution (34) and transferred to aCSF (mM: 126 NaCl, 2.5 KCl, 1.25 Na2PO4, 26 NaHCO3, 10 glucose, 2 CaCl2, 1 MgSO4) supplemented with 500μM ascorbate for 1hr. The recording chamber was perfused with aCSF (31±1°C) at a rate of 2mL/min. For field recordings, recording electrodes filled with aCSF were placed in PL layer V. For whole-cell recordings voltage-clamped at −70mV, mCherry-positive neurons in PL layer V were filled with a potassium-based internal solution (mM: 125 K-gluconate, 4 NaCl, 10 HEPES, 4 MgATP, 0.3 NaGTP, 10 Tris-phosphocreatine). Local glutamate release was elicited with 470nm light to activate ChR2 or via a concentric bipolar stimulating electrode in layer II/III at a rate of 0.05Hz for field and 0.1Hz for whole-cell recordings.
Behavior – Cued and Contextual Fear Extinction
Fear conditioning was performed as described in Supplemental Materials and Methods. Mice were handled for 2 days prior to fear conditioning. Percent time spent freezing was used as a measure of learned fear. The stress-enhanced fear learning (SEFL) model involved an initial day of 10 random foot-shocks delivered over 1 hr in a distinct context.
Compounds
Oxotremorine-M was obtained from Tocris. VU0255035, VU0364572, and VU0453595 were synthesized in-house. For electrophysiology, stock solutions were prepared in diH2O or DMSO and diluted to working concentrations in aCSF (≤0.1% DMSO). For behavior, compounds were prepared in 20% β-cyclodextrin and administered i.p.
Data Analysis
The number of mice in each experiment is denoted by “N” and cells/slices by “n”. Data presented as mean ± standard error (SEM). Statistical analyses performed using GraphPad Prism (CA). A paired/unpaired two-tailed Student’s t-test, one/two-way ANOVA, or repeated measures two-way ANOVA with Bonferroni’s post-test were used where appropriate. Results of statistical analyses are presented in the figure legends.
Results
Muscarinic LTD in the PFC is Input-Specific
Our lab and others previously reported that the cholinergic agonist carbachol induces LTD of extracellular field excitatory postsynaptic potentials (fEPSPs) recorded in layer V in response to electrical stimulation of layer II/III in PL PFC slices (9, 35). We first confirmed that this LTD is induced by the mAChR-selective agonist oxotremorine-M (OxoM) (36) in acute slices of the mouse PFC. Bath application of OxoM (10μM) induced a robust LTD of electrically-evoked fEPSPs measured after drug washout (Fig 1A, E), consistent with our previous carbachol data and confirming that LTD in the PFC can be induced by a more selective mAChR agonist.
Figure 1: Muscarinic LTD in the PFC is Input-Specific.
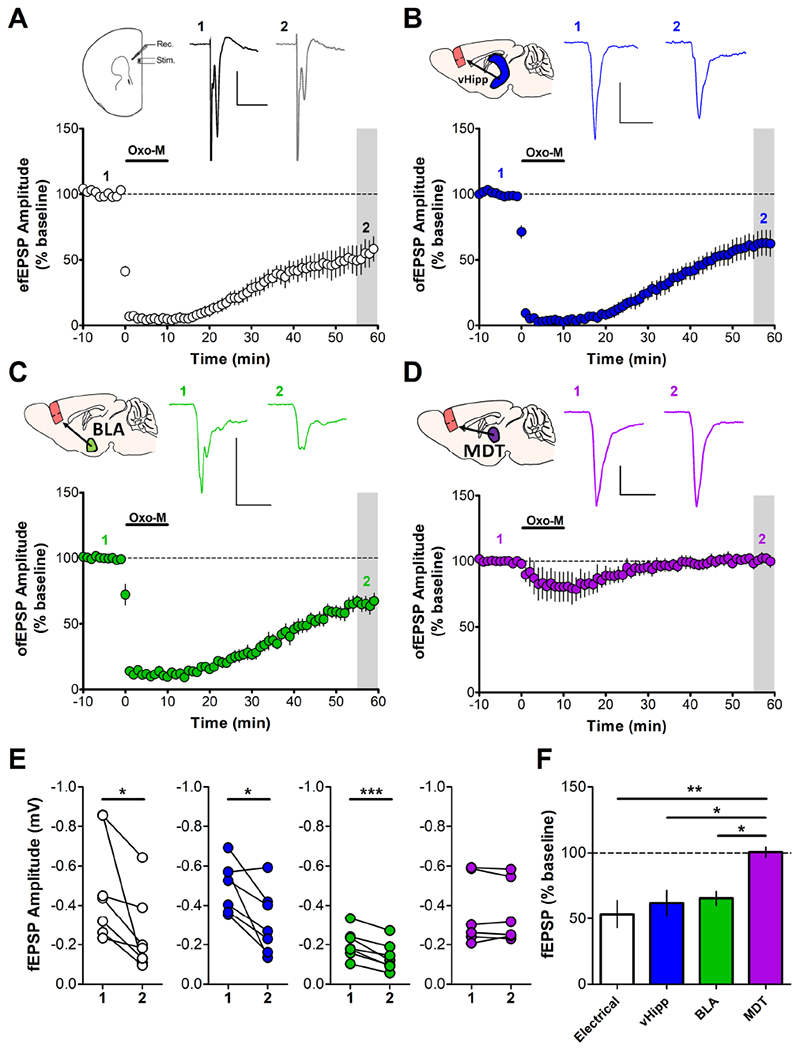
Acute slices of the mouse PFC were prepared 4 weeks after AAV-CaMKIIa-ChR2-eYFP was injected into the vHipp (blue), BLA (green) or MDT (purple). (A) Electrical stimulation of PL layer II/III evoked field excitatory postsynaptic potentials (efEPSPs) recorded in layer V (inset, sample traces). Application of 10μM OxoM induces an acute depression followed by LTD of efEPSPs measured 55-59 min post-drug add. (53.49 ± 10.47%; n = 7) (B) Optical stimulation of afferents from vHipp-ChR2 injected mice with paired pulses of 470nm blue light (1ms pulse duration; 50ms interpulse interval) elicited ofEPSPs that also underwent induction of LTD following bath application of OxoM (10μM). (62.01 ± 9.50%; n = 7) (C) ofEPSPs evoked from stimulation of BLA-ChR2 afferents were also sensitive to OxoM (10μM) and expressed LTD. (65.61 ± 5.28%; n = 7) (D) ofEPSPs evoked in MDT-ChR2 mice exhibited a small acute depression in the presence of OxoM (10μM) but rapidly returned to baseline, not expressing LTD. (100.6 ± 3.72%; n = 6). Sample traces for A-D correspond to baseline (1) and grey shaded area (2). Scale bars: 0.2mV and 20ms. (E) Summary data of change in fEPSP amplitude for each input; 1 = baseline amplitude, 2 = amplitude at 55-59min post-drug add corresponding to the grey shaded regions in A-D. Paired student’s t-test: Electrical, vHipp *p < 0.05, BLA ***p < 0.001, MDT p = 0.778. (F) Summary data of fEPSP amplitude corresponding to grey shaded regions expressed as a percent of baseline compared across inputs. One-way ANOVA: F3,23 = 6.228, p = 0.003. Bonferroni’s post-test: Electrical vs. MDT: ** p < 0.01, vHipp vs MDT and BLA vs MDT: * p < 0.05, Electrical vs. vHipp: p > 0.05; Electrical vs. BLA: p > 0.05; BLA vs. vHipp: p > 0.05.
We then determined whether OxoM would induce LTD at distinct subcortical inputs to the PFC. We used an optogenetic approach whereby we injected mice with virus encoding the expression of eYFP-tagged channelrhodopsin-2 (ChR2) into the afferent region of interest and prepared acute PFC slices 3-4 weeks later. Corroborating previous reports (32), we detected terminals from the vHipp, BLA, and the MDT throughout the PFC (Supplemental Fig. 1). After establishing a stable baseline of optically-evoked fEPSPs (ofEPSPs), bath application of OxoM (10μM) induced LTD of vHipp-evoked ofEPSPs (Fig 1B, E) and BLA-evoked ofEPSPs (Fig 1C, E) but not of MDT-evoked ofEPSPs (Fig 1D, E). The LTD of electrically-evoked fEPSPs and vHipp- and BLA-evoked ofEPSPs were of similar magnitude but were all significantly different from the MDT input (Fig 1F). Together, these data suggest that mAChR LTD of glutamatergic transmission in the PFC exhibits input specificity, and is observed at specific inputs from the BLA and vHipp.
Input-Specific mAChR LTD is mediated by M1 Receptors
Next, we assessed whether M1 mediates mAChR LTD at vHipp-PFC and BLA-PFC synapses. Consistent with prior studies using electrical stimulation (9), OxoM-induced LTD at the vHipp input was blocked in the constant presence of the M1 antagonist VU0255035 (10μM), at a concentration selective for M1 over other mAChR subtypes (9, 21, 37, 38) (Fig 2A,C). Furthermore, we found that bath application of the selective M1 allosteric agonist VU0364572 (20) (30μM) was sufficient to induce LTD at the vHipp-PFC synapse (Fig 2B, C). Similarly, BLA-PFC mAChR LTD was significantly attenuated by VU0255035 (Fig 2D, F) and was induced by the allosteric agonist VU0364572 (Fig 2E, F). This is consistent with the role of M1 in mediating mAChR LTD of electrically-evoked fEPSPs and confirms that M1 is the subtype mediating mAChR LTD at inputs from the vHipp and BLA to the PFC.
Figure 2: Input-Specific mAChR LTD is mediated by M1 Receptors.
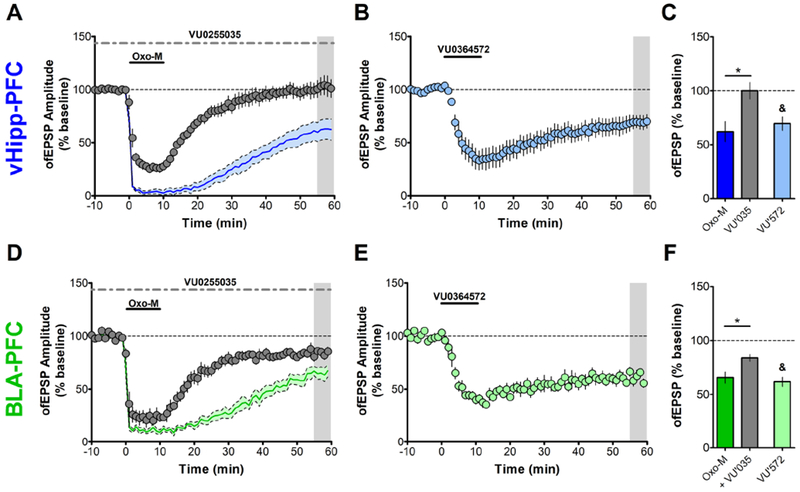
Recordings from vHipp-ChR2 or BLA-ChR2 injected mice. (A) In the constant presence of the selective M1 antagonist VU0255035 (VU’035, 10μM), OxoM (10μM) induced an acute depression of ofEPSPs PL layer V evoked from vHipp afferents but mAChR LTD was blocked. (99.96 ± 7.67%; n = 5). (B) Bath application of the selective M1 allosteric agonist VU0364572 (VU’572, 30μM) for 10 min also induces LTD of ofEPSPs elicited from vHipp afferent stimulation (69.48 ± 6.38%; n = 5). (C) Summary data for vHipp ofEPSP amplitude 55-59 min post-drug add. Unpaired student’s t-test, OxoM vs. OxoM + VU’035: * p < 0.05; paired student’s t-test comparing baseline to shaded area in B: &, p < 0.05. (D) LTD of ofEPSPs evoked from BLA-ChR2 expressing afferents in response to OxoM (10μM) was also blocked in the constant presence of VU’035 (83.86 ± 3.34%; n = 6). (E) Bath application of VU’572 for 10 minutes also induces LTD of ofEPSPs elicited from BLA afferent stimulation (61.71 ± 4.24%; n = 5). (F) Summary data for BLA ofEPSP amplitude 55-59 min post-drug add. Unpaired student’s t-test, OxoM vs. OxoM + VU’035: * p < 0.05; paired student’s t-test comparing baseline to shaded area in E: &, p < 0.05. Shaded time courses in A and D correspond to OxoM alone from Fig 1. Solid colored line represents mean ofEPSP amplitude and grey shaded region around line is ± SEM.
vHipp-PFC mAChR LTD Requires Postsynaptic M1 Receptors
Previously, we reported that mAChR LTD of electrically-evoked fEPSPs correlated with increased inhibition onto layer V pyramidal neurons and that this may contribute to M1 LTD (33). As M1 is expressed on both PFC glutamatergic pyramidal neurons and GABAergic interneurons (39–41), this brings into question the localization of M1 involved in M1 LTD. To address this, we used a viral-mediated knockdown approach allowing for selective deletion of M1 receptors from glutamatergic pyramidal neurons in the PFC (Fig 3A). 5-6 weeks post-injection, we prepared slices to confirm viral expression and observed cell bodies labelled with mCherry and terminals positive for eYFP throughout the PFC (Supplemental Fig. 2).
Figure 3: vHipp-PFC mAChR LTD Requires Postsynaptic M1 Receptors.
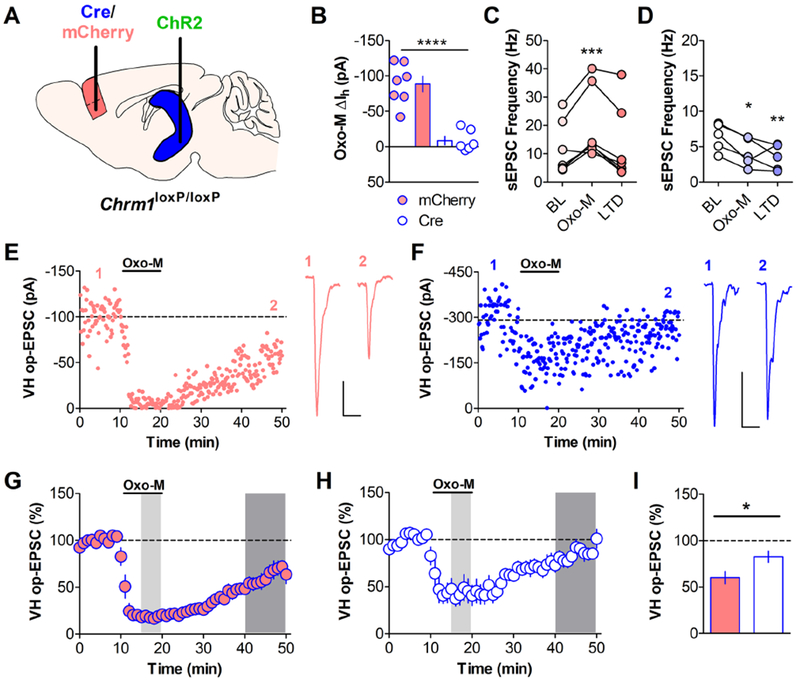
(A) Chrm1loxP/loxP mice were injected with AAV-CaMKIIa-Cre-mCherry (white with blue outline throughout) or AAV-CaMKIIa-mCherry (red with blue outline throughout) into the PFC and co-injected with AAV-CaMKIIa-ChR2-eYFP into the vHipp. Recordings were performed 5-6 weeks post-injection. (B) OxoM (10μM) induced an inward current in neurons from control mCherry-infected mice (−88.55 ± 10.92 pA; n = 7) but failed to elicit an inward current in neurons from Cre-mCherry infected mice (−8.651 ± 6.06 pA; n = 6). (Student’s t-test, mCherry vs. Cre **** p < 0.0001). (C) OxoM (10μM) induced a significant increase in sEPSC frequency recorded before optical stimulation in mCherry neurons. (One-way repeated measures ANOVA, F2,6 = 13.52, p < 0.001, Bonferroni’s post-test *** p < 0.001 baseline (BL) vs OxoM, n = 7). (D) Conversely, OxoM induced a significant decrease in sEPSC frequency in Cre-mCherry neurons. (One-way repeated measures ANOVA, F2,4 = 11.49, p < 0.01, Bonferroni’s post-test * p < 0.05 BL vs OxoM, ** p < 0.01 BL vs LTD, n = 5). (E) A representative experiment for an mCherry-infected neuron (scale bar: 25pA and 25ms) and (F) a Cre-infected neuron (scale bar: 100pA and 25ms). (G) Summary time course for control mCherry mAChR LTD experiments. Bath application of OxoM (10μM) induced a long-term depression of oEPSCs evoked from vHipp-ChR2 terminals in mCherry-infected neurons (60.15 ± 6.67%; n = 7). (H) Summary time course for Cre LTD experiments. LTD of oEPSCs was attenuated in Cre-mCherry infected neurons (82.66 ± 6.13%; n = 6). In both G and H, light shaded areas correspond to the time at which Oxo-M sEPSC measurements were taken for C and D. Dark shaded areas correspond to the time at which LTD sEPSC measurements were taken for C and D and for quantification in I. (I) Summary data for oEPSC amplitude 40-49 min post-OxoM add. Unpaired student’s t-test, * p < 0.05.
Using whole-cell electrophysiology in acute slices, we confirmed the genetic deletion of M1 by monitoring the depolarizing inward current induced by a cholinergic agonist, previously shown to be dependent on postsynaptic M1 receptors (42). In mCherry-positive neurons from CaMKIIα-mCherry infected mice, OxoM (10 μM) induced a depolarizing inward current while in mCherry-positive neurons from CaMKIIα-Cre-mCherry infected mice, OxoM did not cause any change in the holding current (Fig 3B). OxoM caused a significant increase in the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) during OxoM add that returned to baseline levels upon washout (Fig 3C) in control-infected cells. In Cre-infected cells, the OxoM-induced increase in sEPSC frequency was abolished and, interestingly, we observed a significant decrease in sEPSC frequency that persisted following drug washout (Fig 3D) which might be due to activation of other, inhibitory mAChRs (15). These data functionally confirm deletion of M1 from PFC pyramidal cells, validating our genetic approach.
Having confirmed deletion of M1 from pyramidal cells, we then determined whether postsynaptic M1 receptors were required for mAChR LTD at the vHipp-PFC synapse. We selected the vHipp-PFC input based on the complete blockade of LTD by the M1 antagonist (Fig 2A) compared to the significant but incomplete block of BLA-PFC LTD (Fig 2D). Furthermore, to control for the effects of incomplete viral infection on extracellular field recordings (Supplemental Fig. 3), we used whole-cell patch clamp recordings to measure optically-evoked EPSCs (oEPSCs) from vHipp terminals. In mice infected with control virus, OxoM (10 μM) induced an LTD of oEPSCs (Fig 3E, G). Compared to controls, LTD induced by OxoM in mice infected with Cre virus was significantly attenuated (Fig 3F, H, I). These data indicate that postsynaptic M1 mediates mAChR LTD at vHipp-PFC synapses.
M1 Receptor Function is Necessary for Contextual but not Cued Fear Extinction
Together, these data demonstrate that M1 is poised to regulate synaptic transmission at two long-range inputs to the PFC. Given the established role of mAChRs and inputs from the BLA and vHipp in extinction of fear conditioning, we hypothesized that the in vivo relevance of this input-specific modulation may relate to fear extinction. We implemented a five-day fear conditioning protocol to assess the effects of M1 antagonism on both auditory cued and contextual fear extinction (Fig 4A). Mice were conditioned on day 1. During cued extinction on day 2 and context extinction on day 4, mice were administered vehicle (20% β-cyclodextrin) or 3, 10, or 30 mpk VU0255035 i.p. 30 minutes prior to being placed into the extinction context. There was no significant effect of M1 antagonism within the cued fear extinction session (Fig 4B) nor on cued extinction recall on day 3 (Fig 4B). Interestingly, there was a significant effect of M1 antagonism on within-session contextual fear extinction (Fig 4C) and mice administered 30 mpk VU0255035 prior to contextual fear extinction on day 4 displayed significantly higher freezing to the context on recall day 5 compared to vehicle-treated mice (Fig 4C). Importantly, the maximal dose of 30 mpk VU0255035 did not affect freezing in animals that were not exposed to foot-shocks on day 1 (Supplemental Fig. 4). Overall, these data suggest that M1 activation is not required for auditory cued fear extinction but is required for contextual fear extinction.
Figure 4: M1 Receptor Function is Necessary for Contextual but not Cued Fear Extinction.
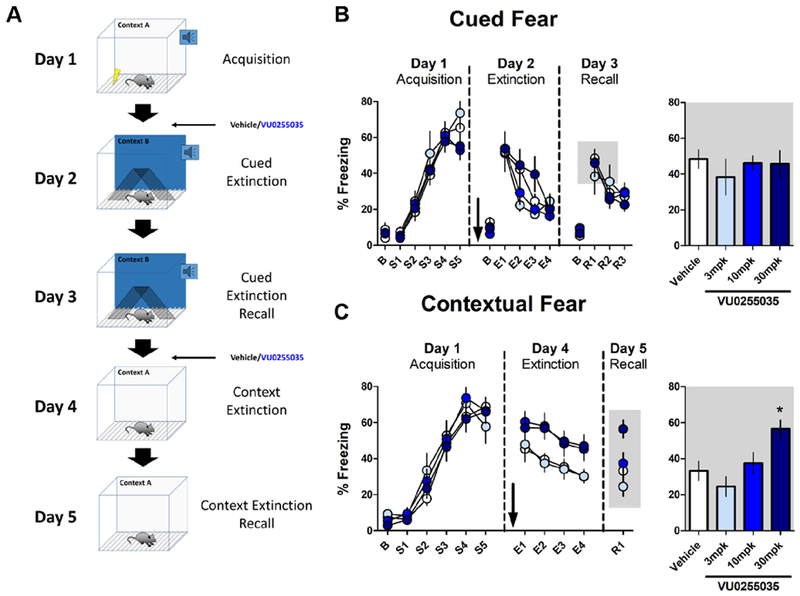
Effect of systemic M1 antagonism on cued and contextual fear extinction in mice. (A) Schematic depicting the training and testing procedure used. Mice were conditioned in Context A with 5 mild footshocks, each preceded by a 30s tone. On day 2, mice were administered the M1 antagonist VU0255035 (3, 10, 30 mpk, i.p.) or vehicle (20% β-cyclodextrin) 30 minutes before being exposed to a series of 12 tones in a novel Context B to assess extinction of auditory cued fear. On day 3, mice were placed back in Context B and exposed to 9 tones to assess consolidation of cued fear extinction. On day 4, mice were again administered VU0255035 or vehicle and placed in Context A for 12 minutes to assess contextual fear extinction. On day 5, mice were placed back in Context A for 3 minutes to assess contextual fear extinction consolidation. (B) At all doses VU0255035 had no effect on auditory cued fear extinction on extinction day 2 (two-way repeated-measures ANOVA, Effect of drug: F3,35 = 0.960, p = 0.423; Effect of tone block: F3,35 = 36.00, p < 0.0001; Interaction, F9,35 = 1.787, p = 0.079) or on recall day 3 (one-way ANOVA, F3,35 = 0.350, p = 0.789). Data for days 2 and 3 are binned by 3 tones and mice were excluded from analysis if baseline freezing was >30%. Bar graph depicts average % freezing to the first three tones on recall day 3, corresponding to the grey shaded box. (N, Veh = 13, 3 mpk = 5, 10 mpk = 12, 30 mpk = 9) (C) Systemic M1 antagonism impairs within-session contextual fear extinction (two-way repeated-measures ANOVA, Effect of drug: F3,39 = 3.663, p = 0.020; Effect of time block: F3,39 = 12.56, p < 0.0001; Interaction: F3,39 = 0.317, p = 0.968) and 30 mpk VU0255035 significantly impaired contextual extinction recall on day 5 (One-way ANOVA, F3,39 = 5.177, p < 0.01; Bonferroni’s post-test, Veh vs. 30 mpk * p < 0.05). Extinction on days 4 and 5 are depicted as 3 min bins. Bar graph depicts R1. (N, Veh = 14, 3 mpk = 7, 10 mpk = 11, 30 mpk = 11).
M1 Potentiation Enhances Fear Extinction in a Model of PTSD
Impaired fear extinction is a hallmark of anxiety-related disorders including PTSD, estimated to affect 3.5% of the US population annually (43). Exposure therapy is one of the most common treatment paradigms for PTSD and shares many similarities with Pavlovian fear extinction used in rodents (44). Pharmacological manipulations that enhance the acquisition and/or consolidation of fear extinction therefore may be beneficial for use in conjunction with exposure therapy. Based on our findings, we hypothesized that enhancing M1 function with a PAM may enhance contextual fear extinction in a rodent model of PTSD.
To test this, we used the extensively validated SEFL model, which produces phenotypes in rodents that mimic PTSD symptoms in the clinical population (43) (Fig 5A). On day 1, mice underwent SEFL conditioning and exhibited greater freezing during fear acquisition in a novel context (context B) on day 2 (Fig 5B) and when exposed to context B on day 3 (Fig 5C). SEFL conditioned mice then received either vehicle (20% β-cyclodextrin) or the M1 PAM VU0453595 (10 mpk) prior to contextual fear extinction in context B on day 3. Pretreatment with VU0453595 had no effect on the expression of contextual fear assessed during the first 3 minutes in context B and no effect on within-session extinction (Fig 5D). When mice were tested for the consolidation of extinction on day 4, PAM-treated mice froze significantly less compared to vehicle-treated mice (Fig 5D), indicating that VU0453595 enhanced the consolidation of contextual fear extinction in SEFL-conditioned mice.
Figure 5: M1 Potentiation Enhances Fear Extinction in a Model of PTSD.
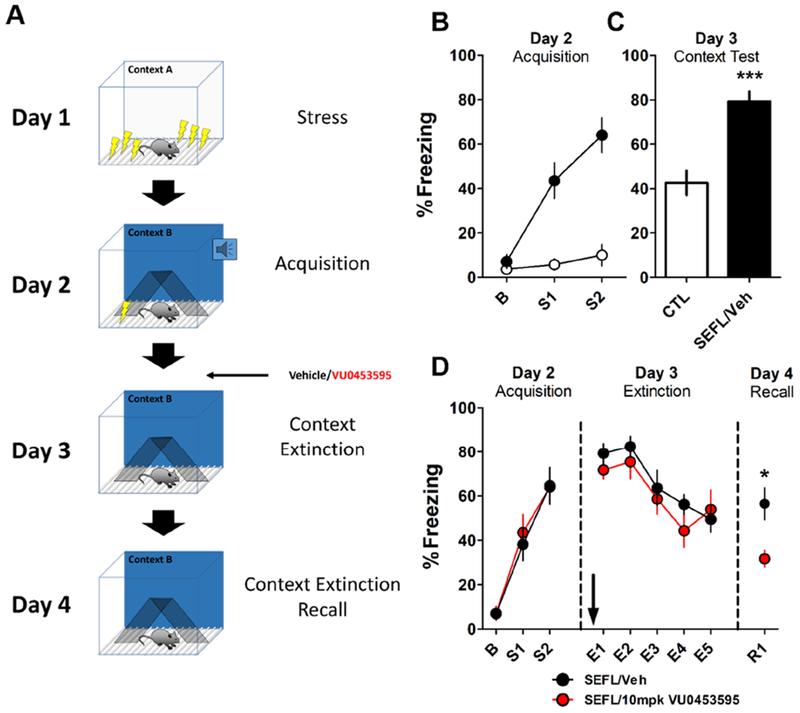
(A) Schematic illustrating the SEFL model and experimental design. On day 1, mice underwent SEFL conditioning in Context A where they received 10 footshocks at random intervals over 1 hour. Control mice were placed in Context A for 1 hour. Days 2, 3, and 4 were performed in a novel Context B. On day 2, mice were conditioned with 2 mild footshocks in Context B. On day 3, SEFL-conditioned mice were administered vehicle (20% β-cyclodextrin) or 10 mpk VU0453595 i.p. 15 min before being placed back in Context B where they underwent a 15 min context extinction session. On day 4, mice were placed back in Context B for 3 min to assess context extinction consolidation. (B) Mice that received SEFL on day 1 froze significantly more on day 2 during acquisition and (C) on day 3. Bar graph depicts first 3 min in Context B on day 3. (Unpaired student’s t-test, ***p < 0.001). (D) Administration of 10 mpk VU0453595 had no effect on within-session extinction on day 3 (Two-way repeated-measures ANOVA: Effect of Drug: F1,18 = 5.033, p = 0.440; Effect of time block: F4,18 = 15.15, p < 0.0001; Interaction: F4,18 = 0.782, p = 0.541) but enhanced consolidation of contextual fear extinction measured on day 4. (Unpaired student’s t-test, *p < 0.05. N, CTL = 8, SEFL/Veh = 10)
Discussion
In the present studies, we found that mAChR activation induces LTD at synapses onto PL PFC layer V from the vHipp and BLA inputs but not from the MDT. Furthermore, we confirmed that M1 mediates LTD at both inputs and that postsynaptic M1 is required for LTD at the vHipp-PFC synapse. This suggests that M1 activation modulates fear-related inputs to the PFC in an input-selective manner. Based on the roles of the vHipp, BLA, and PFC in fear extinction, we further identified M1 as necessary for contextual fear extinction. Finally, we demonstrated that M1 potentiation enhances fear extinction in a rodent model of PTSD, suggesting that M1 PAMs have potential clinical utility in the treatment of PTSD and stress-related disorders.
Dysregulated connectivity of subcortical regions to the PFC is present in multiple psychiatric disorders (45–47). Understanding the functional consequences of this has been a major focus of psychiatric-related research and has been aided by novel circuit-based techniques including optogenetics (48). There have been tremendous advances in establishing the circuitry underlying specific behaviors and how these circuits might be perturbed in psychiatric disorders. However, there is a critical need to identify circuit-specific targets to translate preclinical observations into clinically effective treatments (49). We took advantage of these circuit-based approaches and found that activation of M1 selectively induces LTD at the vHipp and BLA inputs to the PFC, identifying M1 as a potential therapeutic target to modulate these circuits.
Intact communication between the hippocampus, amygdala, and PFC is essential for proper fear extinction in both humans (50) and rodents (51) and is dysregulated in anxiety-related disorders such as PTSD (52). In animal models, BLA and vHipp inputs to the PFC are involved in anxiety-related behaviors (48, 53, 54) and inactivation studies demonstrate that the vHipp, BLA, and PFC are all required for fear extinction (51) while the vHipp-PFC pathway gates fear after extinction learning (29). Thus, it is clear that the vHipp and BLA inputs to the PFC are important for fear extinction and may be disrupted in anxiety-disorder models.
Our observation that M1 activation induces LTD at the vHipp-PFC and BLA-PFC synapses along with work demonstrating that PFC mAChRs are required for fear extinction (18) suggested that these two phenomena are related. Consistently, we found that M1 antagonism impairs contextual fear extinction but had no effect on the extinction of auditory cued fear. Our data does not definitively identify M1 in the PFC as the mediator of these behavioral effects due to technical limitations including that muscarinic LTD measured extracellularly was still intact in Cre-injected Chrm1loxP/loxP mice. This suggests incomplete viral knockdown of M1 therefore testing the necessity of PFC M1 for the observed behavioral effects remains elusive. Nonetheless, our approach identified the involvement of M1 in fear extinction and that an M1 PAM could enhance fear extinction in a model of PTSD, thus translating circuit-based neuroscience to a potential therapeutic mechanism.
Concerning the potential mechanism, the hippocampus communicates contextual information to the PFC via monosynaptic connections from the ventral pole (55). M1 LTD at the vHipp-PFC synapse may therefore reflect a modulation of contextual information flowing into the PFC and be more related to regulation of contextual aspects of fear rather than non-spatial cued fear (56). This is consistent with our observation that M1 antagonism blocks LTD at the vHipp-PFC synapse and impairs contextual fear extinction. Furthermore, single-unit recordings in PL indicate that decreased activity of PL pyramidal neurons corresponds with reduced fear responses (29), consistent with a reduced afferent drive into the PL via an LTD-like mechanism. M1 LTD of vHipp-PL PFC transmission could be required to reduce fear responses during contextual fear extinction by reducing vHipp-mediated excitation of PL neurons. While we identified postsynaptic M1 as necessary for mAChR LTD at the vHipp-PFC synapse, the molecular mechanisms mediating vHipp-PFC M1- LTD are still unknown. Future work investigating signaling downstream of M1 necessary for the induction, expression, and maintenance mechanisms will be instrumental to investigate how this plasticity changes after fear extinction and will identify targets and mechanisms that could improve the treatment of disorders with dysfunctional vHipp-PFC connectivity. M1 also enhances the output of infralimbic (IL) cortex pyramidal neurons and fear extinction correlates with enhanced activity of IL neurons (18, 57) thus M1 PAMs might enhance fear extinction via actions in the IL in addition to LTD in the PL. It is possible that both mechanisms contribute to extinction and investigating the differential involvement of M1 in the PL and IL to fear extinction is an interesting future direction.
vHipp afferents increase feedforward inhibition (FFI), contributing to the decreased activity of PL pyramidal neurons during reduced fear responding during extinction (29). We found that M1 LTD at the vHipp-PFC synapse occurs at excitatory inputs onto PL pyramidal neurons recorded under whole-cell conditions where the contribution of inhibition is negligible. Therefore, vHipp-PL LTD may occur simultaneously with enhanced vHipp-mediated FFI to synergistically reduce the activity of PL pyramidal neurons. M1 activation enhances PFC interneuron activity (41) and an M1-driven increase in FFI may also contribute to fear extinction. Our previous finding that M1 LTD of electrically-evoked fEPSPs correlates with enhanced inhibition onto PL pyramidal neurons (33) may suggest this, and the contribution of muscarinic modulation of inhibition to fear extinction is an interesting future direction as our results do not rule out contributions of both enhanced FFI and M1 LTD mechanisms to fear extinction. M1 is expressed in pyramidal neurons in human cortex (58–60) but M1 in GABAergic interneurons has only been demonstrated in rodent (40, 41) and non-human primate (61) cortex. Therefore, while our results pertaining to M1 in PFC pyramidal neurons are likely relevant to humans, the clinical implications of M1 modulation of inhibitory transmission are unknown and would require identification of M1 in human cortical interneurons.
Systemic and intracortical delivery of the pan-muscarinic antagonist scopolamine impairs the consolidation of cued fear extinction in rats (18). In contrast to these findings, the M1 antagonist VU0255035 did not impair cued fear extinction in the present studies. While M1 mAChR activation promotes cued fear consolidation (62), our findings suggest that M1 is not necessary for cued fear extinction and other muscarinic subtypes may contribute to extinction of cued fear. Our present studies provide insight into this hypothesis. M1 antagonism or genetic deletion does not impair the acute depression of fEPSPs at vHipp/BLA-PFC synapses. This transient depression may be permissive for cued, but not contextual, fear extinction. Additionally, although the M1 antagonist attenuated mAChR LTD at the BLA-PFC synapse, we did not observe a complete block. Other muscarinic receptors such as M4 likely contribute to mAChR LTD at the BLA-PFC synapse and M1-independent depression may be sufficient for cued fear extinction. The involvement of M4 in fear extinction is an intriguing future direction given the aforementioned scopolamine effect and the relatively high expression of M4 in the PFC.
The rodent PL PFC shares connectivity and anatomical similarities to the human dorsal anterior cingulate cortex (dACC) and thus, the LTD we observed could relate to decreased activity of the human dACC observed during fear extinction (63). In an fMRI study, PTSD patients exhibited dACC and amygdala hyperactivity and hippocampal hypoactivity compared to controls during a fear extinction task (52). Hyperactivity of the dACC and amygdala might reflect a deficit in mechanisms similar to mAChR LTD while reductions in hippocampal activity could relate to deficits in the previously described hippocampal LTD-to-LTP switch (64), a reduction in vHipp-mediated FFI, and/or a deficit in vHipp-PFC M1 LTD. The aforementioned functions of M1 suggest it could be a valid therapeutic target to rescue deficient extinction in PTSD and imply together with M1 expression in human cortex that our findings have translational relevance to humans.
M1 potentiation could possibly reduce dACC hyperactivity in PTSD patients via LTD of hyperactive amygdala inputs and shifting vHipp input towards inhibition via enhanced FFI and LTD of excitatory transmission. This hypothesis is consistent with our finding that the M1 PAM VU0453595 enhances contextual fear extinction in the SEFL model. Mimicking the disrupted circuitry in PTSD, rodents exposed to stressors including SEFL exhibit hyperactivity of the PL and BLA and hypoactivity of the hippocampus (13), suggesting these models exhibit excellent face validity with respect to the human disorder. Treatment with an M1 PAM before extinction enhanced the consolidation and recall of contextual fear extinction, suggesting that M1 PAMs may be effective therapeutics to enhance exposure therapy in the clinic. Dysfunctional connectivity between the hippocampus, amygdala, and PFC (46, 65) and impaired fear extinction (66) are present in many psychiatric disorders therefore these results and potential translatability may be relevant to disorders other than PTSD. This is especially exciting as M1 PAMs have entered or completed Phase I trials (see ClinicalTrials.gov Identifiers NCT03220295 and NCT02769065) with schizophrenia and Alzheimer’s disease as intended therapeutic indications. Excitingly, our findings suggest that PTSD might be another promising therapeutic area for these novel drugs.
Altogether, we report that activation of M1 induces LTD of fear-related inputs from the vHipp and BLA to the PFC. This is consistent with previous studies demonstrating mAChR LTD at hippocampal inputs to the PFC (33, 67) and further identifies the BLA, but not the MDT, as another input that expresses this form of synaptic plasticity. We also show that M1 activation is required for contextual fear extinction and that potentiating M1 in vivo with a PAM enhances contextual fear extinction in the SEFL model of PTSD. Our results add M1 LTD at the vHipp and BLA inputs to the extensively studied functions of M1 in the PFC, however future studies are necessary to determine the role of M1-dependent input-specific modulation in other PFC-dependent processes. Overall, these results demonstrate that M1 is poised to regulate fear-related information processing and suggest M1 PAMs could modulate aberrant limbic inputs to the PFC and be useful as adjunct therapeutics to facilitate exposure therapy for PTSD in the clinic.
Supplementary Material
Acknowledgments:
This work was supported by Canadian Institutes for Health Research CIHR DFS146189 (JM), the Vanderbilt International Scholars Program (JM), NIH R01 NS069689 (JJL), NIH R01 MH062646 (PJC), R01 MH073676 (PJC) and R37 NS031373 (PJC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
JM and PJC designed the study and wrote the manuscript. JM, MEJ, and SPM designed and performed the electrophysiology experiments. JM and BJS designed and JM and BLL performed the behavioral experiments. JM and BLL performed the immunofluorescence experiments. KT, DWE, and CWL developed and synthesized VU0255035, VU0364572, and VU0453595. JJL provided the Chrm1loxP/loxP mice. All authors contributed to the preparation of the manuscript. The authors would like to thank Jennifer Zachry and Weimin Peng for their assistance with viral surgeries and colony maintenance and acknowledge the Vanderbilt Murine Neurobehavioral Core.
This work was previously presented in poster form at Neuroscience 2018.
Disclosures:
DWE is an inventor on patents that protect different classes of metabotropic glutamate and muscarinic allosteric modulators. CWL has been funded by the NIH, Johnson and Johnson, Bristol-Myers Squibb, AstraZeneca, Michael J. Fox Foundation, as well as Seaside Therapeutics. He has consulted for AbbVie and received compensation. He is an inventor on patents that protect different classes of metabotropic glutamate and muscarinic receptor allosteric modulators. PJC has been funded by NIH, Michael J. Fox Foundation, Dystonia Medical Research Foundation, CHDI Foundation and others. Over the past three years he has served on the Scientific Advisory Boards for Michael J. Fox Foundation, Stanley Center for Psychiatric Research Broad Institute (MIT/Harvard), Karuna Pharmaceuticals, Lieber Institute for Brain Development, Clinical Mechanism (POCM) and Proof of Concept (POC) Consortium, and Neurobiology Foundation for Schizophrenia and Bipolar Disorder He is an inventor on patents that protect different classes of metabotropic glutamate and muscarinic receptor allosteric modulators. JM, MEJ, SPM, BJS, BL, KT, and JJL report no biomedical financial interests or potential conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hoover WB, Vertes RP (2007): Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct. Funct 212(2): 149–179. [DOI] [PubMed] [Google Scholar]
- 2.Vertes RP (2006): Interactions among the medial prefrontal cortex, hippocampus and midline thalamus in emotional and cognitive processing in the rat. Neuroscience. 142(1): 1–20. [DOI] [PubMed] [Google Scholar]
- 3.Goldman-Rakic PS (1995): Cellular basis of working memory. Neuron. 14(3): 477–485. [DOI] [PubMed] [Google Scholar]
- 4.Gazzaley A, Nobre AC (2012): Top-down modulation: bridging selective attention and working memory. Trends Cogn. Sci. (Regul. Ed.). 16(2): 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller EK, Cohen JD (2001): An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci 24: 167–202. [DOI] [PubMed] [Google Scholar]
- 6.Arnsten AFT (2009): Stress signalling pathways that impair prefrontal cortex structure and function. Nat. Rev. Neurosci 10(6): 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnsten AFT, Jin LE (2014): Molecular influences on working memory circuits in dorsolateral prefrontal cortex. Prog Mol Biol Transl Sci 122: 211–231. [DOI] [PubMed] [Google Scholar]
- 8.Goto Y, Yang CR, Otani S (2010): Functional and dysfunctional synaptic plasticity in prefrontal cortex: roles in psychiatric disorders. Biol. Psychiatry. 67(3): 199–207. [DOI] [PubMed] [Google Scholar]
- 9.Ghoshal A, Rook JM, Dickerson JW, Roop GN, Morrison RD, Jalan-Sakrikar N, et al. (2016): Potentiation of m1 muscarinic receptor reverses plasticity deficits and negative and cognitive symptoms in a schizophrenia mouse model. Neuropsychopharmacology. 41(2): 598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joffe ME, Santiago CI, Engers JL, Lindsley CW, Conn PJ (2017): Metabotropic glutamate receptor subtype 3 gates acute stress-induced dysregulation of amygdalo-cortical function. Mol. Psychiatry [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maren S, Phan KL, Liberzon I (2013): The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat. Rev. Neurosci 14(6): 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chattarji S, Tomar A, Suvrathan A, Ghosh S, Rahman MM (2015): Neighborhood matters: divergent patterns of stress-induced plasticity across the brain. Nat. Neurosci 18(10): 1364–1375. [DOI] [PubMed] [Google Scholar]
- 13.Maren S, Holmes A (2016): Stress and fear extinction. Neuropsychopharmacology. 41(1): 58–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson MA, Fadel JR (2017): Cholinergic regulation of fear learning and extinction. J. Neurosci. Res 95(3): 836–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR (1991): Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J. Neurosci 11(10): 3218–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilcher JJ, Sessions GR, McBride SA (1997): Scopolamine impairs spatial working memory in the radial maze: an analysis by error type and arm choice. Pharmacol. Biochem. Behav 58(2): 449–459. [DOI] [PubMed] [Google Scholar]
- 17.Mirza NR, Stolerman IP (2000): The role of nicotinic and muscarinic acetylcholine receptors in attention. Psychopharmacology. 148(3): 243–250. [DOI] [PubMed] [Google Scholar]
- 18.Santini E, Sepulveda-Orengo M, Porter JT (2012): Muscarinic receptors modulate the intrinsic excitability of infralimbic neurons and consolidation of fear extinction. Neuropsychopharmacology. 37(9): 2047–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navarria A, Wohleb ES, Voleti B, Ota KT, Dutheil S, Lepack AE, et al. (2015): Rapid antidepressant actions of scopolamine: role of medial prefrontal cortex and m1-subtype muscarinic acetylcholine receptors. Neurobiol. Dis 82: 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Digby GJ, Noetzel MJ, Bubser M, Utley TJ, Walker AG, Byun NE, et al. (2012): Novel allosteric agonists of m1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J. Neurosci 32(25): 8532–8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheffler DJ, Williams R, Bridges TM, Xiang Z, Kane AS, Byun NE, et al. (2009): A novel selective muscarinic acetylcholine receptor subtype 1 antagonist reduces seizures without impairing hippocampus-dependent learning. Mol. Pharmacol 76(2): 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, et al. (2009): Selective activation of the m1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. USA. 106(37): 15950–15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marlo JE, Niswender CM, Days EL, Bridges TM, Xiang Y, Rodriguez AL, et al. (2009): Discovery and characterization of novel allosteric potentiators of m1 muscarinic receptors reveals multiple modes of activity. Mol. Pharmacol 75(3): 577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grannan MD, Mielnik CA, Moran SP, Gould RW, Ball J, Lu Z, et al. (2016): Prefrontal cortex-mediated impairments in a genetic model of nmda receptor hypofunction are reversed by the novel m1 pam vu6004256. ACS Chem. Neurosci 7(12): 1706–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moran SP, Dickerson JW, Cho HP, Xiang Z, Maksymetz J, Remke DH, et al. (2018): M1-positive allosteric modulators lacking agonist activity provide the optimal profile for enhancing cognition. Neuropsychopharmacology. 43(8): 1763–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rook JM, Bertron JL, Cho HP, Garcia-Barrantes PM, Moran SP, Maksymetz JT, et al. (2018): A novel m1 pam vu0486846 exerts efficacy in cognition models without displaying agonist activity or cholinergic toxicity. ACS Chem. Neurosci 9(9): 2274–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamilton SE, Loose MD, Qi M, Levey AI, Hille B, McKnight GS, et al. (1997): Disruption of the m1 receptor gene ablates muscarinic receptor-dependent m current regulation and seizure activity in mice. Proc. Natl. Acad. Sci. USA. 94(24): 13311–13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamsler A, McHugh TJ, Gerber D, Huang SY, Tonegawa S (2010): Presynaptic m1 muscarinic receptors are necessary for mglur long-term depression in the hippocampus. Proc. Natl. Acad. Sci. USA. 107(4): 1618–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sotres-Bayon F, Sierra-Mercado D, Pardilla-Delgado E, Quirk GJ (2012): Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron. 76(4): 804–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burgos-Robles A, Vidal-Gonzalez I, Quirk GJ (2009): Sustained conditioned responses in prelimbic prefrontal neurons are correlated with fear expression and extinction failure. J. Neurosci 29(26): 8474–8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker AG, Wenthur CJ, Xiang Z, Rook JM, Emmitte KA, Niswender CM, et al. (2015): Metabotropic glutamate receptor 3 activation is required for long-term depression in medial prefrontal cortex and fear extinction. Proc. Natl. Acad. Sci. USA. 112(4): 1196–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Little JP, Carter AG (2012): Subcellular synaptic connectivity of layer 2 pyramidal neurons in the medial prefrontal cortex. J. Neurosci 32(37): 12808–12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghoshal A, Moran SP, Dickerson JW, Joffe ME, Grueter BA, Xiang Z, et al. (2017): Role of mglu5 receptors and inhibitory neurotransmission in m1 dependent muscarinic ltd in the prefrontal cortex: implications in schizophrenia. ACS Chem. Neurosci 8(10): 2254–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ting JT, Lee BR, Chong P, Soler-Llavina G, Cobbs C, Koch C, et al. (2018): Preparation of acute brain slices using an optimized n-methyl-d-glucamine protective recovery method. J. Vis. Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caruana DA, Warburton EC, Bashir ZI (2011): Induction of activity-dependent ltd requires muscarinic receptor activation in medial prefrontal cortex. J. Neurosci 31(50): 18464–18478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bubser M, Byun N, Wood MR, Jones CK (2012): Muscarinic receptor pharmacology and circuitry for the modulation of cognition. Handb Exp Pharmacol [DOI] [PubMed] [Google Scholar]
- 37.Bell LA, Bell KA, McQuiston AR (2013): Synaptic muscarinic response types in hippocampal ca1 interneurons depend on different levels of presynaptic activity and different muscarinic receptor subtypes. Neuropharmacology. 73: 160–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiang Z, Thompson AD, Jones CK, Lindsley CW, Conn PJ (2012): Roles of the m1 muscarinic acetylcholine receptor subtype in the regulation of basal ganglia function and implications for the treatment of parkinson’s disease. J. Pharmacol. Exp. Ther 340(3): 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamasaki M, Matsui M, Watanabe M (2010): Preferential localization of muscarinic ml receptor on dendritic shaft and spine of cortical pyramidal cells and its anatomical evidence for volume transmission. J. Neurosci 30(12): 4408–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wohleb ES, Wu M, Gerhard DM, Taylor SR, Picciotto MR, Alreja M, et al. (2016): Gaba interneurons mediate the rapid antidepressant-like effects of scopolamine. J. Clin. Invest [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yi F, Ball J, Stoll KE, Satpute VC, Mitchell SM, Pauli JL, et al. (2014): Direct excitation of parvalbumin-positive interneurons by m1 muscarinic acetylcholine receptors: roles in cellular excitability, inhibitory transmission and cognition. J. Physiol. (Lond.). 592(16): 3463–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Kennedy JP, et al. (2009): A selective allosteric potentiator of the m1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J. Neurosci 29(45): 14271–14286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perusini JN, Meyer EM, Long VA, Rau V, Nocera N, Avershal J, et al. (2016): Induction and expression of fear sensitization caused by acute traumatic stress. Neuropsychopharmacology. 41(1): 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Graham BM, Milad MR (2011): The study of fear extinction: implications for anxiety disorders. Am. J. Psychiatry. 168(12): 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Godsil BP, Kiss JP, Spedding M, Jay TM (2013): The hippocampal-prefrontal pathway: the weak link in psychiatric disorders? Eur. Neuropsychopharmacol 23(10): 1165–1181. [DOI] [PubMed] [Google Scholar]
- 46.Ghoshal A, Conn PJ (2015): The hippocampo-prefrontal pathway: a possible therapeutic target for negative and cognitive symptoms of schizophrenia. Future Neurol 10(2): 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rauch SL, Shin LM, Phelps EA (2006): Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research-past, present, and future. Biol. Psychiatry. 60(4): 376–382. [DOI] [PubMed] [Google Scholar]
- 48.Allsop SA, Vander Weele CM, Wichmann R, Tye KM (2014): Optogenetic insights on the relationship between anxiety-related behaviors and social deficits. Front. Behav. Neurosci 8: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordon JA (2016): On being a circuit psychiatrist. Nat. Neurosci 19(11): 1385–1386. [DOI] [PubMed] [Google Scholar]
- 50.Phelps EA, Delgado MR, Nearing KI, LeDoux JE (2004): Extinction learning in humans: role of the amygdala and vmpfc. Neuron. 43(6): 897–905. [DOI] [PubMed] [Google Scholar]
- 51.Sierra-Mercado D, Padilla-Coreano N, Quirk GJ (2011): Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 36(2): 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, et al. (2009): Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol. Psychiatry. 66(12): 1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Felix-Ortiz AC, Burgos-Robles A, Bhagat ND, Leppla CA, Tye KM (2016): Bidirectional modulation of anxiety-related and social behaviors by amygdala projections to the medial prefrontal cortex. Neuroscience. 321: 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Padilla-Coreano N, Bolkan SS, Pierce GM, Blackman DR, Hardin WD, Garcia-Garcia AL, et al. (2016): Direct ventral hippocampal-prefrontal input is required for anxiety-related neural activity and behavior. Neuron. 89(4): 857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sigurdsson T, Duvarci S (2015): Hippocampal-prefrontal interactions in cognition, behavior and psychiatric disease. Front. Syst. Neurosci 9: 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tovote P, Fadok JP, Luthi A (2015): Neuronal circuits for fear and anxiety. Nat. Rev. Neurosci 16(6): 317–331. [DOI] [PubMed] [Google Scholar]
- 57.Santini E, Quirk GJ, Porter JT (2008): Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J. Neurosci 28(15): 4028–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scarr E, Hopper S, Vos V, Seo MS, Everall IP, Aumann TD, et al. (2018): Low levels of muscarinic m1 receptor-positive neurons in cortical layers iii and v in brodmann areas 9 and 17 from individuals with schizophrenia. J. Psychiatry Neurosci 43(5): 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scarr E, Seo MS, Aumann TD, Chana G, Everall IP, Dean B (2016): The distribution of muscarinic m1 receptors in the human hippocampus. J Chem Neuroanat 77: 187–192. [DOI] [PubMed] [Google Scholar]
- 60.Levey AI (1996): Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of alzheimer disease. Proc. Natl. Acad. Sci. USA. 93(24): 13541–13546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Disney AA, Domakonda KV, Aoki C (2006): Differential expression of muscarinic acetylcholine receptors across excitatory and inhibitory cells in visual cortical areas v1 and v2 of the macaque monkey. J. Comp. Neurol 499(1): 49–63. [DOI] [PubMed] [Google Scholar]
- 62.Young MB, Thomas SA (2014): M1-muscarinic receptors promote fear memory consolidation via phospholipase c and the m-current. J. Neurosci 34(5): 1570–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Milad MR, Quirk GJ, Pitman RK, Orr SP, Fischl B, Rauch SL (2007): A role for the human dorsal anterior cingulate cortex in fear expression. Biol. Psychiatry. 62(10): 1191–1194. [DOI] [PubMed] [Google Scholar]
- 64.Stansley BJ, Fisher NM, Gogliotti RG, Lindsley CW, Conn PJ, Niswender CM (2018): Contextual fear extinction induces hippocampal metaplasticity mediated by metabotropic glutamate receptor 5. Cereb. Cortex. 28(12): 4291–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park J, Chun J-W, Park H-J, Kim E, Kim J-J (2018): Involvement of amygdala-prefrontal dysfunction in the influence of negative emotion on the resolution of cognitive conflict in patients with schizophrenia. Brain Behav 8(8): e01064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holt DJ, Lebron-Milad K, Milad MR, Rauch SL, Pitman RK, Orr SP, et al. (2009): Extinction memory is impaired in schizophrenia. Biol. Psychiatry. 65(6): 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang L, Yuan L-L (2009): Activation of m2 muscarinic receptors leads to sustained suppression of hippocampal transmission in the medial prefrontal cortex. J. Physiol. (Lond.). 587(Pt 21): 5139–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.